Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
Cdc42 Promotes ADSC-Derived IPC Induction, Proliferation, And Insulin Secretion Via Wnt/β-Catenin Signaling
Authors Xiao XH, Huang QY ![]() , Qian XL
, Qian XL ![]() , Duan J, Jiao XQ, Wu LY
, Duan J, Jiao XQ, Wu LY ![]() , Huang QY, Li J, Lai XN, Shi YB, Xiong LX
, Huang QY, Li J, Lai XN, Shi YB, Xiong LX ![]()
Received 5 August 2019
Accepted for publication 25 October 2019
Published 13 November 2019 Volume 2019:12 Pages 2325—2339
DOI https://doi.org/10.2147/DMSO.S226055
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Antonio Brunetti
Xing-Hua Xiao,* Qi-Yuan Huang,* Xian-Ling Qian,* Jing Duan, Xue-Qiao Jiao, Long-Yuan Wu, Qing-Yun Huang, Jun Li, Xing-Ning Lai, Yu-Bo Shi, Li-Xia Xiong
Department of Pathophysiology, Medical College, Nanchang University, Nanchang 330006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Li-Xia Xiong
Department of Pathophysiology, Medical College, Nanchang University, 461 Bayi Road, Nanchang 330006, People’s Republic of China
Tel +86-791-8636-0556
Email [email protected]
Purpose: Type 1 diabetes mellitus (T1DM) is characterized by irreversible islet β cell destruction. Accumulative evidence indicated that Cdc42 and Wnt/β-catenin signaling both play a critical role in the pathogenesis and development of T1DM. Further, bio-molecular mechanisms in adipose-derived mesenchymal stem cells (ADSCs)-derived insulin-producing cells (IPCs) remain largely unknown. Our aim was to investigate the underlying mechanism of Cdc42/Wnt/β-catenin pathway in ADSC-derived IPCs, which may provide new insights into the therapeutic strategy for T1DM patients.
Methods: ADSC induction was accomplished with DMSO under high-glucose condition. ML141 (Cdc42 inhibitor) and Wnt-3a (Wnt signaling activator) were administered to ADSCs from day 2 until the induction finished. Morphological changes were determined by an inverted microscope. Dithizone staining was employed to evaluate the induction of ADSC-derived IPCs. qPCR and Western blotting were employed to measure the mRNA and protein expression level of islet cell development-related genes and Wnt signaling-related genes. The proliferation ability of ADSC-derived IPCs was also detected with a cell counting kit (CCK) assay. The expression and secretion of Insulin were detected with immunofluorescence test and enzyme-linked immunosorbent assay (ELISA) respectively.
Results: During induction, morphological characters of ADSCs changed into spindle and round shape, and formed islet-line cell clusters, with brown dithizone–stained cytoplasm. Expression levels of islet cell development-related genes were up-regulated in ADSC-derived IPCs. Wnt-3a promoted Wnt signaling markers and islet cell development-related gene expression at mRNA and protein levels, while ML141 played a negative effect. Wnt-3a promoted ADSC-derived IPC proliferation and glucose-stimulated insulin secretion (GSIS), while ML141 played a negative effect.
Conclusion: Our research demonstrated that DMSO and high-glucose condition can induce ADSCs into IPCs, and Wnt signaling promotes the induction. Cdc42 may promote IPC induction, IPC proliferation and insulin secretion via Wnt/β-catenin pathway, meaning that Cdc42 may be regarded as a potential target in the treatment of T1DM.
Keywords: Cdc42, ML141, ADSCs, IPCs, Wnt signaling, insulin
Introduction
Diabetes mellitus (DM) is a progressive metabolic disease characterized by hyperglycemia. Nowadays, diabetes has become one of the major public health care problems in the world, with millions of people around the world suffering.1 As diabetes progresses, it can permanently damage target organs, causing complications such as diabetic nephropathy, heart failure, stroke, angina, and diabetic retinopathy.2–4 T1DM is characterized by irreversible islet β cell destruction resulted from T-cell-mediated autoimmune disorder, which eventually leads to absolute insulin deficiency.5 Nowadays, diet control, oral hypoglycemic drugs and insulin injection are the most common applied diabetes therapies. However, these regimens failed to maintain sustained blood glucose homeostasis.6 How to regenerate β cells and prevent the autoimmune destruction of remnant β cells is a tough problem. Many scholars consider whole pancreas and islet transplantation as more effective than traditional therapeutic schedule.7,8 Unfortunately, their clinical use is limited due to operation-related risks, lifetime immunosuppression, and the scarcity of organ donors.9 Considering shortages of islet donors, stem cell transplantation provides possibilities of replacing pancreatic β cells, and it has drawn great attention to academia. Considering stem cells can differentiate into insulin-producing cells (IPCs), a previous study suggests that stem cell–derived IPC transplantation is the most promising treatment other than islet transplantation.10 However, previous researchers failed to approach an appropriate source of IPCs without moral conflict, immunogenicity, and tumorigenicity. In recent years, mesenchymal stem cells (MSCs) from different tissues have attracted great attention. It is convenient to obtain MSCs from various tissues, such as skin,11 umbilical cord,12 spleen,13 bone marrow14 and adipose tissue.15 Among them, adipose-derived mesenchymal stem cells (ADSCs) serve as a proper candidate for clinical application, for no moral issues involved. Considering its paracrine effect and potential for differentiation, ADSCs might also be an effective therapeutic target for cell replacement in diabetic patients.
The Rho GTPase protein cell division cycle protein 42 (Cdc42) can activate signaling cascades related to β cell proliferation, insulin secretion, etc.16 Glucose-stimulated insulin secretion (GSIS) consists of two phases: the first phase is fast with a small amount of insulin released; the second phase is more persistent with a higher level of insulin released. Many studies have demonstrated that Cdc42 participates in the second stage of GSIS under the high-glucose condition.17–20 However, there are no published articles that display the link between Cdc42 and ADSC induction into IPCs.
Wnt signaling plays a crucial regulatory role in the development process and tissue homeostasis of multicellular organisms, including cell-specific differentiation, cell proliferation, morphogenesis, and tissue integrity maintenance.21,22 Wnt signaling network is composed of several branches, classified as 1) the classical β-catenin-dependent Wnt pathway; 2) the β-catenin-independent Wnt/planar cell polarity (PCP) pathway; and 3) the non-classical Wnt/calcium pathway.23 Wnt/β-catenin signaling is involved in the genesis of pancreatic islets and the proliferation of pancreatic β cells.24 Therefore, we speculate that Wnt/β-catenin signaling may play vital roles in Cdc42 regulation in ADSC induction.
However, the exact mechanism by which Cdc42 regulates in Wnt/β-catenin signaling in ADSC-derived IPC induction remains unclear. Some relevant reports indicate that Wnt/β-catenin signaling promotes the differentiation of ADSCs into insulin-secreting cells.25 Mechanically, Wnt ligand binds to frizzled (Fz) and low-density lipoprotein receptor-associated protein 5/6 (LRP5/6) receptors. Subsequently, Fz recruits Dv1, leading to intracellular phosphorylation of LRP5/6 terminal. Glycogen synthase kinase 3β (GSK3β) phosphorylation impedes complex formation of casein kinase 1 (CK1), scaffold protein (Axin), tumor suppressor adenomatous polyposis coli (APC) and GSK3β. Unphosphorylated β-catenin accumulates in the cytoplasm and traverses into the nucleus.25 Endogenous GSK3β activity controls in vitro islet β cell growth by feedback inhibition of the insulin receptor/PI3K/Akt signaling pathway.26 In addition, studies have shown that inhibiting GSK3β has a potent stimulatory effect on β cell regeneration in vivo via increasing pancreatic and duodenal homeobox 1 (PDX1) or glucose transporter 2 (GLUT2) expression in ductal cells.27 PDX1 plays crucial roles in the early embryonic pancreatic formation and later maturation of beta-cell function.28 PDX1 protein activated Insulin and neurogenin 3 (Ngn3) by combining with the promoter regions of them, thereby inducing differentiation of induced pluripotent stem cells into islet β cells.29,30 GLUT2 specifically locates on the cell membrane of islet β cell and can regulate the synthesis and secretion of insulin.31 β-Catenin is able to convert a transcription factor (TCF) protein into a transcriptional activator. Besides, silencing transcription factor 7-like 2 (TCF7L2) gene increases apoptosis of pancreatic β cells in islets, with impeded β cell proliferation and insulin secretion.32 Loss of TCF7L2 leads to decreased insulin secretion, indicating that Wnt signaling disorder may cause the susceptibility and pathogenesis of diabetes.33 Recent studies demonstrated a positive effect of Cdc42 in regulating migration, proliferation and differentiation of MSCs.34,35 More importantly, Li et al observed that Cdc42activates APC and GSK3β, downstream of Wnt/β-catenin signaling, via regulating the formation of the Par6/aPKC complex, thereby affecting proliferation and migration of endocardial endothelium cells.36
In this study, we aim to demonstrate whether and how Cdc42 modulates in Wnt/β-catenin signaling in ADSC-derived IPCs and the effects on ADSC-derived IPC proliferation and insulin secretion.
Materials And Methods
Reagents
Anti-Cdc42 antibody was purchased from Abcam (Cambridge, MA, USA) and anti-Dvl-2 antibody was purchased from Cell Signaling Technology (Trask Lane Danvers, MA, USA). Anti-Insulin, anti-Ngn3, anti-NeuroD1, anti-PDX1, anti-non-p-GSK3β, anti-p-GSK3β, anti-non-p-β-catenin, and anti-β-actin antibodies were purchased from Affinity Biosciences (Cincinnati, OH, USA). HPR-labeled anti-rabbit IgG of goat and HPR-labeled anti-rat IgG of goat were purchased from Zhongshan Jinqiao Company (Beijing, China). Recombinant human Wnt-3a was purchased from R&D Inc. (Minneapolis, MN, USA), and Cdc42 inhibitor ML141 (5mg) was purchased from Selleck (Houston, TX, USA). DTZ was obtained from Sigma (St. Louis, MO, USA). Bovine serum albumin (BSA), Triton-100 and DMSO were purchased from Solarbio (Beijing, China). Low-glucose Dulbecco’s modified Eagle’s medium (DMEM) and DMEM containing 4.5 g/L glucose were obtained from Gibco-BRL (Gaithersburg, MD, USA). Wistar rat ADSC basal medium was purchased from Cyagen (USA). Fetal bovine serum (FBS) was purchased from Biological Industries (Cromwell, CT, USA). Penicillin and streptomycin were purchased from Gibco Life Technologies (Carlsbad, CA, USA). Krebs-Ringer bicarbonate HEPES (KRBH) was purchased from PanEra Company (Guangzhou, China). Enzyme-linked immunosorbent assay (ELISA) Kit for Insulin was purchased from Cloud-Clone Corp (Wuhan, China).
Cell Culture
Wistar rat ADSCs were purchased from the American Type Culture Collection (Manassas, VA, USA). ADSCs were cultured in Wistar rat ADSC basal medium supplemented with 10% fetal bovine serum, 1% glutamine and 1% penicillin/streptomycin at 37 °C in a 5% CO2 humidified incubator.
Experiment Groups
To investigate whether ML141 administered into ADSC induction via Wnt/β-catenin signaling, we arranged ADSCs into different groups including control group, control+Wnt3a group, induction group, induction+Wnt-3a group, induction+ML141 group, induction+Wnt-3a+ML141 group. The control group was cultured in DMEM/F12 medium containing 10% FBS for 10 days. ML141 and Wnt-3a were administered into the culture medium on the second day of induction, at a concentration of 10 M and 100ng/mL, respectively. The medium was changed every 2 days. Morphological observation and follow-up protein and mRNA detection were performed during the induction.
Induction Of ADSCs37,38
When ADSCs were cultured to about 80–90% confluence at P3–P5, the culture medium of ADSCs was changed to low-glucose DMEM culture medium with 1% DMSO for 3 days. Then, ADSCs were completely induced with 4.5 g/L D-glucose DMEM culture medium with 10% FBS for 7 days. The induction process was carried out at 37 °C, 5% CO2 and saturated humidity.
Dithizone Staining39
Fifty-milligram DTZ was dissolved in 5 mL DMEM. After filtrating through a 0.22-µm filter, the DTZ solution was stored at −20°C as mother liquor. On day 10 of induction, 100μl DTZ was diluted to 10 mL (phosphate buffered saline) PBS or culture media. After induction, ADSCs were removed from the incubator and were washed 3 times with PBS, and then DTZ (10mg/mL) staining solution was added to the cells in a six-well plate in 2 ml volume. Cells were incubated at 37°C for 15 mins and washed three times with PBS. Cells were analyzed using an inverted microscope (Olympus, Japan) for the detection of Crimson red-stained clusters.
Real-Time RT-qPCR
Reverse transcription into cDNA using RNA as a template with EasyScript One-Step gDNA Removal and cDNA Synthesis SuperMix kit (TransGen Biotech, Beijing, China) was conducted by following the manufacturer’s instruction. The qPCR was determined by the ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using the following conditions: 94°C for 30s and 40 cycles of 94°C for 5s and 62°C for 30s. The qPCR detected the expression of islet cell development-related genes in each group with TransStart Tip Green qPCR SuperMix (+Dye II) kit (TransGen). Specific primer pairs were designed and synthesized by The Beijing Genomics Institute (BGI) including Cdc42, GLUT2, PDX1, Ngn3, GSK-3β, β-Catenin, NeuroD1, TCF7L2, Insulin, GAPDH (Table 1). The software calculates the corresponding CT value of each gene and the RQ value, that is, the 2−ΔΔCt value. The relative expression level of each gene mRNA was calculated using the 2−ΔΔCt value. At least three independent RT-qPCR experiments were performed for statistical analysis.
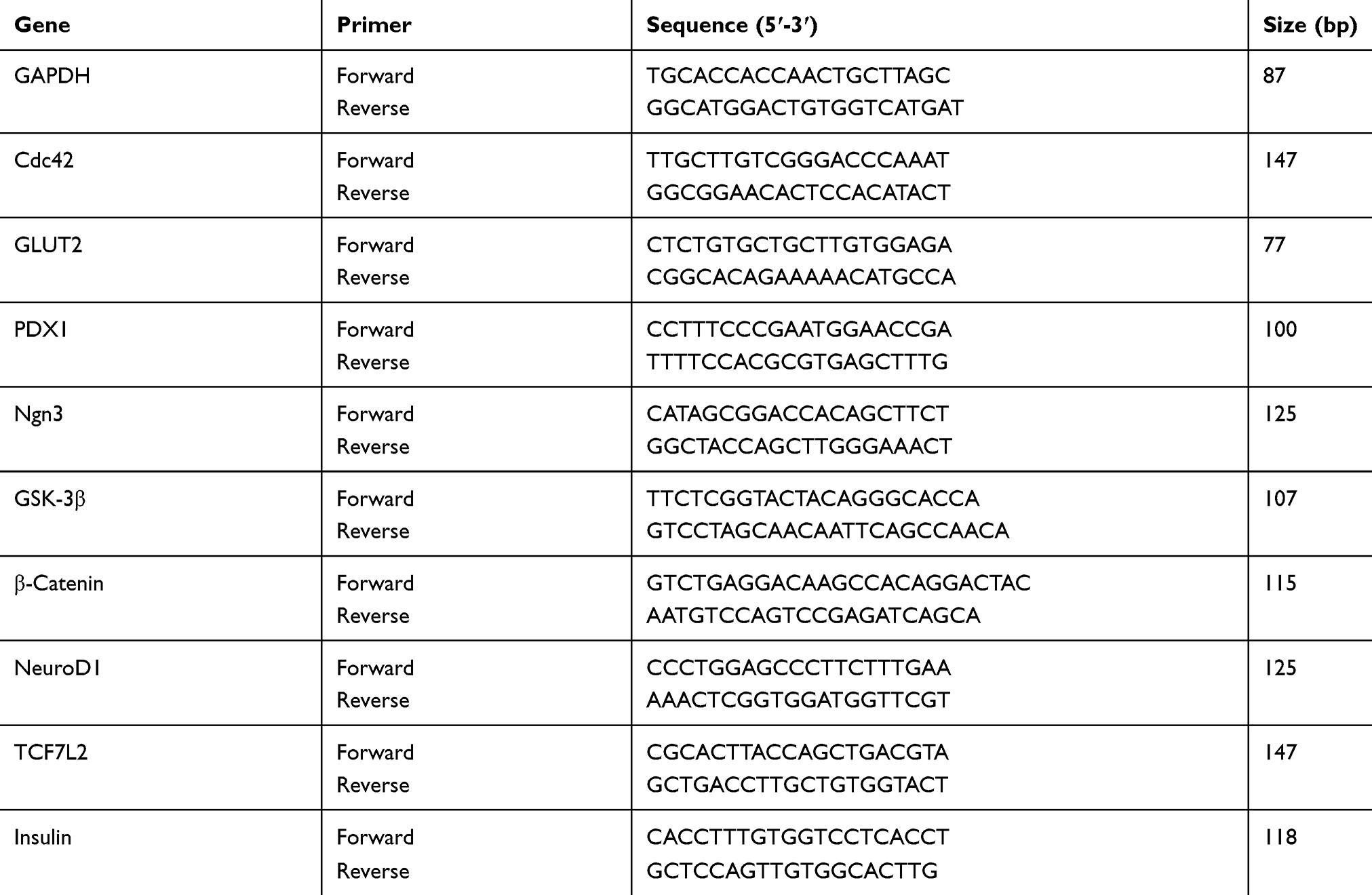 |
Table 1 Details Of The 10 Genes Markers |
Western Blot Assay
Protein concentration was detected by Enhanced BCA Protein Assay Kit (Beyotime, Shanghai, China). The proteins in the lysate were then subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE, Solarbio) for separation and transferred to polyvinylidene difluoride (PVDF) membranes (Merck Millipore, Billerica, MA, USA). After transfection, the membrane was blocked in 5% skim milk powder (Becton, Dickinson, and Company, Franklin Lakes, NJ, USA) and slowly incubated for 2 hrs in the shaker at room temperature. The immunoreactive protein bands were visualized by EasySee Western Blot Kit (TransGen Biotech). Primary antibodies were anti-Cdc42, anti-Dvl-2, anti-Insulin, anti-Ngn3, anti-NeuroD1, anti-PDX1, anti-non-p-GSK3β, anti-p-GSK3β, anti-non-p-β-catenin, and anti-β-actin at a dilution of 1:1000. β-Actin was used as an internal control. All Western blot assays were determined by a luminescent image analyzer (GE Healthcare Bio-Sciences AB, Sweden).
Cell Proliferation Assay
The proliferation rate of IPCs was determined by TransDetect cell counting kit (CCK) (TransGen Biotech) according to the manufacturer’s instruction. On the 10th day after the induction of ADSCs, each group of cells was digested and plated at 5000 cells per well of a 96-well plate, and each group of cells was provided with 4 replicate wells and 1 zero-adjusted well (zero-well plus CCK plus medium). PBS was added to the peripheral wells for containing cell wells and placed in a layer of the incubator. The optical density at 450 nm (OD450) was measured by SpectraMaxParadigm enzyme labelling apparatus (Molecular Devices LLC, Sunnyvale, CA, USA) at 24, 48, and 72hrs, respectively. One hour before the measurement, the cells were washed with PBS, and 10 μL of CCK reagent and 90 μL of the medium were added to each well and then incubated in an incubator for 1 hr. No bubbles were observed in each well during the measurement. The final CCK value is the measured value minus the value of the zeroing well.
Immunofluorescence Staining
After 10 days of ADSC induction, the cells were administered 2 mL of a fixing solution (methanol: acetone = 1:1) per well of a 96-well plate and fixed at room temperature for 30 mins. The fixative was discarded and washed 3 times with PBS at room temperature. Subsequently, the fixative was discarded and washed 3 times with PBS at room temperature. The cells were blocked for 2 hrs with 2 mL of PBS containing 5% BSA and 0.3% Triton-100 per well. And ADSCs were incubated with a primary antibody (insulin) at a dilution of 1:100 overnight at 4°C condition. After being washed with PBS 3 times for 10 mins each time, the sections were incubated with the prepared fluorescent secondary antibody Alesa Fluor 488 at a dilution of 1:250 for 1 hr at 37°C (starting with secondary antibody, avoiding light throughout the follow-up experiment). 40,6-Diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA, USA) was used to stain the nuclei. Images were captured using the Olympus th4-200 microscopy system (Olympus, Tokyo, Japan).
Insulin Secretion Assay
To identify the effects of the Cdc42/Wnt/β-catenin signaling pathway on insulin secretion by ADSCs, Wnt-3a and ML141 were administered into ADSCs. The level of insulin secretion under 3.0 and 20.0 mM glucose stimulation was detected by measuring the amount of secreted insulin in the supernatant. Then, cells were stimulated with Krebs–Ringer bicarbonate HEPES (KRBH; PanEra, Guangzhou, China) containing 3.0 and 20.0 mM glucose for 1 hr. To assess insulin secretion, 5.5×105 ADSCs were inoculated in 6-well plates and cultured for 24 hrs before transfection. After 40 hrs of transfection, the medium was removed. Next, 1 mL KRBH buffer was added to each well and the mixture was incubated for 1 hrs. Thereafter, the KRBH buffer was removed and 1ml KRBH containing 3.0 or 20.0 mM glucose (Solarbio) was added for 1 hr. The levels of insulin were detected by enzyme-linked immunosorbent assay (ELISA) using an ELISA Kit for Insulin (Cloud-Clone Corp., Wuhan, China) according to the manufacturer’s instructions.
Statistical Analysis
All data for this experiment were the results of at least three independent experiments and were presented as mean ± standard deviation (SD). The graphs were made using GraphPad Primer 6 software (GraphPad Software, San Diago, CA, USA), and the experimental data were analyzed by software SPSS 17.0 (SPSS Inc., Chicago, IL, USA). The comparison between the two groups was analyzed by Student’s t-test. The comparison of each index among the groups was analyzed by one-way analysis of variance (ANOVA) method. Statistical significance was set at a p-value of less than 0.05.
Results
Endogenous Cdc42 Protein Expression In ADSCs And Effect Of ML141 In IPCs
Endogenous Cdc42 Protein Expression In ADSCs Is Up-Regulated
To investigate whether Cdc42 affects the differentiation of ADSCs into islet β-like cells, we compared the endogenous Cdc42 expression level between ADSCs and rat islets. The Western blot results showed a higher expression level of Cdc42 in ADSCs compared with that in rat islets (p<0.001) (Figure 1A), indicating that Cdc42 might affect the differentiation of ADSCs into IPCs. To this end, we decided to investigate the effect of Cdc42 by inhibiting the expression of Cdc42 in ADSCs rather than overexpression.
ML141 Suppresses Cdc42 mRNA Expression In IPCs
Considering the overexpression of Cdc42 in ADSCs, we applied ML141, a Cdc42 inhibitor, and investigated its effect on Cdc42 mRNA expression in IPCs. qPCR results showed that, compared with IPCs, Cdc42 mRNA expression decreased significantly in IPCs with ML141 (10μM) (p<0.01) (Figure 1B). These results showed that ML141 administration decreased the Cdc42 mRNA expression level in ADSC-derived IPCs.
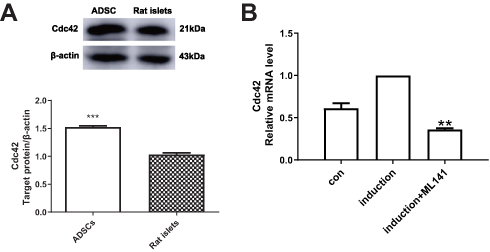 |
Figure 1 (A) Endogenous Cdc42 expression level in ADSCs is up-regulated. Endogenous Cdc42 expression in ADSCs and rat islets was measured by Western blot. (B) ML141 suppresses the Cdc42 expression in IPCs. Cdc42 mRNA expression in control group, induction group and induction+ML141 group were measured by qPCR. Data were shown as the mean ± SD, n = 3. Values were significantly different compared with the corresponding control value at **p<0.01, ***p<0.001. Abbreviations: ADSCs, adipose-derived mesenchymal stem cells; Cdc42, cell division cycle protein 42; con, control group. |
ADSCs Are Induced Into IPC
Morphological Changes During IPC Induction
To investigate the morphological change of ADSCs during the induction, we examined morphological characters of ADSCs with a 10× inverted microscope objective (100× total magnification) on days 3, 6, and 10. As shown in Figure 2, on day 3, control group shaped fibroblast-like (Figure 2A(a)), while the number of ADSCs in the induction group decreased, and cell morphology changed into spindle shape and round shape (Figure 2A(d)). On day 6, the induction group showed spindle shape with distinct cell borders (Figure 2A(e)), while the control group showed a compact arrangement (Figure 2A(b)). On day 10, the induction group began forming islet-line cell clusters (ICCs) (Figure 2A(f)), while the control group compacted more tightly, and no ICCs were observed (Figure 2A(c)).
Dithizone Stain In IPCs
To confirm whether ADSC-derived IPCs were inducted, we stained the induced ADSCs with dithizone (DTZ) for 10 days after the induction. DTZ is a zinc-chelating agent known to stain islet β cells selectively. As shown in Figure 2B, induced ADSCs formed several clusters and contained intracellular brown particles stained by DTZ (Figure 2B(a)), compared to the uninduced control group (Figure 2B(b)). These results indicated that ADSC-derived IPCs were inducted.
To verify whether we successfully inducted ADSCs into IPCs, we employed qPCR to analyze mRNA expression of genes related to pancreatic β cell growth and function, including PDX1, insulin, Ngn3, GLUT2 and neurogenic differentiation 1 (NeuroD1). The results showed that mRNA expression levels of PDX1 (p<0.05), Insulin (p<0.01), Ngn3 (p<0.01) and NeuroD1 (p<0.05) were significantly up-regulated in ADSC-derived IPCs (Figure 2C). These results showed that ADSCs were inducted into IPCs successfully.
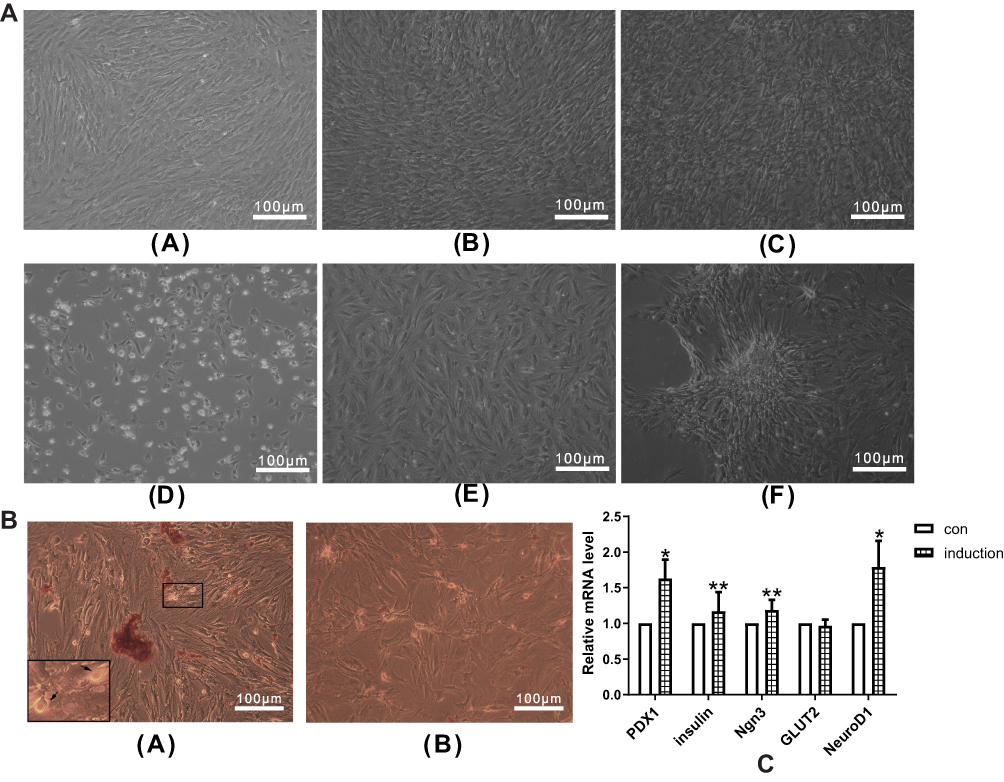 |
Figure 2 (A) Morphological changes of cells during IPC induction. Morphological changes of cells in the control group on days 3 (a), 6 (b) and 10 (c). Morphological changes of cells in the induction group on days 3 (d), 6 (e) and 10 (f). Morphological changes of cells were observed by inverted microscope. (B) ADSCs with DTZ staining. (a) Stained ADSC clusters in the induction group with intracellular brown particles (+) (100×magnification), and the zoom-in figure in the lower left corner (200×magnification). (b) No DTZ stained cells were found in the control group (−) (100×magnification). (c) mRNA expression changes of islet cell development–related genes in IPCs. qPCR was performed to measure the mRNA expression of islet cell development–related genes, including PDX1, Insulin, Ngn3, GLUT2 and NeuroD1. Data were shown as the mean ± SD, n = 3. Values were significantly different compared with the corresponding control value at *p<0.05 and **p<0.01. Abbreviations: ADSCs, adipose-derived mesenchymal stem cells; IPCs, insulin-producing cells, PDX1, pancreatic and duodenal homeobox 1; Ngn3, neurogenin 3; GLUT2, glucose transporter 2; NeuroD1, neurogenic differentiation 1; con, control group. |
Effect Of ML141 And Wnt/β-Catenin Signaling On Induction Of ADSCs Into IPCs
ML141 Negatively Affects Wnt/β-Catenin Signaling In IPCs
To evaluate the effect of ML141 on β-catenin, GSK3β, and TCF7L2 mRNA expression, we performed qPCR to determine mRNA expression of critical genes. As shown in Figure 3A–C, qPCR results showed no significant difference of GSK3β mRNA expression between the induction group and the induction+Wnt-3a group (Figure 3B), while the mRNA expression of non-p-β-catenin (Figure 3A) and TCF7L2 (Figure 3C) both increased (p<0.05). Compared with the induction group, mRNA expression of non-p-β-catenin (Figure 3A) and TCF7L2 (Figure 3C) in the induction+ML141 group both decreased significantly (p<0.01), while GSK3β mRNA expression has no difference (Figure 3B). Pre-treated with Wnt-3a, ML141-administrated IPCs showed significant down-regulation of non-p-β-catenin (Figure 3A) and TCF7L2 (Figure 3C) mRNA expression (p<0.01) and up-regulation of GSK3β mRNA expression (p<0.05) (Figure 3B). These results indicated that ML141 might negatively affect Wnt/β-catenin signaling via down-regulating β-catenin and TCF7L2 mRNA expression and upregulating GSK3β mRNA expression.
To evaluate the effect of ML141 on β-catenin, Dvl2, GSK3β, p-GSK3β and Cdc42 protein expression, we performed Western blot to determine the protein expression of β-catenin, Dvl2, GSK3β, p-GSK3β and Cdc42. As shown in Figure 3D–I, the Western blot results showed no significant difference of Dvl-2 (Figure 3F), phosphorylated (Figure 3G) and unphosphorylated GSK3β (Figure 3H), and Cdc42 (Figure 3I) protein expression between induction group and induction+Wnt-3a group; however, unphosphorylated β-catenin protein expression increased significantly (p<0.05) (Figure 3E). Compared with induction group, protein expression of β-catenin (p<0.01) (Figure 3E), Dvl-2 (p<0.01) (Figure 3F), p-GSK3β (p<0.01) (Figure 3G) and Cdc42 (p<0.01) (Figure 3I) in the induction+ML141 group decreased, while protein expression of non-p-GSK3β increased (p<0.01) (Figure 3H). Pre-treated with Wnt-3a, ML141-administered IPCs showed a significant down-regulation of β-catenin (p<0.01) (Figure 3E), Dvl-2 (p<0.01) (Figure 3F), and p-GSK3β (p<0.05) (Figure 3G) protein expression, while up-regulation of non-p-GSK3β protein expression (p<0.01) (Figure 3H). These results indicated that ML141 might negatively affect Wnt/β-catenin signaling via down-regulating β-catenin, Dvl-2 and p-GSK3β protein expression and up-regulating non-p-GSK3β protein expression.
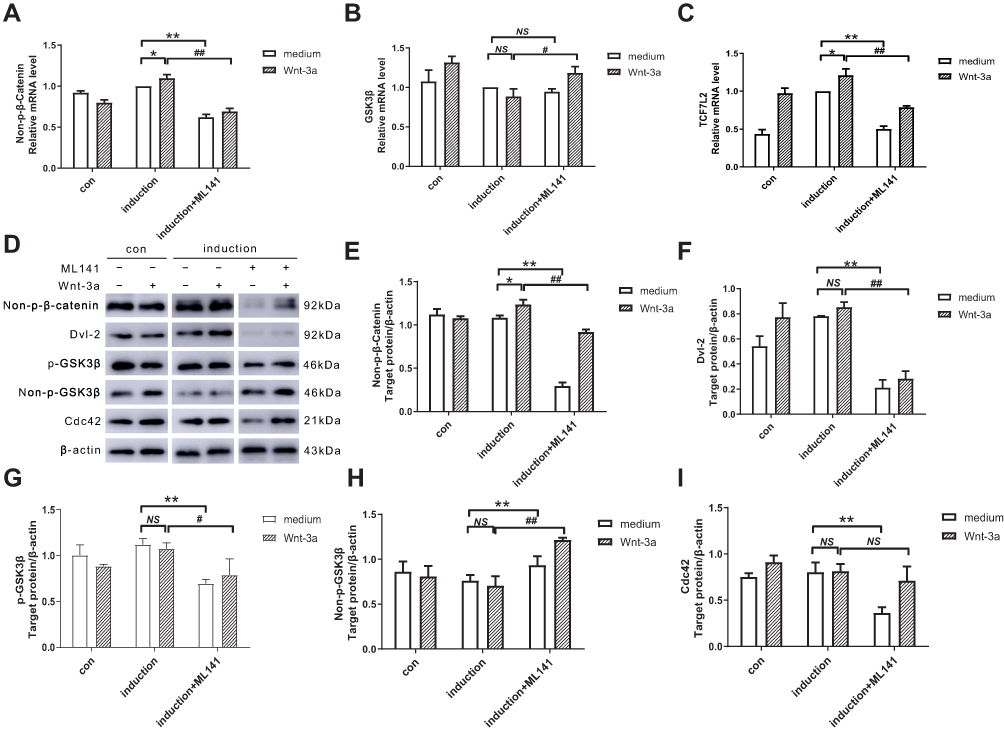 |
Figure 3 (A–C) Effect of ML141 and Wnt-3a on β-catenin, GSK3β, and TCF7L2 mRNA expression. qPCR detected the mRNA expression of (A) Non-p-β-catenin, (B) GSK3β, (C) TCF7L2 in IPCs after induction with Wnt-3a or ML141 administration. (D-I) Effect of ML141 and Wnt-3a on non-p-β-catenin, Dvl-2, p-GSK3β, non-p-GSK3β, and Cdc42 protein expression. Western blot measured the protein expression of (E) non-p-β-catenin, (F) Dvl-2, (G) p-GSK3β, (H) non-p-GSK3β and (I) Cdc42 in IPCs after induction with Wnt-3a or ML141 administration. Data were shown as the mean ± SD, n = 3. Values were significantly different compared with the corresponding control values at **p<0.01, *p<0.05, ##p<0.01 and #p<0.05. Abbreviations: GSK3β, glycogen synthase kinase 3β; p-GSK3β, phosphorylated glycogen synthase kinase 3β; non-p-GSK3β, unphosphorylated glycogen synthase kinase 3β; TCF7L2, transcription factor 7-like 2; Dvl-2, dishevelled homolog 2; IPCs, insulin-producing cells; Cdc42, cell division cycle protein 42; con, control group. |
ML141 Negatively Affects The Expression Of Islet Cell Development-Related Genes In IPCs
Considering that ML141 negatively affects the Wnt/β-catenin signaling, we next focused on the expression of islet cell development-related genes in IPCs. After 10-dayinduction, we performed qPCR to determine mRNA expression of islet cell development-related genes. As shown in Figure 4A–D, compared with the induction group, mRNA expression of Ngn3 (Figure 4B), Insulin (Figure 4C) and GLUT2 (Figure 4D) increased significantly (p<0.01) in induction+Wnt-3a group. However, there was no significant change in PDX1 mRNA expression (Figure 4A). mRNA expression of PDX1 (Figure 4A) decreased significantly (p<0.01) in the induction+ML141 group, while there was no significant change in mRNA expression of Ngn3 (Figure 4B) and Insulin (Figure 4C). Pre-treated with Wnt-3a, ML141-administered IPCs showed significant downregulation of PDX1 (Figure 4A) (p<0.01), Ngn3 (Figure 4B) (p<0.01), Insulin (Figure 4C) (p<0.01), and GLUT2 (Figure 4D) (p<0.01) mRNA expression.
To identify the role of ML141 on the expression of islet cell development–related genes, we applied Western blot and determined the protein expression of PDX1, Ngn3, and Insulin after 10-day induction. As shown in Figure 4E–H, compared with the induction group, Insulin protein expression increased significantly (p<0.05) in the induction+Wnt-3a group (Figure 4H), while there was no significant change in PDX1 (Figure 4F) and Ngn3 (Figure 4G) protein expression. Protein expression of PDX1 (p<0.01) (Figure 4F), Ngn3 (p<0.01) (Figure 4G) and Insulin (p<0.05) (Figure 4H) decreased significantly in the induction+ML141 group. Pre-treated with Wnt-3a, ML141-administered IPCs showed significant down-regulation of PDX1 (p<0.01) (Figure 4F) and Insulin (p<0.05) (Figure 4H) protein expression. These results indicated that Wnt-3a could promote Insulin protein expression in IPCs, while ML141 can suppress the expression of PDX1, Ngn3 and Insulin protein. Thus, ML141 can negatively affect the mRNA and protein expression of islet cell development–related genes in IPCs.
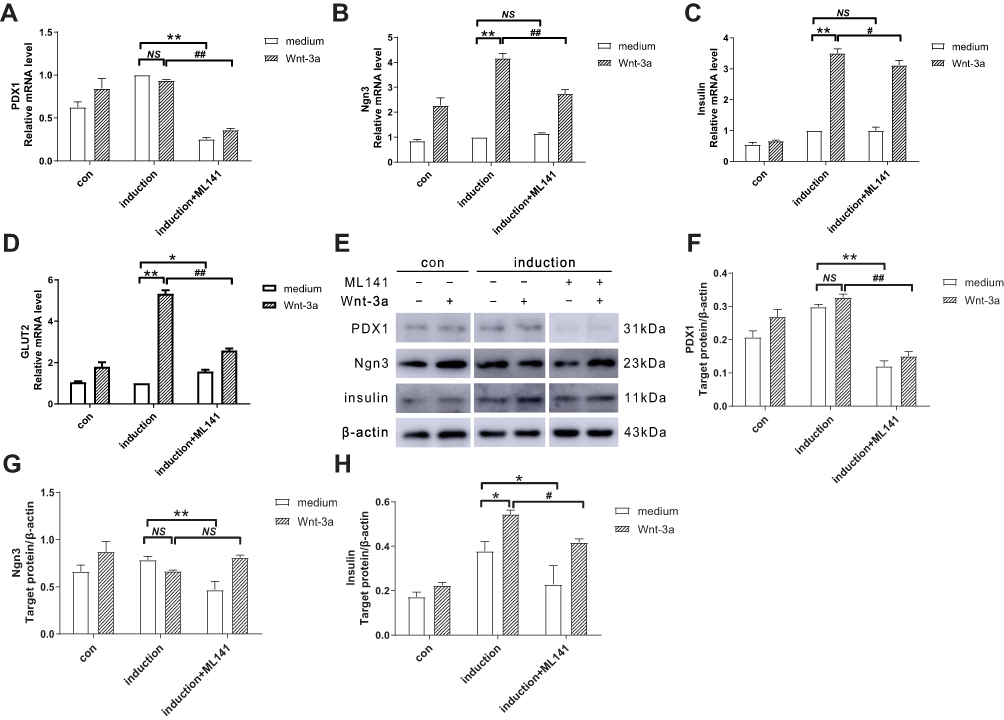 |
Figure 4 (A-D) ML141 suppresses the mRNA expression of PDX1, Ngn3, Insulin and GLUT2. qPCR detected the mRNA expression of (A) PDX1, (B) Ngn3, (C) Insulin and (D) GLUT2 in IPCs. (E-H) ML141 suppresses protein expression of PDX1, Ngn3 and Insulin. Western blot measured the protein expression of (F) PDX1, (G) Ngn3 and (H) Insulin in IPCs. Data were shown as the mean ± SD, n = 3. Values were significantly different compared with the corresponding control value at *p<0.05, **p<0.01, #p<0.05 and ##p<0.01. Abbreviations: PDX1, pancreatic and duodenal homeobox 1; Ngn3, neurogenin 3; GLUT2, glucose transporter 2; con, control group. |
ML141 Inhibits Proliferation Rate And Insulin Expression By ADSC-Derived IPCs
ML141 Inhibits ADSC-Derived IPC Proliferation
To investigate the role of ML141 in IPC proliferation, we performed CCK assay to measure IPC proliferation rate at 24hrs, 48hrs, and 72hrs. As shown in Figure 5A, CCK assay revealed no significant difference between each group after 24hrs. However, we observed a significantly increased proliferation rate in the induction+Wnt-3a group (p<0.01), while a significantly decreased proliferation rate was observed in the induction+ML141 group (p<0.01) after 48hrs and 72hrs, compared to control group. Moreover, the induction+ML141+Wnt-3a group showed a significant inhibited proliferation rate compared to the induction+Wnt-3a group (p<0.01). These results indicated that Wnt-3a could promote IPC proliferation, while ML141 inhibits IPC proliferation. As shown in Figure 5B, we found a significant increased proliferation rate in the control group compared to the induction group (Figure 5A). These results indicated that ADSC-derived IPCs might be limited in cell proliferation.
ML141 Inhibits ADSC-Derived IPC Insulin Expression
To investigate the effect of ML141 on protein expression of Insulin in ADSC-derived IPCs, we employed the immunofluorescence test to detect Insulin protein expression levels in each group. As shown in Figure 5C, IPCs in the induction group expressed Insulin protein (Figure 5C(c)), while there was little protein expression of Insulin in the control group (Figure 5C(a)). Compared with the induction group (Figure 5C(c)), the induction+Wnt-3a group showed an increased protein expression level of Insulin (Figure 5C(d)), while the induction+ML141 group showed a decreased protein expression level of Insulin (Figure 5C(e)). Moreover, compared with induction+Wnt-3a group (Figure 5C(d)), IPCs in induction+Wnt-3a+ML141 group showed lower protein expression level of Insulin (Figure 5C(f)). These results indicated that ADSC-derived IPCs can express Insulin at the protein level, and Wnt-3a can promote Insulin protein expression in ADSC-derived IPCs, while ML141 can suppress it.
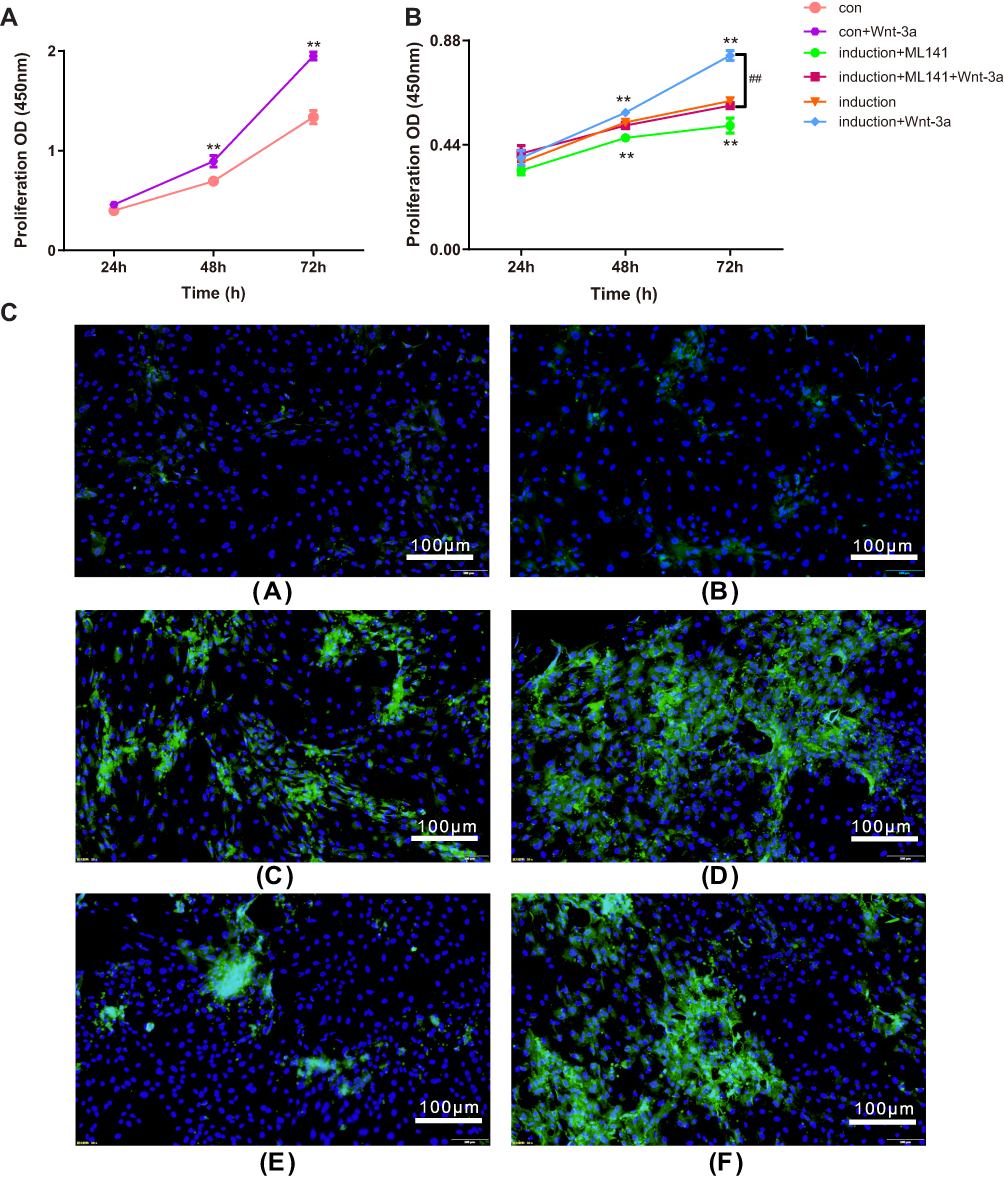 |
Figure 5 (A-B) ML141 inhibits IPC proliferation. CCK assay determined the cell proliferation rate at 24, 48 and 72hrs. (A) Proliferation rate of IPCs in induction group. (B) Proliferation rate of ADSCs in control group. Data were shown as the mean ± SD, n = 3. Values were significantly different compared with the corresponding control value at **p<0.01 and ##p<0.01. (C) ML141 inhibits Insulin protein expression in ADSC-derived IPCs. The Insulin protein expression levels were measured by immunofluorescence test in (a) control group, (b) control+Wnt-3a group, (c) induction group, (d) induction+Wnt-3a group, (e) induction+ML141 group, (f) induction+ML141+Wnt-3a group. The nuclei were labeled with DAPI (blue) and Insulin protein was labeled with the Alesa Fluor 488 (yellowish-green). Abbreviations: IPCs, insulin-producing cells; CCK, cell counting kit; ADSCs, adipose-derived mesenchymal stem cells; DAPI, 4ʹ,6-diamidino-2-phenylindole. |
ML141 Inhibits ADSC-Derived IPC Insulin Secretion In Vitro
To identify the effects of Cdc42/Wnt/β-catenin on insulin secretion by ADSC-derived IPCs, we administered to ADSC-derived IPCs with Wnt-3a and ML141 and then stimulated them by KRBH containing 3.0 and 20.0 mM glucose for 1 hr. The amount of secreted insulin in the supernatant was measured by ELISA (Figure 6). The results showed that under high-glucose stimulation, insulin secretion by ADSC-derived IPCs increased significantly, compared to the control group (p<0.001). ML141 administration into induction group inhibited insulin secretion (p<0.01), and Wnt-3a administration to induction group promoted insulin secretion (p<0.001) under high-glucose stimulation. ML141 administration reversed the effect of the Wnt-3a regarding insulin secretion inhibition under high-glucose stimulation (p<0.001). These in vitro results further indicated that ML141 could inhibit insulin secretion by ADSC-derived IPCs under high-glucose stimulation.
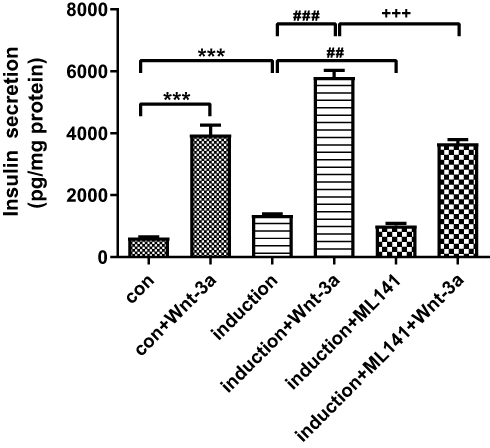 |
Figure 6 ML141 inhibits insulin secretion by ADSC-derived IPCs under high-glucose stimulation. Data were shown as the mean ± SD, n = 3. Values were significantly different compared with the corresponding control value at ##p<0.01, ###p<0.001, ***p<0.001, and +++p<0.001. |
Discussion
Irreversible islet β cell destruction in T1DM resulted from an autoimmune disorder, and reducing impaired β cells and functional β cell regeneration is an ideal treatment for T1DM patients.5 Previous studies have found that MSC-derived IPCs not only effectively improve the function of the pancreas, but also cure complications.40 MSCs are easier to isolate and cultivate than pluripotent stem cell transplants, and they can proliferate without forming teratomas. However, the lack of IPCs is a major obstacle to the diabetes transplantation application.41 ADSCs are a feasible source of IPCs, for the advantages of more convenient acquisition.42–45 Activation of crucial signaling pathways in the induction of ADSCs into IPCs is a suitable way to improve differentiation protocols.46 In summary, we demonstrated for the first time that there exists a direct link between Cdc42 and induction of ADSCs into IPCs; additionally, our main outcome is that Cdc42/Wnt/β-catenin signaling plays a critical role in regulating the induction of ADSCs into IPCs and proliferation and insulin secretion of ADSCs-derived IPCs.
Currently, many studies have demonstrated the positive effect of Cdc42 in regulating migration, proliferation, and differentiation of MSCs.34,35 In this study, we observed the up-regulation of endogenous Cdc42 in ADSCs, compared to rat islets. Thereby, the effect of Cdc42 might not be obvious if we overexpress Cdc42 in the following experiments, and so we selected a novel Cdc42 inhibitor ML141 to inhibit Cdc42 expression.47,48 There are no published data showing the effect of ML141 on the induction of ADSCs into IPCs. According to previous studies, we prepared ML141 (10μM) with 1% DMSO, because ML141 and DMSO at this concentration would not affect ADSC growth. Down-regulated expression of Cdc42 confirmed the Cdc-42-inhibiting effects of ML141. We avoided direct knockout of Cdc42 in ADSCs for reasons as follows: a) it was time-consuming; b) it was hard to ensure cell status; c) long-term culturing would be hindered by growing cell generation; d) proliferation situation would change after more than 10 generations and thus obstructed follow-up studies. In addition, we did not adopt transfecting siRNA to interfere with the Cdc42 expression level because of the particularity of this stem cell induction process. We tried to transfect siRNA into ADSCs, which resulted in the death of a large portion of ADSCs during the induction process. However, explanations for this phenomenon remain unclear. One possible reason may be the cytotoxicity of commonly used transfection liposomes, such as lip2000. The presence of cationic surfactant in cationic liposomes makes the cell membrane unstable and damages the cell membrane, and so cationic liposomes may contribute to the death of ADSCs.
Many differentiation schemes can induce ADSCs into islet β-like cells, and various signaling pathways participate in this process, including Wnt signaling.49,50 After comparing the expression of endogenous Cdc42 protein in ADSCs and rat islets, in accordance with previous studies, we successfully induce ADSCs into IPCs.51–53 Our data indicate that high glucose (4.5 g/L D-glucose) and DMSO (10%) significantly promote the induction of IPCs from ADSCs. Interestingly, after we constructed DTZ staining to further verify the induction of IPCs, some cells in the control group fell off, but the induction group remained. DMSO at excessive concentration would inhibit cell growth. It was possible that DMSO may have pernicious effects on ADSCs and this might have led to the death of control group cells, while induction was already tolerance to DMSO.
The induction of ADSC-derived IPCs may be due to the activation of the islet cell development–related genes in IPCs. The expressions of PDX1,54 Ngn3,53 NeuroD,55 and Insulin56 mRNA were in line with the results of previous studies, indicating that the ADSCs had differentiated toward IPCs. The interaction of PDX1, Ngn3 and V-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA) activated mouse insulin promoter to trans-differentiate pancreatic and exocrine cells into insulin-producing beta-like cells.57,58 Interestingly, our results showed no significant changes in GLUT2 expression before and after the induction. As mentioned before, GLUT2 is a membrane protein expressed in tissues involved in maintaining glucose homeostasis and in cells where glucose sensing is necessary. Similar to PDX1 and Insulin, GLUT2 is one of the islet-specific markers. The reason for this rather contradictory result is still not entirely clear, but a reasonable explanation for this may be as follows: we employ high-glucose (HG) culture medium containing DMSO to induce ADSCs into IPCs. In many experimental models of diabetes, GLUT2 gene expression decreased in islet β cells, and a number of studies have demonstrated that HG condition could be “toxic”, which ended up with inhibited GLUT2 expression of islet β cells.59,60 The possible mechanism may partly rely on the proximal 338 bp of the murine GLUT2 promoter, which contains cis-elements required for the islet-specific expression of GLUT2.61 Transcription regulationmay aim to prevent high ambient glucose from entering islet β cells. In accordance with islet β cell under HG conditions, we consider that this protective regulation (regulation of GLUT2 mRNA) may also exist during the induction of ADSC into IPCs, thereby resulting in an insignificant change of GLUT2 expression in induced ADSC-derived IPCs.
To elucidate the further bio-molecular mechanisms of how Cdc42 plays in ADSC-derived IPCs, we conditionally used Wnt-3a (Wnt signaling activator) and ML141 (Cdc42 inhibitor), respectively. Then, we analyzed the effects of Wnt-3a and ML141 administration on Wnt signaling and the islet cell development–related genes. As shown in this report, Wnt-3a promotes IPC development as a Wnt/β-catenin signaling activator; inhibiting Cdc42 (by ML141) inactivated Wnt signaling, thus impeding IPC development by impairing expression of islet cell development–related genes (Ngn3, Insulin, GLUT2, and PDX10). Furthermore, ML141 may reverse the Wnt-3a-mediated activation of Wnt/β-catenin signaling and IPC development. The above-mentioned findings indicate that Cdc42/Wnt/β-catenin signaling may be important for this differentiation process. Interestingly, pre-treated with Wnt-3a, ML141 administration showed greater reductions than independent ML141 administration, except a non-significant deletion of Cdc42 protein expression. We attribute this unexpected result to the lack of repeats. In addition, when we measured the protein expression of these genes, we found that Wnt-3a administration resulted in enhanced Insulin protein expression, but PDX1 and Ngn3 remained. A reasonable explanation for this unexpected result might be the post-transcriptional modification and effects of some microRNAs.
In the end, our data showed that ML141 administration could inhibit proliferation rate and Insulin expression by ADSC-derived IPCs in vitro; ML141 administration could inhibit insulin secretion in vivo. Therefore, we suggested that Cdc42 might play positive effects on proliferation and GSIS by ADSC-derived IPCs via activating Wnt/β-catenin signaling.
Collectively, according to all of our findings, we may summarize the possible mechanism as shown in Figure 7. Wnt-3a administration activates Wnt/β-catenin signaling starting with Dvl2 recruitment, which promotes GSK3β phosphorylation. Due to the increasing expression of p-GSK3β, the synthesis of the destruction complex decreases. According to previous findings of Na et al, with stimulation of Wnt ligand, non-p-β-catenin may escape ubiquitin-proteasome-dependent degradation and activate its target genes (TCF7L2, PDX1, Ngn3, and Insulin) after translocation to the nucleus.62 Additionally, previous studies on TCF7L2 and PDX1 showed that TCF7L2 may promote β cell regeneration and IPC formation via the JAK2/STAT3 pathway63 and may activate mouse insulin promoter by the interaction of PDX1, Ngn3 and MafA, thus endowing function of trans-differentiated IPCs.57,58 Finally, Wnt-3a administration resulted in promoted proliferation and GSIS in ADSC-derived IPCs. On the contrary, it is also reasonable to speculate that ML141-mediated Wnt signaling inhibition starts with inhibiting Dvl2 recruitment and p-GSK3β expression, and then the destruction complex promotes ubiquitin-proteasome-dependent phosphorylation and degradation of β-catenin. Cdc42 inhibition impedes translocation of β-catenin to the nucleus and islet cell development-related gene expression (TCF7L2, PDX1, Ngn3 and Insulin), thus IPC proliferation and insulin secretion are inhibited. Collectively, we suggest that Cdc42 inhibition may impair proliferation and GSIS of ADSC-derived IPCs via Wnt/β-catenin signaling.
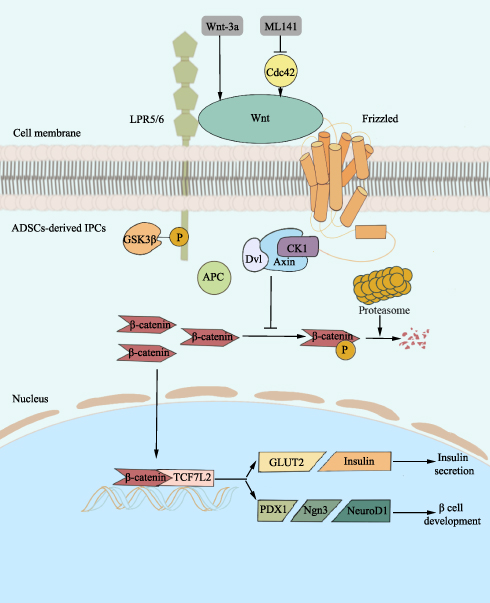 |
Figure 7 The mechanism of Cdc42/Wnt/β-catenin signaling pathway in ADSC-derived IPCs. Wnt-3a administration activates Wnt, which combines with LPR5/6 and Fz, and restores Dvl2 recruitment. GSK3β goes through phosphorylation to impede β-catenin phosphorylation by obstructing the combination of Dvl, GSK3β, Axin, and CK1. As a result, non-p-β-catenin escapes ubiquitin-proteasome-dependent degradation and activates TCF7L2 and downstream genes (PDX1, Ngn3, NeuroD1, GLUT2, and Insulin) after translocation to the nucleus. Eventually, Wnt/β-catenin signaling promotes ADSC-derived IPC development and insulin secretion. Cdc42 inhibition by ML141 leads to down-regulation of Wnt/β-catenin signaling. Therefore, interaction of Dvl, GSK3β, Axin, and CK1 promotes β-catenin ubiquitin-proteasome-dependent phosphorylation and degradation, ending up with inactivated target genes (TCF7L2, PDX1, Ngn3, NeuroD1, and GLUT2), thereby inhibiting ADSC-derived IPC development and insulin secretion. Abbreviations: Cdc42, cell division cycle protein 42; LPR5/6, low-density lipoprotein receptor-associated protein 5/6; Fz, frizzled; GSK3β, glycogen synthase kinase 3β; p-GSK3β, phosphorylated glycogen synthase kinase 3β; Dvl, dishevelled; CK1, casein kinase 1; non-p-β-catenin, unphosphorylated β-catenin; TCF7L2, transcription factor 7-like 2; PDX1, pancreatic and duodenal homeobox 1; Ngn3, neurogenin 3; NeuroD1, neurogenic differentiation 1; GLUT2, glucose transporter 2; ADSCs, adipose-derived mesenchymal stem cells; IPCs, insulin-producing cells. |
In conclusion, our observations indicate that Cdc42/Wnt/β-catenin signaling pathway may play a positive effect on induction, proliferation, and GSIS of ADSC-derived IPCs. Further investigations need to be carried out to illustrate how TCF7L2, PDX1, and Ngn3 modulate ADSC-derived IPC proliferation and to illustrate the therapeutic effect of ADSC-derived IPC transplantation for T1DM patients. Our work clearly has some limitations. Nevertheless, we are confident that our findings could provide further evidence for the role of Cdc42 in the induction and development of ADSC-derived IPCs. Appropriate manipulation of signaling will enhance the efficiency of differentiation of islet β cells and may provide a basis for regarding Cdc42 as a potential target in the therapy for T1DM patients.
Acknowledgments
This research was funded by the National Natural Science Foundation of China (Nos. 31660287). We would like to thank the technical support by Dr. Hui Lin. Xing-Hua Xiao, Qi-Yuan Huang, and Xian-Ling Qian contributed equally as co-first authors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet (London, England). 2014;383(9922):1068–1083. doi:10.1016/S0140-6736(13)62154-6
2. Shao H, Yang S, Fonseca V, Stoecker C, Shi L. Estimating quality of life decrements due to diabetes complications in the United States: the Health Utility Index (HUI) diabetes complication equation. Pharmacoeconomics. 2019. doi:10.1007/s40273-019-00775-8
3. Kawasaki R, Kitano S, Sato Y, et al. Factors associated with non-proliferative diabetic retinopathy in patients with type 1 and type 2 diabetes: the Japan Diabetes Complication and its Prevention prospective study (JDCP study 4). Diabetol Int. 2019;10(1):3–11. doi:10.1007/s13340-018-0357-z
4. Straw S, Witte KK, Kearney MT. Heart failure: a preventable and treatable complication of type 2 diabetes. J Diabetes. 2019. doi:10.1111/jdb.2019.11.issue-7
5. Knip M, Veijola R, Virtane SM, et al. Environmental triggers and determinants of type 1 diabetes. Diabetes. 2005;S125–S136.
6. Fanelli CG, Porcellati F, Pampanelli S, Bolli GB. Insulin therapy and hypoglycaemia: the size of the problem. Diabetes Metab Res Rev. 2004;20(Suppl 2):S32–S42. doi:10.1002/dmrr.514
7. Health Quality O. Pancreas islet transplantation for patients with Type 1 diabetes mellitus: a clinical evidence review. Ont Health Technol Assess Ser. 2015;15(16):1–84.
8. Cefalu WT. American diabetes association-European association for the study of diabetes position statement: due diligence was conducted. Diabetes Care. 2012;35(6):1201–1203. doi:10.2337/dc12-0564
9. El-Badri N, Ghoneim MA. Mesenchymal stem cell therapy in diabetes mellitus: progress and challenges. J Nucleic Acids. 2013;2013:194858. doi:10.1155/2013/194858
10. Zang L, Hao H, Liu J, Li Y, Han W, Mu Y. Mesenchymal stem cell therapy in type 2 diabetes mellitus. Diabetol Metab Syndr. 2017;9:36. doi:10.1186/s13098-017-0233-1
11. Toma JG, Akhavan M, Fernandes KJL, et al. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3(9):778–784. doi:10.1038/ncb0901-778
12. Hankamolsiri W, Manochantr S, Tantrawatpan C, Tantikanlayaporn D, Tapanadechopone P, Kheolamai P. The effects of high glucose on adipogenic and osteogenic differentiation of gestational tissue-derived MSCs. Stem Cells Int. 2016;2016:9674614. doi:10.1155/2016/9674614
13. Kodama S, Davis M, Faustman DL. Regenerative medicine: a radical reappraisal of the spleen. Trends Mol Med. 2005;11(6):271–276. doi:10.1016/j.molmed.2005.04.004
14. Starc N, Ingo D, Conforti A, et al. Biological and functional characterization of bone marrow-derived mesenchymal stromal cells from patients affected by primary immunodeficiency. Sci Rep. 2017;7(1):8153. doi:10.1038/s41598-017-08550-5
15. Mafi P. Adult mesenchymal stem cells and cell surface characterization - a systematic review of the literature. Open Orthop J. 2011;5:253–260. doi:10.2174/1874325001105010253
16. Huang QY, Lai XN, Qian XL, et al. Cdc42: a novel regulator of insulin secretion and diabetes-associated diseases. Int J Mol Sci. 2019;20(1).
17. Yoder SM, Dineen SL, Wang Z, Thurmond DC. YES, a Src family kinase, is a proximal glucose-specific activator of cell division cycle control protein 42 (Cdc42) in pancreatic islet beta cells. J Biol Chem. 2014;289(16):11476–11487. doi:10.1074/jbc.M114.559328
18. Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature. 1984;311(5983):271–273. doi:10.1038/311271a0
19. Tokarz VL, MacDonald PE, Klip A. The cell biology of systemic insulin function. J Cell Biol. 2018;217(7):2273–2289. doi:10.1083/jcb.201802095
20. Kesavan G, Lieven O, Mamidi A, et al. Cdc42/N-WASP signaling links actin dynamics to pancreatic beta cell delamination and differentiation. Development. 2014;141(3):685–696. doi:10.1242/dev.100297
21. Nusse R, Clevers H. Wnt/ beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. doi:10.1016/j.cell.2017.05.016
22. Mattes B, Dang Y, Greicius G, et al. Wnt/PCP controls spreading of Wnt/beta-catenin signals by cytonemes in vertebrates. Elife. 2018;7. doi:10.7554/eLife.36953
23. Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13(12):767–779. doi:10.1038/nrm3470
24. Jin T. The WNT signalling pathway and diabetes mellitus. Diabetologia. 2008;51(10):1771–1780. doi:10.1007/s00125-008-1084-y
25. Wang H, Ren Y, Hu X, et al. Effect of Wnt signaling on the differentiation of islet beta-cells from adipose-derived stem cells. Biomed Res Int. 2017;2017:2501578.
26. Fornoni A, Pileggi A, Molano RD. Inhibition of c-jun N terminal kinase (JNK) improves functional beta cell mass in human islets and leads to AKT and glycogen synthase kinase-3 (GSK-3) phosphorylation. Diabetologia. 2008;51(2):298–308. doi:10.1007/s00125-007-0889-4
27. Figeac F, Uzan B, Faro M, Chelali N, Portha B, Movassat J. Neonatal growth and regeneration of beta-cells are regulated by the Wnt/beta-catenin signaling in normal and diabetic rats. Am J Physiol Endocrinol Metab. 2010;298(2):E245–E256. doi:10.1152/ajpendo.00538.2009
28. Zhu Y, Liu Q, Zhou Z, Ikeda Y. PDX1, Neurogenin-3, and MAFA: critical transcription regulators for beta cell development and regeneration. Stem Cell Res Ther. 2017;8(1):240. doi:10.1186/s13287-017-0694-z
29. Qin Y, Xiao L, Zhan XB, Zhou HX. Pdxl and its role in activating Ngn3 and Pax6 to induce differentiation of iPSCs into islet beta cells. Genet Mol Res. 2015;14(3):8892–8900. doi:10.4238/2015.August.3.12
30. Arcidiacono B, Iiritano S, Chiefari E, et al. Cooperation between HMGA1, PDX-1, and MafA is essential for glucose-induced insulin transcription in pancreatic beta cells. Front Endocrinol (Lausanne). 2015;5:237.
31. Navarro-Tableros V, Fiordelisio T, Hernandez-Cruz A, Hiriart M. Physiological development of insulin secretion, calcium channels, and GLUT2 expression of pancreatic rat beta-cells. Am J Physiol Endocrinol Metab. 2007;292(4):E1018–E1029. doi:10.1152/ajpendo.00457.2006
32. Shu L, Sauter NS, Schulthess FT, Matveyenko AV, Oberholzer J, Maedler K. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes. 2008;57(3):645–653. doi:10.2337/db07-0847
33. Mitchell RK, Mondragon A, Chen L, et al. Selective disruption of Tcf7l2 in the pancreatic beta cell impairs secretory function and lowers beta cell mass. Hum Mol Genet. 2015;24(5):1390–1399. doi:10.1093/hmg/ddu553
34. Xu XP, He HL, Hu SL, et al. Ang II-AT2R increases mesenchymal stem cell migration by signaling through the FAK and RhoA/Cdc42 pathways in vitro. Stem Cell Res Ther. 2017;8(1):164. doi:10.1186/s13287-017-0617-z
35. Lu J, Wang QH, Huang LH, Dong HY, Lin LJ, Tan JM. Correlation of CDC42 activity with cell proliferation and palmitate-mediated cell death in human umbilical cord Wharton’s jelly derived mesenchymal stromal cells. Stem Cells Dev. 2017;26(17):1283–1292. doi:10.1089/scd.2017.0032
36. Li J, Miao L, Zhao C, et al. CDC42 is required for epicardial and pro-epicardial development by mediating FGF receptor trafficking to the plasma membrane. Development. 2017;144(9):1635–1647. doi:10.1242/dev.147173
37. Wang H, Ren Y, Hu X, et al. Effect of Wnt signaling on the differentiation of islet -cells from adipose-derived stem cells. Biomed Res Int. 2017;2017:2501578.
38. Oh S-H, Muzzonigro TM, Bae S-H. Adult bone marrow-derived cells trans-differentiating into insulin-producing cells for the treatment of type I diabetes. Lab Invest. 2004;84(5):607–617. doi:10.1038/labinvest.3700074
39. Conget JI, Sarri Y, González-Clemente JM. Deleterious effect of dithizone-DMSO staining on insulin secretion in rat and human pancreatic islets. Pancreas. 1994;9(2):157–160. doi:10.1097/00006676-199403000-00003
40. Hirabaru M, Kuroki T, Adachi T, et al. A Method for Performing Islet Transplantation Using Tissue-Engineered Sheets of Islets and Mesenchymal Stem Cells. Tissue Eng Part C Methods. 2015;21(12):1205–1215.
41. Elham H, Mahmoud H. The effect of pancreas islet-releasing factors on the direction of embryonic stem cells towards Pdx1 expressing cells. Appl Biochem Biotechnol. 2018;186(2):371–383. doi:10.1007/s12010-018-2733-3
42. Guo QS, Zhu MY, Wang L, et al. Combined transfection of the three transcriptional factors, PDX-1, NeuroD1, and MafA, causes differentiation of bone marrow mesenchymal stem cells into insulin-producing cells. Exp Diabetes Res. 2012;2012:672013.
43. Wang H, Yang Y, Ho G, et al. Programming of human umbilical cord mesenchymal stem cells in vitro to promote pancreatic gene expression. Mol Med Rep. 2013;8(3):769–774. doi:10.3892/mmr.2013.1598
44. Zhang X, Qin J, Wang X, et al. Netrin-1 improves adipose-derived stem cell proliferation, migration, and treatment effect in type 2 diabetic mice with sciatic denervation. Stem Cell Res Ther. 2018;9(1):285. doi:10.1186/s13287-018-1020-0
45. Thakkar UG, Vanikar AV, Trivedi HL. Should we practice stem cell therapy for type 1 diabetes mellitus as precision medicine? Cytotherapy. 2017;19(5):574–576. doi:10.1016/j.jcyt.2017.02.001
46. Hashemitabar M, Heidari E. Redefining the signaling pathways from pluripotency to pancreas development: in vitro β-cell differentiation. J Cell Physiol. 2019;234(6):7811–7827. doi:10.1002/jcp.v234.6
47. Surviladze Z, Waller A, Strouse JJ, et al. A potent and selective inhibitor of Cdc42 GTPase. In: Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (US); 2010.
48. Chen H-Y, Yang YM, Stevens BM. Inhibition of redox/Fyn/c-Cbl pathway function by Cdc42 controls tumour initiation capacity and tamoxifen sensitivity in basal-like breast cancer cells. EMBO Mol Med. 2013;5(5):723–736. doi:10.1002/emmm.v5.5
49. Ikemoto T, Feng R, Shimada M, et al. A new 2-step acceleration protocol using a histone deacetylase inhibitor to generate insulin-producing cells from adipose-derived mesenchymal stem cells. Pancreas. 2018;47(4):477–481. doi:10.1097/MPA.0000000000001017
50. Bahrebar M, Soleimani M, Karimi MH, Vahdati A, Yaghobi R. Generation of islet-like cell aggregates from human adipose tissue-derived stem cells by lentiviral overexpression of PDX-1. Int J Organ Transplant Med. 2015;6(2):61–76.
51. Wang R, Zhang D, Zhang T, et al. The differentiation of human MSCs derived from adipose and amniotic tissues into insulin-producing cells, induced by PEI@Fe3O4 nanoparticles-mediated NRSF and SHH silencing. Int J Mol Med. 2018;42(5):2831–2838. doi:10.3892/ijmm.2018.3827
52. Dayer D, Tabandeh MR, Moghimipour E, et al. MafA overexpression: a new efficient protocol for in vitro differentiation of adipose-derived mesenchymal stem cells into functional insulin-producing cells. Cell J. 2019;21(2):169–178. doi:10.22074/cellj.2019.5669
53. Dayer D, Tabar MH, Moghimipour E, et al. Sonic hedgehog pathway suppression and reactivation accelerates differentiation of rat adipose-derived mesenchymal stromal cells toward insulin-producing cells. Cytotherapy. 2017;19(8):937–946. doi:10.1016/j.jcyt.2017.05.003
54. Fazili A, Gholami S, Minaie Zangi B, Seyedjafari E, Gholami M. In vivo differentiation of mesenchymal stem cells into insulin producing cells on electrospun Poly-L-Lactide acid scaffolds coated with matricaria chamomilla L. Oil. Cell J. 2016;18(3):310–321.
55. Chandra V, G S, Muthyala S. Islet-like cell aggregates generated from human adipose tissue derived stem cells ameliorate experimental diabetes in mice. PLoS One. 2011;6(6):e20615. doi:10.1371/journal.pone.0020615
56. Boroujeni ZN. Insulin producing cells established using non-integrated lentiviral vector harboring PDX1 gene. World J Stem Cells. 2013;5(4):217–228. doi:10.4252/wjsc.v5.i4.217
57. Akinci E, Banga A, Greder LV, Dutton JR, Slack JM. Reprogramming of pancreatic exocrine cells towards a beta (beta) cell character using Pdx1, Ngn3 and MafA. Biochem J. 2012;442(3):539–550. doi:10.1042/BJ20111678
58. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455(7213):627–632. doi:10.1038/nature07314
59. Reimer MK, Ahrén B. Altered beta-cell distribution of pdx-1 and GLUT-2 after a short-term challenge with a high-fat diet in C57BL/6J mice. Diabetes. 2002;S138–S143.
60. Thorens B, Wu YJ, Leahy JL. The loss of GLUT2 expression by glucose-unresponsive beta cells of db/db mice is reversible and is induced by the diabetic environment. J Clin Invest. 1992;90(1):77–85. doi:10.1172/JCI115858
61. Waeber G. A 338-bp proximal fragment of the glucose transporter type 2 (GLUT2) promoter drives reporter gene expression in the pancreatic islets of transgenic mice. Mol Cell Endocrinol. 1995;114:205–215. doi:10.1016/0303-7207(95)96801-N
62. Chen N, Wang J. Wnt/β-catenin signaling and obesity. Front Physiol. 2018;9:792. doi:10.3389/fphys.2018.00792
63. Shu L, Zien K, Gutjahr G, et al. TCF7L2 promotes beta cell regeneration in human and mouse pancreas. Diabetologia. 2012;55(12):3296–3307. doi:10.1007/s00125-012-2693-z
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.