Back to Journals » OncoTargets and Therapy » Volume 12
CDK7 inhibitor suppresses tumor progression through blocking the cell cycle at the G2/M phase and inhibiting transcriptional activity in cervical cancer
Authors Zhong S, Zhang Y, Yin X, Di W
Received 22 November 2018
Accepted for publication 18 February 2019
Published 22 March 2019 Volume 2019:12 Pages 2137—2147
DOI https://doi.org/10.2147/OTT.S195655
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Shanshan Zhong,1,2,* Yi Zhang,1,2,* Xia Yin,1,2 Wen Di1–3
1Department of Obstetrics and Gynecology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, People’s Republic of China; 2Shanghai Key Laboratory of Gynecologic Oncology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, People’s Republic of China; 3State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, People’s Republic of China
*These authors contributed equally to this work
Background: The disordered cell cycle and dysregulated expression of numerous oncogenes involved in tumor-relevant processes are highly related to the tumorigenesis of cervical cancer. Cyclin-dependent kinase 7 (CDK7) constitutes the indispensable catalytic subunit of CDK-activating kinase (CAK), which is required for both cell cycle transition and transcriptional regulation. However, research regarding the antitumor effects of CDK7 inhibition in cervical cancer remains unclear.
Purpose: Our study aims to explore the antineoplastic effects of the CDK7 inhibitor THZ1 in cervical cancer cells and to find a potential agent for cervical cancer treatment.
Methods: The CRISPR-Cas9 system was used to knock down CDK7. The Cell Counting Kit-8 (CCK-8) assay was used to detect the cell viability after CDK7 depletion and THZ1 treatment. Western blot was employed to detect protein expression. The expression levels of mRNA were assayed through qRT-PCR. Flow cytometry analysis was used to assay the apoptotic cells and cell cycle distribution. Gene expression microarray analysis was used to identify the differential expression of the genes. Subcutaneous xenograft mouse model was performed to test the antineoplastic effects of THZ1 in vivo.
Results: We revealed that the genetic depletion of CDK7 using the CRISPR-Cas9 system exhibited great cell growth inhibition in cervical cancer cell lines, consistent with the effects of CDK7 blocking using THZ1. Cervical cancer cells were highly sensitive to THZ1 treatment, and a low concentration of THZ1 could induce substantial cell apoptosis. THZ1 specifically perturbed the phosphorylation of cell cycle regulator CDK1 and decreased the expression of cyclin B1, leading to a cell cycle blockage at the G2/M phase and inducing cell growth inhibition. The gene expression microarray analysis showed that massive oncogene transcripts, especially those associated with tumorigenesis, were preferential suppressed after THZ1 treatment. The qRT-PCR confirmed that several essential oncogenes in tumorigenesis (c-MYC, hTERT, RAD51, and BCL-2) and HPV viral oncogenes (E6 and E7) were preferentially repressed by THZ1. Moreover, THZ1 exhibited substantial antineoplastic effects against cervical cancer in vivo without inducing obvious side effects.
Conclusion: These findings indicated that the CDK7 inhibitor THZ1 is a potential option in cervical cancer treatment owing to its ability to inhibit cell cycle progression and transcriptional activity.
Keywords: CDK7, cervical cancer, cell cycle, THZ1, transcriptional regulation
Introduction
In 2018, cervical cancer ranked fourth for both cancer incidence and mortality among females worldwide.1 With the increased application of cervical cancer screening and human papillomavirus (HPV) vaccinations, cervical cancer incidence rates and death rates have exhibited a discernible reduction in most high-income countries.1–3 However, attributed to the increasing prevalence of HPV among young adults, cervical cancer incidence and mortality among 20–39-year-olds have increased worldwide.4 Notably, little progress has been made in overall survival for cervical cancer owing to the minimally chemosensitive and limited effective treatment strategies for patients with advanced cervical cancer, whose 5-year survival rate is less than 20%.2,5,6 Therefore, investigating novel antitumor strategies is a critical area of research.
Cell cycle disturbance is one of the main characteristics of cervical cancer cells, as it leads to abnormal regulation of the cell cycle progression, which results in cell proliferation and apoptosis disorders.7 Numerous studies have observed many special histone modifications in cervical cancer cells, facilitating enhancer activation, which recruits excessive transcription factors and cofactors to the enhancers, consequently activating transcriptional processes.8–11 As a result, cervical cancer cells exhibited remarkable different patterns of oncogene expression compared with normal human tissues, especially genes associated with transcription, cell cycle, and DNA repair processes.12,13 Recently, a vast number of studies has revealed that oncogenic transcription in cancer cells is highly associated with super-enhancers, which are particularly vulnerable to the perturbation of transcriptional regulation.14–18
CDK7 is a major member of the cyclin-dependent kinase family, together with cyclin H and MAT1, forming a functional CDK-activating kinase (CAK), which regulates the cell cycle by phosphorylating other CDKs.19,20 CDK7 is also a major component of transcription factor II H (TFIIH), which activates transcriptional initiation and elongation by phosphorylating serine 5 (S5), serine 7 (S7), and serine 2 (S2) of the carboxy-terminal domain (CTD) of RNA polymerase II (Pol II).21–23 In this study, we focused on THZ1, a CDK7 inhibitor, which has an acrylamide moiety that can inhibit the activity of CDK7 by selectively reacting with the cysteine 312 (C312) residues of CDK7 outside the kinase domain.24–26 Recent studies have indicated that THZ1 possesses powerful antineoplastic activity in several cancers, including triple-negative breast cancer,27 small cell lung cancer,24 osteosarcoma,28 ovarian cancer,16 esophageal squamous cell carcinoma,17 and T-cell acute lymphoblastic leukemia.27 However, research regarding the effects of THZ1 in cervical cancer remains unclear.
In this study, we treated cervical cancer cells with THZ1 and found that THZ1 exhibited a potent antineoplastic effect on cervical cancer cells. Low-dose THZ1 treatment led to a profound inhibition of cell proliferation and induction of cell apoptosis. Further research revealed that THZ1 exhibits cell cycle inhibition and transcription repression, especially of the essential oncogene transcription in tumorigenesis. These findings indicate that CDK7 inhibitor THZ1 is a promising potential drug for cervical cancer treatment.
Materials and methods
Cell culture and reagents
All cervical cancer cell lines (HeLa, SiHa, and C33A) and human embryonic kidney cells 293T were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China) in 2016. The cells were maintained in DMEM/high glucose medium (HyClone, South Logan, UT, USA) containing 10% FBS (Gibco, Gaithersburg, MD, USA) and 1% penicillin/streptomycin (Sigma Aldrich, St Louis, MO, USA). THZ1 was purchased from MedChem Express (Monmouth Junction, NJ, USA) and dissolved in DMSO (Sigma-Aldrich) as a 10 mM stock solution and stored at −80°C.
CRISPR-Cas9 system-mediated knockdown of CDK7
The transfer plasmids (lentiCRISPRv2) and packaging plasmids (delta8.9 and VSVG) were gifts from the Skate Key Laboratory of Oncogenes and Related Genes (Shanghai Jiao Tong University, People’s Republic of China). To generate lentiviral sgRNA plasmids, a pair of annealed oligonucleotides was cloned into lentiCRISPRv2 plasmids. The forward sequence of sgEGFP was CAC CGG AAG TTC GAG GGC GAC ACC C and the reverse sequence was AAA CGG GTG TCG CCC TCG AAC TTC C. The forward sequence of sgCDK7-1 was CAC CGT GAG AAG CTG GAC TTC CTT G and the reverse sequence was AAA CCA AGG AAG TCC AGC TTC TCA C. The forward sequence of sgCDK7-2 was CAC CGA GCT CCA AAT AGT AAC TCG G and the reverse sequence was AAA CCC GAG TTA CTA TTT GGA GCT C. The lentiviral sgRNA plasmids and packaging plasmids were co-transfected into human embryonic kidney cells 293T cells using lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instruction. They were filtered using a Millex-HV Syringe Filter Unit with a 0.45 μm pore size hydrophilic polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). Then, cervical cancer cells were cultured in supernatants containing lentivirus together with 8 mg/mL polybrene (Sigma-Aldrich) for 24 hours, changing the supernatants for fresh DMEM/high glucose medium containing 10% FBS. After transduction for 72 hours, the transfected cells were selected with 2.5 μg/mL puromycin.
Cell Counting Kit-8 (CCK-8) cell viability assay
The cervical cancer cells were seeded in 96-well plates at a density of 5,000 cells per well and treated with various concentrations of THZ1. At indicated time-points, cells were incubated with 10 μL CCK-8 reagent (Dojindo, Kumamoto, Japan) in each well for 1 hour. Then the absorbance was measured using a microplate reader (Thermo Scientific, Waltham, MA, USA) at 450 nm. Concentration–response curves were created using GraphPad Prism 6.0 (La Jolla, CA, USA).
Western blot analysis
Protein was extracted using RIPA buffer (Sangon Biotech, Shanghai, People’s Republic of China) containing proteinase inhibitors (MedChem Express) and phosphatase inhibitors (MedChem Express). The protein concentration was measured using the Pierce BCA Protein Assay Kit (Thermo Scientific). The following antibodies were used for Western blot: anti-CDK7 (Cell Signaling Technology, Danvers, MA, USA), anti-α-tubulin (Cell Signaling Technology), anti-poly(ADP-ribose) polymerase (anti-PARP) (Cell Signaling Technology), anti-cleaved PARP (Cell Signaling Technology), anti-RNA Pol II (Abcam, Cambridge, UK), anti-phospho RNA Pol II (Ser2) (Abcam), anti-phospho RNA Pol II (Ser5) (Abcam), anti-phospho RNA Pol II (Ser7) (Millipore), anti-CDK1 (Abcam), and anti-CDK1 (phospho T161) (Abcam).
Cell cycle and apoptosis analysis
A cell cycle analysis was performed 24 hours after the THZ1 treatment. Each sample was collected, and cell suspensions of approximately 1×106 cells were generated. Then, 75% ethanol (1 mL) was added to each cell suspension. The cells were fixed at 4°C overnight. Then, 500 μL PI/RNase Staining Buffer solution (BD Biosciences, Franklin Lakes, NJ, USA) was added to the cells for 30 minutes, and the cells were assessed using the BD Accuri C6 plus flow cytometer (BD Biosciences). Cell apoptosis was assayed using the fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit (BD Biosciences) following the manufacturer’s instructions. The apoptosis rate was measured using flow cytometry analysis (Beckman Coulter FC500MPL; Brea, CA, USA).
Gene expression microarray analysis
Total RNA was extracted using TRIZOL (Invitrogen) after treatment with THZ1 or DMSO for 6 hours. The RNA was quantified using a NanoDrop ND-2000 (Thermo Scientific). Then the integrity of the RNA was assessed using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). The Agilent SurePrint G3 Human Gene Expression v3 Microarray (8*60K) was used to detect the gene expression in this experiment. Microarray hybridization was performed according to the manufacturer’s protocols. The microarrays were scanned using an Agilent Scanner G2505C (Agilent Technologies). A basic analysis of the raw data was performed using Genespring software (Agilent Technologies). For further analysis, fold change ≥2.0 and P-value ≤0.05 (calculated using a t-test) were used to identify the differential expression of the genes. A gene ontology analysis was performed using the fisher-text function of an R program. The gene expression microarray data have been deposited in NCBI GEO profiles (https://www.ncbi.nlm.nih.gov/geoprofiles/) under the accession number GSE125543.
RNA extraction and qRT-PCR
The RNA was extracted using TRIZOL (Invitrogen) according to the manufacturer’s protocols. The RNA concentration was measured using NanoDrop ND-1000 (Thermo Scientific). Reverse transcription was performed to convert mRNA to cDNA using the PrimeScript RT Reagent Kit (Takara, Otsu, Japan). PCR was performed using QuantStudio™ Test Development Software (Life Technologies) through SYBR Green Ex TaqTM II (Takara). The data were analyzed using the 2−ΔΔCT method. The sequences of primers were as follows: CDK7, forward: GGC ACA CCA ACT GAG GAA CA, reverse: AGT CGT CTC CTG CTG CAC TG; c-MYC, forward: CGA GGA GAA TGT CAA GAG GCG AAC, reverse: GCT TGG ACG GAC AGG ATG TAT GC; hTERT, forward: CGT ACT GCG TGC GTC GGT ATG, reverse: ACG GCT GGA GGT CTG TCA AGG; RAD51, forward: AGC TGT GTG ACC GTC TGA GT; reverse: AGG CCC TGC TGA CCA TAC AT; BCL-2, forward: GTG GAT GAC TGA GTA CCT GAA CCG, reverse: AGA GTC TTC AGA GAC AGC CAG GAG; HPV18 E6, forward: AAC CGT TGA ATC CAG CAG AA, reverse: GAG TCG TTC CTG TCG TGC TC; HPV18 E7, forward: CGA ACC ACA ACG TCA CAC AA, reverse: GGA ATG CTC GAA GGT CGT CT; HPV16 E6, forward: AAG TTA CCA CAG TTA TGC ACA GAG C, reverse: AAG TCA TAT ACC TCA CGT CGC AGT; HPV16 E7, forward: CAG CTC AGA GGA GGA GGA TGA, reverse: GTA CGC ACA ACC GAA GCG TA. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Sangon Biotech (B661104).
Tumor xenograft studies
All animal experiments followed the guidelines of the Institutional Animal Care and Use Committee of Ren Ji Hospital, and all animal protocols were approved by the same committee. HeLa cells (5×106) were subcutaneously injected into the dorsal flanks in 5-week-old female BALB/c mice. When the tumor volumes reached 100–150 mm3 (volume = (length × width2)/2), the mice were randomly separated into two groups (five mice per group) and treated with 10 mg/kg THZ1 or vehicle (10% DMSO in 5% glucose) that was administered intraperitoneally twice daily. The volumes of the tumors were measured every 4 days for 28 days. At the end of the experiments, we calculated the tumor volumes and weights of each group for tumor growth analysis. The tumors were fixed in formalin and embedded in paraffin, cut into 4 μm sections, stained with H&E, and incubated with the Ki67 antibody (1:200) (Servicebio, Wuhan, People’s Republic of China) for tumor proliferation analysis. The tumor apoptosis was measured by TUNEL staining using an In Situ Cell Death Detection Kit (Roche, Basel, Switzerland). The sections were observed through microscopy (DM 2500; Leica, Wetzlar, Germany).
Statistical analysis
All the experiments were performed in triplicate. The data were analyzed by two-sided Student’s t-test using IBM SPSS Statistics version 21. A P-value <0.05 was considered statistically significant.
Results
CDK7 knockdown induces cell growth inhibition in cervical cancer cells
CDK7 is a major molecular in cellular biological processes, as it plays a role in the regulation of both cell cycle progression and transcription. Here, we tried to investigate the potential critical role of CDK7 in cervical cancer cells.
First, we chose three cervical cancer cell lines for research: HPV18-positive cell line HeLa, HPV16-positive cell line SiHa, and HPV-negative cell line C33A. We employed two independent CRISPR-Cas9 systems to knock down CDK7 in cervical cancer cell lines, which led to a substantial reduction of CDK7 at both the RNA and protein levels (Figure 1A and B). Strikingly, the downregulation of CDK7 demonstrated significant growth inhibition compared to their respective non-targeting controls in cervical cancer cells (Figure 1C). These results suggest that CDK7 plays an essential role in cervical cancer survival.
 | Figure 1 CDK7 knockdown exhibited cell growth inhibition in cervical cancer cells. |
Antineoplastic effects of CDK7 inhibitor THZ1 in cervical cancer cell lines
We presented a covalent CDK7 inhibitor THZ1, which selectively inhibits the function of CDK7 by modifying the C312 residue of the kinase domain,26 and we probed the antitumor effect of THZ1 in cervical cancer cells.
First, we performed concentration–response experiments on cervical cancer cells. As a result, all three cervical cancer cell lines were highly sensitive to THZ1, with half-maximum inhibitory concentration (IC50) values of less than 150 nM, exhibiting time and dose dependency on THZ1 treatment (Figure 2A).
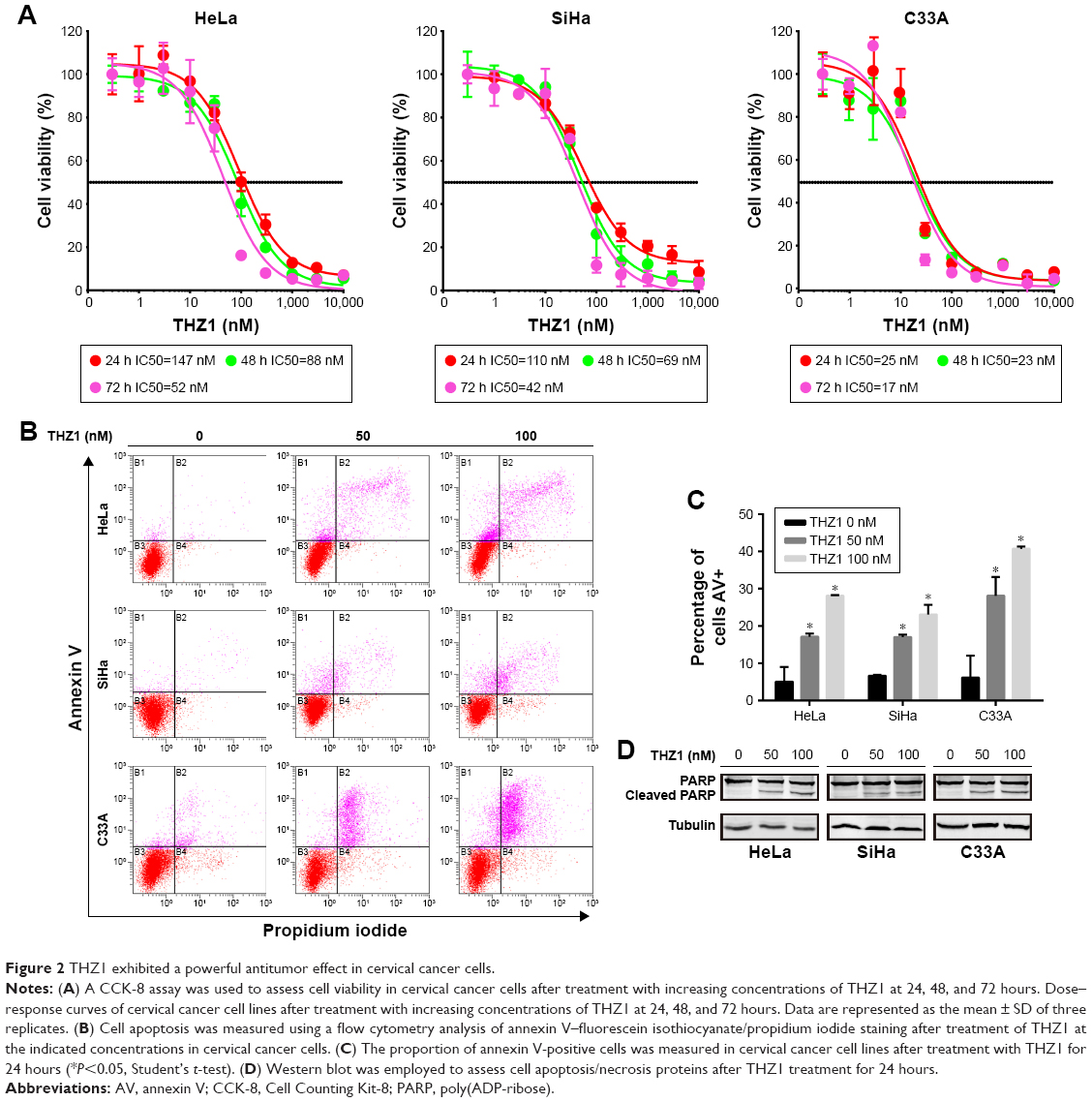 | Figure 2 THZ1 exhibited a powerful antitumor effect in cervical cancer cells. |
Moreover, the inhibition of CDK7 caused cell apoptosis in cervical cancer cell lines, as demonstrated by a flow cytometry analysis measuring the population of annexin V-positive cells after 24-hour treatment with THZ1 (Figure 2B and C). Furthermore, we observed an increase in cell apoptosis/necrosis in a dose-dependent manner, as assessed through a cleaved PARP Western blot analysis (Figure 2D). Collectively, the inhibition of CDK7 with THZ1 had significant antitumor effects in cervical cancer cells.
Cell cycle arrest and growth inhibition following THZ1 treatment
Cell cycle disturbance and uncontrolled proliferation have an immense influence on cervical cancer immortalization and transformation.29 As the vital component of the CAK complex, CDK7 phosphorylates other CDKs involved in cell cycle transition.19 Therefore, we sought to confirm whether blockage of CDK7 affects cell cycle progression in cervical cancer cell lines.
To further test the effect of THZ1 on cell proliferation, we performed a cell cycle analysis using flow cytometry in cervical cancer cell lines. As a result, CDK7 inhibition via THZ1 caused cell cycle redistribution in these cells. Specifically, a conspicuous increase in the G2/M phase and a concomitant decrease in the S phase in cervical cancer cells were observed after THZ1 treatment (Figure 3A). CDK1 and cyclin B1 are indispensable cell cycle regulators required for the G2/M transition.19,30 Western blot revealed that THZ1 led to a striking reduction in cyclin B1 and phospho-CDK1 in a dose-dependent manner in the cervical cancer cells (Figure 3B). Next, we performed proliferation assays in the cervical cancer cell lines and observed significant inhibition of cell proliferation after THZ1 treatment (Figure 3C). Overall, the above data demonstrated that THZ1 inhibited cell cycle progression, causing cell proliferation inhibition.
 | Figure 3 THZ1 inhibited CDK1 phosphorylation and decreased cyclin B1, causing a cell cycle blockage at the G2M phase. |
Widespread inhibition of gene transcription by THZ1 in cervical cancer cells
The dysregulation of gene expression is thought to stimulate tumorigenesis through eliciting a plethora of downstream effects in cervical cancer cells. CDK7 regulates transcriptional processes through recognizing and phosphorylating the CTD of RNA Pol II at S2, S5, and S7.21 Indeed, the cells that received THZ1 treatment showed a striking reduction in the phosphorylation of RNA Pol II at S2, S5, and S7, in a dose-dependent manner (Figure 4A). Next, we examined the effect of CDK7 inhibition on gene expression profiles by performing a gene expression microarray. As expected, treatment with 100 nM THZ1 for 6 hours resulted in a dramatic decrease in global RNA levels (Figure 4B).
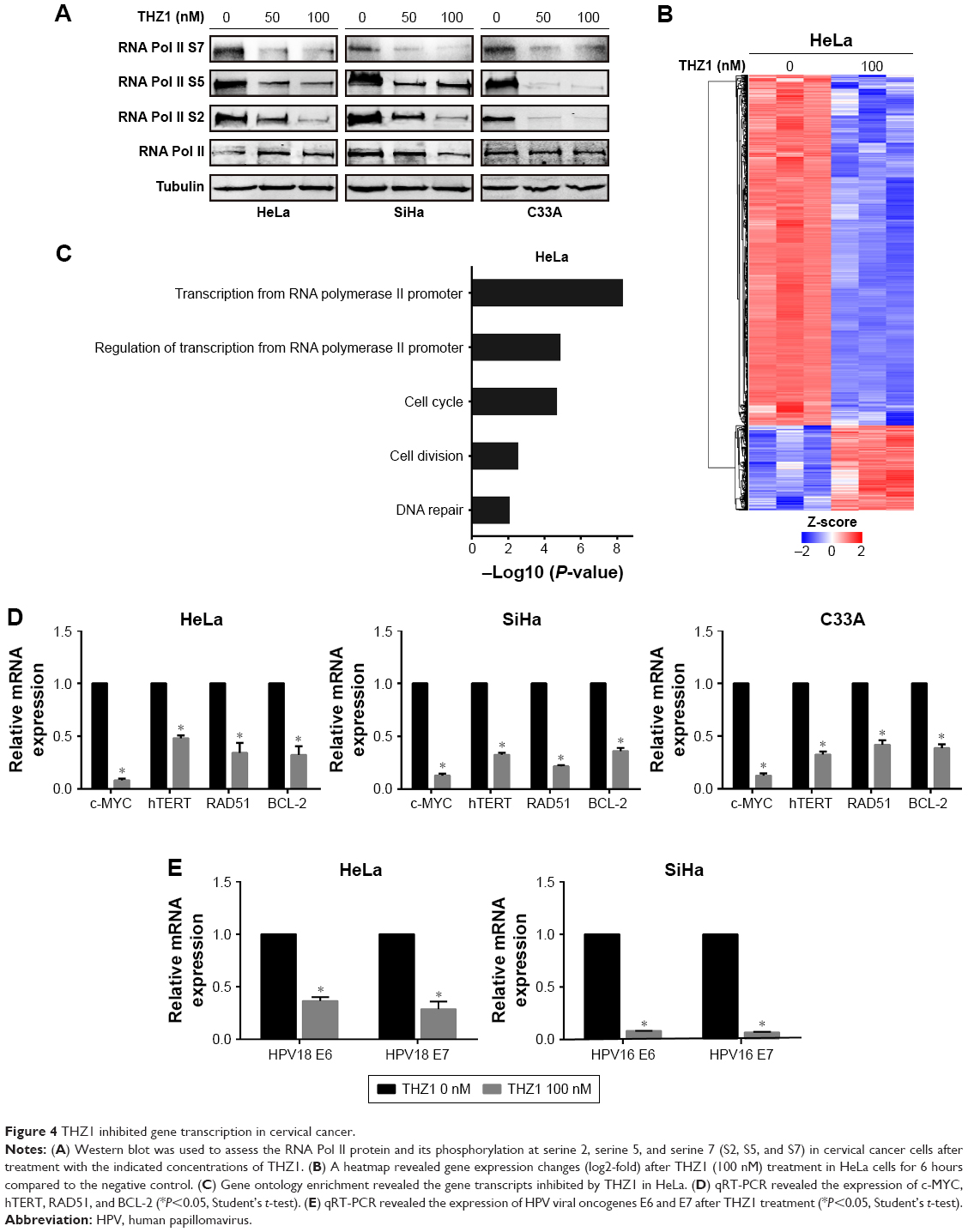 | Figure 4 THZ1 inhibited gene transcription in cervical cancer. |
According to the previous sequencing obtained from HeLa cells, DNA repair, proliferation (cell cycle phase), and transcription (RNA processing) genes are highly expressed in HeLa.12 Surprisingly, gene ontology analysis of these core transcripts inhibited by THZ1 revealed a significant enrichment of genes involved in transcriptional regulation, proliferation, and DNA repair processes (Figure 4C). For example, the main oncogenes in the development of tumorigenesis, c-MYC (transcription),31,32 hTERT (cell proliferation),33,34 BCL-2 (cell apoptosis),35,36 and RAD51 (DNA repair),37 which play a crucial role in the development of cervical cancer, were significantly downregulated after the THZ1 treatment (Figure 4D). In addition, THZ1 exhibited a profound reduction in the transcription of HPV viral oncogenes E6 and E7 in HeLa and SiHa cell lines (Figure 4E). Taken together, these data demonstrate that targeting CDK7 using THZ1 led to a widespread transcriptional downregulation in the cervical cancer cells. In particular, the actively transcribed genes and vial oncogenes in the cervical cancer cells were greatly inhibited by THZ1.
Antineoplastic effect of THZ1 in a cervical cancer subcutaneous xenograft mouse model
To further investigate the antitumor effect of THZ1 in cervical cancer, we assessed the in vivo therapeutic potential of THZ1 in xenotransplantation models, in which each mouse harbored one explant formed by HeLa cells. When the tumors reached 100–150 mm3 in size, the mice were randomized into two groups (five mice per group). The mice were treated with the vehicle and the THZ1 twice daily (10 mg/kg) for 28 days. Strikingly, the mice receiving THZ1 treatment exhibited obvious tumor repression compared with the vehicle-treated mice (Figure 5A). We next compared the change in tumor size and weight between THZ1 and vehicle treatment. In contrast, tumors derived from THZ1-treated mice displayed potent growth inhibition (Figure 5B and C). Moreover, immunohistochemical analysis revealed a dramatic decrease in the cell proliferation marker ki67, and a TUNEL assay showed an increase in the cell apoptosis of the THZ1-treated tumors (Figure 5D). Importantly, no significant side effects were observed during THZ1 treatment. Collectively, these results demonstrated that THZ1 exhibited substantial antineoplastic effects against cervical cancer in vivo.
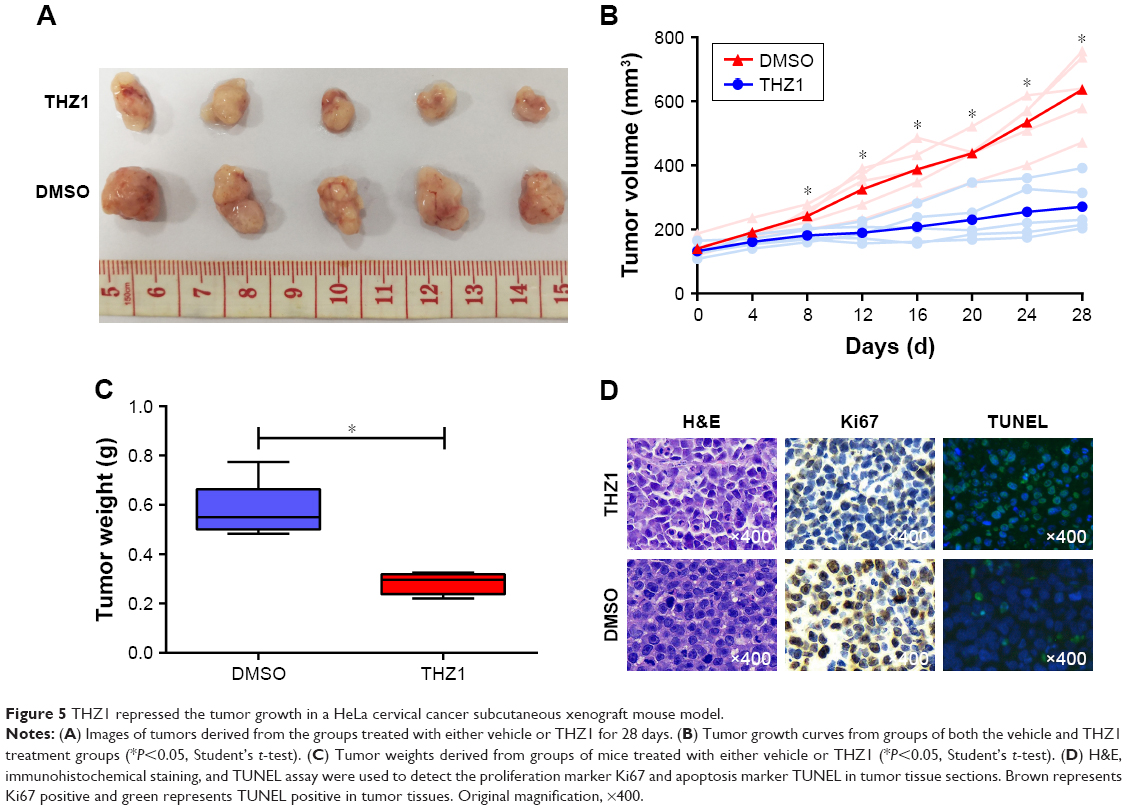 | Figure 5 THZ1 repressed the tumor growth in a HeLa cervical cancer subcutaneous xenograft mouse model. |
Discussion
The incidence and mortality of cervical cancer exhibit considerable geographic differences. In total, 80% of diagnoses and 85% of deaths occur in developing countries where both vaccination and screening programs are not widely available.38,39 The majority of patients with cervical cancer in developing countries are often not identified until the disease is advanced, and these patients often have limited access to adequate treatment. CDK7 forms a kinase module with both CAK and TFIIH, which are responsible for cell cycle and transcription regulation, respectively. In this study, we demonstrated that blockage of the CDK7 function may provide an attractive therapeutic method for treating cervical cancer by simultaneously inhibiting cell cycle progression and transcriptional activity.
We treated cervical cancer cell lines with the CDK7 inhibitor THZ1, and all of these cell lines were highly sensitive to THZ1. THZ1 exhibited potent anti-tumor effects against cervical cancer cell lines, and induced cell growth inhibition and apoptosis in a dose-dependent manner. Moreover, THZ1 strongly repressed tumor growth in subcutaneous cervical cancer mouse models without inducing obvious side effects. Further studies indicated that CDK7 inhibition by THZ1 decreased the cell cycle checkpoint mediator cyclin B1 and the phosphorylation of CDK1, inducing cell cycle arrest at the G2/M phase and inhibiting cell proliferation.
Previous studies identified that the integration of viral genes into host genes was crucial for tumorigenesis, given that viral proteins interact with other epigenetic modulators that dictate post-translational modifications on histones, upregulating viral oncogenes and many crucial cell oncogenes, especially genes associated with transcription, cell cycle, and DNA repair processes.9,40–42 For example, some essential oncogenes during tumor processes, including a crucial transcription factor c-MYC,31,32 a main catalytic subunit of telomerase hTERT,33,34 a cell apoptosis regulator protein BCL-2,35,36 and a DNA damage repair protein RAD51,37 are closely associated with tumorigenesis and highly upregulated in cervical cancer. Earlier studies have demonstrated that overactivation and upregulation of oncogenes in tumor cells were likely associated with super-enhancers and were more sensitive to perturbation of associated enhancer-binding transcriptional regulators.14–16 Our microarray analysis demonstrated that low-dose THZ1 treatment leads to the transcriptional inhibition of a set of oncogenes related to cell cycle, DNA repair process, and transcriptional regulation, which are highly upregulated in cervical cancer cells. qRT-PCR confirmed that these crucial oncogenes, including c-MYC, hTERT, RAD51, and BCL-2, were downregulated after THZ1 treatment. In addition, the key viral oncogenes E6 and E7, which are of crucial importance for cervical cancer tumorigenesis, were also repressed after THZ1 treatment.
In conclusion, the above results suggest that the antineoplastic effects of THZ1 in cervical cancer are most likely caused by cell cycle arrest and global oncogene transcription repression. Compared with the previous targeted drugs, which exclusively target one of the molecules in tumorigenesis pathways, CDK7 inhibition may provide a more effective therapeutic strategy by suppressing widespread oncogene transcription as well as blocking the cell cycle in cervical cancer.
Acknowledgment
We gratefully acknowledge Shanghai Key Laboratory of Gynecologic Oncology and the financial support from National Key R&D Program of China (No 2016YFC1302900) and Key Discipline Project of Shanghai Municipal Commission of Health and Family Planning (No 15GWZK0701).
Disclosure
The authors report no conflicts of interest in this work.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. | ||
Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J Natl Cancer Inst. 2017;109(9). doi:10.1093/jnci/djx007 | ||
Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC. Human papillomavirus and cervical cancer. Lancet. 2013;382(9895):889–899. | ||
Fidler MM, Gupta S, Soerjomataram I, Ferlay J, Steliarova-Foucher E, Bray F. Cancer incidence and mortality among young adults aged 20–39 years worldwide in 2012: a population-based study. Lancet Oncol. 2017;18(12):1579–1589. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Barra F, Lorusso D, Leone Roberti Maggiore U, et al. Investigational drugs for the treatment of cervical cancer. Expert Opin Investig Drugs. 2017;26(4):389–402. | ||
de Freitas AC, Coimbra EC, Leitao MC. Molecular targets of HPV oncoproteins: potential biomarkers for cervical carcinogenesis. Biochim Biophys Acta. 2014;1845(2):91–103. | ||
Dooley KE, Warburton A, McBride AA. Tandemly integrated HPV16 can form a Brd4-dependent super-enhancer-like element that drives transcription of viral oncogenes. mBio. 2016;7(5):e01446–16. | ||
Chen X, Loo JX, Shi X, et al. E6 protein expressed by high-risk HPV activates super-enhancers of the EGFR and c-MET oncogenes by destabilizing the histone demethylase KDM5C. Cancer Res. 2018;78(6):1418–1430. | ||
Warburton A, Redmond CJ, Dooley KE, et al. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLoS Genet. 2018;14(1):e1007179. | ||
Durzynska J, Lesniewicz K, Poreba E. Human papillomaviruses in epigenetic regulations. Mutat Res Rev Mutat Res. 2017;772:36–50. doi:10.1016/j.mrrev.2016.09.006 | ||
Landry JJ, Pyl PT, Rausch T, et al. The genomic and transcriptomic landscape of a HeLa cell line. G3 (Bethesda). 2013;3(8):1213–1224. | ||
Kuner R, Vogt M, Sultmann H, et al. Identification of cellular targets for the human papillomavirus E6 and E7 oncogenes by RNA interference and transcriptome analyses. J Mol Med. 2007;85(11):1253–1262. doi:10.1007/s00109-007-0230-1 | ||
Sengupta S, George RE. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer. 2017;3(4):269–281. doi:10.1016/j.trecan.2017.03.006 | ||
Chipumuro E, Marco E, Christensen CL, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159(5):1126–1139. doi:10.1016/j.cell.2014.10.024 | ||
Zhang Z, Peng H, Wang X, et al. Preclinical efficacy and molecular mechanism of targeting CDK7-dependent transcriptional addiction in ovarian cancer. Mol Cancer Ther. 2017;16(9):1739–1750. doi:10.1158/1535-7163.MCT-17-0078 | ||
Jiang YY, Lin DC, Mayakonda A, et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut. 2017;66(8):1358–1368. doi:10.1136/gutjnl-2016-311818 | ||
Nagaraja S, Vitanza NA, Woo PJ, et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell. 2017;31(5):635–652 e636. doi:10.1016/j.ccell.2017.03.011 | ||
Larochelle S, Merrick KA, Terret ME, et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25(6):839–850. doi:10.1016/j.molcel.2007.02.003 | ||
Lolli G, Johnson LN. CAK-cyclin-dependent activating kinase: a key kinase in cell cycle control and a target for drugs? Cell Cycle. 2005;4(4):572–577. | ||
Larochelle S, Amat R, Glover-Cutter K, et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012;19(11):1108–1115. doi:10.1038/nsmb.2399 | ||
Coin F, Egly JM. Revisiting the function of CDK7 in transcription by virtue of a recently described TFIIH kinase inhibitor. Mol Cell. 2015;59(4):513–514. | ||
Compe E, Egly JM. Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu Rev Biochem. 2016;85:265–290. | ||
Christensen CL, Kwiatkowski N, Abraham BJ, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26(6):909–922. | ||
Kwiatkowski N, Zhang T, Rahl PB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511(7511):616–620. | ||
Cao K, Shilatifard A. Inhibit globally, act locally: CDK7 inhibitors in cancer therapy. Cancer Cell. 2014;26(2):158–159. | ||
Wang Y, Zhang T, Kwiatkowski N, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163(1):174–186. | ||
Chen D, Zhao Z, Huang Z, et al. Super enhancer inhibitors suppress MYC driven transcriptional amplification and tumor progression in osteosarcoma. Bone Res. 2018;6:11. | ||
Senapati R, Senapati NN, Dwibedi B. Molecular mechanisms of HPV mediated neoplastic progression. Infect Agent Cancer. 2016;11:59. | ||
Larochelle S, Pandur J, Fisher RP, Salz HK, Suter B. Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes Dev. 1998;12(3):370–381. | ||
Li T, Tang L, Bian D, Jia Y, Huang X, Zhang X. Detection of hTERC and c-MYC genes in cervical epithelial exfoliated cells for cervical cancer screening. Int J Mol Med. 2014;33(5):1289–1297. | ||
Zhu D, Jiang XH, Jiang YH, et al. Amplification and overexpression of TP63 and MYC as biomarkers for transition of cervical intraepithelial neoplasia to cervical cancer. Int J Gynecol Cancer. 2014;24(4):643–648. | ||
Zejnullahu VA, Zejnullahu VA, Josifovska S, Vukovik N, Pakovski K, Panov S. Correlation of hTERT expression with cervical cytological abnormalities and human papillomavirus infection. Prilozi. 2017;38(3):143–151. | ||
Colebatch AJ, Dobrovic A, Cooper WA. TERT gene: its function and dysregulation in cancer. J Clin Pathol. Epub 2019 Jan 29. | ||
Shukla S, Dass J, Pujani M. p53 and bcl2 expression in malignant and premalignant lesions of uterine cervix and their correlation with human papilloma virus 16 and 18. South Asian J Cancer. 2014;3(1):48–53. | ||
Yilmaz A, Alp E, Onen HI, Menevse S. Reduced BCL2 and CCND1 mRNA expression in human cervical cancer HeLa cells treated with a combination of everolimus and paclitaxel. Contemp Oncol (Poznan, Poland). 2016;20(1):28–32. | ||
Chen Q, Cai D, Li M, Wu X. The homologous recombination protein RAD51 is a promising therapeutic target for cervical carcinoma. Oncol Rep. 2017;38(2):767–774. | ||
Kent A. HPV vaccination and testing. Rev Obstet Gynecol. 2010;3(1):33–34. | ||
Small W Jr, Bacon MA, Bajaj A, et al. Cervical cancer: a global health crisis. Cancer. 2017;123(13):2404–2412. | ||
Peta E, Sinigaglia A, Masi G, et al. HPV16 E6 and E7 upregulate the histone lysine demethylase KDM2B through the c-MYC/miR-146a-5p axys. Oncogene. 2018;37(12):1654–1668. | ||
Ott M, Warburton A, Redmond CJ, et al. HPV integration hijacks and multimerizes a cellular enhancer to generate a viral-cellular super-enhancer that drives high viral oncogene expression. PLoS Genet. 2018;14(1):e1007179. | ||
McLaughlin-Drubin ME, Crum CP, Munger K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc Natl Acad Sci U S A. 2011;108(5):2130–2135. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.