Back to Journals » Clinical Interventions in Aging » Volume 10
Causative factors for formation of toxic islet amyloid polypeptide oligomer in type 2 diabetes mellitus
Received 31 August 2015
Accepted for publication 6 October 2015
Published 19 November 2015 Volume 2015:10 Pages 1873—1879
DOI https://doi.org/10.2147/CIA.S95297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Hye Rin Jeong, Seong Soo A An
Department of Bionano Technology, Gachon Medical Research Institute, Gachon University, Gyeonggi-do, Republic of Korea
Abstract: Human islet amyloid polypeptide (h-IAPP) is a peptide hormone that is synthesized and cosecreted with insulin from insulin-secreting pancreatic β-cells. Recently, h-IAPP was proposed to be the main component responsible for the cytotoxic pancreatic amyloid deposits in patients with type 2 diabetes mellitus (T2DM). Since the causative factors of IAPP (or amylin) oligomer aggregation are not fully understood, this review will discuss the various forms of h-IAPP aggregation. Not all forms of IAPP aggregates trigger the destruction of β-cell function and loss of β-cell mass; however, toxic oligomers do trigger these events. Once these toxic oligomers form under abnormal metabolic conditions in T2DM, they can lead to cell disruption by inducing cell membrane destabilization. In this review, the various factors that have been shown to induce toxic IAPP oligomer formation will be presented, as well as the potential mechanism of oligomer and fibril formation from pro-IAPPs. Initially, pro-IAPPs undergo enzymatic reactions to produce the IAPP monomers, which can then develop into oligomers and fibrils. By this mechanism, toxic oligomers could be generated by diverse pathway components. Thus, the interconnections between factors that influence amyloid aggregation (eg, absence of PC2 enzyme, deamidation, reduction of disulfide bonds, environmental factors in the cell, genetic mutations, copper metal ions, and heparin) will be presented. Hence, this review will aid in understanding the fundamental causative factors contributing to IAPP oligomer formation and support studies for investigating novel T2DM therapeutic approaches, such as the development of inhibitory agents for preventing oligomerization at the early stages of diabetic pathology.
Keywords: amyloid aggregation, causative factor, IAPP, islet amyloid polypeptide, toxic oligomer, T2DM, type 2 diabetes mellitus
Introduction
Type 2 diabetes mellitus (T2DM) is caused by the reduction of β-cell mass and accumulation of islet amyloid polypeptide (IAPP, or amylin) in the pancreatic islets, leading to insulin resistance.1 Islet amyloid deposition could affect and occur in up to 80% of diabetic patients.2 IAPP, a 4 kDa peptide hormone composed of 37 amino acids (Figure 1), is synthesized and cosecreted with insulin from pancreatic β-cells. As a glucomodulatory hormone, IAPP is responsible for slowing down gastric emptying and regulating glucose levels. It inhibits the effects of insulin as well as arginine-stimulated glucagon release by α-cells.3,4 In addition, IAPP is responsible for other physiological functions such as sleep and appetite regulation via the gut–brain axis, and it functions as a growth factor to maintain β-cell mass.5 The incidence of T2DM is considered to increase with age because of the reduced proliferative capacity and increased apoptosis rate of β-cells, which can also lead to higher rates of IAPP aggregation.6 The IAPP monomer appears to show normal biological activity in healthy β-cells. Moreover, oligomers can regularly form and degrade in a normal cell system. However, unlike normal monomers and oligomers, toxic oligomers can cause membrane-perforating toxicity, which is a decisive factor that is indicative of the cell cytotoxicity in diabetes models. IAPP oligomers can be generated via the amyloid fibril off-pathway in the fibril assembly process. However, the exact structure and role of toxic IAPP oligomers have been difficult to establish thus far. The toxic amyloid-β oligomer has a cross-β structure,7 and it is considered that the IAPP oligomer that is ultimately cytotoxic to pancreatic β-cells would share similar structural elements.8 On the bright side, rifampicin has been reported to prevent the formation of human-IAPP (h-IAPP) fibrils. However, rifampicin did not seem to be able to control the production of toxic h-IAPP oligomers from the off-pathway and could therefore not prevent its cytotoxic effects.9 Currently, the causative factors of the conversion of soluble IAPP into insoluble fibrils are unknown. However, it is known that these factors lead to the destruction of β-cell function to the point where they cannot properly respond to insulin.2
 | Figure 1 Schematic diagram of the primary sequence of IAPP. |
The final stage of cell disruption involves membrane destabilization and disruption. Hence, it is important to understand how amyloidogenic peptides cause cell membrane disruption. The “carpet” and “barrel-stave” models are used to represent the mechanisms of cell membrane disruption. In the carpet model, monomers interrupt the membrane through nonspecific binding, by dissolving into the membrane in a detergent-like manner and exceeding the concentration threshold value of the membrane.10 In the barrel-stave model, after assembling on the membrane surface, peptides insert themselves into the membrane, forming channels or pores.11 Thus, membrane disruption from barrier deformation is observed in these two models. In particular, both models reveal how high cytotoxicity can result from cell disruption induced by a toxic IAPP oligomer. Further in-depth studies with these models are needed to characterize the pore generation induced by h-IAPP, identify the cytotoxic mechanisms, screen for the causative factors that initiate toxic oligomer formation, and ultimately identify how these causative factors can be inhibited.
Factors that cause oligomer aggregation
Environmental condition of the cell
Amyloidogenic protein aggregation can be induced by several factors within the cellular environment. IAPP or amylin, as a potential amyloidogenic protein, was shown to interact with the lipid membrane in one of the pathogenic cytometabolic pathways.12 IAPP fibrillization was found to play a role in the cytotoxicity in vitro by disrupting the negatively charged phospholipid membrane.13 In the same context, it was suggested that a high-fat diet could increase the lipid level, thereby increasing the possibility of oligomer penetration or membrane disruption through increased oligomer–lipid binding. As another possibility, the hydrophobic region of IAPP could be exposed in protein misfolding, which would implicate it in the fibrillation process.14 Changes in pH were reported to affect the conformation and aggregation patterns of IAPP, where the alkaline pH of 8.8 was suggested to be more favorable for aggregate formation than the acidic condition of pH 4.0.15,16 Likewise, chemical modification, salt concentrations, natural ligands, racemization, isomerization, deamidation, oxidation, lipid oxidation, and glycation are factors that could affect the stability of IAPP.2
IAPP sequence
Genetic mutations of IAPP
According to a previous study, a naturally occurring missense mutation (S20G) of the human IAPP gene is associated with early onset or more severe types of T2DM.17 The S20G mutation results in increased hydrophobicity and amyloidogenic characteristics of IAPP, which could increase its fibrillogenic potential.18 In addition, S20G IAPP showed a nearly twofold increased rate of the formation of amyloid fibrils, resulting in more than threefold greater aggregation and consequently higher cytotoxicity than the wild-type protein.18 F15, an aromatic residue in IAPP that conserves its hydrophobicity, has been suggested to play a significant role in the amyloid biosynthesis pathway.19 In an in silico study, an F15L mutation generated from a single-point mutation, which altered the α-helix and β-sheet propensities of the protein, resulted in rapid amyloid formation.20 In another in silico study, the Y37L and F23L IAPP mutations resulted in decreased rates of amyloid fibril formation.20 The replacement of tyrosine (Y) with leucine (L) resulted in greater flexibility of the C-terminus, with loss of the steric zipper interactions, and the F23L mutation, in which phenylalanine (F) was replaced with leucine (L), showed slower amyloid formation.21 Overall, the rate of aggregate formation was reduced significantly in these two mutations compared to other mutants and the wild-type protein.21 Interestingly, the single point mutants G24P and I26P also showed potential for inhibiting amyloid aggregation.22–24 A list of the genetic mutations of IAPP is shown in Figure 2. Mutations by displacement of one amino acid residue could greatly affect the rate and property of amyloid formation. These aforementioned studies described suggest a close correlation for balancing the reversal or the rate of fibril formation in reducing or increasing cytotoxicity.
 | Figure 2 List of genetic mutations of IAPP determined from in silico studies, and naturally occurring (eg, S20G) mutations. |
Comparison of IAPP sequences among species and establishment of transgenic rodent models
IAPP residues at the N-terminus and C-terminus are conserved in mammals, whereas the amyloidogenic core region is species specific (Figure 3). Sequence homology has been found between primate and human IAPPs, and these peptides form islet amyloids that lead to the development of T2DM. In contrast, the presence of three proline residues in rat and mouse IAPPs renders the protein water soluble, thereby giving it nonamyloidogenic properties by providing a water-soluble environment; as a consequence, T2DM does not typically occur in rodents.25,26 Since the cytotoxicity of IAPP seems to be dependent on its propensity for oligomer formation, the prevention of fibril formation by the proline residues of IAPP 20–29 in the rat and mouse is very likely to be the cause of their reduced IAPP cytotoxicity, which is hardly detected in these species.2
 | Figure 3 Alignment of IAPP amino acid sequences from different species. |
Since pancreatic β-cell apoptosis and the induction of diabetes have been confirmed in h-IAPP transgenic rat and mouse models, it is a viable hypothesis that amyloids are associated with the induction and progression of diabetes.27 Indeed, an h-IAPP transgenic model formed toxic IAPP oligomers that eventually generated endoplasmic reticulum stress-induced apoptosis and T2DM characteristics such as hyperglycemia, impaired insulin secretion, insulin resistance, and hyperglucagonemia.28,29
IAPP processing
IAPP is encoded by an 89-residue coding sequence that produces the 67-amino acid pro-IAPP residue. Mature IAPP is then generated by the following process. First, pro-IAPP is formed after cleavage of the 22-amino acid signal peptide before it undergoes proteolysis and posttranslational modification. Sixteen amino acids at the C-terminus are removed by proprotein convertase 1/3 (PC1/3), followed by truncation of eleven amino acids at the N-terminus by proprotein convertase 2 (PC2).30 Then, the C-terminus carboxypeptidase E removes the terminal lysine and arginine residues,31 and peptidylglycine α-amidating monooxygenase (PAM) adds an amine group to the C-terminus. Finally, a disulfide bond is generated between the cysteine 2 and cysteine 7 residues to produce the mature IAPP protein made up of 37 amino acids (IAPP sequence: KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY).32 Both an intact intramolecular disulfide bond and C-terminal amidation are required for IAPP to have normal biological activity. Therefore, the factors that prevent the formation of the disulfide bond and/or cause deamidation, as well as the absence of PC2, contribute to the aggregation of IAPP.
Absence of PC2
Impaired processing of pro-IAPP can affect amyloid formation in T2DM. The first enzyme in the pro-IAPP processing pathway is PC1/3, which plays a crucial role in removing the amino acids of the C-terminus. In the absence of PC1/3, PC2 takes over at both the N-terminal and C-terminal cleavage sites as part of normal posttranslational modification.33 In contrast, because PC2 usually only functions after PC1/3 in the pro-IAPP processing pathway, its absence creates a block at the level of the pro-IAPP convertase intermediate (NH2-terminally extended pro-IAPP). This intermediate acts as a critical initiating factor to induce intracellular and extracellular amyloid formation, and finally induces β-cell death.34,35
Deamidation
Deamidation is an important factor in amyloid formation and affects protein structure, folding, stability, and aggregation.36,37 Deamidation of the side chain at Asn and Gln is a spontaneous reaction and a nonenzymatic posttranslational modification.38 There are six Asn residues and one Gln residue in the IAPP sequence that can be subject to deamidation effects. In addition, for IAPP to properly perform its biological function, it should have some form of C-terminal amidation. Thus, once the C-terminal amide group is exposed to deamidation, the protein structure will be affected. Deamidation may influence the protein structure and stability by adding a negative charge, and thus change the amyloid formation kinetics such that unmodified IAPP (deamidated form) accelerates amyloid formation, creating the seeds for aggregation.39 In other words, if an impurity is produced by deamidation, it induces amyloid formation, thereby affecting the purified peptide’s secondary structure and aggregation behavior.40
Reduction of disulfide bond formation
In one study, the intramolecular disulfide bond of C2–C7 was shown to be the critical site for the formation of IAPP fibrils; however, the role of the intrinsic disulfide bond in the IAPP monomer structure as well as the specific mechanism of aggregation remain unclear.41 The N-loop that is generated by the disulfide bond (C2–C7) of IAPP is highly conserved in the rigid structure.42,43 Although the N-loop is not involved in direct molecular interactions, it nevertheless affects the IAPP aggregation kinetics indirectly, since N-loop removal was found to change the mass/length distribution and kinetics of the h-IAPP fiber.44,45 The absence of the disulfide bond decreases the extent of the helix at the N-terminal region, but favors random coiling and β-sheet formation, which can affect the protein kinetics such as reducing the rate of IAPP aggregation by secondary nucleation.41 In fact, early oligomerization is connected to helix formation. The S–S bridge, which stabilizes the N-terminal helix of h-IAPP, collapses the protein into an amorphous aggregate with less β-sheets. These factors were shown to facilitate the initialization of h-IAPP aggregation, encoded at the monomeric level.41,46,47 Likewise, the disulfide bond determines the morphology of the fibril (eg, stabilizing the amyloid fibril in the folded state), and also plays a role in limitation by topologically restraining the polypeptide during amyloid fibril arrangement.48 Therefore, the disulfide bond may ultimately reduce the toxicity of the amyloid fibril.49
Copper(II) ion effect
Copper ions have been associated with many diseases.50 Furthermore, calcium and zinc ions were shown to be involved in maintaining the native structure of IAPP, by inhibiting amyloid aggregation.51,52 Although the precise impact of copper ions on IAPP is still a matter of debate,53 copper metabolism is thought to be associated with the pathological mechanism of amyloidosis in diabetes.54 In one previous study, copper inhibited IAPP fibrillization,52 whereas in another study, copper contributed to h-IAPP aggregation and cytotoxic oligomer formation.55 Copper can possibly interact with the C2, C7, H18, and Y37 regions in the IAPP sequence.55 These copper–IAPP complexes have metallopeptide complex structures with low aggregation potential56 and may instead produce granular oligomers, which are the main cause of increased copper-mediated h-IAPP cytotoxicity. Granular oligomers are expected to show the characteristics of toxic oligomers, inducing membrane destabilization and, ultimately, cell apoptosis.55,57,58 In addition, copper-promoted reactive oxygen species (ROS) generation, such as H2O2, and mitochondrial disruption affect the degeneration of islet cells, leading to the production of caspase-3 and poly (ADP-ribose) polymerase (PARP), which in turn promote apoptosis.59–61 Furthermore, noncomplexed copper ions (ie, free copper ions) also show toxicity and can easily turn into reactive copper ions that generate cell-damaging ROS.56 Therefore, treatment with a copper-chelating agent has a beneficial effect against the pathogenesis of T2DM and diabetes complications, by lowering the copper levels, and hence the ROS levels.62
Heparin
The heparin-binding property of amyloid β-protein is dependent on the state of aggregation of this amyloidogenic protein since heparin does not interact with nonfibrillar forms.63 A similar heparin study was conducted with IAPP. Heparin enhanced IAPP fibrillization, and the binding property was found to be dependent on the length of the heparin polysaccharide fragment and the aggregation state. The negatively charged heparin helix essentially binds with the positively charged N-terminal cross β-sheet of the IAPP fibril.64 Short heparin fragments of 2–8 saccharides showed a better protective effect against cell cytotoxicity than fragments of 20 saccharide or longer, which exhibited no protective function.65
Conclusion
A wide range of diabetes-specific biomarkers has been identified. However, it appears that it is the unusual protein properties induced by such factors and the effect of protein aggregation on β-cell apoptosis that ultimately induce the development of diabetes. In particular, IAPP oligomers generated from IAPP monomers lead to β-cell-specific toxicity, ultimately causing diabetes. This review presented the known factors that appear to trigger IAPP aggregation. Figure 4 demonstrates how pro-IAPP decomposes into the IAPP monomer through the posttranslational process, and how these IAPP monomers can create and organize into toxic IAPP oligomers in β-cells. As illustrated in Figure 4, the mechanistic factors that influence IAPP aggregation include the absence of PC2 enzyme, deamidation, reduction of disulfide bonds, environmental factors in the cell, genetic mutations, copper metal ions, and heparin. Other factors can also affect amyloid aggregation. Further research on these factors could ultimately aid in the inhibition of amyloid aggregation, which would be a breakthrough for the treatment of diabetes. To date, lack of knowledge about the toxic IAPP oligomer, the ultimate causative factors for reduction of β-cell mass, and the cause of cell death has hindered the ability in treating diabetes and limiting its progression, even in the prediabetic stage, and for selecting or developing novel therapeutic agents. Therefore, determining the mechanism by which the formation of toxic IAPP oligomers leads to T2DM should be an important research goal with the aim of elucidating the pathogenesis of this disorder and establishing new treatments.
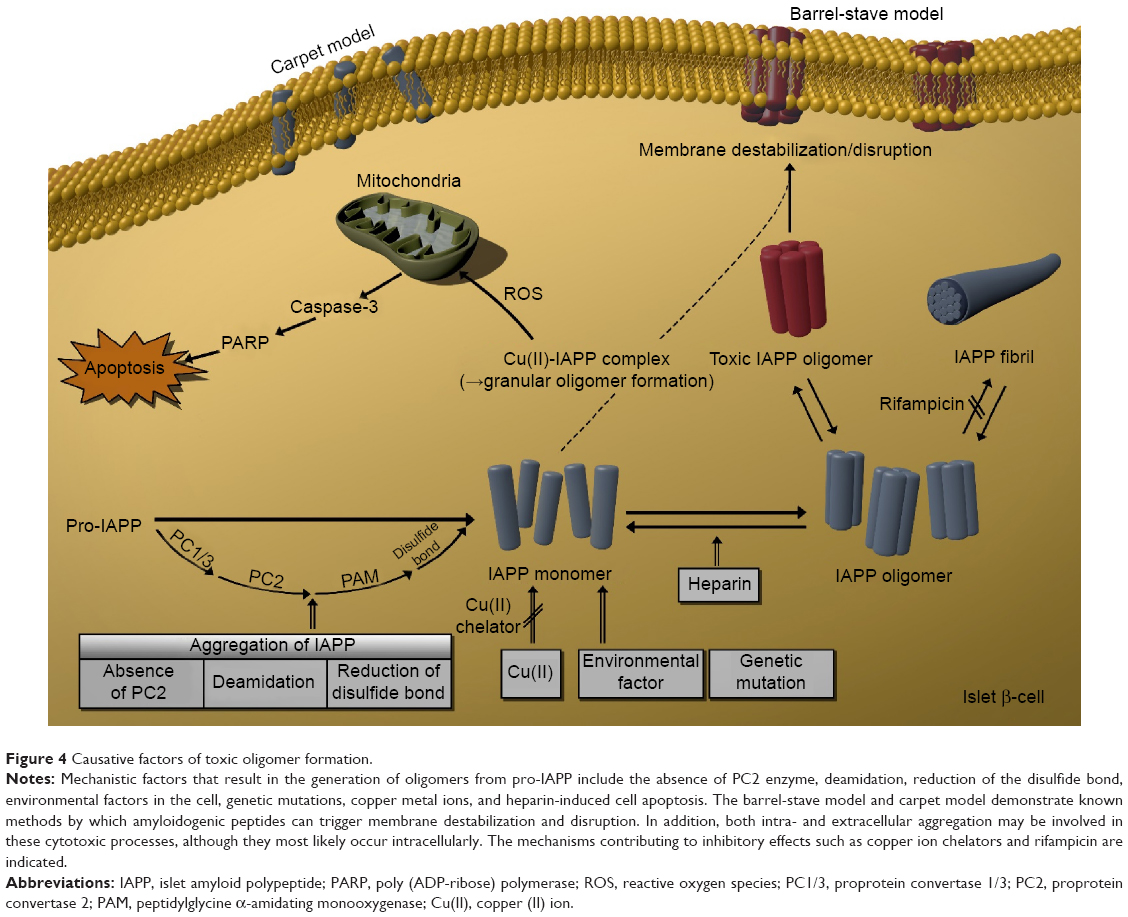 | Figure 4 Causative factors of toxic oligomer formation. |
Acknowledgments
This research was supported by the Industrial Core Technology Development Program (grant number 10049051, Development of bench-top automatic immunoassay system with intelligent quality control features for screening cancer or chronic diseases in local clinical setting) funded by the Ministry of Trade, Industry and Energy (MI, South Korea) and and the Korea Health Technology R&D Project (HI14C3331) through the Korea Health Industry Development Institute (KHIDI), Korea Ministry of Health & Welfare.
Disclosure
The authors report no conflicts of interest in this work.
References
Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz P. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Pathol Immunopathol Res. 1985;4(2):110–125. | ||
Clark A, Nilsson M. Islet amyloid: a complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia. 2004;47(2):157–169. | ||
Nguyen PT, Andraka N, De Carufel CA, Bourgault S. Mechanistic contributions of biological cofactors in islet amyloid polypeptide amyloidogenesis. J Diabetes Res. 2015. | ||
Young A, Gedulin B, Vine W, Percy A, Rink T. Gastric emptying is accelerated in diabetic BB rats and is slowed by subcutaneous injections of amylin. Diabetologia. 1995;38(6):642–648. | ||
Wookey PJ, Lutz TA, Andrikopoulos S. Amylin in the periphery II: an updated mini-review. Sci World J. 2006;6:1642–1655. | ||
Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging (Albany NY). 2011;3(6):565. | ||
Stroud JC, Liu C, Teng PK, Eisenberg D. Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc Natl Acad Sci. 2012;109(20):7717–7722. | ||
Wiltzius JJ, Sievers SA, Sawaya MR, et al. Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin). Protein Sci. 2008;17(9):1467–1474. | ||
Meier JJ, Kayed R, Lin C-Y, et al. Inhibition of human IAPP fibril formation does not prevent β-cell death: evidence for distinct actions of oligomers and fibrils of human IAPP. Am J Physiol Endocrinol Metab. 2006;291(6):E1317–E1324. | ||
Pouny Y, Rapaport D, Mor A, Nicolas P, Shai Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogs with phospholipid membranes. Biochemistry. 1992;31(49):12416–12423. | ||
Ojcius DM, Young JDE. Cytolytic pore-forming proteins and peptides: is there a common structural motif? Trends Biochem Sci. 1991;16:225–229. | ||
Jayasinghe SA, Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44(36):12113–12119. | ||
Domanov YA, Kinnunen PK. Islet amyloid polypeptide forms rigid lipid–protein amyloid fibrils on supported phospholipid bilayers. J Mol Biol. 2008;376(1):42–54. | ||
Ahmad E, Ahmad A, Singh S, Arshad M, Khan AH, Khan RH. A mechanistic approach for islet amyloid polypeptide aggregation to develop anti-amyloidogenic agents for type-2 diabetes. Biochimie. 2011;93(5):793–805. | ||
Abedini A, Raleigh DP. The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry. 2005;44(49):16284–16291. | ||
Jha S, Snell JM, Sheftic SR, et al. pH dependence of amylin fibrillization. Biochemistry. 2014;53(2):300–310. | ||
Sakagashira S, Sanke T, Hanabusa T, et al. Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes. 1996;45(9):1279–1281. | ||
Sakagashira S, Hiddinga HJ, Tateishi K, et al. S20G mutant amylin exhibits increased in vitro amyloidogenicity and increased intracellular cytotoxicity compared to wild-type amylin. Am J Pathol. 2000;157(6):2101–2109. | ||
Marek P, Abedini A, Song B, et al. Aromatic interactions are not required for amyloid fibril formation by islet amyloid polypeptide but do influence the rate of fibril formation and fibril morphology. Biochemistry. 2007;46(11):3255–3261. | ||
Tu L-H, Raleigh DP. Role of aromatic interactions in amyloid formation by islet amyloid polypeptide. Biochemistry. 2013;52(2):333–342. | ||
Bernhardt NA, Berhanu WM, Hansmann UH. Mutations and seeding of amylin fibril-like oligomers. J Phys Chem B. 2013;117(50):16076–16085. | ||
Abedini A, Meng F, Raleigh DP. A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrillization inhibitor. J Am Chem Soc. 2007;129(37):11300–11301. | ||
Meng F, Raleigh DP, Abedini A. Combination of kinetically selected inhibitors in trans leads to highly effective inhibition of amyloid formation. J Am Chem Soc. 2010;132(41):14340–14342. | ||
Miller C, Zerze GlH, Mittal J. Molecular simulations indicate marked differences in the structure of amylin mutants, correlated with known aggregation propensity. J Phys Chem B. 2013;117(50):16066–16075. | ||
Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev. 2008;29(3):303–316. | ||
Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci. 1990;87(13):5036–5040. | ||
Butler AE, Jang J, Gurlo T, Carty MD, Soeller WC, Butler PC. Diabetes due to a progressive defect in β-cell mass in rats transgenic for human islet amyloid polypeptide (HIP rat) a new model for type 2 diabetes. Diabetes. 2004;53(6):1509–1516. | ||
Matveyenko AV, Butler PC. β-Cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type 2 diabetes. Diabetes. 2006;55(7):2106–2114. | ||
Huang C-J, Lin C-Y, Haataja L, et al. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress–mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56(8):2016–2027. | ||
Sanke T, Bell G, Sample C, Rubenstein A, Steiner D. An islet amyloid peptide is derived from an 89-amino acid precursor by proteolytic processing. J Biol Chem. 1988;263(33):17243–17246. | ||
Marzban L, Soukhatcheva G, Verchere CB. Role of carboxypeptidase E in processing of pro-islet amyloid polypeptide in β-cells. Endocrinology. 2005;146(4):1808–1817. | ||
Roberts A, Leighton B, Todd J, et al. Molecular and functional characterization of amylin, a peptide associated with type 2 diabetes mellitus. Proc Natl Acad Sci. 1989;86(24):9662–9666. | ||
Marzban L, Trigo-Gonzalez G, Zhu X, et al. Role of β-cell prohormone convertase (PC) 1/3 in processing of pro-islet amyloid polypeptide. Diabetes. 2004;53(1):141–148. | ||
Marzban L, Rhodes CJ, Steiner DF, Haataja L, Halban PA, Verchere CB. Impaired NH2-terminal processing of human proislet amyloid polypeptide by the prohormone convertase PC2 leads to amyloid formation and cell death. Diabetes. 2006;55(8):2192–2201. | ||
Wang J, Xu J, Finnerty J, Furuta M, Steiner DF, Verchere CB. The prohormone convertase enzyme 2 (PC2) is essential for processing pro-islet amyloid polypeptide at the NH2-terminal cleavage site. Diabetes. 2001;50(3):534–539. | ||
Takata T, Oxford JT, Demeler B, Lampi KJ. Deamidation destabilizes and triggers aggregation of a lens protein, βA3-crystallin. Protein Sci. 2008;17(9):1565–1575. | ||
Nilsson MR, Dobson CM. Chemical modification of insulin in amyloid fibrils. Protein Sci. 2003;12(11):2637–2641. | ||
Bischoff R, Kolbe HV. Deamidation of asparagine and glutamine residues in proteins and peptides: structural determinants and analytical methodology. J Chromatogr B Biomed Sci Appl. 1994;662(2):261–278. | ||
Dunkelberger EB, Buchanan LE, Marek P, Cao P, Raleigh DP, Zanni MT. Deamidation accelerates amyloid formation and alters amylin fiber structure. J Am Chem Soc. 2012;134(30):12658–12667. | ||
Nilsson MR, Driscoll M, Raleigh DP. Low levels of asparagine deamidation can have a dramatic effect on aggregation of amyloidogenic peptides: implications for the study of amyloid formation. Protein Sci. 2002;11(2):342–349. | ||
Laghaei R, Mousseau N, Wei G. Effect of the disulfide bond on the monomeric structure of human amylin studied by combined Hamiltonian and temperature replica exchange molecular dynamics simulations. J Phys Chem B. 2010;114(20):7071–7077. | ||
Vaiana SM, Best RB, Yau W-M, Eaton WA, Hofrichter J. Evidence for a partially structured state of the amylin monomer. Biophys J. 2009;97(11):2948–2957. | ||
Cope SM, Shinde S, Best RB, Giovanna G, Vaiana SM. Cyclic N terminal fragment of amylin forms non amyloid fibers: implications for intra-and inter-molecular interactions in amylin. Biophys J. 2013;104(2):389a–390a. | ||
Goldsbury C, Goldie K, Pellaud J, et al. Amyloid fibril formation from full-length and fragments of amylin. J Struct Biol. 2000;130(2):352–362. | ||
Yonemoto IT, Kroon GJ, Dyson HJ, Balch WE, Kelly JW. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47(37):9900–9910. | ||
Koo BW, Miranker AD. Contribution of the intrinsic disulfide to the assembly mechanism of islet amyloid. Protein Sci. 2005;14(1):231–239. | ||
Abedini A, Raleigh DP. A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys Biol. 2009;6(1):015005. | ||
Mossuto MF, Bolognesi B, Guixer B, et al. Disulfide bonds reduce the toxicity of the amyloid fibrils formed by an extracellular protein. Angew Chem Int Ed. 2011;50(31):7048–7051. | ||
Monsellier E, Ramazzotti M, Taddei N, Chiti F. Aggregation propensity of the human proteome. PLoS Comput Biol. 2008;4(10):e1000199. | ||
Uriu-Adams JY, Keen CL. Copper, oxidative stress, and human health. Mol Aspects Med. 2005;26(4–5):268–298. | ||
Brender JR, Hartman K, Nanga RPR, et al. Role of zinc in human islet amyloid polypeptide aggregation. J Am Chem Soc. 2010;132(26):8973–8983. | ||
Ward B, Walker K, Exley C. Copper (II) inhibits the formation of amylin amyloid in vitro. J Inorg Biochem. 2008;102(2):371–375. | ||
Sinopoli A, Magrì A, Milardi D, et al. The role of copper (II) in the aggregation of human amylin. Metallomics. 2014;6(10):1841–1852. | ||
Drew SC, Masters CL, Barnham KJ. Alanine-2 carbonyl is an oxygen ligand in Cu2+ coordination of Alzheimer’s disease amyloid-β peptide – relevance to N-terminally truncated forms. J Am Chem Soc. 2009;131(25):8760–8761. | ||
Yu Y-P, Lei P, Hu J, Wu W-H, Zhao Y-F, Li Y-M. Copper-induced cytotoxicity: reactive oxygen species or islet amyloid polypeptide oligomer formation. Chem Commun. 2010;46(37):6909–6911. | ||
Lee EC, Ha E, Singh S, et al. Copper (II)–human amylin complex protects pancreatic cells from amylin toxicity. Phys Chem Chem Phys. 2013;15(30):12558–12571. | ||
Mirzabekov TA, Lin M-C, Kagan BL. Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem. 1996;271(4):1988–1992. | ||
Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48(3):491–498. | ||
Ma L, Li X, Wang Y, Zheng W, Chen T. Cu (II) inhibits hIAPP fibrillation and promotes hIAPP-induced beta cell apoptosis through induction of ROS-mediated mitochondrial dysfunction. J Inorg Biochem. 2014;140:143–152. | ||
Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757(5):509–517. | ||
Masad A, Hayes L, Tabner BJ, et al. Copper-mediated formation of hydrogen peroxide from the amylin peptide: a novel mechanism for degeneration of islet cells in type-2 diabetes mellitus? FEBS Lett. 2007;581(18):3489–3493. | ||
Tanaka A, Kaneto H, Miyatsuka T, et al. Role of copper ion in the pathogenesis of type 2 diabetes. Endocr J. 2009;56(5):699–706. | ||
Watson DJ, Lander AD, Selkoe DJ. Heparin-binding properties of the amyloidogenic peptides Aβ and amylin dependence on aggregation state and inhibition by congo red. J Biol Chem. 1997;272(50):31617–31624. | ||
Jha S, Patil SM, Gibson J, Nelson CE, Alder NN, Alexandrescu AT. Mechanism of amylin fibrillization enhancement by heparin. J Biol Chem. 2011;286(26):22894–22904. | ||
Alexandrescu AT, Jha S, Patil SM, Gibson J, Alder NN, Nelson CE. Effects of heparin on amylin fibrillization. Biophys J. 2012;102(3):243a. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.