")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Case Report: A Novel MVK Missense Mutation in the Sporadic Porokeratosis Ptychotropica in China
Authors Mei Q, Xing F, Yin Y, Yuan C
Received 10 February 2023
Accepted for publication 10 May 2023
Published 23 May 2023 Volume 2023:16 Pages 1325—1329
DOI https://doi.org/10.2147/CCID.S408016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Qin Mei,1 Fengling Xing,2 Yue Yin,2 Chengda Yuan2
1Department of Dermatology, Dermatology Hospital of Fuzhou, Fuzhou, Fujian, People’s Republic of China; 2Department of Dermatology, Hangzhou TCM Hospital Affiliated to Zhejiang Chinese Medical University, Hangzhou, Zhejiang, People’s Republic of China
Correspondence: Chengda Yuan, Department of Dermatology, Hangzhou TCM Hospital Affiliated to Zhejiang Chinese Medical University, TiYuChang Road 453th, Hangzhou, 310007, People’s Republic of China, Tel +8613336116910, Fax +86-571-85827534, Email [email protected]
Abstract: Porokeratosis ptychotropica (PPt) is a rare type of porokeratosis (PK) characterized by pruritic, reddish-brownish verrucous papules, and plaques usually around genital area or buttocks. Here, a case of a 70-year-old woman who was diagnosed as PPt was reported. The patient suffered from severe pruritic papules and plaques in the buttock region and pubis for 4 years. The skin lesions were giant, well-defined brown plaques with many satellite papules scattered around. Both clinical manifestations and histopathological features supported the diagnosis of PPt. In review of the identified mutation was found in patients with disseminated superficial actinic porokeratosis (DSAP) combined with PPt, while its unclear in PPt. To investigate the hypothesis that the variant reported in the present case report may played as an independent “likely pathogenic factor” of PPt. Consequently, a de novo missense pathogenic mutation in the MVK gene was identified in this case. Unexpectedly, it is a first report of a novel MVK mutation in sporadic PPt. This rare case suggested an isogenetic background between PPt and DSAP, which may help to explore the underlying pathogenesis of PPt.
Keywords: PPt, MVK, genetic testing, missense mutations, mevalonate pathway
Introduction
Porokeratosis ptychotropica (PPt) is a rare variant of porokeratosis (PK) that was first described in 1995, characterized by pruritic, symmetrical, reddish-brownish verrucous papules, and plaques most commonly on the buttocks.1 Zhang et al have performed massively parallel sequencing and exonic CNV screening of 134 different PK patients, which identified four mutated genes (MVK, PMVK, MVD, and FDPS) in the mevalonate pathway.2 As a rare form of PK, the exact pathogenesis of PPt is not known, especially the genetic predisposition. Here, we present a sporadic Chinese PPt case identified with novel compound heterozygous missense mutations of MVK, which might support a unique phenotype mutation associated with PPt. This article reviewed literatures on the mutation of MVK gene as causal gene involved in the PPt, and summarized the clinical features and potential treatment target.
Case Presentation
A 70-year-old Chinese woman presented with a 4-year history of progressive itching brown hyperkeratotic papules and plaques on her gluteal cleft and buttocks (Figure 1A). The lesion begins as reddish-brownish, bean-sized papules scattered on her left buttock. Gradually, these satellite lesions involved her perianal region and right buttock, and eventually became aggregated and coalesced into large plaques with well-defined and slightly elevated borders, and with severe itching. Diagnosis of PPt in this case was made by an experienced dermatologist, and was confirmed by dermoscopy and histological findings of involved skin (Figure 1B and C). Previously, PK can be identified as a skin-specific auto-inflammatory disease, which was often inherited and linked to ultraviolet light exposure and immunosuppression.2 As an uncommon variant of PK, the pathogenesis of this disease remains unclear. In this present study, the patient was otherwise healthy and did not have any risk factor for PK. That is to say both her medical examination and family medical history were normal.
 |
Figure 1 Clinical manifestation and histopathological findings of Porokeratosis Ptychotropica. (A) Clinical features show “butterfly shaped” verrucous plaque on buttocks with multiple satellite papules. (B) Dermoscopic view (X10) of the skin lesion, similar to keratotic ridge: annular, brown-yellowish hyperkeratotic ridge (arrows) demarcating a central area with whitish scales and some globular vessels. (C) A histological hallmark of all variants of porokeratosis, cornoid lamella (CL) as a “column” overlying a zone of the epidermis (cornoid lamella, marked as red asterisk). Dyskeratotic cells and focal hypogranulosis were present in the epidermis underlying the CL. Here, localized subcutaneous inflammatory cells infiltration suggested an eruptive pruritic PPt (H&E staining, X100). |
The case report written informed consent was obtained from the patient, and the study was carried out after approval by the ethics committee of Hangzhou Hospital of Traditional Chinese Medicine and conducted according to the Declaration of Helsinki principles. After the patient and other participants signed informed consent for MVK gene testing, peripheral blood samples were taken from the patient and her brother and son. All exons and flanking intronic sequences of MVK were amplified by PCR, and the PCR products were purified and sequenced on an automated sequencer (ABI 3730xl; Applied Biosystems, Foster City, CA, USA). Sequence analysis showed a heterozygous C>A transition at nucleotide 64 in exon 2 of MVK in the patient (Figure 2), which was not detected in her brother and son or in 100 unrelated healthy controls.
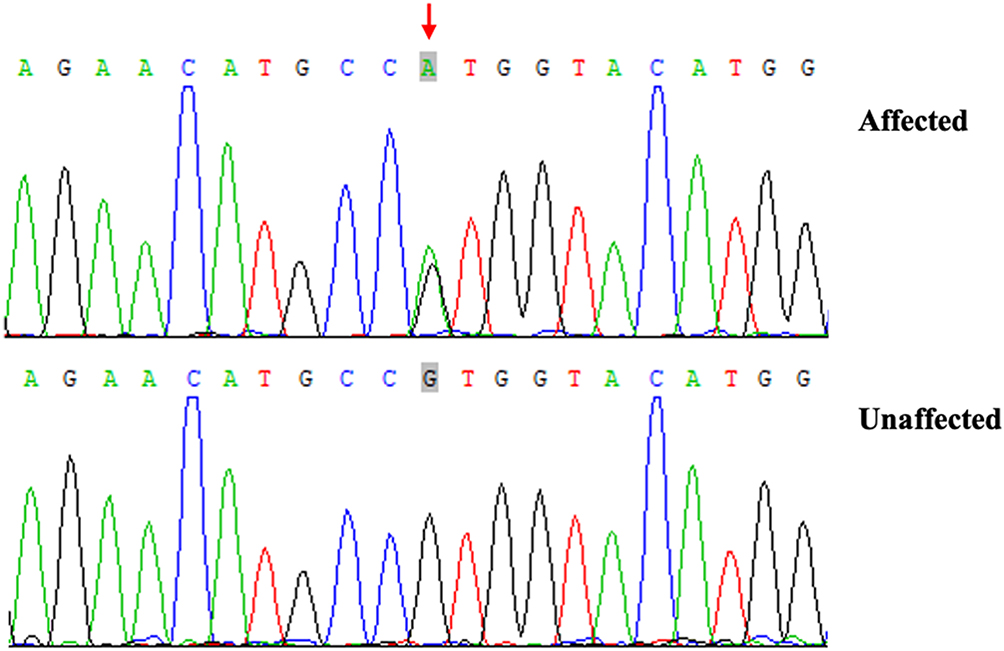 |
Figure 2 Sanger sequencing chromatograms of proband (patient with PP, affected) and normal control (unaffected) at the c.64 G>A mutation site indicated by arrow (NM_000431: exon2:c.G64A:p.V22M). |
After the mutation was found, the consulting dermatologist conducted an interview with her family history again: there was no cancer history and radiation therapy in parent’s side. Even the porokeratosis has a low risk of transformation to skin cancer. While no malignant degeneration was found in Zhang’s research.1 Given the reported poor response to treatment of PPt, sun protection, emollients, and a regular follow-up is recommended.
Discussion
PPt is characterized by symmetrical reddish-brownish verrucous plaques localized on the perianal cleft and buttocks region, usually forming a butterfly-shaped appearance and accompanied by stubborn itching. Histologically, the key change to differentiate PPt from the classic cases is that the cornoid lamellae (CL) can be seen throughout the lesion, while in other variants, the CL is just located at the periphery.3 We summarized 56 patients whose detailed clinical data were available. It is indicated that PPt is generally prevalent in Asia and affected more men than women. The onset of PPt ranged from 1 year to 84 years (mean age 32 years). Disease duration ranged from 1 month to 23 years. The lesions mainly involved the buttocks and gluteal fold, and diameter of lesions ranged from 2 mm to 20 cm.
Previously, 5 cases of PPt coexisting with DSAP have been reported in the English literature, 3 of them had family history, and MVK gene mutation was found in two families (Table 1). Happle4 proposed that somatic recombination may explain the coexistence of multiple forms of linear porokeratosis and DSAP. But in sporadic cases, only our patient found a novel MVK missense mutations (c.64G>A, p.V22M, not in mutational spectrum of MVK), premature generation of termination signal leads to domain deletion, which may be the cause of disease.
 |
Table 1 Clinical Features of Patients with Coexisting DSAP and PPt |
MVK gene mutations have been identified as vital causal mutation both in familial and sporadic PK patients, and have been reported mainly in DSAP. Mutations in MVK gene are known to cause the keratin disorder, and people suffering from such a problem develop hardened skin lesions. However, such mechanical research in PPt is almost a gap and needs to further explore. Presently, research observed the correlation of genotypes and several clinical features in PPt patients, which might be helpful to confirm the diagnosis. The result showed that giant plaque-type PPt with lesion diameters at least 5 cm appeared to be a unique phenotype related to MVK mutations, and this feature was observed in 50% (19/38) of index patients.2 Additionally, patients with MVK mutations generally showed the widest range of phenotypes in terms of both the number and size of lesions. Furthermore, when the homozygotic form of the disorder occurs in skin, some patients firstly presented with untypical lesions of genitogluteal porokeratosis,5 the appearance may be earlier and more severe in the pubis and buttock region with verrucous lesions of this individual.
There is no known cure for PPt, and the outcomes of treatment are generally disappointing. The symptoms may be alleviated via following measures. The systemic administration of retinoids or hydroxyurea,6 and the topical treatment by photodynamic therapy, laser therapy and cryotherapy, or gents like corticosteroids, imiquimod, 5-fluorouracil, vitamin D3 analogs, salicylic acid and α-hydroxyl acids have shown efficacy, but the lesions commonly recur after treatment. However, mounting evidences suggest that many patients with porokeratosis may harbor postzygotic mosaic somatic mutations in enzymes of the mevalonate biosynthetic pathway, that is the MVK gene mutation.7 In the human body, the rate-limiting step of the mevalonate pathway is catalyzed by hydroxymethylglutaryl coenzyme A reductase (HMG-CoA), leading to the formation of mevalonic acid, and numerous reports have been account for HMG-CoA reductase as a therapeutic target.8 Therefore, this study would provide new insight into the mechanism of action of these inhibitors or other preceding steps in the mevalonate biosynthetic pathway may represent a future therapeutic approach of PK. However, the result and hypothesis reported herein should be considered in the light of some limitations. Until recently, a larger sample and a verified deduction were not available.
Conclusion
PPt is an unusual type of PK, and likely be misdiagnosed as other skin diseases. Besides, there is a risk of malignant skin transformation and poor response. Recognition of clinical features and diagnose of PPt under the guidance of genetic testing may help to initiate early treatment. In this case, de novo MVK missense variant was identified in PPt, which encodes an enzyme that is involved in producing chemicals called isoprenoids. It is supposed that human isoprenoid synthase enzymes could be as therapeutic targets of PK with MVK gene mutations.
Ethics Statement
The publications of images were included with the patient’s consent.
Consent Statement
The authors certify that the patient has given her informed consent for case details and images to be published. Institutional approval is required for this case study.
Acknowledgments
We thank the patient and her family members who participated in this study.
Funding
This work was supported by The Construction Fund of Medical Key Disciplines of Hangzhou (2020SJZDXK03).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Lucker GP, Happle R, Steijlen PM. An unusual case of porokeratosis involving the natal cleft: porokeratosis ptychotropica? Br J Dermatol. 1995;132:150–151. doi:10.1111/j.1365-2133
2. Zhang Z, Li C, Wu F, et al. Genomic variations of the mevalonate pathway in porokeratosis. Elife. 2015;4:e6322. doi:10.7554/eLife.06322
3. Liu W, Liu JW, Ma DL. Porokeratosis Ptychotropica. JAMA Dermatol. 2019;155(7):845. doi:10.1001/jamadermatol.2019.0602
4. Happle R. Dohi Memorial Lecture. New aspects of cutaneous mosaicism. J Dermatol. 2002;29:681–692. doi:10.1111/j.1346-8138.2002.tb00204.x
5. Murase J, Gilliam Anita C. Disseminated superficial actinic porokeratosis co-existing with linear and verrucous porokeratosis in an elderly woman: update on the genetics and clinical expression of porokeratosis. J Am Acad Dermatol. 2010;63:886–891. doi:10.1016/j.jaad.2009.07.038
6. Maurizio R, Davide R, Silvia AV, et al. Disseminated superficial actinic porokeratosis following hydroxyurea treatment: a case report. Australas J Dermatol. 2023;64(1):e72–e75. doi:10.1111/ajd.13943
7. Atzmony L, Khan Habib M, Lim Young H, et al. Second-Hit, postzygotic PMVK and MVD mutations in linear porokeratosis. JAMA Dermatol. 2019;155:548–555. doi:10.1001/jamadermatol.2019.0016
8. Park J, Matralis Alexions N, Berghuis Albert M, et al. Human isoprenoid synthase enzymes as therapeutic targets. Front Chem. 2014;2:e50. doi:10.3389/fchem.2014.00050
9. Peng JM, Xiao XM, Chen JW, et al. Novel mutation in MVK gene for co-occurrence of disseminated superficial actinic porokeratosis with porokeratosis ptychotropica. J Dermatol. 2021;48:e137–e139. doi:10.1111/1346-8138.15748
10. Xu HJ, Wen GD. Mixed porokeratosis with a novel mevalonate kinase gene mutation: a case report. World J Clin Cases. 2022;10(14):4528–4534.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.