Back to Journals » Journal of Pain Research » Volume 18
Causal Relationships Between Neuropathic Pain and Alzheimer’s Disease: A Multi-Omics Mendelian Randomization Study with Exploratory Evidence of a Potential Protective Role of Diabetic Neuropathy
Authors Li X, Zhang C, Liu D, Shang H, Wang K, Lin X, Zheng J ![]()
Received 17 July 2025
Accepted for publication 26 November 2025
Published 6 December 2025 Volume 2025:18 Pages 6527—6544
DOI https://doi.org/10.2147/JPR.S554433
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor King Hei Stanley Lam
Xiao Li,1,2,* Chi Zhang,1,2,* Di Liu,2 Hui Shang,1,3 Kejia Wang,2 Xianjian Lin,2 Junyi Zheng2
1The First Clinical Medical School, Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China; 2Department of Anesthesiology, the First Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China; 3Shenzhen Hospital (Futian), Guangzhou University of Chinese Medicine, Shenzhen, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Junyi Zheng, Department of Anesthesiology, the First Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong, People’s Republic of China, Email [email protected]
Background: Neuropathic pain (NP) frequently co-occurs with Alzheimer’s disease (AD), yet the causal relationship and underlying molecular mechanisms between the two remain unclear, necessitating further investigation to elucidate their intrinsic connection.
Methods: This study employed a bidirectional two-sample Mendelian randomisation (MR) approach to systematically analyse the association between six NP subtypes and AD. Concurrently, functional annotation and transcriptomic analysis were conducted using the GTEx v10 and GEO GSE156184 databases to explore potential molecular mechanisms.
Results: The study revealed a significant inverse causal effect of diabetic neuropathy (DN) on AD risk (OR=0.86, 95% CI: 0.77~0.95, PIVW=0.0043), with sensitivity analyses confirming the robustness of this finding. Further analysis indicated that DN-associated SNPs regulate four tissue-specific genes including FAM200A and GPC2. These genes exhibit differential expression in the DN transcriptome and are significantly enriched in key pathways such as mitochondrial function and autophagy.
Conclusion: This study provides the first evidence that DN may exert a protective effect against AD by regulating the aforementioned tissue-specific genes and associated pathways. This finding challenges the conventional understanding that chronic pain exacerbates AD and offers novel potential targets for developing therapeutic strategies. However, due to population limitations in the study, further experimental validation remains necessary.
Keywords: neuropathic pain, Alzheimer disease, Mendelian randomization analysis, diabetic neuropathies, transcriptomics
Background
Neuropathic pain (NP) is common chronic condition characterized by persistent and irreversible pain resulting from pathological changes or diseases affecting the peripheral or central somatosensory nervous system.1–3 Based on the anatomical location of the lesion, it is generally classified into peripheral neuropathic pain, involving the peripheral sensory nervous system, and central NP, affecting the central somatosensory system.3 The etiology of NP is heterogeneous and includes trauma (eg, spinal cord injury, carpal tunnel syndrome), metabolic disorders (eg, diabetic peripheral neuropathy), viral infections (eg, postherpetic neuralgia, HIV-associated neuropathy), autoimmune diseases (eg, multiple sclerosis), and neurotoxic chemotherapeutic agents.1,4 Most patients experience either continuous or intermittent spontaneous pain. The underlying mechanisms are complex and remain incompletely elucidated, and there are currently no definitive diagnostic criteria, standardized methods, or specific biomarkers for the objective assessment of NP.4 Epidemiological data indicate that prevalence of neuropathic pain in general population is approximately 7%~8%, accounting for 20%~25% of all chronic pain cases.1,4
Diabetic neuropathy (DN), a metabolic subtype of NP, is one of the most common complications of diabetes. It is estimated that the prevalence of DN among diabetic patients worldwide is about 19.2%~40%. Its pathological features include peripheral nerve axonal degeneration, demyelination, and impaired nerve conduction velocity.5 Compared to other NP subtypes (such as traumatic or infectious neuropathies), the pathogenesis of DN is closely associated with metabolic dysregulation factors. Factors such as hyperglycemia-induced oxidative stress, accumulation of advanced glycation end products, and activation of the renin-angiotensin system contribute to its development.6–8These unique pathological characteristics suggest that DN may share a distinct connection with Alzheimer’s disease (AD) that differs from other NP subtypes.
AD is a progressive neurodegenerative disorder characterized by the gradual loss of memory and cognitive function.9 AD is the leading cause of global dementia. As of 2024, approximately 55 million people worldwide are affected by AD, with this number projected to rise to 139 million by 2050 (https://www.alzint.org/about/dementia-facts-figures/). AD’s core pathogenesis involves extracellular β-amyloid (Aβ) plaque deposition, intracellular hyperphosphorylated tau neurofibrillary tangles, and neuroinflammation-induced neuronal loss. Clinical diagnosis follows the 2011 National Institute on Aging-Alzheimer’s Association (NIA-AA) criteria, integrating clinical symptoms (progressive memory decline and executive dysfunction), neuroimaging findings (hippocampal atrophy and positive Aβ positron emission tomography), and cerebrospinal fluid biomarkers (reduced Aβ42/Aβ40 ratio and elevated phosphorylated tau).10 In patients with AD, the interplay between pain and cognitive impairment is particularly prominent and is frequently accompanied by comorbid chronic pain.11,12 Non-cancer chronic pain conditions are significantly positively associated with AD and related dementias.13 In recent years, research on the association between NP and AD has increasingly demonstrated subtype specificity. For example, a prospective cohort study (n = 120,000) found that non-metabolic NP subtypes (such as idiopathic neuropathy) were associated with a 1.18-fold increased risk of AD (95% confidence interval: 1.05~1.32), whereas DN showed a potential inverse association with AD.14 These inconsistent findings underscore the need for dedicated research into the causal relationship between DN and AD, rather than broadly examining the association between NP and AD.
In AD, the hippocampus is among the earliest brain regions to undergo pathological and functional alteration,15 as a key component of the limbic system, the hippocampus plays a critical role in both cognitive processing and pain modulation.16 Certain regions affected by neurodegeneration in Alzheimer’s disease anatomically overlap with brain networks involved in pain processing, particularly those selectively impacting key areas of the medial pain pathway.17 Studies have shown that microglial activation is strongly associated with the onset and progression of NP, as peripheral nerve injury can markedly activate microglia within the central nervous system, thereby inducing synaptic reorganization and functional changes that lead to heightened pain sensitivity and disturbances in the emotional and cognitive processes related to pain.18 The accumulation of β-amyloid (Aβ) plaques and hyperphosphorylated tau aggregates—central neuropathological features of AD—has been shown to activate microglia, which in turn trigger pro-inflammatory responses and contribute to both tau pathology and its subsequent propagation within the brain.19–22 Collectively, the interaction between neurons and microglia is considered critical for the development of chronic pain and neuroinflammation.23 Although a bidirectional relationship between NP and AD has been proposed, the underlying mechanisms and causal links remain unclear.
MR is a genetic epidemiological method used to infer causal relationships between modifiable risk factors (exposures) and disease (outcomes) based on genetic variants.24 By leveraging the random allocation of alleles during meiosis, MR ensures that genotypes are independent of disease status and environmental confounders, thereby minimizing bias from confounding and reverse causation.25 To validate MR study findings and delve deeper into molecular mechanisms, this research integrated three key approaches: First, expression quantitative trait locus (eQTL) analysis identifies single nucleotide polymorphisms (SNPs) regulating gene expression. Its core principle establishes links between genetic variation and target gene expression levels, thereby revealing potential “genotype - expression - phenotype” causal chain;26 second, the Genotype-Tissue Expressions (GTEx v10) project data, a large-scale public database covering genotype and gene expression data from 50 human tissues (eg, brain, kidney) of 1,738 donors, providing reference for annotating tissue-specific eQTLs;27 third, transcriptome analysis identifies differentially expressed genes (DEGs) by comparing gene expression differences between case and control groups, reflecting functional alterations under pathological conditions.28 However, the potential causal relationship between NP and AD has not yet been systematically investigated using MR. Therefore, in our study, we employed bidirectional MR to explore the causal associations between six common subtypes of NP and AD. Additionally, eQTL analysis and transcriptomic data were integrated to validate the MR findings. These results may enhance our understanding of the shared pathophysiological mechanisms between the two conditions and provide insights into novel therapeutic strategies.
Methods
MR Analysis
We applied two-sample MR approach to reflect potential causal relationship linking NP and AD. Within the MR framework, six subtypes of NP were treated as exposures, with AD as outcome, and SNPs were utilized as instrumental variables (IVs). Validity of two-sample MR is grounded on three core assumptions: (I) the relevance assumption, which requires the IVs to have a strong association with the exposure (NP); (II) the independence assumption, stating that the IVs should be unrelated to confounding variables; and (III) the exclusion restriction assumption, which holds that IVs influence outcome (AD) solely through their impact on exposure, without alternative causal pathways (Figure 1).
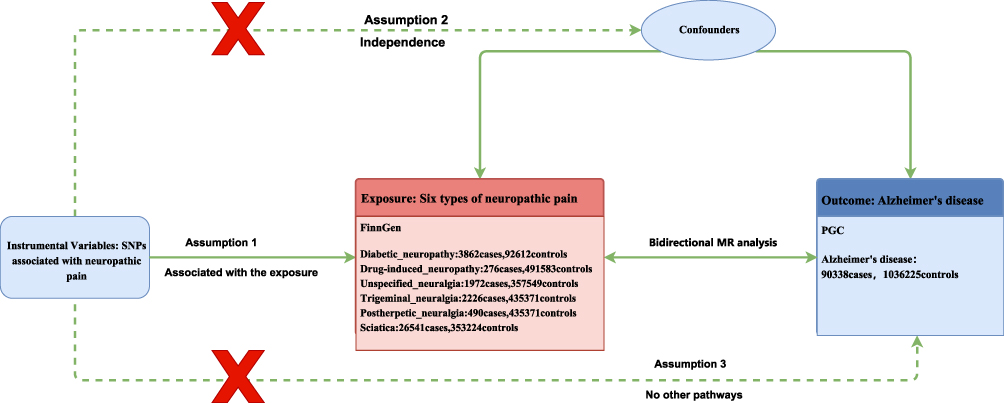 |
Figure 1 Bidirectional Mendelian Randomization Framework: Neuropathic Pain and Alzheimer’s Disease. |
We acquired genome-wide association (GWAS) summary data for six subtypes of NP, including diabetic neuropathy (DN), drug-induced neuropathy, unspecified neuralgia, trigeminal neuralgia, postherpetic neuralgia, and sciatica, from the FinnGen R12 database (https://r12.finngen.fi/), which primarily consists of individuals of European ancestry.29 SNP data associated with AD risk were derived from large-scale GWAS that applied stringent criteria for the definition and diagnosis of AD.30 GWAS primarily employ the 2011 NIA-AA diagnostic criteria, which integrate clinical symptoms (progressive memory decline, executive dysfunction), neuroimaging findings (hippocampal atrophy, positive Aβ positron emission tomography scans), and cerebrospinal fluid biomarkers (reduced Aβ42/Aβ40 ratio, elevated phosphorylated tau protein). This multidimensional approach ensures the accuracy of AD case diagnoses.10 The original GWAS dataset was obtained from the Psychiatric Genomics Consortium (PGC; https://pgc.unc.edu/for-researchers/download-results/), comprising total sample size of 1,126,563 individuals, including 90,338 cases and 1,036,255 controls, all of European ancestry (Table S1). The GWAS dataset for DN included 3,862 cases and 92,612 controls.
In the MR analysis, IVs were used to reflect potential causal relationships about exposures and outcomes. SNPs associated with DN and sciatica were initially selected as IVs using genome-wide threshold of P < 5 × 10−8. For remaining four types of NP, namely drug-induced neuropathy, unspecified neuralgia, trigeminal neuralgia, and postherpetic neuralgia, a more lenient significance threshold of P < 5 × 10−6 was applied because the number of genome-wide significant SNPs was limited. To ensure independence among SNPs, linkage disequilibrium (LD) pruning was performed with r2 <0.001 and a linkage disequilibrium distance >10,000 kb. Besides, we calculated the F statistic for each SNP using the formula F=β2/SE2—where β (regression coefficient) and SE (standard error) were derived from NP’s GWAS meta-analysis data, specifically reflecting the association strength and sampling error between each SNP and its corresponding NP subtype (eg, DN, trigeminal neuralgia).31 This study employed five MR analysis methods (MR-Egger regression, weighted median method, inverse variance weighting (IVW), simple model method, weighted model method) to systematically evaluate the potential causal relationship between each NP subtype and AD. Among these, the IVW method was selected as the primary analysis strategy, primarily because: when core MR assumptions (eg, no horizontal pleiotropy, strong association between instrumental variables and exposure) hold, IVW statistically outperforms other methods by variance-weighted integration of all valid instrumental variables’ association effects. Furthermore, this choice aligns with methodological selections in comparable MR studies (eg, analyses linking metabolic disorders to neurodegenerative diseases), facilitating cross-study comparability of results.32 To assess the stability of the results, multiple sensitivity analyses were conducted. Heterogeneity among the selected SNPs was evaluated through Cochrane’s Q test, where P < 0.05 was considered to reflect significant heterogeneity.33 Additionally, we employed MR-Egger intercept to assess possibility of directional pleiotropy, while leave-one-out analysis was carried out to determine effect of each individual SNP and to test the robustness of the causal estimates.34,35 All MR analyses were conducted with TwoSampleMR package under R version 4.3.2.
eQTL Annotation Analysis
We employed expression quantitative trait loci (eQTL) data obtained from 50 human tissues included in version 10 of Genotype-Tissue Expression (GTEx) project,36 which provides a comprehensive view of tissue-specific gene expression differences. In establishing SNP-gene associations, we focused on identifying genes significantly associated with the SNPs identified through MR analysis. A stringent threshold of q-value < 0.05 was employed to manage the false discovery rate, thereby ensuring the statistical robustness of our findings. Ultimately, 15 SNPs were identified as being significantly correlated with gene expression levels. These SNPs serve as important genetic markers for exploring gene regulatory mechanisms and disease associations, highlighting the critical role of eQTL analysis in elucidating gene function and advancing genetic association studies.
Transcriptomic Differential Expression Analysis
We retrieved transcriptomic data relevant to DN originating from the Gene Expression Omnibus (GEO) database37 (accession number: GSE156184). In this dataset, stromal cells derived from normal human prostate tissue (PSC27) were cultured in complete stromal medium supplemented with 10% fetal bovine serum until approximately 1,000,000 cells were obtained. Cellular senescence was induced by treating the cells with 50 μg/mL bleomycin (BLEO) for 12 hours. Afterward, the medium was replaced and 100 μM α-lipoic acid (ALA) was added to suppress the senescence-associated secretory phenotype (SASP). The original aim of the experiment was to evaluate the in vitro therapeutic potential of ALA—an antioxidant with no sedative properties—on human senescent cells. Although prior evidence supports the efficacy of ALA in improving diabetic neuropathy, this study further investigated its potential application in aging-related conditions. For transcriptomic analysis, RNA-seq data from three biological replicates of both the control (CTRL) and BLEO-treated groups, which served as a model for DN, were analyzed to detect differentially expressed genes between pre- and post-neuropathic conditions.
We employed HISAT238 to align paired-end RNA-seq reads to human reference genome hg38 (GRCh37). The resulting alignment files were processed using SAMtools,39 and gene-level expression counts were obtained using featureCounts.40 We subsequently performed differential expression analysis with the DESeq2 package,41 which enabled the identification of genes exhibiting significant expression changes related to DN. This study employs “adjusted P < 0.05 and |log2 fold change (FC)| > 1” as the screening criteria for differentially expressed genes, based on the following: ① |log2FC| > 1 indicates at least a twofold difference in gene expression between the case and control groups (≥2-fold upregulation or ≤0.5-fold downregulation). This threshold balances statistical significance with biological relevance, preventing the inclusion of genes with minor expression changes lacking functional significance; ② This criterion is the default threshold recommended by DESeq2 software and is widely adopted in DN-related transcriptomics studies, ensuring methodological consistency of results.39,42,43 In conjunction with analysis of the bidirectional relationship between NP and AD, two genes were identified in each direction whose expression levels were significantly upregulated following the onset of DN. Further investigation of these differentially expressed genes may help uncover the potential molecular links between DN, pain, and AD, thereby providing a theoretical foundation for the development of targeted therapeutic strategies. Raw gene count data used for DESeq2 differential expression analysis were derived from the transcriptomic dataset and are provided in Table S2.
Gene Ontology (GO) Enrichment Analysis
Gene Ontology (GO) enrichment analysis was applied to systematically characterize and elucidate the biological functions of the candidate gene sets identified in this study.44 Based on the SNP–eQTL association analysis, 15 key genes (FDR < 0.05) were subjected to functional analysis across the three GO domains: Molecular Function (MF), Biological Process (BP), and Cellular Component (CC), using the GO TermFinder tool (v1.4.3). Hypergeometric tests were applied during the analysis, and multiple testing correction was performed using Benjamini–Hochberg procedure, with significance defined as an adjusted p-value (q-value) < 0.05. Similarly, the genes significantly upregulated and downregulated in the transcriptomic differential expression analysis were analyzed separately using the same approach.
Statistical Analysis
All statistical analyses were conducted using R (version 4.3.2). The significance thresholds, statistical tests, and software packages employed for each analysis are summarized below.
For Mendelian Randomization Analysis
The IVW method served as the primary analysis for causal inference. A significance threshold of P < 0.05 was applied for the IVW result. The strength of instrumental variables was assessed using the F-statistic, with F < 10 indicating a weak instrument. Heterogeneity was evaluated using Cochran’s Q test (P < 0.05 indicating significance), and potential horizontal pleiotropy was assessed via the MR-Egger intercept test (P < 0.05 indicating significance). All MR analyses were performed using the TwoSampleMR package.
eQTL Annotation Analysis
A FDR-adjusted q-value < 0.05 was used as the significance threshold for identifying SNP-gene associations.
Transcriptomic Differential Expression Analysis
Differential gene expression analysis was performed using DESeq2. Genes with an adjusted P-value < 0.05 and |log2FC| > 1 were considered significantly differentially expressed.
Gene Ontology Enrichment Analysis
Enrichment of GO terms was evaluated using a hypergeometric test with Benjamini-Hochberg correction. Terms with an adjusted q-value < 0.05 were deemed significantly enriched. The analysis was implemented with the GO TermFinder tool (v1.4.3). According to the relevant national regulations and the review of our Ethics Committee, the study met the requirements for exemption from ethical review.
Results
MR Analysis Results
MR analysis revealed distinct causal relationship between DN and AD among six subtypes of NP.
In the two-sample MR framework, SNPs were used as IVs, NP served as exposure, and AD was defined as outcome. According to IVW method with significance threshold of P < 0.05, DN was found to have a protective effect against AD, with odds ratio (OR) of 0.86 and 95% confidence interval (CI) of 0.77~0.95 (Figure 2).
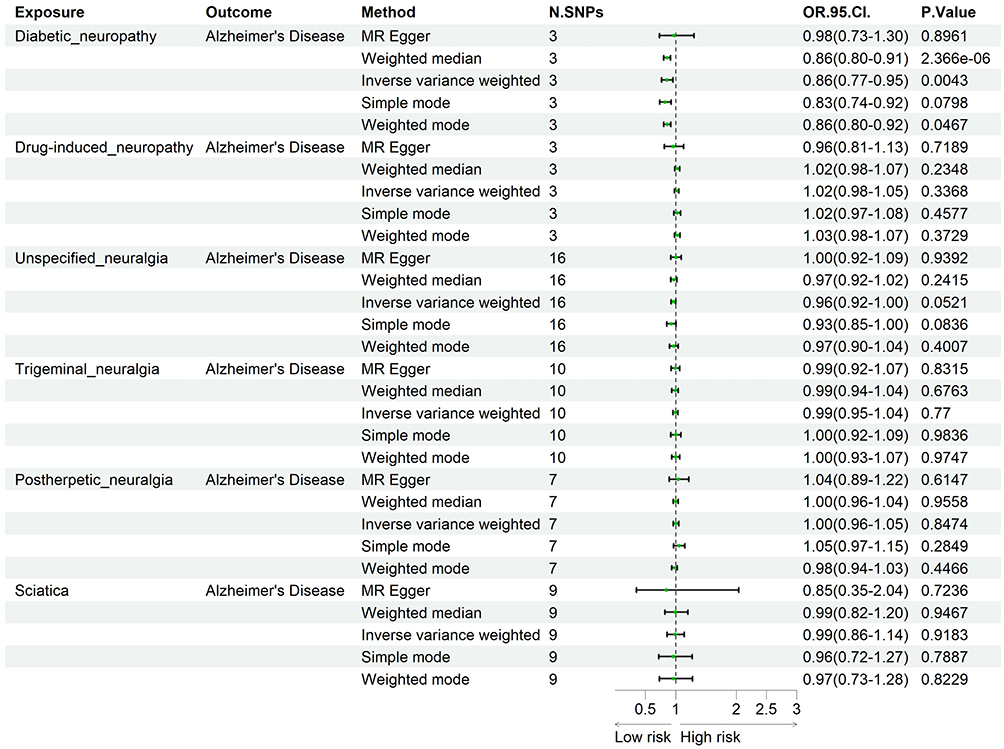 |
Figure 2 Forest plot with neuropathic pain as exposure. |
MR analysis demonstrated a causal association whereby increased risk of DN was linked to a decreased risk of AD, as supported by multiple analytical approaches (PMR-Egger = 0.8961, Pweighted-median = 2.37×10−6, PIVW = 0.0043, Psimple mode = 0.0798, Pweighted mode = 0.0467; Figure 2, Table S3). Assessment of heterogeneity showed Cochran’s QMR-Egger = 3.6867 (P = 0.0582) and QIVW= 6.8416 (P = 0.0327). Since P < 0.05 indicated potential heterogeneity, a random-effects IVW model was adopted, which yielded consistent and robust results in comparison with the fixed-effects model (Table S3). Furthermore, MR-Egger regression was conducted to evaluate potential horizontal pleiotropy, and the intercept test produced a P-value of 0.5154, indicating no substantial pleiotropic bias among the instrumental variables.
In reverse MR analysis, with AD considered as exposure and NP as outcome, we observed a marginally significant association between AD and sciatica (OR = 0.97, 95% CI: 0.95~1.00; PMR-Egger = 0.1353, Pweighted median = 0.2881, PIVW = 0.0178, Psimple mode = 0.4104, Pweighted mode = 0.1224), suggesting a potentially weak protective effect. Heterogeneity and horizontal pleiotropy tests were not statistically significant (Cochran’s Q test P = 0.5461; MR-Egger intercept P = 0.5587) (Figure 3, Table S3). Comprehensive Mendelian randomization results can be found in Tables S1 and S3–S7, as well as Figure S1–S4.
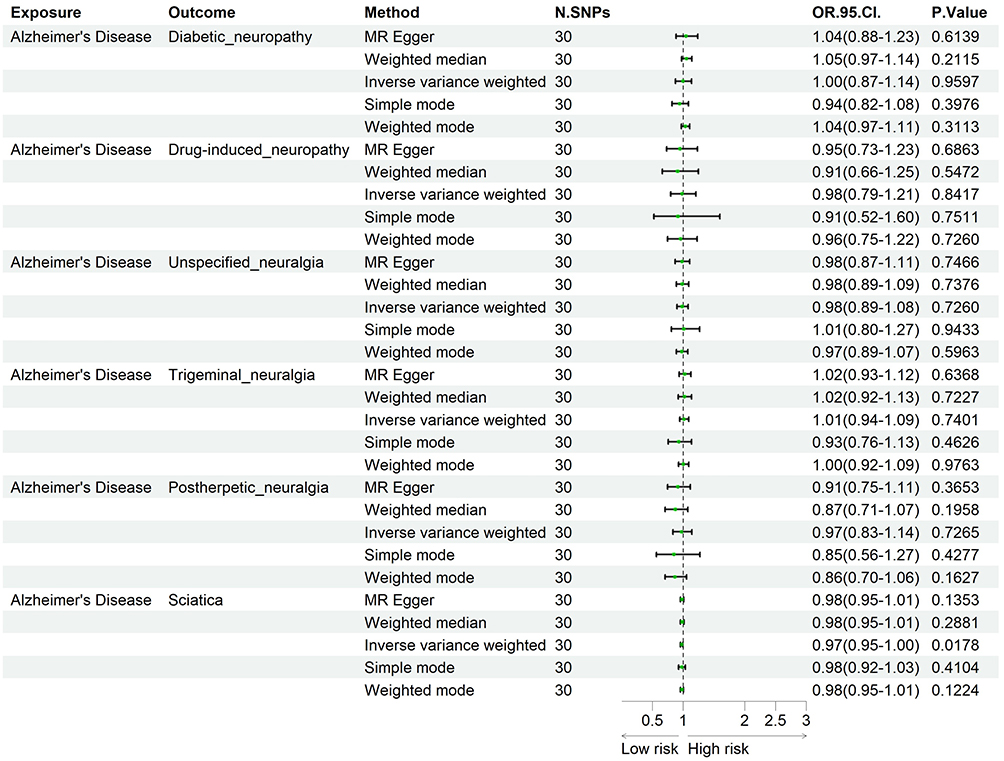 |
Figure 3 Forest plot with Alzheimer’s disease as exposure. |
eQTL Analysis Results
As shown in Table 1, SNPs associated with the broader phenotype of NP—the context for our specific focus on DN—exert eQTL effects primarily in the cerebellum and peripheral tissues. For instance, rs9789699 regulates the expression of ANXA4 in the cerebellum, a finding relevant to DN as it suggests that painful diabetic states may influence cognition and motor coordination through cerebellar mechanisms. Other SNPs, such as rs2811465 and rs9267659, modulate the expression of RHO and CYP21A2, respectively, implicating pathways related to immune and metabolic processes that are highly pertinent to the pathogenesis of DN. These findings indicate that genetic factors contributing to NP may influence AD pathology indirectly via systemic metabolic and immune regulation, a mechanism that likely underlies the DN-AD relationship.
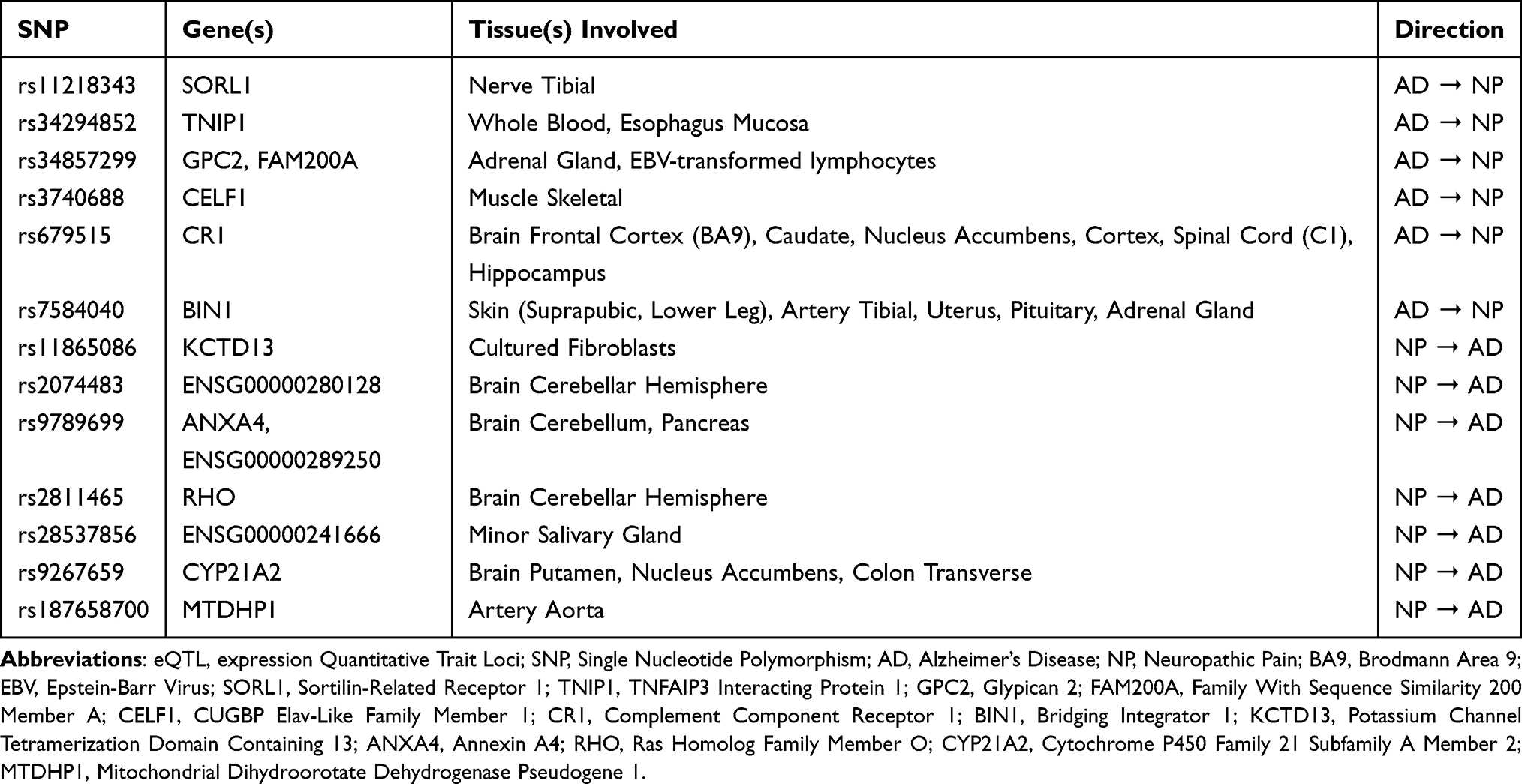 |
Table 1 Bidirectional eQTL-Linked Causal SNPs Between Alzheimer’s Disease and Neuropathic Pain |
Conversely, AD-associated SNPs show significant eQTL effects in both central and peripheral nervous system tissues, regulating expression of genes involved in neuronal function and inflammatory responses. For example, rs11218343 modulates SORL1 expression in the tibial nerve, suggesting that the genetic predisposition to AD may contribute to the pathogenesis of peripheral neuropathies, including DN, through peripheral neural pathways. Additionally, rs679515 is associated with the expression of CR1 across multiple brain regions, including the frontal cortex, hippocampus, and spinal cord, highlighting the role of central immune modulation in the interaction between AD and pain conditions such as DN. Furthermore, rs7584040 influences the expression of BIN1 in peripheral tissues such as the skin and endocrine organs, supporting the hypothesis that AD-related genetic variants may affect susceptibility to painful neuropathies like DN via systemic and neuroinflammatory pathways.
Results of Differential Gene Expression Analysis
Based on the GSE156184 dataset (a model of DN), transcriptomic differential expression analysis identified a total of 3,414 significantly upregulated genes and 1,762 significantly downregulated genes. The distribution of these genes is presented in Figure 4. In accordance with the findings from MR and eQTL annotation, candidate genes with potential causal relevance were marked on the volcano plot using purple diamond symbols. Under bleomycin (BLEO) treatment conditions, which model fibrotic neuropathic damage akin to DN, we observed a significant upregulation of FAM200A and GPC2. These genes, identified through our NP-focused analysis, are causal candidates implicated in the relationship between AD and NP, and thus are of direct relevance to understanding the AD-DN link. In the reverse direction, where NP potentially influences AD, the expression levels of CYP21A2 and MTDHP1 were also markedly elevated in this DN model. These differentially expressed genes may participate in molecular response pathways induced by DN and potentially mediate the shared pathological mechanisms between NP and AD, with specific implications for DN. Detailed results are provided in Table S8.
 |
Figure 4 Transcriptomic landscape of diabetic neuropathy-related genes. Volcano plot shows differential gene expression between bleomycin-treated (BLEO, diabetic neuropathy model) and control cells. Significantly up-regulated (blue) and down-regulated (yellow) genes are defined by |Log2FC| > 1 and FDR < 0.05 (dashed lines). Purple diamonds highlight candidate genes (FAM200A, GPC2, CYP21A2, MTDHP1) identified through MR and eQTL analyses. Numerical values represent Log2 fold change and FDR for labeled genes. |
Results of GO Analysis
To systematically investigate potential biological functions of differentially expressed genes (DEGs) in context of DN and its association with AD, we performed GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses. The results are presented on Figures 5 and 6.
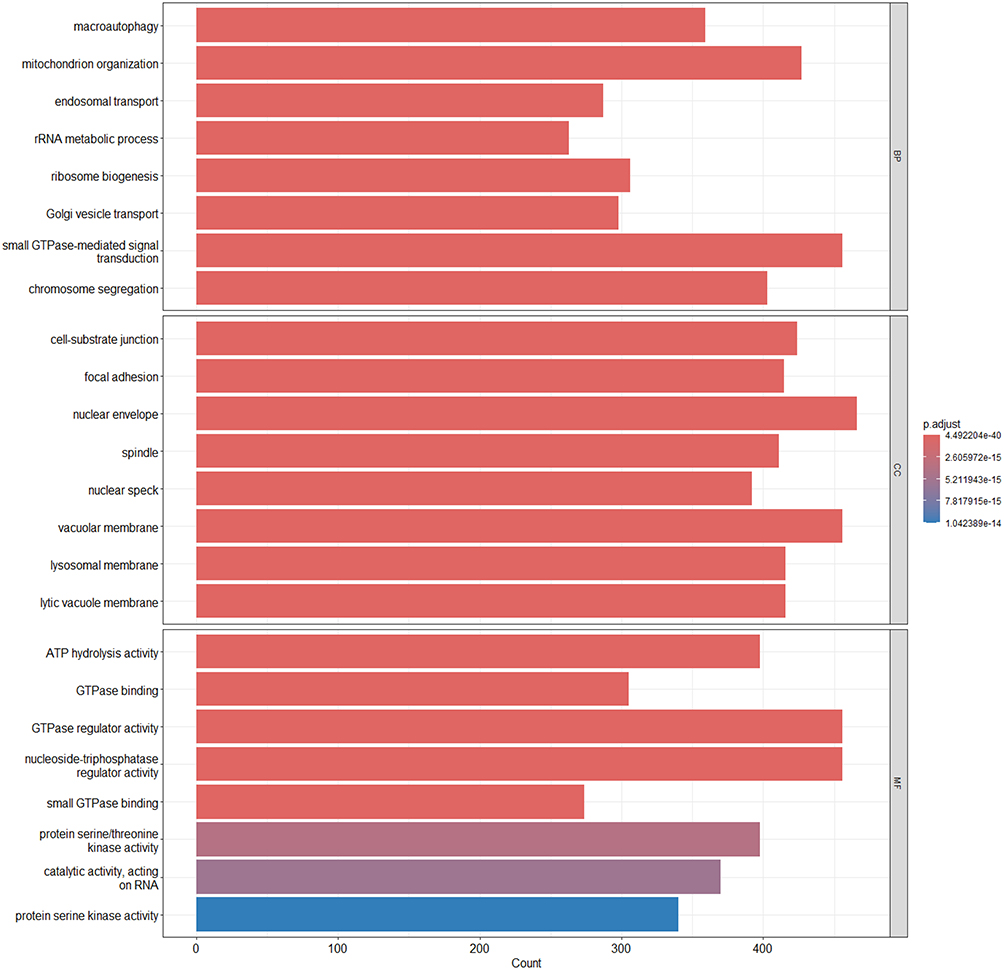 |
Figure 5 GO enrichment analysis of differentially expressed genes. GO terms with significant enrichment among DEGs were categorized into Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). The x-axis indicates the number of genes enriched in each term, and the color gradient reflects the adjusted p-value (p.adjust), with darker colors indicating higher statistical significance. |
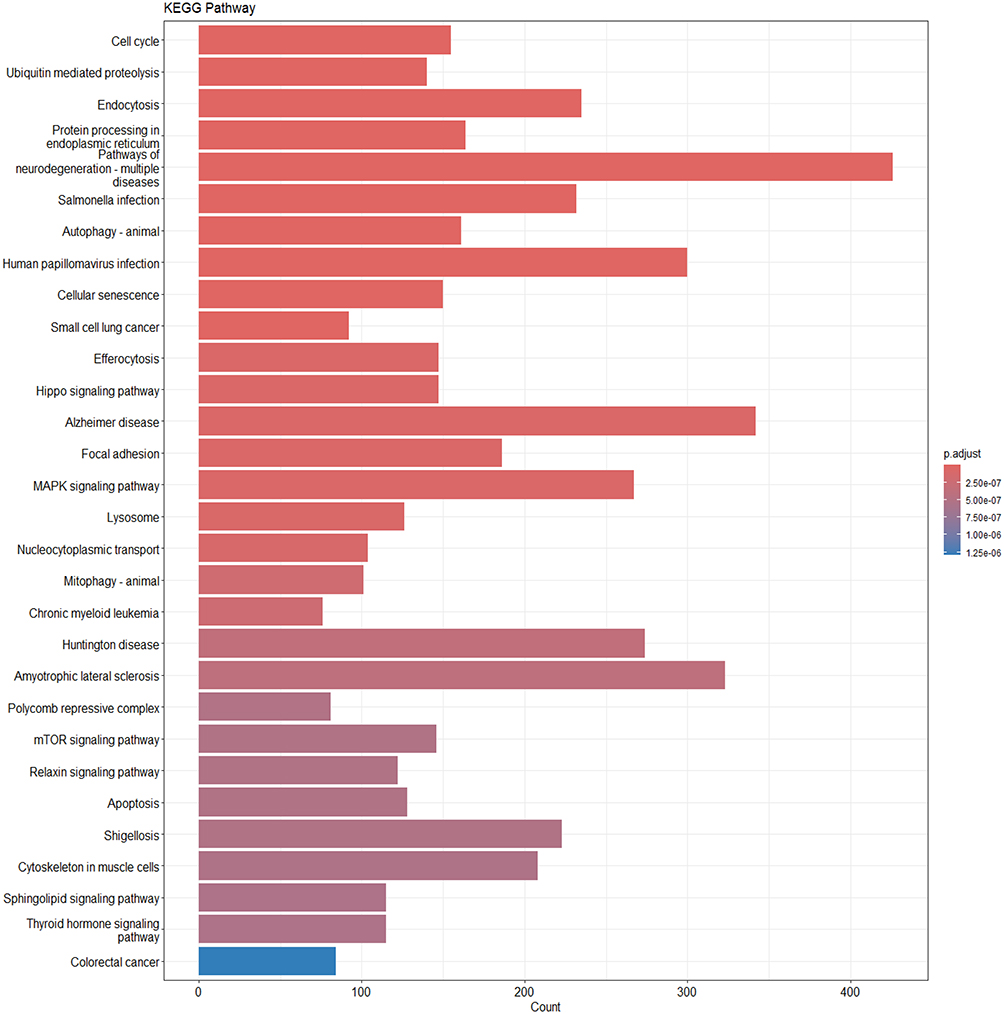 |
Figure 6 Differentially expressed genes underwent KEGG pathway enrichment analysis. The x-axis represents the number of genes involved in each pathway, while the color gradient denotes the adjusted p-value. |
The GO enrichment analysis revealed that DEGs were enriched in several BP, including macroautophagy, mitochondrial organization, and rRNA metabolic process. These terms suggest that the identified genes may participate in maintaining mitochondrial homeostasis, protein degradation, and cellular stress responses, thereby contributing to neural stability.
In CC category, DEGs were predominantly found enriched in structures associated with cellular adhesion and membrane-related systems, including cell-substrate junctions, focal adhesions, lysosomal membranes, and vacuolar membranes. This indicates that these genes are likely localized to cytoskeletal and membrane-bound organelles and may be involved in cell adhesion, material transport, and metabolic regulation.
In MF category, DEGs were predominantly enriched in protein kinase activity, such as protein serine/threonine kinase activity, as well as in GTPase-related functions, including GTPase binding and GTPase regulator activity. These findings indicate that the genes are probably engaged in various signaling pathways governing the cell cycle, immune functions, and synaptic activity. Detailed enrichment results for all six GO subclasses are shown in Tables S9–S14.
KEGG pathway analysis showed that DEGs were broadly associated with pathways related to neurodegenerative diseases and inflammation. The most significantly enriched terms included pathways of neurodegeneration–multiple diseases, AD, Huntington disease, and amyotrophic lateral sclerosis, suggesting that these genes may play shared roles across a range of neurological disorders. In addition, DEGs were mapped to several key signaling pathways, such as MAPK, Hippo, and mTOR pathways, which are known to regulate cell proliferation, apoptosis, and autophagy. These pathways may contribute to neuronal repair processes and inflammatory responses. Notably, pathways related to endoplasmic reticulum protein processing and autophagy also showed significant enrichment. This suggests that pathways associated with protein homeostasis and cellular clearance may play crucial roles in the overlap between DN and AD. These findings support the hypothesis that metabolic and proteostatic regulation are potential mediators linking the two disease processes.
Discussion
This study represents first systematic exploration of the potential causal association and underlying molecular mechanisms between NP and AD by integrating bidirectional MR analysis, eQTL annotation, and transcriptomic differential expression analysis. Among the six common NPs analysed, we found a potential causally protective relationship between DN and AD, an anomalous finding that breaks the traditional paradigm that chronic pain commonly exacerbates AD.12 To further assess the biological plausibility of this association, we incorporated eQTL data from the GTEx project and transcriptome data from the GEO Public Repository. We found that the SNPs mediating this causal linkage were strongly associated with the expression of several genes involved in metabolic regulation and neuroinflammatory pathways, and that the specific metabolic environment of DN may generate compensatory biological pathways that coincidentally mitigate AD risk.
Among the six common types of NP analyzed, we identified a significant protective causal effect of DN on the risk of AD. We further integrated transcriptomic data to analyze the expression patterns of key genes. Based on existing literature and disease-related mechanisms, we propose that FAM200A, GPC2, CYP21A2, and MTDHP1 may have potential functional relevance in both AD and DN. The biological function of FAM200A remains unclear, but it may be involved in post-translational protein modification or cellular stress responses, potentially participating in protein quality control or stress-related signaling pathways,45–47 which could influence the progression of AD. Notably, the potential cross-talk of FAM200A in AD and DN can be explained by mechanisms regulating protein homeostasis. Although autophagy has been implicated in the pathogenesis of DN, it may also promote the clearance of misfolded proteins (eg, Aβ and tau) from the brain, which may have a protective effect on the pathology of AD.48 In AD, clearance of abnormally folded Aβ and tau proteins relies on the ubiquitin-proteasome system (UPS) and autophagy pathways.49 In DN, a high-sugar environment induces accumulation of misfolded proteins within Schwann cells. If FAM200A indeed participates in post-translational modifications (eg, phosphorylation or ubiquitination), it may exert protective effects by enhancing “abnormal protein clearance efficiency” in both diseases. This hypothesis requires subsequent validation through Co-IP (co-immunoprecipitation) to confirm its interactions with autophagy-related proteins (eg, LC3, p62). If FAM200A is implicated in antioxidant defense or the metabolism of advanced glycation end-products, it may also play a role in DN. GPC2, a member of the glypican family, regulates signaling pathways such as Wnt and FGF, and is involved in neurodevelopment and synaptic plasticity.50–52 It may contribute to AD pathogenesis by modulating Wnt signaling activity, thereby affecting tau phosphorylation or synaptic stability. Additionally, it may influence Aβ clearance and modulate neuroinflammation and glial cell function. In the context of DN, GPC2 may promote axonal regeneration or suppress inflammation by regulating Wnt signaling pathway. The bidirectional regulatory properties of the GPC2-regulated Wnt pathway in AD and DN are key to its involvement in cross-talk mechanisms. In AD, Wnt pathway inhibition leads to glycogen synthase kinase 3β (GSK-3β) activation, thereby exacerbating tau phosphorylation.53 In DN, activation of the Wnt pathway promotes Schwann cell proliferation and axonal regeneration, thereby alleviating nerve injury.54 In this study, the differential expression of GPC2 may exert a protective effect against AD by simultaneously suppressing AD pathology and promoting DN nerve repair through upregulating Wnt pathway activity. CYP21A2 is a steroid synthase involved in adrenal hormone metabolism, and its dysfunction may cause hormonal imbalance,55–58 which might exacerbate neuroinflammation or oxidative stress associated with AD, and could also potentially worsen insulin resistance or vascular damage related to DN through disrupted hormone metabolism.59,60 MTDHP1 may participate in folate metabolism or mitochondrial function, indicating that mitochondrial homeostasis might have a pivotal role in the connection between DN and AD. These genes might influence both conditions through intersecting mechanisms such as protein metabolism and clearance, regulation of metabolism and inflammation, and signaling pathway interactions.61–63 Future studies employing gene editing, proteomics, and multi-omics approaches are required to validate their specific functions and potential as therapeutic targets. Meanwhile, to provide a more intuitive representation of the proposed mechanisms, we constructed a schematic diagram (Figure 7) to systematically illustrate the potential roles of FAM200A, GPC2, CYP21A2, and MTDHP1 in the context of AD and DN.
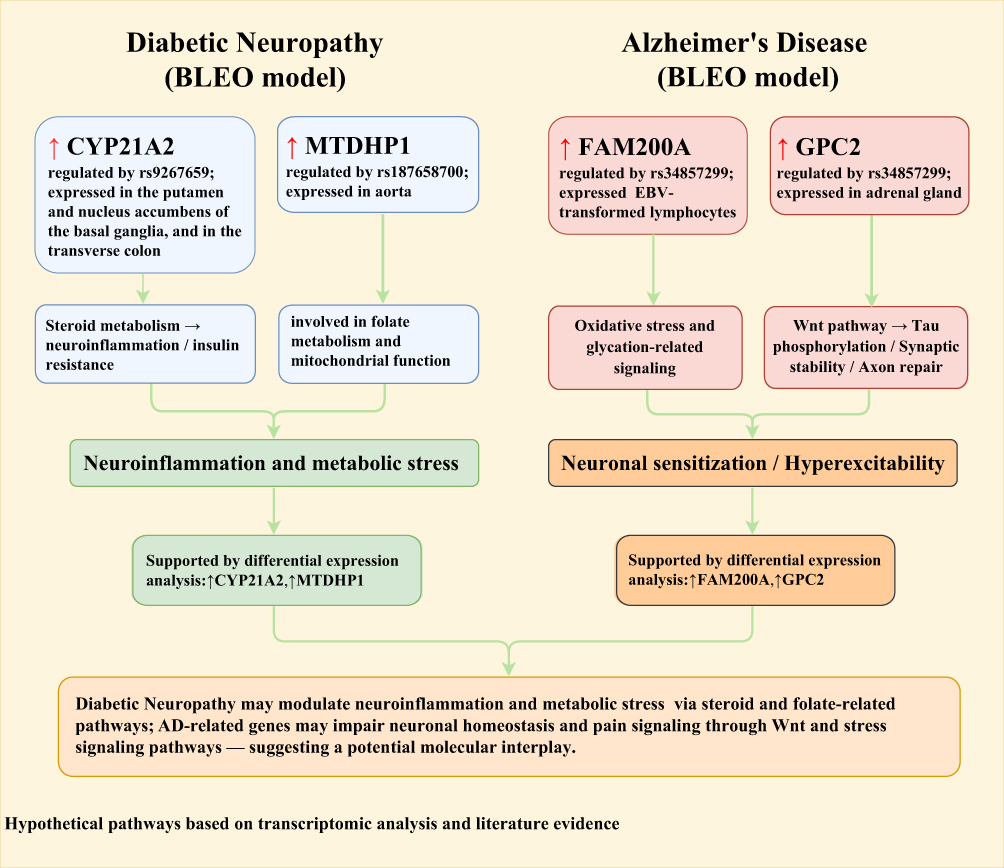 |
Figure 7 Proposed molecular links between diabetic neuropathy and Alzheimer’s disease from transcriptomics. |
We conducted GO and KEGG pathway enrichment analyses on DEGs, which demonstrated their participation in various aspects of cellular function. GO analysis revealed that candidate genes exhibited enrichment in BP categories, including ribosome biogenesis, autophagy, vesicle transport, and mitochondrial regulation, suggesting their central involvement in processes related to protein synthesis, degradation, and energy metabolism. Several key steps of autophagy, such as autophagosome closure and lysosomal fusion, are regulated by proteins including GORASP2 and mATG9, which are involved in vesicle maturation and trafficking, respectively.64,65 Mitochondrial dysfunction is considered a critical factor in the pathogenesis of AD and is closely associated with impaired energy metabolism and oxidative stress.66 The GO analysis results indicating “enrichment in autophagy and mitochondrial regulation pathways” in this study align closely with the classic “energy metabolism-neurodegeneration” theory in the AD field. In AD patients, reduced activity of mitochondrial respiratory chain complexes in neuronal mitochondria leads to insufficient ATP production, while increased release of reactive oxygen species (ROS) further damages mitochondrial DNA.67 Abnormal autophagy pathways lead to impaired clearance of damaged mitochondria (mitophagy), forming a vicious cycle of “mitochondrial damage - ROS accumulation - autophagy suppression.” In DN, similar mitochondrial dysfunction and autophagy defects also result in reduced survival capacity of Schwann cells.68 This finding that abnormalities in the mitochondrial-autophagy axis may represent a core pathological pathway shared by AD and DN also provides the fundamental basis for the cross-regulatory role of DEGs in this study. In CC category, candidate genes were involved in structures such as nuclear envelope, cytoskeleton, and adhesion junctions, highlighting their essential roles in maintaining cellular structural integrity and in transmission of genetic information. For example, the nuclear envelope and nuclear speckles are involved not only in nucleocytoplasmic transport but also in the regulation of RNA splicing and stress responses.69 The results in MF category indicated that the associated genes are implicated in regulating ATP and GTP metabolism, small GTPase signaling, and protein kinase phosphorylation, forming a molecular network that links energy conversion to signal transduction. Small GTPases are activated through GTP/GDP exchange and play broad roles in vesicle trafficking, inflammatory responses, and the regulation of cell polarity.70,71
KEGG pathway analysis additionally indicated that the candidate genes might have essential roles in maintaining cellular homeostasis and influencing disease progression. Notably, pathways such as cell cycle, Hippo, and mTOR are jointly involved in maintaining the dynamic balance between cell proliferation and apoptosis, and their dysregulation is closely associated with neurodegeneration and metabolic abnormalities.72,73 Ubiquitin-mediated protein degradation, autophagy, and lysosomal pathways together form a protein quality control network essential for removing misfolded proteins and damaged organelles, the dysfunction of which is a major pathological feature of AD and other neurodegenerative disorders.74–77 Additionally, the MAPK, Wnt, and hormone signaling pathways are involved in regulating neural plasticity, metabolic homeostasis, and stress responses, suggesting the importance of the metabolism-neural axis in the overlapping mechanisms between DN and AD.72,73,78 These pathways form a complex regulatory network through modifications such as ubiquitination and phosphorylation, as well as molecular switches like GTPases, reflecting an integrated pathological process from metabolic regulation to neurodegeneration.
Considering the significant protective effect of DN on AD revealed by bidirectional MR analysis, we propose that DN may confer potential neuroprotective effects through various metabolic and inflammatory regulatory pathways. This observation challenges the traditional notion that metabolic disorders universally worsen neurodegenerative pathology and suggests that certain diabetes subtypes, particularly those complicated by neuropathy, may involve protective mechanisms. From a clinical translation perspective, the discovery of DN’s protective effect holds significant practical implications. Current early screening for AD primarily relies on cognitive function scales and cerebrospinal fluid biomarkers (such as the Aβ42/Aβ40 ratio and p-tau181).79 However, this study suggests that DN-related indicators (such as peripheral nerve conduction velocity and pain scores) can serve as complementary markers for AD risk assessment. For instance, in diabetic patients with DN, if the severity of peripheral nerve damage correlates inversely with AD biomarker levels (requiring validation in subsequent cohort studies), DN-related clinical indicators could be integrated into AD risk stratification models. This approach would facilitate precision management for patients with metabolic-neurological comorbidities. Clinically, this study offers a novel perspective for the precise screening of high-risk populations, particularly by identifying changes in pain sensitivity and peripheral nerve function during the prodromal phase of AD. Such insights may facilitate early intervention to some extent. In future studies, we will combine single-cell transcriptomics, spatial omics, and in vivo functional experiments to further validate the potential roles of key genes in regulating neuroinflammation and preventing cognitive impairment. This will not only expand our understanding of the interaction mechanisms between metabolic disorders and neurodegenerative diseases but also provide theoretical support and strategic guidance for clinical identification of high-risk populations and the development of multidimensional intervention targets. The clinical significance of our finding is not so much that induction of DN is a therapeutic strategy for AD, but rather that the molecular pathways activated in the DN state may reveal novel, neuroprotective targets for pharmacological treatment. For example, agonists of the protective pathways identified here could be explored without inducing the deleterious effects of full-blown neuropathy (eg, enhancing specific aspects of autophagy or mitochondrial biogenesis). In addition, evaluating these identified genes or pathways could help stratify the relative risk of ADD in diabetic patients, adding a new dimension to precision medicine. Future studies should prioritise in vivo and in vitro functional validation of these candidate genes (eg, FAM200A, GPC2) to ultimately determine their role in the DN-AD axis.
Although we systematically revealed the potential causal relationship and molecular mechanisms between DN and AD by integrating MR, eQTL, and transcriptomic data, several limitations remain. First, eQTL data used were mainly derived from healthy tissue samples in the GTEx database, which may not fully capture the true transcriptional regulatory features under disease conditions. Second, although the transcriptomic data were obtained from publicly available high-quality datasets in GEO database, sample heterogeneity and differences in experimental conditions, such as the bleomycin-induced model used in this study, may affect the generalizability of the differentially expressed genes identified. Furthermore, although MR analysis can partially avoid confounding factors and support causal inference, it cannot replace biological experimental validation. Additionally, the GWAS data used were based on individuals of European ancestry, which may limit generalizability to other populations. Therefore, functional experiments at the cellular or animal level are still needed to further clarify the specific roles of candidate genes in neuropathological processes.
Conclusion
This study systematically evaluated the causal relationships between six common types of NP and AD using bidirectional two-sample MR analysis. We observed notable inverse relationship between DN and the risk of AD, indicating a possible protective effect. By integrating eQTL annotation and transcriptomic data, we identified several key genes regulated by DN-associated SNPs that are specifically expressed in central and peripheral tissues, including FAM200A, GPC2, CYP21A2, and MTDHP1. These genes are enriched in pathways related to mitochondrial function, autophagy regulation, and inflammation, which may mediate the impact of DN on AD risk. This study is the first to construct a potential molecular map linking DN and AD from the perspectives of genetic causality, gene regulation, and functional pathways, providing new evidence to understand the intersecting mechanisms between NP and neurodegenerative diseases.
Abbreviations
NP, Neuropathic Pain; AD, Alzheimer’s Disease; DN, Diabetic Neuropathy; MR, Mendelian Randomization; SNPs, Single Nucleotide Polymorphisms; Aβ, β-amyloid; NIA-AA, National Institute on Aging-Alzheimer’s Association; IVs, Instrumental Variables; IVW, Inverse Variance Weighted; GWAS, Genome-Wide Association Study; LD, Linkage Disequilibrium; Eqtl, Expression Quantitative Trait Locus; GTEx, Genotype-Tissue Expression; GEO, Gene Expression Omnibus; BLEO, Bleomycin; ALA, α-Lipoic Acid; SASP, Senescence-Associated Secretory Phenotype; DEGs, Differentially Expressed Genes; SE, Standard Error; CTRL, Control; GO, Gene Ontology; MF, Molecular Function; BP, Biological Process; CC, Cellular Component; KEGG, Kyoto Encyclopedia of Genes and Genomes; OR, Odds Ratio; CI, Confidence Interval; UPS, Ubiquitin-Proteasome System; Co-IP, Co-Immunoprecipitation; GSK-3β, Glycogen Synthase Kinase 3β; ROS, Reactive Oxygen Species; BA9, Brodmann Area 9; EBV, Epstein-Barr Virus; SORL1, Sortilin-Related Receptor 1; TNIP1, TNFAIP3 Interacting Protein 1; GPC2, Glypican 2; FAM200A, Family With Sequence Similarity 200 Member A; CELF1, CUGBP Elav-Like Family Member 1; CR1, Complement Component Receptor 1; BIN1, Bridging Integrator 1; KCTD13, Potassium Channel Tetramerization Domain Containing 13; ANXA4, Annexin A4; RHO, Ras Homolog Family Member O; CYP21A2, Cytochrome P450 Family 21 Subfamily A Member 2; MTDHP1, Mitochondrial Dihydroorotate Dehydrogenase Pseudogene 1.
Data Sharing Statement
All original data and findings described in this study are available within the article, and any additional questions can be addressed to the corresponding authors.
Ethics Approval and Consent to Participate
Ethical approval was not applicable because this study used data from publicly available databases.
All data in this study are from Finngen (https://r12.finngen.fi/), PGC (https://pgc.unc.edu/for-researchers/download-results/), and DN-related transcriptomic data from GEO (accession: GSE156184). We protect participant privacy (no individual identification; GEO data follows its guidelines), use data only for scientific research (no commercial use; PGC permission needed for commercial purposes if required), and comply with human/genetic research rules. No fetal risk tests will be developed; tests for children/adults are experimental, with acknowledgment of non-genetic factors in phenotypes.
Data will not be cross-posted to third parties (official websites are definitive). Relevant Finngen, PGC, and GEO publications will be cited; PGC’s scientific priorities (per Fort Lauderdale Principles) are respected for pre-publication data.
Acknowledgments
We thank all participants and investigators of the publicly available datasets that made this research possible. Summary-level GWAS data for neuropathic pain phenotypes were obtained from the FinnGen consortium (Release R12), and Alzheimer’s disease summary statistics were provided by the Psychiatric Genomics Consortium (PGC). Expression quantitative trait loci (eQTLs) data were derived from the Genotype-Tissue Expression (GTEx) project (v10 release), which was supported by multiple NIH institutes. Transcriptomic data used for differential expression analysis were obtained from the Gene Expression Omnibus (GEO), accession number GSE156184. We acknowledge the open-access nature of these resources and are grateful to all researchers who contributed to these invaluable datasets.
Authors’ Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors declare no competing interests.
References
1. Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010;9(8):807–819. doi:10.1016/S1474-4422(10)70143-5
2. Pan H, Liu CX, Zhu HJ, Zhang G-F. Immune cells mediate the effects of gut microbiota on neuropathic pain: a Mendelian randomization study. J Headache Pain. 2024;25(1):196. doi:10.1186/s10194-024-01906-z
3. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. 2021;101(1):259–301. doi:10.1152/physrev.00045.2019
4. Thouaye M, Yalcin I. Neuropathic pain: from actual pharmacological treatments to new therapeutic horizons. Pharmacol Ther. 2023;251:108546. doi:10.1016/j.pharmthera.2023.108546
5. Ezzatvar Y, García-Hermoso A. Global estimates of diabetes-related amputations incidence in 2010–2020: a systematic review and meta-analysis. Diabet Res Clin Prac. 2023;195. doi:10.1016/j.diabres.2022.110194
6. L Q, L K, C Y, et al. Oxidative stress in diabetic peripheral neuropathy: pathway and mechanism-based treatment. Mol Neurobiol. 2023;60(8). doi:10.1007/s12035-023-03342-7
7. Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi:10.1038/35008121
8. Vinik AI, Nevoret ML, Casellini C, Parson H. Diabetic neuropathy. Endocrinol Metab Clin North Am. 2013;42(4):747–787. doi:10.1016/j.ecl.2013.06.001
9. Ds K, A H, Rc P, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7(1). doi:10.1038/s41572-021-00269-y
10. Gm M, Ds K, C H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the national institute on aging-alzheimer’s association workgroups on diagnostic guidelines for alzheimer’s disease. Alzheimer’s Dement. 2011;7(3). doi:10.1016/j.jalz.2011.03.005
11. Cao S, Fisher DW, Yu T, Dong H. The link between chronic pain and Alzheimer’s disease. J Neuroinflamm. 2019;16(1):204. doi:10.1186/s12974-019-1608-z
12. Yao K, Wang S, Xu Z, et al. Mechanisms of comorbidity between Alzheimer’s disease and pain. Alzheimer’s Dement. 2025;21(2):e14605. doi:10.1002/alz.14605
13. Non-cancer chronic pain conditions and risk for incident Alzheimer’s disease and related dementias in community-dwelling older adults: a population-based retrospective cohort study of united states medicare beneficiaries, 2001–2013. Available from: https://www.mdpi.com/1660-4601/17/15/5454.
14. K S, S U, Ke I. Non-cancer chronic pain conditions and risk for incident Alzheimer’s disease and related dementias in community-dwelling older adults: a population-based retrospective cohort study of United States medicare beneficiaries, 2001-2013. Int J Environ Res Public Health. 2020;17(15). doi:10.3390/ijerph17155454
15. The relationship between adult hippocampal neurogenesis and cognitive impairment in Alzheimer’s disease - Geigenmüller - 2024 - Alzheimer’s & Dementia - Wiley online library. doi:10.1002/alz.14179.
16. Innes KE, Sambamoorthi U. The potential contribution of chronic pain and common chronic pain conditions to subsequent cognitive decline, new onset cognitive impairment, and incident dementia: a systematic review and conceptual model for future research. J Alzheimers Dis. 2020;78(3):1177–1195. doi:10.3233/JAD-200960
17. Uddin M, Mamun AA, Sumsuzzman D, et al. Emerging promise of cannabinoids for the management of pain and associated neuropathological alterations in Alzheimer’s disease. Front Pharmacol. 2020;11:1097. doi:10.3389/fphar.2020.01097
18. Inoue K, Tsuda M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci. 2018;19(3):138–152. doi:10.1038/nrn.2018.2
19. Rostagno AA. Pathogenesis of Alzheimer’s disease. Inter J Mole Sci. 2023;24(1):107. doi:10.3390/ijms24010107
20. Uddin MS, Kabir MT, Rahman MM, Mathew B, Shah MA, Ashraf GM. TV 3326 for Alzheimer’s dementia: a novel multimodal ChE and MAO inhibitors to mitigate Alzheimer’s-like neuropathology. J Pharm Pharmacol. 2020;72(8):1001–1012. doi:10.1111/jphp.13244
21. Gold M, El Khoury J. β-amyloid, microglia and the inflammasome in Alzheimer’s disease. Semin Immunopathol. 2015;37(6):607–611. doi:10.1007/s00281-015-0518-0
22. Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138(6):1738–1755. doi:10.1093/brain/awv081
23. Karavis MY, Siafaka I, Vadalouca A, Georgoudis G. Role of microglia in neuropathic pain. Cureus. 2023;15(8):e43555. doi:10.7759/cureus.43555
24. Sekula P, Del Greco MF, Pattaro C, Köttgen A. Mendelian randomization as an approach to assess causality using observational data. JASN. 2016;27(11):3253–3265. doi:10.1681/ASN.2016010098
25. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–R98. doi:10.1093/hmg/ddu328
26. J R, Z X, H T, et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 2017;18(1). doi:10.1186/s13059-016-1142-6
27. Lonsdale J, Thomas J, Salvatore M, et al. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi:10.1038/ng.2653
28. W Z, G M, S M. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1). doi:10.1038/nrg2484
29. Kurki MI, Karjalainen J, Palta P, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–518. doi:10.1038/s41586-022-05473-8
30. Wightman DP, Jansen IE, Savage JE, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53(9):1276–1282. doi:10.1038/s41588-021-00921-z
31. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol. 2015;44(2). doi:10.1093/ije/dyv080
32. B S, B A, Sg T. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7). doi:10.1002/gepi.21758
33. Bowden J, Del Greco MF, Minelli C, et al. Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. Int J Epidemiol. 2019;48(3):728–742. doi:10.1093/ije/dyy258
34. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–389. doi:10.1007/s10654-017-0255-x
35. H G, T K, DS G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLOS Genetics. 2017;13(11). doi:10.1371/journal.pgen.1007081
36. Aguet F, Anand S, Ardlie KG, The GTEx Consortium. The GTEx consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–1330. doi:10.1126/science.aaz1776
37. Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol. 2016;1418:93–110. doi:10.1007/978-1-4939-3578-9_5
38. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915. doi:10.1038/s41587-019-0201-4
39. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi:10.1093/bioinformatics/btp352
40. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features | bioinformatics | oxford academic. Available from: https://academic.oup.com/bioinformatics/article/30/7/923/232889.
41. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Abstract Europe PMC. Available from: https://europepmc.org/article/PMC/4302049.
42. K D, L B, Sl S. HISAT: a fast spliced aligner with low memory requirements. Nature Methods. 2015;12(4). doi:10.1038/nmeth.3317
43. Mi L, H W, A S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15(12). doi:10.1186/s13059-014-0550-8
44. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
45. Fan S, Liu Y, Lin Z, et al. ZNF655 promotes the progression of hepatocellular carcinoma through PSMB8. Cell Biol Int. 2023;47(9):1535–1546. doi:10.1002/cbin.12050
46. Xu N, Wu YP, Ke ZB, et al. Identification of key DNA methylation-driven genes in prostate adenocarcinoma: an integrative analysis of TCGA methylation data. J Transl Med. 2019;17(1):311. doi:10.1186/s12967-019-2065-2
47. Ak M, Kahraman A, Arnold FM, et al. Clinicopathological and genomic profiles of atypical fibroxanthoma and pleomorphic dermal sarcoma identify overlapping signatures with a high mutational burden. Genes. 2021;12(7):974. doi:10.3390/genes12070974
48. Ning Y, Zhang Y, Jiang T, et al. LRP1-mediated p-tau propagation contributes to cognitive impairment after chronic neuropathic pain in rats. Neurosci Res. 2025;212:84–96. doi:10.1016/j.neures.2024.12.005
49. Fm M, F A, C A, et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron. 2017;93(5). doi:10.1016/j.neuron.2017.01.022
50. Chen G, Luo D, Zhong N, et al. GPC2 is a potential diagnostic, immunological, and prognostic biomarker in pan-cancer. Front Immunol. 2022:13. doi:10.3389/fimmu.2022.857308
51. Bosse KR, Raman P, Zhu Z, et al. Identification of GPC2 as an oncoprotein and candidate immunotherapeutic target in high-risk neuroblastoma. Cancer Cell. 2017;32(3):295–309.e12. doi:10.1016/j.ccell.2017.08.003
52. Wang R, Lin X. GPC2 as a diagnostic and prognostic marker regulated progression of colorectal cancer. Arab J Gastroenterol. 2024;25(1):51–57. doi:10.1016/j.ajg.2023.11.006
53. Ct Z, W J, Wy W. Wnt signaling in synaptogenesis of Alzheimer’s disease. Ibrain. 2023;9(3). doi:10.1002/ibra.12130
54. Yl T, Ky N, Ry K, P G, Sm C. Melatonin prevents oxidative stress-induced mitochondrial dysfunction and apoptosis in high glucose-treated schwann cells via upregulation of Bcl2, NF-κB, mTOR, wnt signalling pathways. Antioxid. 2019;8(7). doi:10.3390/antiox8070198
55. Kleinle S, Lang R, Fischer GF, et al. Duplications of the functional CYP21A2 gene are primarily restricted to Q318X alleles: evidence for a founder effect. J Clin Endocrinol Metab. 2009;94(10):3954–3958. doi:10.1210/jc.2009-0487
56. Tsai LP, Lee HH. Analysis of CYP21A1P and the duplicated CYP21A2 genes. Gene. 2012;506(1):261–262. doi:10.1016/j.gene.2012.06.045
57. Simonetti L, Bruque CD, Fernández CS, et al. CYP21A2 mutation update: comprehensive analysis of databases and published genetic variants. Hum Mutat. 2018;39(1):5–22. doi:10.1002/humu.23351
58. Parajes S, Quinterio C, Domínguez F, Loidi L. A simple and robust quantitative PCR assay to determine CYP21A2 gene dose in the diagnosis of 21-hydroxylase deficiency. Clin Chem. 2007;53(9):1577–1584. doi:10.1373/clinchem.2007.087361
59. Chen Cardenas SM, El-Kaissi S, Jarad O, Liaqat M, Korbonits M, Hamrahian AH. Unusual combination of MEN-1 and the contiguous gene deletion syndrome of CAH and ehlers-danlos syndrome (CAH-X). J Endocr Soc. 2020;4(8):bvaa077. doi:10.1210/jendso/bvaa077
60. Rabbani B, Moghadam MA, Esmaeili S, Rabbani A, Akbari B, Mahdieh N. Pancreatitis as a main consequence of APOC2-related hypertriglyceridemia: the role of nonsense and frameshift variants. Int J Genomics. 2024;2024:6653857. doi:10.1155/2024/6653857
61. Nazki FH, Sameer AS, Ganaie BA. Folate: metabolism, genes, polymorphisms and the associated diseases. Gene. 2014;533(1):11–20. doi:10.1016/j.gene.2013.09.063
62. Hinterberger M, Fischer P. Folate and Alzheimer: when time matters. J Neural Transm. 2013;120(1):211–224. doi:10.1007/s00702-012-0822-y
63. Wang D, Zhai JX, Liu DW. Serum folate, vitamin B12 levels and diabetic peripheral neuropathy in type 2 diabetes: a meta-analysis. Mol Cell Endocrinol. 2017;443:72–79. doi:10.1016/j.mce.2017.01.006
64. Xing Y, Huang L, Jian Y, et al. GORASP2 promotes phagophore closure and autophagosome maturation into autolysosomes. Autophagy. 2025;21(1):37–53. doi:10.1080/15548627.2024.2375785
65. Zhou C, Ma K, Gao R, et al. Regulation of mATG9 trafficking by src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017;27(2):184–201. doi:10.1038/cr.2016.146
66. D R, S P, M B, P A, Dh R. Recent advances in molecular pathways and therapeutic implications targeting mitochondrial dysfunction for Alzheimer’s disease. Mol Neurobiol. 2022;59(1). doi:10.1007/s12035-021-02612-6
67. T E, Ct M. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145(4). doi:10.1016/j.neuroscience.2006.10.056
68. W X, Z Y, A Y, L B. Regulatory effects of astragaloside IV on hyperglycemia-induced mitophagy in schwann cells. Evid-Based Complement Altern Med. 2022;2022. doi:10.1155/2022/7864308
69. Ahn H, Yu J, Ryu K, et al. Single-molecule analysis reveals that IPMK enhances the DNA-binding activity of the transcription factor SRF. Nucleic Acids Research. 2025;53(1):gkae1281. doi:10.1093/nar/gkae1281
70. Wolff DW, Bianchi-Smiraglia A, Nikiforov MA. Compartmentalization and regulation of GTP in control of cellular phenotypes. Trends Mol Med. 2022;28(9):758–769. doi:10.1016/j.molmed.2022.05.012
71. Kr K, Jp R, R I. Regulation of the small GTPase ras and its relevance to human disease. Methods Mol Biol. 2021;2262. doi:10.1007/978-1-0716-1190-6_2
72. Tikkanen R, Nikolic-Paterson DJ. Mitogen-activated protein kinases: functions in signal transduction and human diseases. Int J Mol Sci. 2019;20(19):4844. doi:10.3390/ijms20194844
73. Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–6251. doi:10.1038/onc.2008.301
74. Alto NM, Orth K. Subversion of cell signaling by pathogens. Cold Spring Harb Perspect Biol. 2012;4(9):a006114. doi:10.1101/cshperspect.a006114
75. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19(3):175–191. doi:10.1038/nrm.2017.107
76. Yin H, Wu H, Chen Y, et al. The therapeutic and pathogenic role of autophagy in autoimmune diseases. Front Immunol. 2018;9:1512. doi:10.3389/fimmu.2018.01512
77. Yin Z, Pascual C, Klionsky DJ. Autophagy: machinery and regulation. Microb Cell. 2016;3(12):588–596. doi:10.15698/mic2016.12.546
78. McCarthy N. Rising to the challenge. Nat Rev Cancer. 2013;13(6):378. doi:10.1038/nrc3530
79. Cr J, Da B, B K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018;14(4). doi:10.1016/j.jalz.2018.02.018
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.