Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 19
Causal Association Between Ankylosing Spondylitis and Hidradenitis Suppurativa: A Twosample Bidirectional Mendelian Randomization Study
Authors Chang G, Zhang J, Chen B, Li X, Guo W, Wang H, Zhang L
Received 5 November 2025
Accepted for publication 10 March 2026
Published 25 March 2026 Volume 2026:19 579441
DOI https://doi.org/10.2147/CCID.S579441
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Rungsima Wanitphakdeedecha
Guizhen Chang,1,2,* Jun Zhang,3,* Baojiang Chen,2 Xing Li,2 Weicong Guo,2 Huan Wang,2 Litao Zhang3
1Graduate School of Tianjin Medical University, Tianjin Medical University, Tianjin, People’s Republic of China; 2Department of Dermatology, Tianjin University TEDA Hospital, Tianjin, People’s Republic of China; 3Department of Dermatology, Affiliated Hospital of Tianjin Academy of Traditional Chinese Medicine, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Litao Zhang, Email [email protected]
Background: Ankylosing spondylitis (AS) and hidradenitis suppurativa (HS) represent two chronic inflammatory diseases that share similar underlying immunological characteristics. Observational studies suggest a potential association, but the causal relationship remains underexplored.
Objective: To evaluate bidirectional causal links between AS and HS using Mendelian randomization (MR).
Methods: We implemented two-sample bidirectional MR analyses, utilizing summary statistics obtained from genome-wide association study (GWAS) datasets. Genetic instruments for AS (ncase=9069, ncontrol=1550) and HS (ncases=409, ncontrols=211,139) were selected under stringent criteria (p< 5e-5 LD clumping: r2< 1e-2, clumping window=1e4 kb). Primary analyses used inverse-variance weighted (IVW) regression, supplemented by MR-Egger, weighted median, and sensitivity analyses.
Results: Genetically predicted AS significantly increased HS risk (IVW OR=3.49, 95% CI=1.43– 8.52, P=0.006), with no evidence of reverse causation (HS→AS: OR=1.00, 95% CI=0.9998– 1.00, P=0.816). To validate the stability of our estimates, sensitivity analyses were performed. The MR-Egger intercept and Cochran’s Q test yielded nonsignificant P values (both P> 0.05), supporting the robustness of our findings.
Conclusion: This investigation provides genetic evidence indicating a causal relationship between ankylosing spondylitis and an increased risk of hidradenitis suppurativa, underscoring a shared etiological pathway, likely involving the IL-17/IL-23 axis. No significant causal effect was observed from HS to AS within the limitations of the current data.
Keywords: ankylosing spondylitis, hidradenitis suppurativa, Mendelian randomization study
Introduction
Ankylosing spondylitis (AS) is an immune-mediated inflammatory arthritis, and it falls within the broader category of spondyloarthropathies (SpA), a group that comprises reactive arthritis and enteropathic arthritis.1 In addition to affecting the skeletal system, AS can also involve various extra-articular organs. A variety of extraarticular features are commonly observed in patients with AS. These include inflammatory bowel disease (affecting up to 50% of patients), acute anterior uveitis (identified in 25–35% of cases), and psoriasis (seen in approximately 10% of individuals).1
Hidradenitis suppurativa (HS), also referred to as acne inversa, is a long-standing inflammatory disorder affecting the skin. This condition typically presents with persistent or recurrent lesions, including double-headed comedones, tender indurated papules and nodules, as well as interconnected draining sinus tracts. Moreover, hypertrophic or atrophic scars often develop as residual sequelae of chronic inflammation. HS lesions predominantly localize to intertriginous areas and sites rich in apocrine glands. Typical affected locations encompass the axillae, groin, perianal region, perineum, and inframammary regions. Typically, HS typically manifests in adolescent and young adult individuals. The condition has a population prevalence of approximately 1–4%, and women are disproportionately affected relative to men.2
Emerging evidence indicates that SpA and HS have overlapping clinical manifestations, shared genetic susceptibility, and similar immunological profiles, as well as common underlying immunopathological targets and comparable treatment strategies.3,4 Clinically, targeted biologic therapy is now the mainstay of treatment of moderate to severe HS. The anti-TNF-α antibody adalimumab and the anti-IL-17 secukinumab, which are the only two biologics approved for HS by the US Food and Drug Administration and Medicines Agency, show moderate efficacy of approximately 50% in placebo-controlled studies.5,6 Similarly, they are the mainstay of treatment of AS.7,8 A narrative review of the literature revealed that the occurrence rate of spondyloarthritis, its subtype AS and RA, In HS, the prevalence of spondyloarthritis is presently found to be higher in comparison with that noted in the general population (0.93–28.2% versus 0.2–1.6%).9,10 Based on the above researches, it seems that AS and HS are strongly correlated. Nevertheless, conclusive evidence supporting a causal link between AS and HS remains lacking, largely due to the presence of unadjusted confounding factors and reverse causality bias inherent to observational research designs.
By using genetic variants as instrumental variables, Mendelian randomization (MR) offers an effective approach to minimize confounding bias that is frequently present in observational investigations, thereby facilitating robust causal inference. Distinct genotypes lead to different intermediate phenotypes. When such a phenotype serves as an individual’s exposure status, the correlation between genotype and disease status can capture the causal effect of the exposure variable on the clinical outcome. This framework is unaffected by conventional confounders or reverse causality, owing to the random allocation of alleles during meiosis.
Materials and Methods
In the present two-sample MR analysis, multiple single nucleotide polymorphisms (SNPs) were selected as genetic tool. Three key assumptions needed to be satisfied (Figure 1): first, the genetic instruments are robustly associated with the exposure; second, the instrumental variables are not related to confounding factors; and third, genetic variants exert an effect on the outcome only through the exposure pathway.11 Bidirectional causal relationships between AS and HS were further assessed using MR approaches.
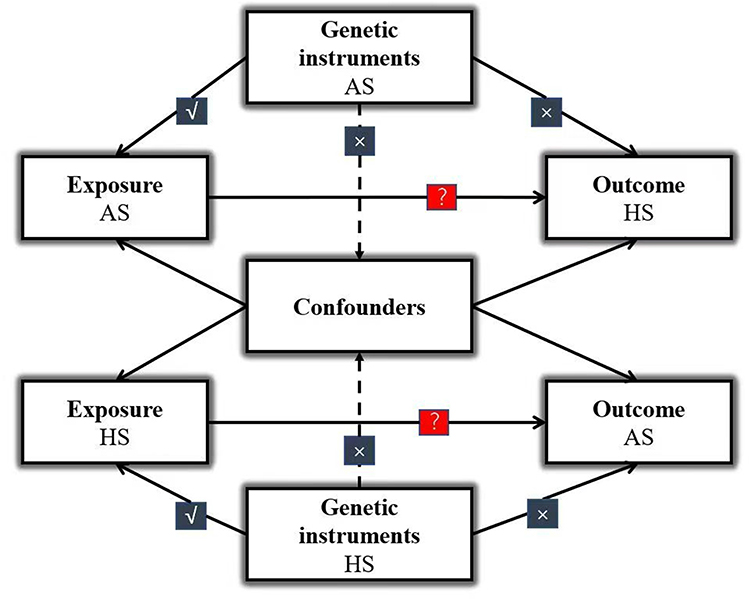 |
Figure 1 Schematic overview of the core assumptions underlying Mendelian randomization. Solid lines represent SNPs associated with the exposure that influence the outcome exclusively through this exposure, while dashed lines indicate SNPs that are independent of confounders between the exposure and outcome. √: predicts; ×: no direct effect; ?: Causality has yet to be established. Abbreviations: AS, ankylosing spondylitis; HS, hidradenitis suppurativa. |
Data Sources
We searched publicly available genome-wide association study (GWAS) databases, including IEU openGWAS and the GWAS Catalog, to identify and extract appropriate datasets. All data included in this study were obtained from public repositories, this case is exempt from approval based on national legislation guidelines, such as item 1 and 2 of Article 32 of the
Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects dated February 18, 2023, China.
Details regarding the summary statistics utilized in this work are presented in Table 1. To ensure sufficient instrument strength in bidirectional MR analysis, we adopted distinct dataset combinations for opposing directions. The AS→HS estimation utilized GWAS summary statistics from datasets ebi-a-GCST00552912 (for xposure) and finn-b-L12_HIDRADENITISSUP (for outcome), whereas the reverse HS→AS analysis employed datasets finn-b-L12_HIDRADENITISSUP (exposure) and ukb-b-18194 (outcome). This adjustment was necessitated by the complete absence of valid instrumental variables (F-statistic >10) when pairing dataset ebi-a-GCST005529 (exposure) with dataset finn-b-L12_HIDRADENITISSUP (outcome), likely attributable to incomplete SNP coverage in dataset E for trait B-related loci.
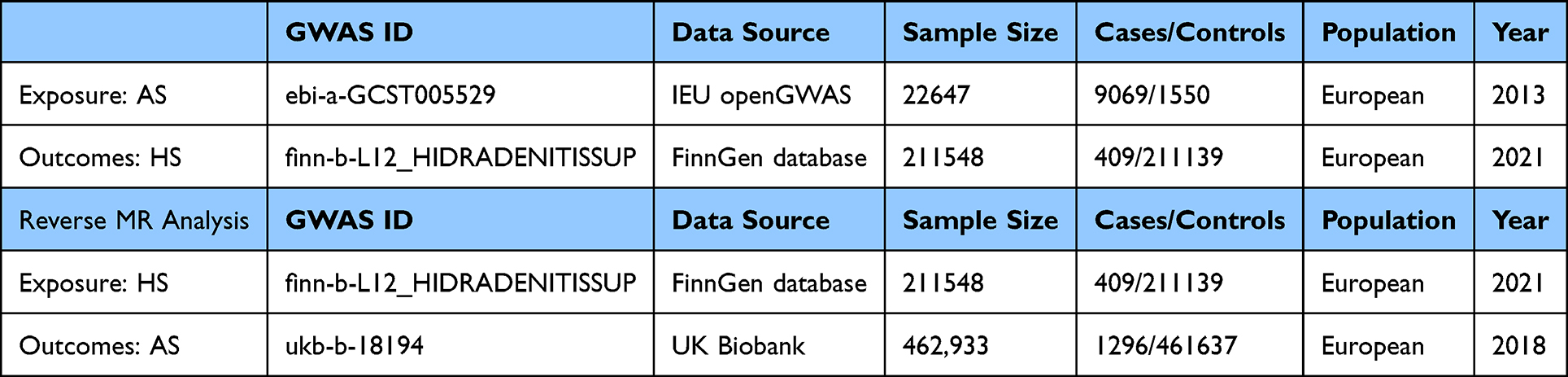 |
Table 1 Comprehensive Details Regarding the GWAS Summary Statistics |
SNP Selection
The most significant and independent SNPs were chosen for the main analysis under the thresholds of p<5e-5 and linkage disequilibrium (LD) r2<1e-3.13 For the exposure analysis of AS, 51 SNPs were associated with AS, and a total of 9 SNPs linked to HS were identified. Given the relatively lenient significance threshold employed, we evaluated the possibility of weak instrument bias using the F statistic, with F > 10 indicating sufficient instrument strength. To avoid any potential confounding links between SNPs and the outcome, the selected variants were further cross-referenced against the NCBI genotype-phenotype database using a threshold of p < 5e-5. Before MR analysis, exposure and outcome datasets were harmonized by aligning SNPs to the same effect allele. 49 independent SNPs were employed as genetic instruments for AS after removal of rs2596501 and rs2517655 due to correlation with other selected variants. Details on IVS are provided in the Supplementary Tables 1 and 2.
MR Estimates
Two‑directional Mendelian randomization analyses were conducted. The first analysis estimated the genetically predicted causal effect of AS on HS, and the second evaluated the reverse causal effect of HS on AS. The primary MR method was the inverse‑variance weighted (IVW) model, which treats all genetic variants as valid instrumental variables. IVW estimates were derived using unconstrained weighted least‑squares regression, with the regression slope representing the causal effect.14 We also applied weighted median estimation and MR‑Egger regression to assess causal relationships under various model specifications.
The weighted median method can provide an unbiased causal estimate when at least 50% of the instrumental variables are valid.15 MR‑Egger regression allows for an intercept term to account for horizontal pleiotropy, yielding more robust estimates even if some instrumental variables are invalid.16 We assessed the reliability and stability of the results using heterogeneity testing, pleiotropy evaluation, and sensitivity analyses.
Heterogeneity among instrumental variables was examined using Cochrane’s Q statistic; a p-value < 0.05 indicated significant heterogeneity, and a random‑effects model was then used. Otherwise, a fixed‑effects model was applied.17 A leave‑one‑out sensitivity analysis was performed by sequentially removing each instrumental variable to test the consistency of MR results.15 The MR‑PRESSO test was used to detect horizontal pleiotropy. The MR‑PRESSO outlier test evaluates the deviation of each SNP from the fitted regression line; larger deviations suggest a higher probability that the SNP is an outlier requiring exclusion.18
The intercept from MR‑Egger regression can be used to detect and adjust for directional pleiotropy. However, MR‑Egger generally yields wider confidence intervals and is therefore considered a supplementary approach to IVW. Accordingly, MR‑Egger was used in cases with significant pleiotropy, and MR‑PRESSO was employed to detect and remove outliers. In the absence of major pleiotropy or outliers, IVW results were used as the primary findings.
We performed all analyses utilizing the two-sample MR package (v0.6.11) and MR-PRESSO package (v1.0) implemented in R programming language (version 4.1.3).
Results
Causal Impact of AS on the Risk of Hidradenitis Suppurativa
Following the implementation of strict exclusion criteria, we detected no evidence of weak instrument bias in our analysis, with F-statistics greater than 10 for AS. No outlier SNPs were identified in the MR-PRESSO analysis. No significant heterogeneity was detected in the Cochran’s Q test for AS (p > 0.05). We therefore applied the fixed-effects model within the IVW framework. Our analysis provided robust evidence supporting a significant causal relationship between AS and HS (IVW OR=3.49, 95% CI=1.43–8.52, P=0.006, Figure 2A). Estimate of IVW were consistent with those obtained from the weighted median method. No significant indication of horizontal pleiotropy was detected by pleiotropy assessment, as demonstrated by a nonsignificant MR‑Egger intercept (p > 0.05), supporting the reliability of IVW estimates for causal inference. Comprehensive outcomes of the robustness checks are summarized in Table 2. The leave-one-out plot is shown in Figure 2B, and the funnel plot is presented in Supplementary Figure 1.
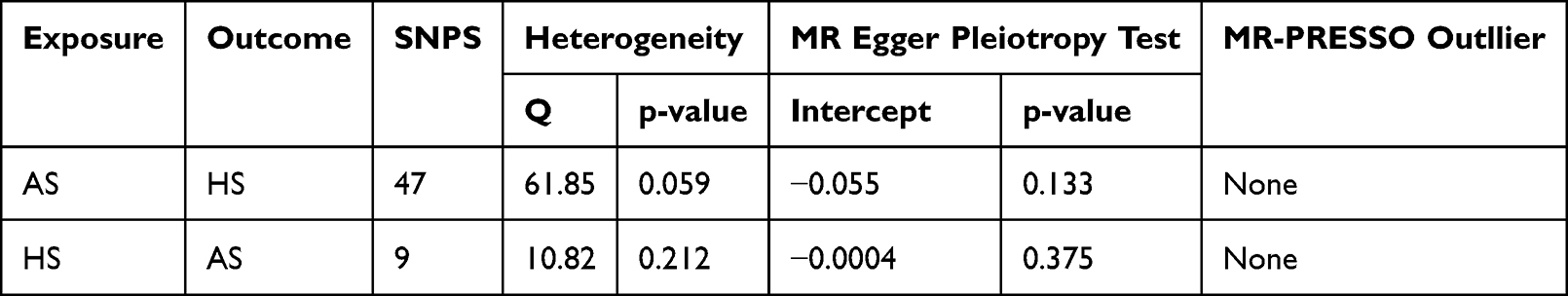 |
Table 2 MR Sensitivity Analyses |
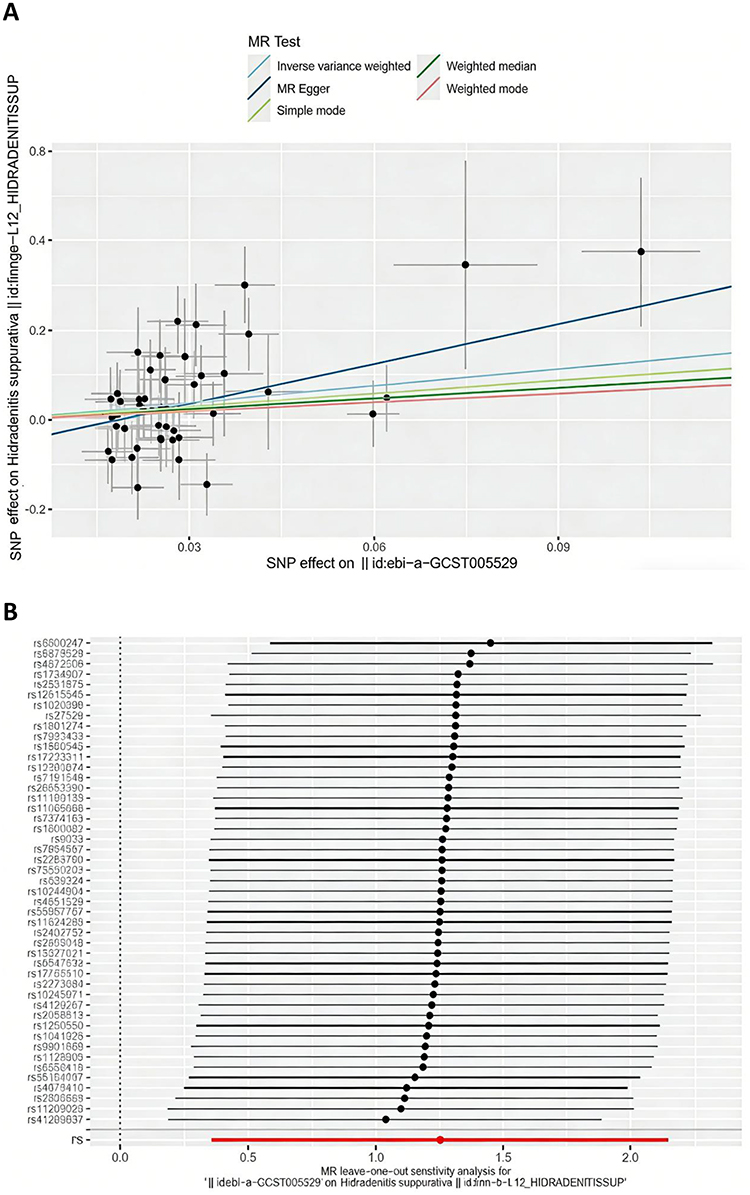 |
Figure 2 (A) The scatter plot generate from the Mendelian randomization (MR) analysis, where the slope of each line represents the MR effect calculated using the IVW model. The scatter plot illustrating the genetically predicted causal effect of AS on HS; (B) The plots of MR and leave-one-out analyses of a genetically causal association between AS on HS. |
Causal Effects of Hidradenitis Suppurativa on AS
We further performed reverse-directional analysis with HS as the exposure and AS as the outcome. The F-statistic for all instrumental variables exceeded 10, indicating no evidence of weak instrument bias. All MR-Egger regression tests yielded non-significant results (pleiotropy test p > 0.05), suggesting no directional pleiotropy. The MR-PRESSO approach did not identify any outlier single-nucleotide polymorphisms(SNPs). Accordingly, fixed-effects models were applied for the AS group. Analysis using the IVW method revealed no causal effect of HS on AS (OR=1.00, 95% CI: 0.9998–1.00, P=0.816). The forest plot illustrating the genetically predicted causal effect of HS on AS is presented in Figure 3A. Detailed results of sensitivity analysis are summarized in Table 2, the leave-one-out plot is displayed in Figure 3B, and the funnel plot is provided in Supplementary Figure 2.
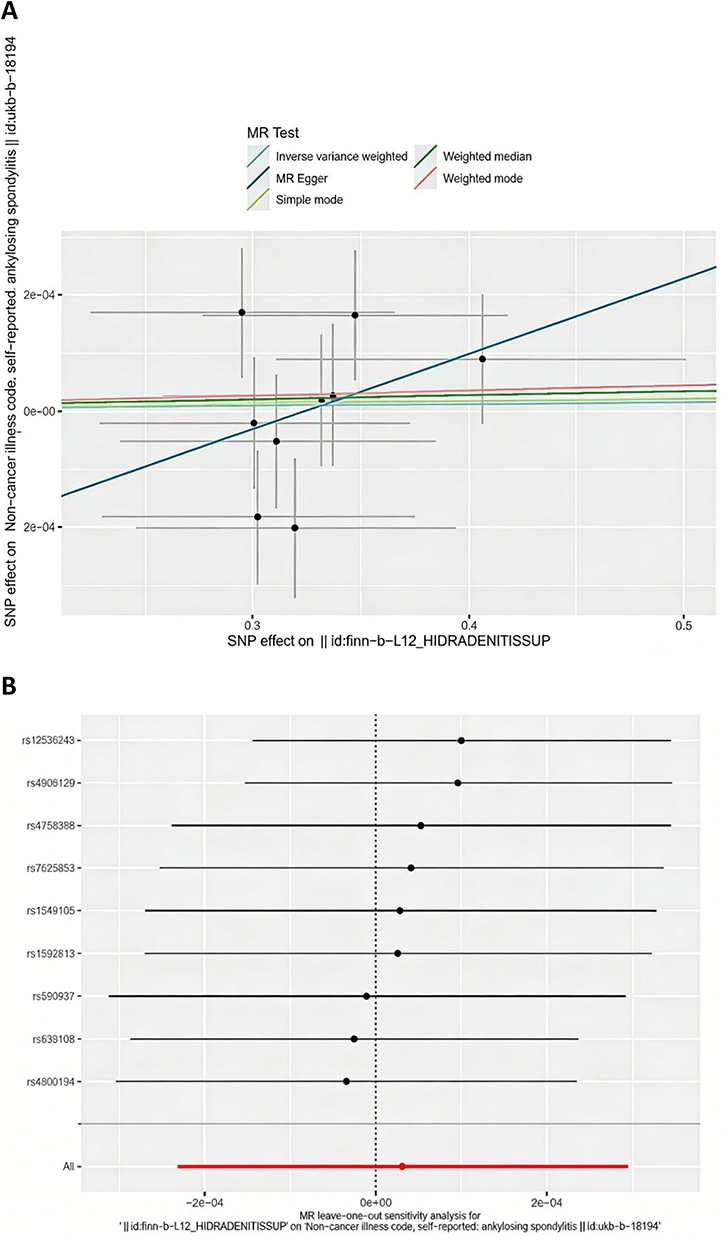 |
Figure 3 (A) The scatter plot generate from the Mendelian randomization (MR) analysis, where the slope of each line represents the MR effect calculated using the IVW model. The scatter plot illustrating the genetically predicted causal effect of HS on AS; (B) The plots of MR and leave-one-out analyses of a genetically causal association between HS on AS. |
Figure 4 shows forest plots assessing the genetically predicted causal associations between AS and HS in both directions.
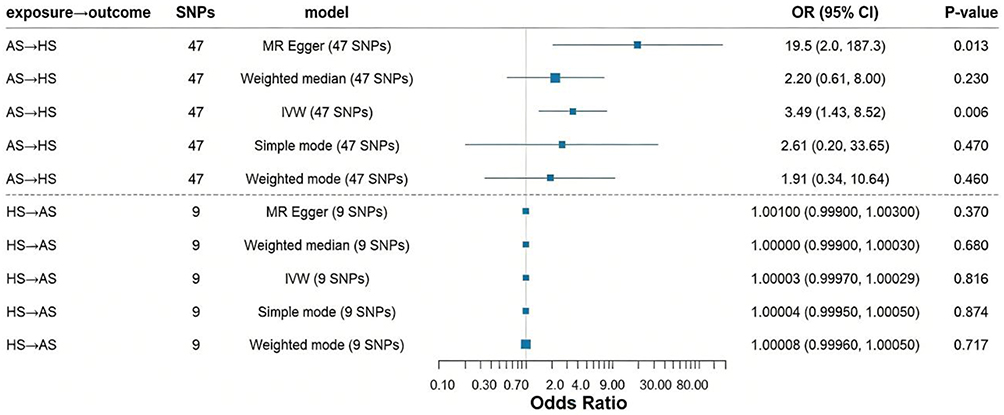 |
Figure 4 For the effect of AS on HS and HS on AS, causal effect estimates are expressed as OR and 95% CI. Abbreviations: AS, Ankylosing Spondylitis; HS, hidradenitis suppurativa. |
Discussion
Using pooled GWAS data, the present study comprehensively explored the causal relationship between AS and HS. To the best of our knowledge, this is the first MR study to investigate the bidirectional causal association between AS and HS. Our bidirectional MR analysis provides robust genetic evidence supporting a unidirectional causal relationship between ankylosing spondylitis (AS) and hidradenitis suppurativa (HS).
Previous studies have reported that 2–28% of patients with HS exhibit axial involvement, while approximately 10% of individuals with axSpA develop HS. The temporal correlation between these conditions suggests a potential shared immunological mechanism.19,20 In the majority of SpA patients, HS occurs before the clinical diagnosis of SpA, with frequent axial involvement. Notably, these two conditions are both strongly associated with HLA‑B27 positivity.21. In essence, AS represents the advanced radiographic stage within the axSpA disease spectrum, sharing identical clinical manifestations and genetic susceptibility (HLA-B27 association), which seemed there is a strong correlation between AS and HS. Our findings align with cohort studies reporting higher HS prevalence in AS populations,20 but extend these observations by establishing temporality and directionality. As patients with axial SpA who present concurrent HS tend to display more severe disease activity,22 systematic evaluation for HS in individuals diagnosed with SpA carries substantial clinical significance. Meanwhile, a considerable number of patients diagnosed with hidradenitis suppurativa (HS) have reported symptoms indicative of inflammatory arthritis.9,21 In studies involving larger patient cohorts, the estimated prevalence of inflammatory arthritis ranges from 0.8% to 5.2%.9 Specifically, the odds ratio for spondyloarthritis is 2.1 (95% confidence interval [CI]: 1.4–3.2), while that for rheumatoid arthritis is 2.0 (95% CI: 1.3–3.0).10 Our results differ somewhat from previous estimates. These discrepancies may be attributed to variations in study design and analytical methodologies. Observational analyses are susceptible to clinical confounding factors that can distort both exposure and outcome assessments, thereby limiting reliable causal inference. Even when a significant association is detected in observational studies, this does not necessarily indicate a direct causal relationship. By contrast, Mendelian randomization is largely unaffected by such confounders and thus enables more robust causal evaluation.
This unidirectional causal relationship may be linked to their pathogenesis. Recent transcriptome‑based investigations have yielded robust support for the notion that shared pathological processes underlying these conditions are associated with the accumulation of neutrophils and macrophages within inflamed tissue sites, accompanied by elevated expression levels of proinflammatory cytokines (IL-1α, IL-1β, IL-17A/F, IL-23, and TNF-α), activation of the JAK–STAT signaling pathway, and genetic variants associated with B-cell costimulation, such as CXCR5.23 In lesional skin specimens from patients with HS, the frequency of CD4+ T cells expressing IL-17 and TNF was significantly elevated. Notably, anti-TNF therapy led to a marked reduction in the number of IL-17-expressing CD4+ T cells within HS skin lesions.24
The AS→HS causality may be mediated by shared genetic architectures in IL-17/IL-23-related pathways. Our sensitivity analysis showing attenuated causality after excluding IL-23R region variants (rs11209026) supports this hypothesis. Mechanistically, HLA-B27 misfolding in AS could induce ER stress and IL-23 overproduction,25 which primes γδ T cells to secrete IL-17A – a cytokine directly implicated in both axial joint inflammation and HS-associated follicular hyperkeratosis. The null HS→AS relationship suggests that HS-related genetic variants (eg, NCSTN mutations affecting γ-secretase26) primarily drive local skin barrier defects rather than systemic autoimmunity This compartmentalization contrasts with psoriasis-associated variants (eg, IL23R), which frequently exhibit pleiotropic effects on both cutaneous and joint inflammation.26
Clinically, our results advocate for dermatologic surveillance in AS management. The increased HS risk per AS genetic predisposition suggests that early recognition of HS in AS patients could prevent disease progression through targeted immunomodulation. Notably, the two biologic medications authorized for the treatment of HS include adalimumab (anti-TNF-α) and secukinumab (anti-IL-17) by the U.S. Food and Drug Administration,5,6 supporting biological plausibility of our genetic findings.
Our analysis was limited by potential horizontal pleiotropy despite rigorous MR-Egger regression (intercept p=0.055). Future studies should validate causality using drug-target MR with AS-related biologics (eg, IL-17 inhibitor gene scores) as instruments. Moreover, the European ancestry of GWAS data may limit generalizability. In addition, although we found strong genetic evidence for a causal effect of AS on HS, and our analysis predicts that approximately 2–10% of HS cases observed in an AS patient cohort are attributable to the causal effect of AS itself, based on the genetic evidence from MR (Supplementary Note 1), the estimated odds ratio (OR=3.49) was accompanied by a wide confidence interval, This suggests that the precise magnitude of the risk increase requires more precise quantification in future, larger-scale studies. Nonetheless, the present result robustly supports the shared etiological pathways between the two diseases. In the reverse MR analysis (HS→AS), we utilized GWAS data for HS from the FinnGen consortium (409 cases). The relatively small number of cases may limit the statistical power to detect weak to moderate genetic associations. Therefore, the null finding of no causal effect of HS on AS could partly result from insufficient power rather than definitively indicate a lack of biological association. Future replication using larger GWAS datasets for HS is warranted to confirm this finding. We utilized different dataset combinations for the forward (IEU/FinnGen) and reverse (FinnGen/UK Biobank) analyses, the core reason for using different data sources in the forward and reverse analyses was to ensure that both analyses were computationally and scientifically feasible. When attempting the reverse MR analysis (HS → AS) using the same combination as in the forward direction (ie, FinnGen HS data as the exposure), we encountered a critical issue: the relatively small number of HS cases (n=409) in FinnGen resulted in a GWAS with insufficient power to identify a robust set of genetic instruments. After applying standard MR quality control steps (eg, genome-wide significance threshold, clumping for independence), no single-nucleotide polymorphisms (SNPs) remained as valid instrumental variables (IVs). An MR analysis cannot be performed with zero IVs. Therefore, to meaningfully test the reverse causal hypothesis, we were compelled to seek a larger GWAS for HS as the exposure source, which led us to the UK Biobank summary statistics. This approach prioritizes analytical validity (having valid IVs) and statistical independence in each direction over symmetric data sources, which is a common and rigorous practice in bidirectional MR when data resources are asymmetric.
In conclusion, Our study establishes AS as an independent risk factor for HS development. For dermatologists, this implies: HS patients with comorbid AS may represent a distinct subtype requiring combined rheumatologic-dermatologic management. Existing AS therapies (eg, TNF/IL-17 inhibitors) could be prioritized for HS patients with elevated inflammatory markers. Future RCTs should test this targeted treatment approach. AS patients, especially those with HLA-B27 positivity or elevated inflammatory markers (CRP/IL-17), should undergo routine dermatologic evaluation for early HS signs (eg, axillary/inguinal nodules). HS patients without axial symptoms need not routinely screen for AS, given the absence of reverse causation.
Data Sharing Statement
The datasets utilized in the present study are available in public online repositories. Detailed information regarding the repository(ies) and corresponding accession number(s) is provided in the main article or Supplementary Materials.
Acknowledgments
Guizhen Chang and Jun Zhang are co-first authors for this study. The authors appreciate the IIBDGC Consortium and FINNGEN Consortium for providing access to the GWAS datasets.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received support from Binhai New Area Health Commission of Tianjin Municipality (No. 2024BWKQ01).
Disclosure
The authors confirm that there are no known conflicts of interest associated with this publication.
References
1. Wenker KJ, Quint JM. Ankylosing Spondylitis. In: StatPearls. Treasure Island (FL): StatPearls Publishing LLC; 2025.
2. Ingram JR. The epidemiology of hidradenitis suppurativa. Br J Dermatol. 2020;183(6):990–10. doi:10.1111/bjd.19435
3. Garbayo-Salmons P, Moreno Martínez-Losa M, Exposito-Serrano V, et al. Insights into new-onset arthritis in patients with hidradenitis suppurativa. Acta Derm Venereol. 2024;104:adv40145. doi:10.2340/actadv.v104.40145
4. Gisondi P, Montalto E, Curic T, et al. Certolizumab pegol in severe hidradenitis suppurativa in pregnancy: a case report. SAGE Open Med Case Rep. 2025;13. doi:10.1177/2050313X241311374
5. Kimball AB, Jemec GBE, Alavi A, et al. Secukinumab in moderate-to-severe hidradenitis suppurativa (SUNSHINE and SUNRISE): week 16 and week 52 results of two identical, multicentre, randomised, placebo-controlled, double-blind Phase 3 trials. Lancet. 2023;401(10378):747–761. doi:10.1016/S0140-6736(23)00022-3
6. Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. New Engl J Med. 2016;375(5):422–434. doi:10.1056/NEJMoa1504370
7. Sukhanova AM, Gilavian MA, Melnik EV, et al. An overview of adalimumab therapy for ankylosing spondylitis. Curr Rheumatol Rev. 2024;20(5):501–513. doi:10.2174/0115733971289295240223095751
8. Saran A, Nishizaki D, Lippman SM, et al. Interleukin-17: a pleiotropic cytokine implicated in inflammatory, infectious, and malignant disorders. Cytokine Growth Factor Rev. 2025;83:35–44. doi:10.1016/j.cytogfr.2025.01.002
9. Hanna N, Silverberg OM, Reaume M, et al. Incidence, prevalence, and predictors of inflammatory arthritis in patients with hidradenitis suppurativa: a systematic review and meta-analysis. Int J Dermatol. 2022;61(9):1069–1079. doi:10.1111/ijd.15860
10. Almuhanna N, Finstad A, Alhusayen R. Association between hidradenitis suppurativa and inflammatory arthritis: a systematic review and meta-analysis. Dermatology. 2021;237(5):740–747. doi:10.1159/000514582
11. Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601. doi:10.1136/bmj.k601
12. Cortes A, Hadler J, Pointon JP, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. 2013;45(7):730–738.
13. Ference BA, Majeed F, Penumetcha R, et al. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial Mendelian randomization study. J Am Coll Cardiol. 2015;65(15):1552–1561. doi:10.1016/j.jacc.2015.02.020
14. Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in Mendelian randomization: comparison of allele score and summarized data methods. Stat Med. 2016;35(11):1880–1906. doi:10.1002/sim.6835
15. Bowden J, Davey Smith G, Haycock PC, et al. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–314. doi:10.1002/gepi.21965
16. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–389. doi:10.1007/s10654-017-0255-x
17. Bowden J, Hemani G, Davey Smith G. Invited commentary: detecting individual and global horizontal pleiotropy in Mendelian randomization-a job for the humble heterogeneity statistic? Am J Epidemiol. 2018;187(12):2681–2685. doi:10.1093/aje/kwy185
18. Chen WT, Chi CC. Association of hidradenitis suppurativa with inflammatory bowel disease: a systematic review and meta-analysis. JAMA Dermatol. 2019;155(9):1022–1027. doi:10.1001/jamadermatol.2019.0891
19. Fauconier M, Reguiai Z, Barbe C, et al. Association between hidradenitis suppurativa and spondyloarthritis. Joint Bone Spine. 2018;85(5):593–597. doi:10.1016/j.jbspin.2017.09.005
20. Rondags A, Arends S, Wink FR, et al. High prevalence of hidradenitis suppurativa symptoms in axial spondyloarthritis patients: a possible new extra-articular manifestation. Semin Arthritis Rheum. 2019;48(4):611–617. doi:10.1016/j.semarthrit.2018.03.010
21. Richette P, Molto A, Viguier M, et al. Hidradenitis suppurativa associated with spondyloarthritis -- results from a multicenter national prospective study. J Rheumatol. 2014;41(3):490–494. doi:10.3899/jrheum.130977
22. Lee JH, Kwon HS, Jung HM, et al. Prevalence and comorbidities associated with hidradenitis suppurativa in Korea: a nationwide population-based study. J Eur Acad Dermatol Venereol. 2018;32(10):1784–1790. doi:10.1111/jdv.15071
23. Rumberger BE, Boarder EL, Owens SL, et al. Transcriptomic analysis of hidradenitis suppurativa skin suggests roles for multiple inflammatory pathways in disease pathogenesis. Inflammation Res. 2020;69(10):967–973. doi:10.1007/s00011-020-01381-7
24. Meier K, Schloegl A, Poddubnyy D, et al. Skin manifestations in spondyloarthritis. Ther Adv Musculoskelet Dis. 2020;12:1759720x20975915. doi:10.1177/1759720X20975915
25. Jah N, Jobart-Malfait A, Ermoza K, et al. HLA-B27 subtypes predisposing to ankylosing spondylitis accumulate in an endoplasmic reticulum-derived compartment apart from the peptide-loading complex. Arthritis Rheumatol. 2020;72(9):1534–1546. doi:10.1002/art.41281
26. Oliveira CB, Romo-Tena J, Patino-Martinez E, et al. Neutrophil extracellular traps activate Notch-γ-secretase signaling in hidradenitis suppurativa. J Allergy Clin Immunol. 2025;155(1):188–198. doi:10.1016/j.jaci.2024.09.001
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.