")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 8
Cathelicidin suppresses colon cancer development by inhibition of cancer associated fibroblasts
Authors Cheng M, Ho S, Yoo JH, Tran D, Bakirtzi K, Su B, Tran D, Kubota Y, Ichikawa R, Koon HW
Received 10 July 2014
Accepted for publication 8 October 2014
Published 17 December 2014 Volume 2015:8 Pages 13—29
DOI https://doi.org/10.2147/CEG.S70906
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Andreas M. Kaiser
Michelle Cheng,1,* Samantha Ho,1,* Jun Hwan Yoo,1,2,* Deanna Hoang-Yen Tran,1,* Kyriaki Bakirtzi,1 Bowei Su,1 Diana Hoang-Ngoc Tran,1 Yuzu Kubota,1 Ryan Ichikawa,1 Hon Wai Koon1
1Center for Inflammatory Bowel Diseases, Division of Digestive Diseases, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, USA; 2Digestive Disease Center, CHA University Bundang Medical Center, Seongnam, Republic of Korea
*These authors share co-first authorship
Background: Cathelicidin (LL-37 in humans and mCRAMP in mice) represents a family of endogenous antimicrobial and anti-inflammatory peptides. Cancer-associated fibroblasts can promote the proliferation of colon cancer cells and growth of colon cancer tumors.
Methods: We examined the role of cathelicidin in the development of colon cancer, using subcutaneous human HT-29 colon-cancer-cell-derived tumor model in nude mice and azoxymethane- and dextran sulfate-mediated colon cancer model in C57BL/6 mice. We also determined the indirect antitumoral mechanism of cathelicidin via the inhibition of epithelial–mesenchymal transition (EMT) of colon cancer cells and fibroblast-supported colon cancer cell proliferation.
Results: Intravenous administration of cathelicidin expressing adeno-associated virus significantly reduced the size of tumors, tumor-derived collagen expression, and tumor-derived fibroblast expression in HT-29-derived subcutaneous tumors in nude mice. Enema administration of the mouse cathelicidin peptide significantly reduced the size and number of colonic tumors in azoxymethane- and dextran sulfate-treated mice without inducing apoptosis in tumors and the adjacent normal colonic tissues. Cathelicidin inhibited the collagen expression and vimentin-positive fibroblast expression in colonic tumors. Cathelicidin did not directly affect HT-29 cell viability, but did significantly reduce tumor growth factor-ß1-induced EMT of colon cancer cells. Media conditioned by the human colonic CCD-18Co fibroblasts promoted human colon cancer HT-29 cell proliferation. Cathelicidin pretreatment inhibited colon cancer cell proliferation mediated by media conditioned by human colonic CCD-18Co fibroblasts. Cathelicidin disrupted tubulin distribution in colonic fibroblasts. Disruption of tubulin in fibroblasts reduced fibroblast-supported colon cancer cell proliferation.
Conclusion: Cathelicidin effectively inhibits colon cancer development by interfering with EMT and fibroblast-supported colon cancer cell proliferation.
Keywords: colon cancer, epithelial–mesenchymal transition, fibroblasts
Introduction
Cathelicidin is a family of peptides with established antimicrobial and anti-inflammatory functions.1 Previous reports have shown that cathelicidin peptide administration via enema ameliorates dextran sulfate (DSS)-mediated colitis and Clostridium difficile-mediated intestinal inflammation.2,3 Bacterial vector or DNA plasmid-mediated expression of mouse cathelicidin can also confer similar anti-inflammatory effects in a DSS model of mouse colitis.4,5 The anti-inflammatory effects of cathelicidin may partially depend on its ability to neutralize lipopolysaccharide.6 An alternative mechanism for the anti-inflammatory effects of cathelicidin is that it may inhibit C. difficile toxin A- and B-mediated tumor necrosis factor alpha (TNFα) expression in human peripheral blood monocytes via suppression of the nuclear factor kappa-light-chain-enhancer of activated B cell-dependent pathway.2 The anti-inflammatory effects of cathelicidin can also be explained by how the human version LL-37 can inhibit lipoteichoic acid-induced TNFα and interleukin-6 (IL-6) production in macrophages via suppressing p38 and Akt pathways.7
According to information from American Cancer Society (http://www.cancer.org/cancer/colonandrectumcancer/detailedguide/colorectal-cancer-key-statistics), colorectal cancer is the third most common cancer in both sexes. It is also the third most common cause of cancer deaths in the United States. Despite recent medical advancement, many colorectal cancers are undiagnosed until late stages. The rate of treatment success and survival declines with advancing stages, and new solutions and medical therapies are still being actively sought.
The expression of cathelicidin in different cancer tumors is very diverse.8,9 LL-37 expression is increased in breast, ovarian, and lung cancers,10–12 but it is decreased in colorectal cancer.13 Cathelicidin can also suppress gastric cancer cell proliferation via a pathway mediated by the bone morphogenetic protein.14 The role of cathelicidin in colorectal cancer is still being investigated, but its antitumoral mechanism is still not fully understood.
Recent reports have shown that endogenous cathelicidin expression modulates azoxymethane (AOM)-mediated colon cancer in mice.13 Endogenous cathelicidin expression is downregulated in human colon tumors and may be unable to confer protection against colon cancer development.13 Cathelicidin and its analog FK-16 can induce apoptosis in human colon cancer HCT116 cells via a p53-dependent mechanism.13,15 However, other cathelicidin analogs such as FF/CAP18 and Ceragenins CSA-13 can inhibit HCT116 cell proliferation without relying on the p53-dependent mechanism in vitro.16,17 All available evidence suggests that cathelicidin may become a novel therapeutic approach against colon cancer. However, the antitumoral mechanism of cathelicidin in colon cancer development has not been fully elucidated.
From the findings that cancer-associated fibroblasts (CAFs) promote cell proliferation of colon cancer cells,18 it is possible that cathelicidin may inhibit colon cancer indirectly. We hypothesize that cathelicidin indirectly inhibits colon tumor growth in vivo. We have determined that cathelicidin overexpression and cathelicidin peptide administration via enema can inhibit subcutaneous colon cancer tumor xenograft growth in nude mice and colonic tumor growth in AOM- and DSS-treated mice, respectively. Furthermore, we explored whether cathelicidin-mediated inhibition of fibroblasts indirectly reduces colon cancer cell proliferation. These findings provide a novel scientific basis of cathelicidin-mediated therapy of colorectal cancer.
Materials and methods
Cell culture
HT-29 human colon cancer cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen, Carlsbad, CA, USA) containing 10% fetal calf serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen). CCD-18Co human colon cancer cells were cultured in Minimum Essential Medium (MEM) (Invitrogen) containing 10% fetal calf serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen).19 All cultured cells were purchased from American Type Culture Collection (Manassas, VA, USA).
Adeno-associated viral vector of cathelicidin expression
The human cathelicidin CAMP gene overexpressing the adeno-associated virus (AAV) was generated by Vector Biolabs, Inc. (Philadelphia, PA, USA). The construct carries the full complementary DNA (cDNA) sequence of human cathelicidin CAMP and hemagglutinin (HA) tag sequence, ie, CAMP-HA-AAV. The control AAV carries only the HA sequence but no CAMP sequence, ie, HA-AAV.
Subcutaneous tumor in nude mouse model
Human colon cancer HT-29 cells (1×106 cells) in Hank’s Balanced Salt solution (100 μL) were injected under the skin of the left and right flanks of nude mice (Stock number #002019; Jackson Laboratories, Sacramento, CA, USA). The injected nude mice were housed in the University of California Los Angeles (UCLA) animal facility (Division of Laboratory Animal Medicine) under standard conditions with a 12-hour light period and a 12-hour dark period per day at 25°C (room temperature). They were housed in disposable plastic cages with high-efficient particulate air (HEPA)-filtered air circulation, autoclaved bedding, standard animal chow, and sterile water ad libitum.2,20 HA-AAV and CAMP-HA-AAV (4×1012 genomic copies in 100 μL) were injected into nude mice intravenously via tail veins under transient isoflurane anesthesia. Subcutaneous tumors were collected for analyses on day 28. The experimental details are described in Figure 1A. Each group consisted of ten mice per group. Each mouse carried two subcutaneous tumors (left and right flanks).
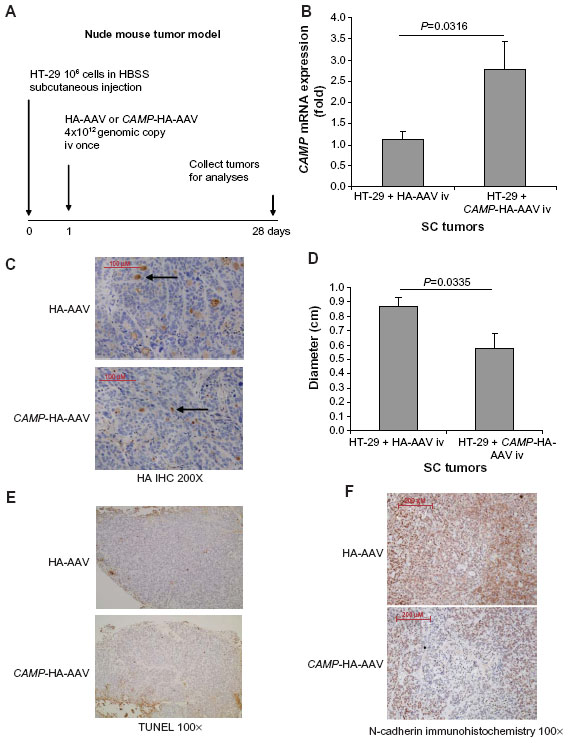 | Figure 1 Intravenous cathelicidin expressing AAV administration inhibited EMT in HT-29-derived subcutaneous tumors in nude mice. (A) Experimental plan. (B) Human cathelicidin mRNA expression in subcutaneous tumors. The CAMP-HA-AAV expressed human cathelicidin mRNA in tumors. (C) Immunohistochemistry of HA tag. AAV-mediated infected cells were stained by HA antibody and appeared brown. The extent of infection was similar among tumors from the HA-AAV and CAMP-HA-AAV groups. (D) Diameters of subcutaneous tumors. Intravenous CAMP-HA-AAV significantly reduced subcutaneous tumor diameter in nude mice. (E) TUNEL staining. Apoptotic cells should be staining with intense brown color. There was no apoptosis in the cathelicidin-expressing and control groups. (F) Immunohistochemistry of N-cadherin. N-cadherin-positive cells were identified by brown color staining. Intravenous cathelicidin-expressing AAVs reduced mesenchymal cell marker N-cadherin expression in the subcutaneous tumors. |
In vivo colon cancer model
Eight-week-old (C57BL/6J) female mice (Stock number #000664; Jackson Laboratories) were housed in the UCLA animal facility (Division of Laboratory Animal Medicine) under standard conditions with a 12-hour light period and a 12-hour dark period per day at 25°C (room temperature). They were housed in disposable plastic cages with HEPA-filtered air circulation, autoclaved bedding, standard animal chow, and sterile water ad libitum.2,20 They were given a single intraperitoneal injection of AOM (10 mg/kg body weight; Sigma, St Louis, MO, USA). Seven days later, 2% DSS ~40,000 molecular weight (#42867; Sigma) was given in the drinking water for 5 days after which the mice were switched to normal drinking water until the end of the experiment for three cycles as previously described.21 Specifically, mice were given an AOM injection on day 0, and were provided with water on days 0–6, DSS on days 7–13, water on days 14–20, DSS on days 21–27, water on days 28–34, DSS on days 35–41, and water from day 42 until the end of experiment on week 18. Mouse cathelicidin-related antimicrobial peptide (mCRAMP) (10 mg/kg body weight synthesized from American Peptide, Inc. [Sunnyvale, CA, USA]) was administered every 2 days between weeks 10 and 18 via enema. The mice were oriented head-down and tail-up for 10 seconds to prevent leakage. The enema injection was given via the insertion of 3-cm PE10 catheter attached to a 25-G needle into the rectum without anesthesia. The colonic tumors and adjacent normal tissues were obtained for analyses. The experimental details are described in Figure 2A. The animal studies were approved by the Institutional Animal Research Committee of UCLA. Each group had ten to twelve mice.
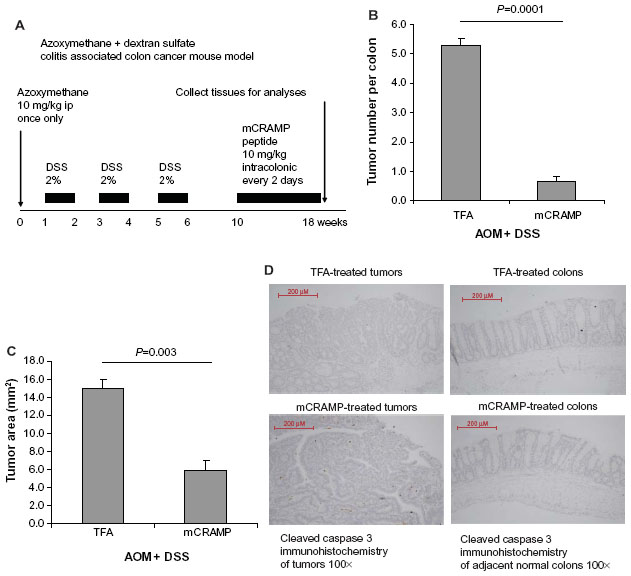 | Figure 2 Cathelicidin peptide administration via enema inhibited colitis-associated colonic tumor development. (A) Experimental plan of colitis-associated colon cancer mouse model. (B) Tumor number per colon. (C) Average colonic tumor area. Mouse cathelicidin mCRAMP administration via enema significantly reduced colonic tumor number and colonic tumor size of mice treated with AOM and DSS. (D) TUNEL staining. Apoptotic cells should be stained with intense brown color. There was no apoptosis in colonic tumors and adjacent normal colonic tissues of the cathelicidin-treated and control groups. |
Immunohistochemistry and TUNEL staining
Tumor tissues were fixed in 4% paraformaldehyde and embedded in paraffin. After incubation with blocking buffer, sections were incubated with a rabbit anti-N-cadherin antibody (#GTX112733; Genetex, Irvine, CA, USA; 1:1000 dilution), goat polyclonal anti-vimentin antibody (sc-7557; Santa Cruz Biotechnology, Dallas, TX, USA; 1:50 dilution), or anti-HA-Tag antibody (#3724; Cell Signaling Technology, Inc., Danvers, MA, USA; 1:500 dilution) overnight at 4°C. After washing, sections were incubated with donkey anti-goat IgG or bovine anti-rabbit IgG, and the slides were stained with an ABC kit for color development (Santa Cruz, sc-2018). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining and immunohistochemistry were carried out with assistance from the Translational Pathology Core Laboratory (TPCL) of UCLA. Images were analyzed with a Zeiss AX10 microscope.
Masson trichrome staining
Staining of collagen deposition of colonic tissue sections was performed with the Masson Trichrome staining kit (HT-10) and Bouin solution (HT-10132) as previously described.19
Real-time reverse transcription polymerase chain reaction
The total RNA from colonic tumors in colitis associated colon cancer model, adjacent normal colonic tissues in colitis-associated colon cancer model, healthy normal control tissue, subcutaneous tumors in nude mice, and cultured cells were extracted by RNA mini kit (#74106; Qiagen RNeasy kit, Valencia, CA, USA). The RNA was converted to cDNA by iScript cDNA Synthesis kit (170-8891; Bio-Rad, Hercules, CA, USA). The messenger RNA (mRNA) expression was determined using their respective inventoried real-time primer sets obtained from Applied Biosystems (Thermo Fisher Scientific, Waltham, MA, USA) (Table S1) and iTaq Universal Probe Supermix (#172-5135; Bio-Rad) according to the manufacturer’s instructions. Levels were normalized to equal levels of human 18S or mouse GAPDH mRNA, and results were expressed as fold induction compared to their respective control, as previously described.20
Western blot analyses
Cells were lysed in 1× blue loading buffer (#7722; Cell Signaling Technology, Inc.). Equal amounts of cell extracts were fractioned by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and proteins were transferred onto nitrocellulose membranes (400 mA for 1.5 hours; Bio-Rad). Membranes were blocked in 5% nonfat milk in TBST (50 mM Tris, pH 7.5, 0.15 M NaCl, 0.05% Tween 20), and then incubated with antibodies against various proteins: β-tubulin #2128 (Cell Signaling Technology, Inc.); GAPDH sc25778 (Santa Cruz Biotechnology), E-cadherin #3195 (Cell Signaling Technology, Inc.), N-cadherin #GTX112733, Twist1 #GTX121924, and Slug #GTX127310 (Genetex, South San Francisco, CA, USA). Horseradish peroxidase-labeled anti-rabbit antibody (#7074; Cell Signaling Technology, Inc.) was detected by chemiluminescence (Fisher Scientific, Pittsburgh, PA, USA). Signals were captured by a luminescent image analyzer (LAS4000; Fujifilm, Tokyo, Japan).
Human colonic fibroblast-modulated colon cancer cell proliferation
The human colonic CCD-18Co fibroblasts in serum-free DMEM were first incubated with LL-37 and trifluoroacetic acid (TFA) 0.1% (LL-37 vehicle solution) for 6 hours to influence the cell proliferation mediator expression mechanism. Afterward, the CCD-18Co fibroblasts were further incubated with fresh media without LL-37 or TFA for 24 hours. The purpose of this media change was to ensure that TFA or LL-37 did not exist in HT-29 cell culture. The 24-hour incubation period with TFA/LL-37-free media provided sufficient time for the fibroblasts to secrete cell-proliferation-stimulating factors into the conditioned media. The resulting media contained no cathelicidin, and thus it was shown that cathelicidin would not directly influence HT-29 cell viability. The medium conditioned by CCD-18Co fibroblasts was then transferred to HT-29 cell culture and incubated for up to 72 hours. Cell proliferation of HT-29 cells was determined by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assays.
Cell viability MTS assay
HT-29 and CCD-18Co cells, seeded on 96-well plates (106 cells/plate) were treated with TFA 0.1% or LL-37 1–10 μM. After 24 hours, 20 μL of CellTiter AQueous One solution (MTS tetrazolium compound, G3580; Promega, Fitchburg, WI, USA) was added to each well and incubated at 37°C for 1 hour. Absorbance at 490 nm (indicating cell viability) was measured using a 96-well plate reader as previously described.22
Tubulin tracker staining
CCD-18Co fibroblasts (2×105 cells/well) were seeded to chamber slides and were exposed to TFA 0.1% or LL-37 for 24 hours. The cells were incubated with a tubulin tracker (T34075; Invitrogen) for 45 minutes, followed by Hoechst 33342 (1 drop per well) (H3570; Invitrogen) according to manufacturer’s instructions. The stained cells were immediately covered and viewed under a Zeiss AX10 confocal microscope.
Immunoprecipitation
CCD-18Co fibroblasts (2×105 cells/well) were seeded to chamber slides and were exposed to TFA 0.1% or LL-37 for 24 hours. The cell lysate was immunoprecipitated with anti-β-tubulin antibody (#sc-9104; Santa Cruz Biotechnology) using the Pierce classic IP kit #26146 (Fisher Scientific) according to manufacturer’s instructions. The lysate was eluted and used for Western blot analyses and LL-37 ELISA (enzyme-linked immunosorbent assay) (HK321; Hycult Biotech, Plymouth Meeting, PA).
Statistical analyses
Quantitative results were expressed with error bars as mean ± standard error of the mean (SEM). Results were analyzed using the Prism professional statistics software program (Graphpad, San Diego, CA, USA). Unpaired Student’s t-tests were used for intergroup comparisons. Only P-values of statistically significant differences are shown in the figures, and results with P<0.05 are regarded as statistically significant.
Results
Viral expression of cathelicidin inhibits subcutaneous tumor development in nude mice
To further understand the role of cathelicidin in the inhibition of tumoral development, human cathelicidin CAMP gene-expressing AAV was injected intravenously to nude mice implanted with human colon cancer HT-29-derived subcutaneous tumors (Figure 1A). Human cathelicidin CAMP gene mRNA expression was induced in the subcutaneous tumors of the nude mice with CAMP-expressing AAV injection, and was compared with those with HA-expressing control AAV injection (Figure 1B). Equal AAV infection efficiency was demonstrated in both CAMP-HA-AAV and control HA-AAV groups as observed by HA-tag immunohistochemistry (Figure 1C). CAMP-expressing group showed significantly reduced diameter of subcutaneous tumors compared to HA-AAV-expressing control group (Figure 1D). However, cathelicidin overexpression did not lead to apoptosis, as reflected by weak TUNEL staining (Figure 1E). This suggests that cathelicidin inhibits tumor development without inducing apoptosis.
Cathelicidin inhibits cancer-associated fibroblasts
Since cathelicidin fails to induce cancer cell apoptosis directly in vivo, we believe that cathelicidin can mediate an indirect pathway to inhibit colon tumor development. CAFs can facilitate cancer cell proliferation and tumor development.18 Some CAFs are developed from colon cancer cells via epithelial–mesenchymal transition (EMT). N-cadherin immunohistochemistry identified cells with mesenchymal characteristics in the subcutaneous HT-29-derived tumors in nude mice (Figure 1F). CAMP-expressing group showed reduced N-cadherin expression of subcutaneous tumors compared to HA-AAV-expressing control group (Figure 1F), indicating cathelicidin-mediated inhibition of EMT. CAFs express collagen,23 and we therefore determined tumoral collagen expression to reflect CAF activity. Using Masson Trichrome staining, prominent collagen deposition was found in the subcutaneous tumors in nude mice (Figure 3A). CAMP-expressing group showed significantly reduced tumoral collagen deposition compared to HA-AAV-expressing control group (Figure 3A). Alternatively, CAMP-expressing group had significantly lower human collagen COL1A2 mRNA expression than HA-AAV-expressing control group (Figure 3B). This reflects that the human-cell-derived CAFs in the subcutaneous tumors contribute to the tumoral collagen deposition.
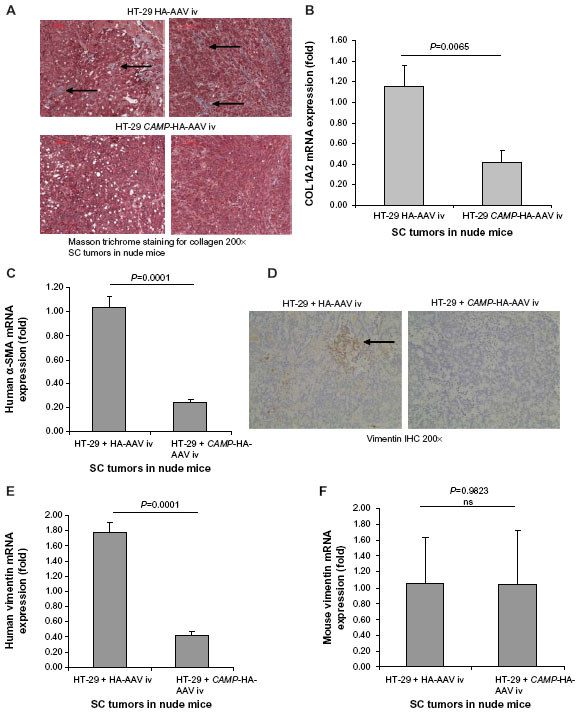 | Figure 3 Intravenous cathelicidin-expressing AAV administration inhibited human cell-derived fibroblasts in HT-29 derived subcutaneous tumors in nude mice. (A) Collagen deposition (blue color staining indicated by arrows) of subcutaneous tumors in nude mice was identified by Masson Trichrome staining. Intravenous cathelicidin-expressing AAVs reduced collagen deposition in tumors. (B) Human collagen COL1A2 mRNA expression in subcutaneous tumors of nude mouse model. Intravenous cathelicidin-expressing AAVs significantly reduced human-cell-derived fibroblast secretion product collagen in the subcutaneous tumors. (C) Human fibroblast marker α-SMA mRNA expression in subcutaneous tumors. Cathelicidin treatment led to significant reduction of human-cancer-cell-derived mesenchymal cells. (D) Immunohistochemistry of total (human + mouse) vimentin in subcutaneous tumors. Cathelicidin treatment led to reduced protein expression of vimentin in tumors. (E) Human fibroblast marker vimentin mRNA expression in subcutaneous tumors of nude mice. (F) Mouse vimentin mRNA expression in subcutaneous tumors of nude mice. Cathelicidin-expressing AAV infection led to significant reduction of human but not mouse vimentin mRNA expression in the subcutaneous tumors. |
Colon cancer cells undergoing EMT express alpha smooth muscle actin (α-SMA) and vimentin (mesenchymal cell marker) that resembles fibroblast characteristics.24,25 In the subcutaneous tumors of nude mice, the cathelicidin-expressing group had significantly reduced tumoral α-SMA mRNA expression (Figure 3C) as well as vimentin protein expression (Figure 3D), suggesting inhibition of EMT and CAF generation. There was a significant reduction of only human vimentin (HT-29-cell-derived), but not mouse vimentin (host) mRNA expression in the cathelicidin-expressing group (Figure 3E and F). This suggests that cathelicidin inhibits CAF generation from the human colon cancer cells but does not inhibit the mouse fibroblast infiltration of the host.
Administration of cathelicidin peptide inhibited colon cancer development in mice
To confirm whether cathelicidin administration can be an effective therapeutic approach against colon tumor development in vivo, we administered mouse cathelicidin peptide via enema to a colonic cancer mouse model as described in Figure 2A. The mouse cathelicidin dose and enema administration approach were similar to the protocol used to treat DSS-induced colitis in mice.3 We found that mouse cathelicidin administration during the last 8 weeks of the experimental period significantly reduced the tumor number per colon and average tumor area in mice (Figure 2B and C). Similar to the nude mouse model (Figure 1E), cathelicidin did not trigger apoptosis in both colonic tumors and adjacent normal colonic tissues as shown by TUNEL staining (Figure 2D).
We also found prominent collagen deposition in the colonic tumors of mice as identified by Masson Trichrome staining (Figure 4A). Mouse cathelicidin peptide administration via enema substantially reduced collagen deposition in the colonic tumors (Figure 4A). Alternatively, the increased collagen COL1A2 mRNA expression in the colonic tumors was significantly reduced to low levels comparable to colonic tissues from normal control mice (Figure 4B). Normal control mice refer to C57BL/6 mice without AOM and DSS treatment. These normal control mice did not develop colonic tumors.
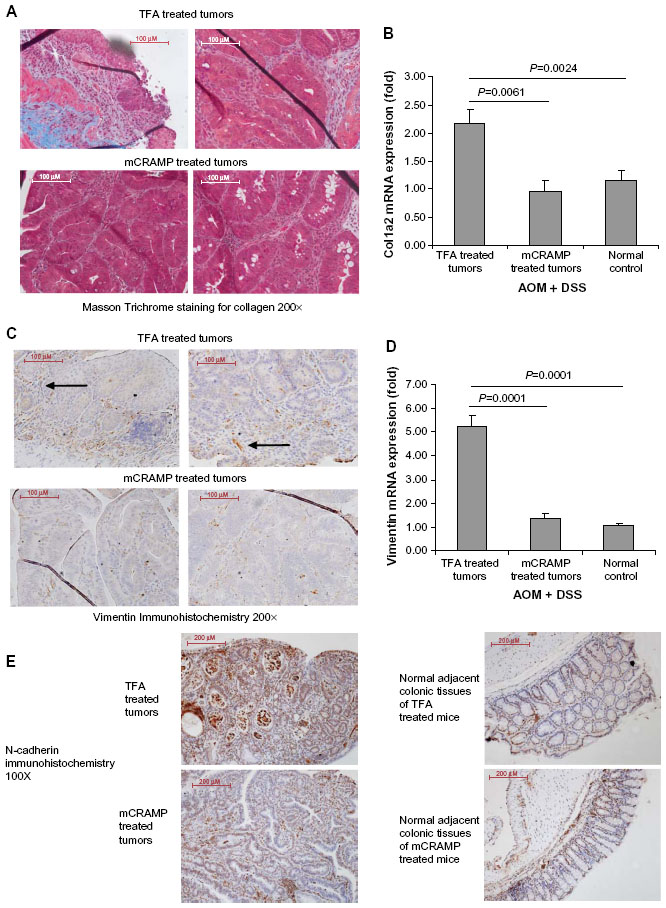 | Figure 4 Cathelicidin reduced collagen expression and fibroblast infiltration in colonic tumors. (A) Collagen deposition of colonic tumors was identified by Masson Trichrome staining (blue color). Cathelicidin peptide administration reduced collagen deposition in colonic tumors. (B) Mouse collagen COL1A2 mRNA expression in colonic tumors of AOM+DSS-treated mice. Cathelicidin peptide administration significantly reduced the fibroblast secretion product collagen in colonic tumors. (C) Immunohistochemistry of vimentin. Vimentin-positive cells (arrows) were identified by brown color staining. Mouse cathelicidin mCRAMP administration reduced tumoral vimentin protein expression. (D) Vimentin mRNA expression in colonic tumors. Cathelicidin reduced tumoral vimentin mRNA expression. (E) Immunohistochemistry of N-cadherin. N-cadherin-positive cells were identified by brown color staining. Mouse-cathelicidin mCRAMP administration reduced tumoral N-cadherin protein expression. N-cadherin protein expression in adjacent normal regions was not affected by cathelicidin treatment. |
Immunohistochemistry identified high numbers of vimentin-positive fibroblasts in the colonic tumors (Figure 4C). Cathelicidin peptide administration considerably reduced vimentin-positive fibroblast infiltration in the colonic tumors (Figure 4C). The increased tumoral vimentin mRNA expression was significantly reduced to normal levels (Figure 4D). Also, N-cadherin immunohistochemistry identified a large number of cells with mesenchymal characteristics in the colonic tumors (Figure 4D left panel). Cathelicidin peptide administration reduced N-cadherin signal in colonic tumors (Figure 4E left panel). N-cadherin signal level in normal adjacent colonic tissues was low and not affected by cathelicidin treatment (Figure 4E right panel). These suggest that cathelicidin inhibits EMT and generation of fibroblast-like cells in colonic tumors.
Cathelicidin inhibits fibroblasts-supported colon cancer cell proliferation via reduced fibroblast activity
Although cathelicidin had been shown to inhibit certain colon cancer cells such as HCT116,13 cathelicidin LL-37 does not affect HT-29 cell viability within 24 hours of incubation (Figure 5A). This suggests that cathelicidin does not affect colon cancer proliferation directly but may modulate the tumor microenvironment.
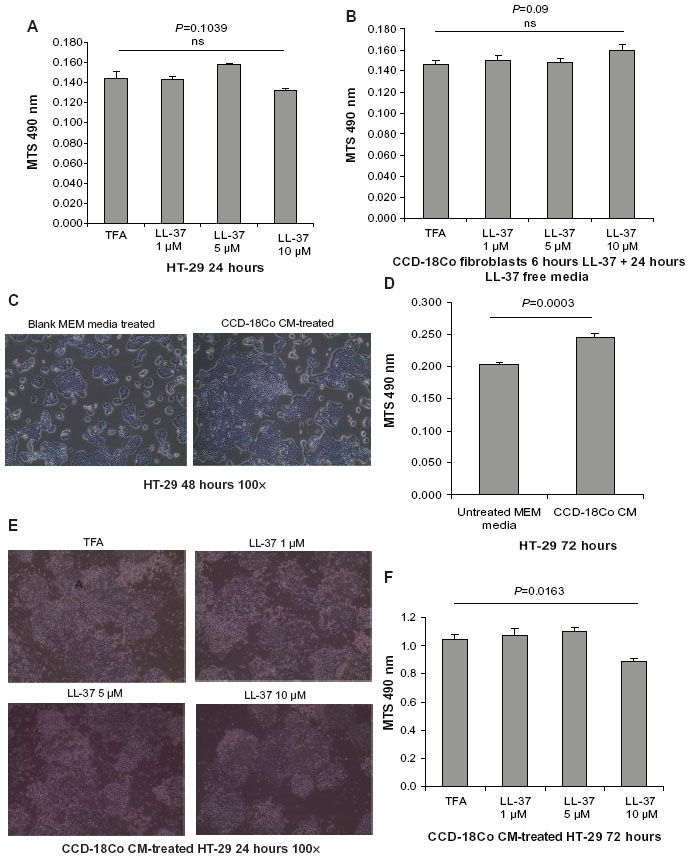 | Figure 5 Cathelicidin modulated fibroblast-stimulated colon cancer cell proliferation. (A) HT-29 cells were treated with trifluoroacetic acid (TFA) 0.1% or LL-37 for 24 hours. Cell viability was determined by MTS assays. LL-37 up to 10 μM did not affect cell viability of HT-29 cells. (B) CCD-18Co fibroblasts were treated with TFA 0.1% or LL-37 for 6 hours, followed by incubation with serum-free MEM without TFA or LL-37 for 24 hours. Cell viability was determined by MTS assays. LL-37 up to 10 μM did not affect cell viability of CCD-18Co cells. (C) Serum-free CCD-18Co-cell-conditioned media (1×105 cells/mL media) or untreated cell-free and serum-free MEM were used to treat HT-29 cells for 48 hours. Cell confluence was evaluated by light microscopy at 100× magnification. HT-29 cells treated with CCD-18Co-conditioned media had higher cell confluence than those treated with blank MEM. (D) Serum-free CCD-18Co-conditioned media (1×105 cells/mL media) or untreated cell-free and serum-free MEM were used to treat HT-29 cells for 72 hours. HT-29 cell proliferation was determined by MTS assays. Fibroblast-conditioned media significantly increased HT-29 cell proliferation. (E) CCD-18Co fibroblasts were first pretreated with TFA 0.1% or LL-37 for 6 hours, followed by incubation with new media without TFA or LL-37 for 24 hours. The conditioned media were used to treat HT-29 cells for 24 hours. HT-29 cell confluence was slightly reduced by cathelicidin-treated fibroblast-conditioned media. (F) CCD-18Co fibroblasts were first pretreated with TFA 0.1% or LL-37 for 6 hours, followed by incubation with new media without TFA or LL-37 for 24 hours. The conditioned media were used to treat HT-29 cells for 72 hours. HT-29 cell proliferation was significantly reduced by conditioned media from cathelicidin-pretreated fibroblasts. Results are representative of three independent experiments. |
We believe that cathelicidin may affect fibroblast activity and its cell proliferation stimulating mediator production, which further influences the colon cancer cell proliferation. To mimic the interactions of CAFs and colon cancer cells, we incubated the HT-29 colon cancer cells with media conditioned by CCD-18Co colonic fibroblasts. LL-37 treatment did not affect cell viability of the CCD-18Co fibroblasts (Figure 5B). Interestingly, the conditioned media of human colonic CCD-18Co fibroblasts increased HT-29 cell confluence and significantly promoted HT-29 cell proliferation by 20% (Figure 5C and D). This suggests that fibroblasts produce growth mediator(s) and promote colon cancer cell proliferation, consistent with previous reports.24,25 To determine the role of cathelicidin in this mechanism, we first treated CCD-18Co fibroblasts with cathelicidin to inhibit the cell proliferation mediator expression mechanism, followed by media change to remove cathelicidin in the media and incubation with cathelicidin-free media for secretion of cell proliferation stimulating mediator(s). HT-29 cell proliferation in cathelicidin-pretreated fibroblast conditioned media was significantly suppressed by 14% with noticeable reduction of HT-29 cell confluence (Figure 5E and F). This suggests that cathelicidin interfered with fibroblast-derived growth mediator expression and indirectly suppressed colon cancer cell proliferation.
Cathelicidin suppresses EMT in colon cancer cells
Fibroblasts and cells with mesenchymal phenotype express vimentin.18,26 TGF-β1 can induce EMT in colon cancer cells with the expression of vimentin and α-SMA.27 Cathelicidin inhibited TGF-β1-induced expression of α-SMA and vimentin in HT-29 colon cancer cells, suggesting the inhibition of transition from an epithelial cell phenotype to a mesenchymal cell phenotype ( Figure 6A and B). In addition, exposure of epithelial HT-29 colon cancer cells to TGF-β1 for 48 hours induced EMT as characterized by partial change to spindle fibroblast-like cell shape and loss of colony size ( Figure 6C). Co-incubation with human cathelicidin LL-37 (10 μM) prevented TGF-β1-induced change of cell shape and maintained the relatively large HT-29 cell colony size ( Figure 6C). In the EMT process, N-cadherin Twist and Slug expressions increased, while E-cadherin expression decreased.26,28 Similarly, exposure of epithelial HT-29 cells to TGF-β1 for 72 hours increased N-cadherin protein expression, indicating mesenchymal phenotype transition ( Figure 6D). Co-incubation with LL-37 (10 μM) suppressed TGF-β1-induced N-cadherin expression, indicating the inhibition of EMT ( Figure 6D). However, TGF-β1 exposure did not change the basal E-cadherin protein expression, suggesting the preservation of some epithelial cell phenotype ( Figure 6D). LL-37 mildly reduced EMT molecule Twist1 and Slug protein expression in the presence of TGF-β1 but did not alter E-cadherin protein expression ( Figure 6D). Inhibition of EMT may reduce the availability of mesenchymal cells with fibroblast phenotypes that support colon cancer cell proliferation.
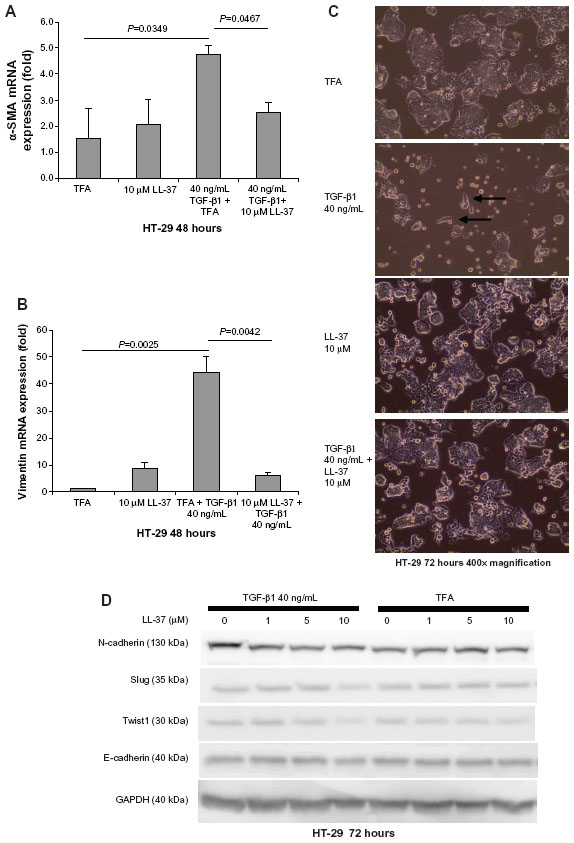 | Figure 6 Cathelicidin inhibits EMT and associated cell morphology changes of HT-29 cells. (A and B) HT-29 cells were treated with TGF-β1 (40 ng/mL) with or without LL-37 for 48 hours. TGF-β1-induced α-SMA and vimentin mRNA expression was significantly inhibited by LL-37. (C) HT-29 cells were treated with TGF-β1 (40 ng/mL) with or without LL-37 for 72 hours. TGF-β reduced HT-29 cell colony cell size, and some cells changed into a fibroblast-like spindle shape (see arrows) which was reversed by the co-incubation with LL-37. Results are representative of three independent experiments. (D) HT-29 cells were treated with TGF-β1 (40 ng/mL) with or without LL-37 for 72 hours. Protein expression of EMT mediators was determined by Western blot analyses. TGF-β1 increased N-cadherin but not the Slug, Twist1, or E-cadherin protein expression. LL-37 (10 μM) reduced N-cadherin, Slug, and Twist1 but not E-cadherin protein expression in the presence of TGF-β1. Results are representative of two independent experiments. |
Cathelicidin-mediated inhibition of EMT and fibroblasts is cytoskeleton related
Tubulin forms the basic structure of microtubules.29 Taxol® (Bristol-Myers Squibb, NY, USA) (Pacitaxel, generic name) binds to tubulin,30 stabilizes microtubules, and interferes with many cellular processes.29 Using the tubulin tracker staining method, Taxol treatment disrupted tubulin distribution in HT-29 cells as a positive control group ( Figure 7A). Taxol also inhibits EMT and TGF-β1 signaling in various human cancer cells.31–33 We found that, similar to cathelicidin, co-incubation of HT-29 colon cancer cells with Taxol abolished TGF-β1-induced α-SMA mRNA expression, ie, EMT inhibition ( Figure 7B).
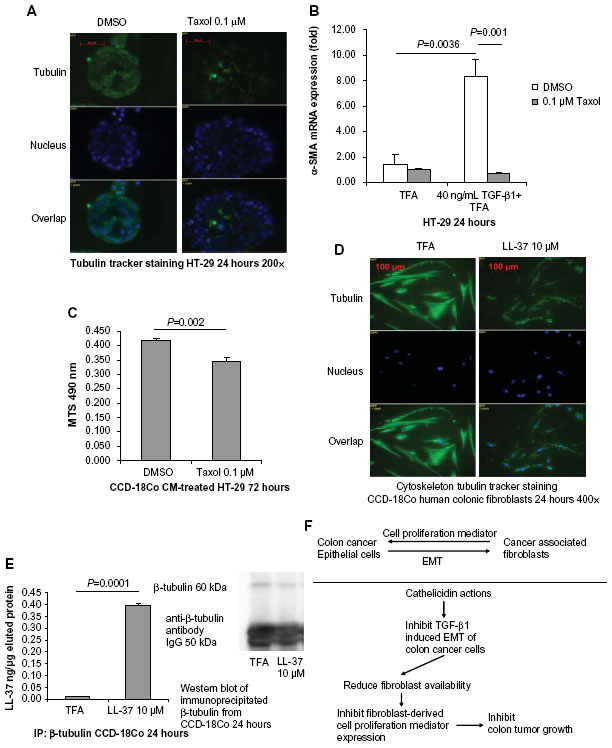 | Figure 7 Cathelicidin-mediated inhibition of EMT and fibroblast-supported colon cancer cell proliferation may be cytoskeleton-mediated. (A) Tubulin tracker staining with nuclear staining in human cancer HT-29 cells. HT-29 cells were incubated with DMSO or Taxol (0.1 μM) for 24 hours. Taxol reduced tubulin expression in fibroblasts. (B) HT-29 cells were treated with TFA or TGF-β1 (40 ng/mL) for 24 hours. Some groups were treated with DMSO or Taxol (0.1 μM). α-SMA mRNA expression was determined by real-time RT-PCR. TGF-β1-induced α-SMA mRNA expression was abolished by Taxol. (C) CCD-18Co fibroblasts were first pretreated with DMSO or Taxol (0.1 μM) for 6 hours, followed by incubation with new media without DMSO or Taxol for 24 hours. The conditioned media were used to treat HT-29 cells for 72 hours. HT-29 cell proliferation was significantly reduced by the conditioned media from Taxol-pretreated fibroblasts. (D) Tubulin tracker staining with nuclear staining in human colonic CCD-18Co fibroblasts. Cathelicidin (10 μM) reduced tubulin expression in fibroblasts. (E) Immunoprecipitation of β-tubulin and LL-37. CCD-18Co fibroblasts were treated with TFA 0.1% or LL-37 10 μM for 24 hours. Increased yield of LL-37 in the immunoprecipitated complex indicated that LL-37 co-immunoprecipitated with β-tubulin. Western blot analyses of the immunoprecipitated protein complex. The amounts of immunoprecipitated β-tubulin of the control and LL-37-treated groups were the same. (F) Summary of the roles of cathelicidin in colon cancer development. |
Cell proliferation of HT-29 cells was significantly reduced by the conditioned media from Taxol-pretreated CCD-18Co fibroblasts ( Figure 7C), suggesting that the disruption of cytoskeleton in fibroblasts was sufficient to affect fibroblast-supported colon cancer cell proliferation. (Note: Taxol was removed from CCD-18Co fibroblast culture in the media change step. No Taxol was carried over to HT-29 cell culture. Such Taxol treatment condition did not affect CCD-18Co cell viability [Figure S1].)
LL-37 disrupted the tubulin distribution in human colonic CCD-18Co fibroblasts (Figure 7D). As demonstrated by immunoprecipitation, we found that the LL-37 molecule was bound with β-tubulin in CCD-18Co fibroblasts (Figure 7E). Similar to Taxol, cathelicidin may disrupt cytoskeleton distribution and other cellular activities by binding with tubulin molecules.
Discussion
A previous report suggested that cathelicidin inhibits colon cancer cell proliferation and induces apoptosis in vitro.13 Here we showed that cathelicidin could be a novel therapeutic approach against colon cancer in vivo (Figure 2). This is the first report in the literature showing the effective inhibition of colonic tumor development by cathelicidin with indirect mechanisms. We discovered several cathelicidin-mediated mechanisms, including cytoskeleton disruption and inhibition of EMT, that are linked to inhibiting indirect fibroblast-stimulated colon cancer cell proliferation.
Administration of mouse cathelicidin mCRAMP peptide via enema significantly reduced the number and area of colonic tumors in the AOM+DSS model (Figure 2B and C). In nude mice, cathelicidin specifically inhibited tumoral vimentin but not mouse host fibroblast vimentin. This suggests a good safety profile. Consistent with the findings in nude mice (Figure 3D and E), administration of mCRAMP peptide significantly reduced the fibroblast marker vimentin protein and mRNA expression in the tumors of an in vivo colon cancer model (AOM+DSS) (Figure 4C and D). As vimentin expression in tumor stroma is associated with short survival period of patients with colon cancer,34 we speculate that cathelicidin-mediated inhibition of tumoral vimentin expression may confer better survival.
There are several reports showing the induction of apoptosis mediated by cathelicidin (and its mimic compounds) in colon cancer cells in vitro.13,15–17 However, mCRAMP administration via enema did not affect the viability marker p21 mRNA expression in colonic tumors and adjacent normal colonic tissues in the AOM+DSS mouse model (data not shown). Cathelicidin did not trigger apoptosis (TUNEL staining) in colonic tumors and adjacent normal tissues (Figure 2D), suggesting that it inhibits colon cancer development without causing cell death in tumors and/or adjacent normal tissues. There was no sign of apoptosis (TUNEL staining) in the subcutaneous tumors of nude mice (Figure 1E). Cathelicidin (up to 10 μM) also failed to inhibit human colon cancer HT-29 cell viability within 24 hours (Figure 5A) and human normal colonic epithelial NCM460 cell viability within 48 hours.2 Apoptosis and direct inhibition of cell viability may not be a mainstay mechanism of the antitumoral effects of cathelicidin in vivo. This may be beneficial to the clinical safety of cathelicidin as an antitumoral agent because the risk of nonspecific damage to nontumoral tissues is low.
As colonic tumors develop and progress, some colon cancer cells change from an epithelial phenotype to a mesenchymal phenotype.35 It is not surprising to observe the existence of CAFs in and around the tumors. As fibroblasts secrete extracellular matrices such as collagen, we used collagen expression to evaluate the activity of CAFs. Cathelicidin peptide administration significantly reduced tumoral mouse collagen COL1A2 mRNA and protein expression in the AOM+DSS colon cancer model (Figure 4A and B). Intravenous administration of cathelicidin expressing AAV significantly reduced tumoral human collagen deposition (Figure 3A) and COL1A2 mRNA expression (Figure 3B) in the subcutaneous nude mouse colonic tumor model. This indicates cathelicidin-mediated specific inhibition of human colon cancer-cell-derived fibroblasts. Both intravenous administration of cathelicidin expressing AAV and enema administration of cathelicidin peptide reduced tumoral N-cadherin expression (Figure 1F and 4E). LL-37 also reduced TGF-β1-induced N-cadherin expression in HT-29 cells (Figure 6D). All these indicate that cathelicidin can inhibit EMT in vivo and in vitro. Inhibition of EMT may reduce the conversion from epithelial colon cancer cells to mesenchymal CAFs.
Co-culture of CAFs with colon cancer cells has been shown to promote colon cancer cell proliferation in a paracrine manner and HT-29-cell-derived subcutaneous tumor growth.24 We found that CCD-18Co fibroblasts supported HT-29 cell proliferation (Figure 5C and D). Keratinocyte growth factor (KGF) has been shown to mediate CCD-18Co fibroblast-supported HT-29 cell proliferation.36 Our data did not observe increased KGF mRNA expression in AOM+DSS-treated colonic tumors (data not shown), and another report has shown that the KGF expression in colon tumor CAFs and cancer cells was only moderate.37 IL-6 has been proposed to mediate bone-marrow-derived fibroblast-supported colon tumorigenesis.38 However, cathelicidin mCRAMP administration via enema failed to alter IL-6 mRNA expression in AOM+DSS-treated tumors and adjacent normal tissues (data not shown). This again shows that cathelicidin does not target the host fibroblasts. The blockade of hepatocyte growth factor by neutralizing antibody also failed to alter CCD-18Co fibroblast-supported HT-29 cell proliferation (data not shown). Although the exact fibroblast-derived cell proliferation mediator is yet to be identified despite our attempts, our experiments demonstrate that pretreatment of fibroblasts with cathelicidin can significantly inhibit fibroblast-supported colon cancer cell proliferation in vitro (Figure 5E and F).
To further understand the mechanism of cathelicidin-mediated inhibition of fibroblast activity, we found that the cathelicidin protein bound to tubulin protein and disrupted cytoskeletal tubulin distribution in CCD-18Co fibroblasts (Figure 7D and E). Both cathelicidin (Figure 7D) and Taxol (Figure 7A)30 are capable of binding to tubulin molecules. The pattern of tubulin disruption and inhibition of fibroblast-supported colon cancer cell proliferation was comparable to the effects of Taxol (Figure 7A and C). Since there is no known way to reverse cathelicidin-mediated cytoskeleton disruption, we speculate that cathelicidin can indirectly inhibit fibroblast-supported colon cancer cell proliferation via disruption of the fibroblast cytoskeleton.
Interestingly, both cathelicidin and Taxol also disrupted tubulin distribution in HT-29 colon cancer cells (Figures S2 and 7A). (Note: TGF-β1 did not affect tubulin distribution in HT-29 cells [Figure S2].) As Taxol abolished TGF-β1-induced α-SMA expression of HT-29 cells (Figure 7B), ie, EMT inhibition, we speculate that the disruption of tubulin distribution may be involved in cathelicidin-mediated inhibition of TGF-β1-induced expression of mesenchymal fibroblast markers (α-SMA and vimentin) mRNA expression in HT-29 colon cancer cells. The inhibition of colon cancer cell EMT to mesenchymal phenotype may subsequently reduce the generation of tumor-derived CAFs in the tumors. Again, cathelicidin inhibited tumoral human vimentin but not mouse host vimentin in the subcutaneous tumors of nude mice (Figure 3E and F), suggesting that nonspecific elimination of normal fibroblasts is unlikely. We believe that cathelicidin-mediated inhibition of tumor-derived CAF availability (with their activities and subsequent growth mediator production) limits the promotion of tumoral development in vivo.
Cathelicidin may be a promising future therapeutic solution against colon cancer. However, cathelicidin peptide can be easily degraded in body fluids or blood, making it difficult to be directly used in a clinical setting. However, ectopic expression of cathelicidin by a probiotic bacterial vector or a cathelicidin-encoding plasmid may deliver cathelicidin to intestines or stomach for extended periods of time.4,39,40 These approaches have been successful in treating DSS colitis and Helicobacter pylori-mediated gastritis in mice and may become a viable way to prevent or inhibit colon cancer tumors in intestine.
In conclusion, cathelicidin inhibits colon cancer development in multiple pathways including 1) inhibition of EMT of colon cancer cells and 2) inhibition of fibroblast-mediated colon cancer proliferation. A concise summary of our findings is shown in Figure 7F.
Acknowledgments
This work was supported by a Pilot and Feasibility Study grant from UCLA-CURE Center, a Career Development Award from the Crohn’s and Colitis Foundation of America (#2691), K01DK084256 from the National Institutes of Health to Hon Wai Koon and student research fellowship from the Crohn’s and Colitis Foundation of America to Samantha Ho (#3831), Michelle Cheng (#287244), and Deanna Tran (#324000).
Disclosure
The authors declare no conflict of interest in this work.
References
Ho S, Pothoulakis C, Koon HW. Antimicrobial peptides and colitis. Curr Pharm Des. 2012;19:40–47. | |
Hing TC, Ho S, Shih DQ, et al. The antimicrobial peptide cathelicidin modulates Clostridium difficile-associated colitis and toxin A-mediated enteritis in mice. Gut. 2012;62:1295–1305. | |
Tai EK, Wu WK, Wong HP, Lam EK, Yu L, Cho CH. A new role for cathelicidin in ulcerative colitis in mice. Exp Biol Med (Maywood). 2007;232:799–808. | |
Tai EK, Wu WK, Wang XJ, et al. Intrarectal administration of mCRAMP-encoding plasmid reverses exacerbated colitis in Cnlp(–/–) mice. Gene Ther. 2012;20:187–193. | |
Wong CC, Zhang L, Li ZJ, et al. Protective effects of cathelicidin-encoding Lactococcus lactis in murine ulcerative colitis. J Gastroenterol Hepatol. 2012;27:1205–1212. | |
Scott A, Weldon S, Buchanan PJ, et al. Evaluation of the ability of LL-37 to neutralise LPS in vitro and ex vivo. PLoS One. 2011;6:e26525. | |
Ruan Y, Shen T, Wang Y, Hou M, Li J, Sun T. Antimicrobial peptide LL-37 attenuates LTA induced inflammatory effect in macrophages. Int Immunopharmacol. 2013;15:575–580. | |
Wu WK, Sung JJ, Cheng AS, et al. The Janus face of cathelicidin in tumorigenesis. Curr Med Chem. 2014;21:2392–2400. | |
Wu WK, Wang G, Coffelt SB, et al. Emerging roles of the host defense peptide LL-37 in human cancer and its potential therapeutic applications. Int J Cancer. 2010;127:1741–1747. | |
Heilborn JD, Nilsson MF, Jimenez CI, et al. Antimicrobial protein hCAP18/LL-37 is highly expressed in breast cancer and is a putative growth factor for epithelial cells. Int J Cancer. 2005;114:713–719. | |
Coffelt SB, Waterman RS, Florez L, et al. Ovarian cancers overexpress the antimicrobial protein hCAP-18 and its derivative LL-37 increases ovarian cancer cell proliferation and invasion. Int J Cancer. 2008;122:1030–1039. | |
von Haussen J, Koczulla R, Shaykhiev R, et al. The host defence peptide LL-37/hCAP-18 is a growth factor for lung cancer cells. Lung Cancer. 2008;59:12–23. | |
Ren SX, Cheng AS, To KF, et al. Host immune defense peptide LL-37 activates caspase-independent apoptosis and suppresses colon cancer. Cancer Res. 2012;72:6512–6523. | |
Wu WK, Sung JJ, To KF, et al. The host defense peptide LL-37 activates the tumor-suppressing bone morphogenetic protein signaling via inhibition of proteasome in gastric cancer cells. J Cell Physiol. 2010;223:178–186. | |
Ren SX, Shen J, Cheng AS, et al. FK-16 derived from the anticancer peptide LL-37 induces caspase-independent apoptosis and autophagic cell death in colon cancer cells. PLoS One. 2013;8:e63641. | |
Kuroda K, Fukuda T, Okumura K, et al. Ceragenin CSA-13 induces cell cycle arrest and antiproliferative effects in wild-type and p53 null mutant HCT116 colon cancer cells. Anticancer Drugs. 2013;24:826–834. | |
Kuroda K, Fukuda T, Yoneyama H, et al. Anti-proliferative effect of an analogue of the LL-37 peptide in the colon cancer derived cell line HCT116 p53+/+ and p53. Oncol Rep. 2012;28:829–834. | |
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. | |
Koon HW, Shih D, Karagiannides I, et al. Substance P modulates colitis-associated fibrosis. Am J Pathol. 2010;177:2300–2309. | |
Koon HW, Shih DQ, Chen J, et al. Cathelicidin signaling via the Toll-like receptor protects against colitis in mice. Gastroenterology. 2011;141(1852–1863):e1–e3. | |
Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. | |
Koon HW, Zhao D, Na X, Moyer MP, Pothoulakis C. Metalloproteinases and transforming growth factor-alpha mediate substance P-induced mitogen-activated protein kinase activation and proliferation in human colonocytes. J Biol Chem. 2004;279:45519–45527. | |
Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116:1955–1962. | |
Duckworth CA, Clyde D, Worthley DL, Wang TC, Varro A, Pritchard DM. Progastrin-induced secretion of insulin-like growth factor 2 from colonic myofibroblasts stimulates colonic epithelial proliferation in mice. Gastroenterology. 2013;145(197–208):e3. | |
Herrera A, Herrera M, Alba-Castellon L, et al. Protumorigenic effects of Snail-expression fibroblasts on colon cancer cells. Int J Cancer. 2014;134(12):2984–2990. | |
Todosi AM, Gavrilescu MM, Anitei GM, Filip B, Scripcariu V. Colon cancer at the molecular level – usefulness of epithelial-mesenchymal transition analysis. Rev Med Chir Soc Med Nat Iasi. 2012;116:1106–1111. | |
Cai ZG, Zhang SM, Zhang H, Zhou YY, Wu HB, Xu XP. Aberrant expression of microRNAs involved in epithelial-mesenchymal transition of HT-29 cell line. Cell Biol Int. 2013;37:669–674. | |
Katoh Y, Katoh M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review). Int J Mol Med. 2008;22:271–275. | |
Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10:194–204. | |
Amos LA, Lowe J. How Taxol stabilises microtubule structure. Chem Biol. 1999;6:R65–R69. | |
Hirose A, Tajima H, Ohta T, et al. Low-dose paclitaxel inhibits the induction of epidermal-mesenchymal transition in the human cholangiocarcinoma CCKS-1 cell line. Oncol Lett. 2013;6:915–920. | |
Tsukada T, Fushida S, Harada S, et al. Low-dose paclitaxel modulates tumour fibrosis in gastric cancer. Int J Oncol. 2013;42:1167–1174. | |
Wang C, Song X, Li Y, et al. Low-dose paclitaxel ameliorates pulmonary fibrosis by suppressing TGF-beta1/Smad3 pathway via miR-140 upregulation. PLoS One. 2013;8:e70725. | |
Ngan CY, Yamamoto H, Seshimo I, et al. Quantitative evaluation of vimentin expression in tumour stroma of colorectal cancer. Br J Cancer. 2007;96:986–992. | |
Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1:482–497. | |
Visco V, Bava FA, d’Alessandro F, Cavallini M, Ziparo V, Torrisi MR. Human colon fibroblasts induce differentiation and proliferation of intestinal epithelial cells through the direct paracrine action of keratinocyte growth factor. J Cell Physiol. 2009;220:204–213. | |
Watanabe M, Ishiwata T, Nishigai K, Moriyama Y, Asano G. Overexpression of keratinocyte growth factor in cancer cells and enterochromaffin cells in human colorectal cancer. Pathol Int. 2000;50:363–372. | |
Zhu L, Cheng X, Ding Y, et al. Bone marrow-derived myofibroblasts promote colon tumorigenesis through the IL-6/JAK2/STAT3 pathway. Cancer Lett. 2013;343:80–89. | |
Ahluwalia A, Tarnawski AS. Cathelicidin gene therapy: a new therapeutic option in ulcerative colitis and beyond? Gene Ther. 2012;20:119–120. | |
Zhang L, Yu J, Wong CC, et al. Cathelicidin protects against Helicobacter pylori colonization and the associated gastritis in mice. Gene Ther. 2012;20:751–760. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.