Back to Journals » OncoTargets and Therapy » Volume 13
Cas9 Mediated Correction of β-catenin Mutation and Restoring the Expression of Protein Phosphorylation in Colon Cancer HCT-116 Cells Decrease Cell Proliferation in vitro and Hamper Tumor Growth in Mice in vivo
Authors Li Y, Li X, Qu J, Luo D ![]() , Hu Z
, Hu Z ![]()
Received 31 July 2019
Accepted for publication 17 December 2019
Published 6 January 2020 Volume 2020:13 Pages 17—29
DOI https://doi.org/10.2147/OTT.S225556
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Yanlan Li,1,2,* Xiangning Li,1,3,* Jiayao Qu,1,3 Dixian Luo,1,3 Zheng Hu1,3
1Translational Medicine Institute, the First People’s Hospital of Chenzhou Affiliated to University of South China, Hunan 432000, People’s Republic of China; 2Hunan Province Key Laboratory of Tumor Cellular and Molecular Pathology, Cancer Research Institute, University of South China, Hunan 421001, People’s Republic of China; 3National & Local Joint Engineering Laboratory for High-Through Molecular Diagnosis Technology, The First People’s Hospital of Chenzhou, Hunan 432000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zheng Hu; Dixian Luo
Translational Medicine Institute, National & Local Joint Engineering Laboratory for High-Through Molecular Diagnosis Technology, The First People’s Hospital of Chenzhou, Hunan 432000, People’s Republic of China
Tel/Fax +86 735 2343902
Email [email protected]; [email protected]
Purpose: Colorectal cancer (CRC) is one of the major contributors to cancer mortality and morbidity. Finding strategies to fight against CRC is urgently required. Mutations in driver genes of APC or β-catenin play an important role in the occurrence and progression of CRC. In the present study, we jointly apply CRISPR/Cas9-sgRNA system and Single-stranded oligodeoxynucleotide (ssODN) as templates to correct a heterozygous ΔTCT deletion mutation of β-catenin present in a colon cancer cell line HCT-116. This method provides a potential strategy in gene therapy for cancer.
Methods: A Cas9/β-catenin-sgRNA-eGFP co-expression vector was constructed and co-transfected with ssODN into HCT-116 cells. Mutation-corrected single-cell clones were sorted by FACS and judged by TA cloning and DNA sequencing. Effects of CRISPR/Cas9-mediated correction were tested by real-time quantitative PCR, Western blotting, CCK8, EDU dyeing and cell-plated clones. Moreover, the growth of cell clones derived tumors was analyzed at nude mice xenografts.
Results: CRISPR/Cas9-mediated β-catenin mutation correction resulted in the presence of TCT sequence and the re-expression of phosphorylation β-catenin at Ser45, which restored the normal function of phosphorylation β-catenin including reduction of the transportation of nuclear β-catenin and the expression of downstream c-myc, survivin. Significantly reduced cell growth was observed in β-catenin mutation-corrected cells. Mice xenografted with mutation-corrected HCT-116 cells showed significantly smaller tumor size than uncorrected xenografts.
Conclusion: The data of this study documented that correction of the driven mutation by the combination of CRISPR/Cas9 and ssODN could greatly remedy the biological behavior of the cancer cell line, suggesting a potential application of this strategy in gene therapy of cancer.
Keywords: CRISPR/Cas9, ssODN, targeted gene editing, β-catenin, colon cancer
Introduction
Colorectal cancer is one of the common malignant tumors of the digestive tract and is a leading cause of cancer mortality worldwide.1,2 Several risk factors associated with CRC have been identified including aberrantly activated Wnt/β-catenin signaling pathway due to adenomatous polyposis coli (APC) or β-catenin mutation.3–6 Though alteration of APC is found in approximately 70% of CRC patients, several studies reported that β-catenin also has oncogenic activity in CRC cells. Activating mutations lead to accumulation of β-catenin in the cytoplasm and nuclear transportation to form transcriptional activation complex with the T cell transcription factor/lymphoid enhancer factor (TCF/LEF) and then up-regulate downstream genes expression to drive tumor formation and progression. Correcting these mutations can restore β-catenin phosphorylation-degradation cascade, in turn, inhibit activation of transcription of Wnt targeted gene and formation of the tumor.
Recent studies showed that the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) and single-guide RNA (sgRNA) system has emerged as a powerful gene-editing tool, which can be used to correct gene mutations in many cell lines and offer considerable advantages over earlier genome editing tools, such as ZFN and TALENs.7–9 Under the guidance of sgRNA, Cas9 nuclease can be programmed to cleave the target DNA to a site-specific double-strand break (DSB) and initiate non-homologous end joining (NHEJ) or homology-directed repair (HDR).10 With a donor template, the precise HDR of DSB can engineer genomic DNA both in vivo and in vitro. ssODNs can be used as donor templates to improve HDR repair in cells.11–13
In the present study, we applied CRISPR/Cas9 and ssODN to correct a heterozygous ΔTCT deletion mutation of β-catenin gene in colon cancer HCT-116 cells. This ΔTCT deletion mutation is responsible for encoding the 45th serine (Ser45) at the N-terminal region of the protein. We performed functional studies in vitro and in vivo to determine whether the wild-type function of β-catenin Ser45 phosphorylation was restored following mutation correction. The results showed that mutation-corrected single-cell clone had decreased growth rate and related to the formation of tumors in a smaller size. Our data demonstrated that a combination of CRISPR/Cas9 and ssODN provided a new therapeutic strategy for genetic disorder disease.
Materials and Methods
Reagents, Oligonucleotides, and Primers for Vector Construction
Oligonucleotides used for annealing and primers used for PCR were synthesized by GIGA Biotechnology (Guangzhou, China). The endonucleases were obtained from New England Biolabs Inc. (Ipswich, MA, USA), and DNA purification kits were purchased from Tiangen Co. (Beijing, China). ssODN used for transfection studies were synthesized by GenScript (Nanjing, China), and were dissolved in 10 mM Tris buffer (pH 7.6) to a final concentration of 100 μM.
Establishment of MCF-7-GFP-Mut Stable Cell Line
The human cell lines 293T and MCF-7 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA).14 In order to construct a GFP silent mutation cell line, the triplet TCA in GFP gene coding sequence was mutated to stop the code of TGA (Fig. S1A). The full-length sequence of mutated GFP was cloned into lentivirus vector pSIN-EF1-IRES-puromycin, and co-transfected with auxiliary pSPAX2 and pMD2.G plasmids into 293T cells to generate lentivirus. Following lentivirus infection, MCF-7-GFP-Mut cell clones were screened out by puromycin, and the positive cell clones were verified by DNA sequencing and used for the experiments (Fig. S1A and B).
Cell Transfection and Detection of Correction of GFP Silent Mutation
MCF-7-GFP-mut cells were seeded into 6-well plate before transfection. After 24hrs, 3.0 μL GFP ssODN (sense or antisense ssODN) and 3.0 μg CRISPR/Cas9-sgRNA vector were transfected into MCF-7-GFP-mut cell line using Lipofectamin2000 according to the manual. The vector of CRISPR/Cas9-sgRNA was designed to specific target GFP mutation sequence (Fig. S1A). After 48 hrs, cells were harvested and divided into three parts for detection of correction of GFP silent mutation. The first portion was used to analyze the rate of GFP-positive cells by fluorescence-activated cell sorting (FACS).The second portion was used for DNA extraction and DNA sequencing. The third part was seeded into a 6-cm cell culture dish to expend for further assays of fluorescence microscopy and Western blot. The experiment was repeated three times.
Construction of Cas9-GFP-U6-sgRNA Vector for Targeting β-catenin ΔTCT Ser45 Deletion Mutation
The cloning cohesive sites, sgRNAs targeted to β-catenin ΔTCT Ser45 deletion sequences were obtained by annealing two synthesized complementary oligonucleotides. Then, sgRNAs were cloned into the BbsI-digested gRNA expression vector pUC57-U6-sgRNA. Next, a pair of primers with the cloning cohesive sites Xho I and Xba I (Table S1) were used to amplify the U6-sgRNA expression frame from pUC57-U6-sgRNA, and the sgRNA of β-catenin ΔTCT Ser45 sequence was inserted into the locus in front of PolyA of the pCS2-3×FLAG-SpCas9-PolyA-IERS-GFP to construct a sgRNA and Cas9 co-expression vector, pCS2-Cas9-polyA-IRES-GFP-U6-sgRNA (β-catenin ΔTCT Ser45).
Selection of β-catenin ΔTCT Ser45 Mutation-Corrected HCT-116 Clones
The human cell line HCT-116 was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA).15 HCT-116 cells were seeded in a 6-well plate at a density of 8.0×105 cells/well for 24 hrs before transfection. Cells were transfected by Lipfectamine2000 according to the manual (Invitrogen) using the following programs: 3.0 μg pCS2-Cas9-GFP/sgRNA plasmid and 0.3 pmol ssODN were added to 250 μL optimum medium and 10 μL transfection reagent (lipfectamine2000) was added to 250 μL optimum medium, respectively. Then, the medium was mixed and incubated at room temperature for 20 min, and finally added to the cell culture medium. Regular medium was replaced after 5hrs. After 48 hrs, cells were harvested and rinsed with 1×PBS, and GFP-positive cells were sorted out by FACS using FACS Aria (BD Biosciences, USA), and plated single-cell in 96-well plates and incubated at 37°C for 2 weeks. Selected clones were expanded for further experiments.
DNA Isolation, TA Cloning, and Sequencing
Genomic DNA was extracted from the cultured cells using a DNA isolation kit (Tiangen, Beijing, China). Briefly, targeted gene sequences were amplified using premix LA Taq (Takara). The amplicons were separated using agarose gel electrophoresis and the target bands were extracted and purified using the GEL/PCR Purification Kit (Tiangen, Beijing, China). A spectrophotometer is used to measure the DNA concentration. For TA cloning ligation, DNA fragment (0.1–0.3 pmol) was mixed with pMD18-T vector (Takara) and incubated at 16°C overnight. After TA cloning, 20 TA clones were randomly picked up for DNA sequencing, and the disruption rate (%) was calculated based on the results of DNA sequencing.
T7E1 Assay
To analyze the disruption rate of Cas9-sgRNA targeting the GFP-mut, the method of T7E1 assay was used as described previously.16,17 In short, after Cas9-sgRNA vector targeting GFP-mut transfection into MCF-7-GFP-mut cells, cellular DNA was extracted and used as templates to amplify the fragment harboring target sites by PCR. Then, the PCR product was denatured, reannealed, and digested with T7 endonuclease I (New England BioLabs Inc, Ipswich, MA, USA). Finally, the digestion production was analyzed by agarose gel electrophoresis and the cleaved fragment was observed by Gel imager. The disruption rate was calculated as described previously.16,17
PCR Amplification and Mutation Detection
For detection of mutation correction of β-catenin ΔTCT Ser45, we designed two pairs of primers flanking the β-catenin ΔTCT Ser45 site (Table S1, Supplementary information). A total of 100 ng genomic DNA of each sample was used for PCR. PCR was carried out using Q5TM Hot Start High-Fidelity 2×Master Mix (M0494L, New England BioLabs, Ipswich, MA, USA) with the following cycling condition: 98°C for 1 min, then 32 cycles of 98°C for 10 s, 59°C for 20 s and 72°C for 30 s, and a final cycle of 72°C for 3 min. The mutation and mutation correction of β-catenin were confirmed by DNA sequencing.
CCK8 Assay, EDU Dyeing and Clonogenic Assay
For CCK8 assay, HCT-116 cells and HCT-116 corrected clone cells were seeded into 96-well cell culture plates (5000 cells/well). After 24, 48, and 72 hrs, CCK8 solution (Beyotime, China) was added for an additional 4 hrs at 37°C. Absorbance at 450 nm was determined for each well using a microplate enzyme-linked immunosorbent assay reader.
For EDU dyeing, HCT-116 cells and HCT-116 corrected clone cells were seeded into 96-well cell culture plates (5000 cells/well). After 24 hrs, 25 µM 5-ethynyl-2ʹ-deoxyuridine (EDU) was added for an additional 2 hrs at 37°C. Then, the cells stain with Cell-LightTM Apollo Stain Kit (RIBOBIO, Guangzhou, China) after washed with PBS and fixed with 4% formaldehyde for 30 mins. Before stained with Hoechst (RIBOBIO, Guangzhou, China) for 15mins, the cells were permeabilized with 0.5% Triton X-100 (Solarbio, Beijing, China) for 15 min. Images were obtained using a fluorescent microscopy (BX-51, Olympus Corporation, Tokyo, Japan).
For clonogenic assay, cells were plated onto 6-well plates (500 cells/well). Plated cells were cultured in 5% CO2 with 95% humidity at 37°C for 14 days. The plates were then fixed with 4% formaldehyde and stained with Giemsa. The colonies containing more than 50 cells were counted using ImageQuantTL7.0 Image Analysis Software (GE Healthcare, Buckinghamshire, UK).
Quantitative Real-Time PCR
Total RNA was isolated using a RNeasy Mini kit (QIAGEN) according to the manufacturer’s instructions. A total of 2 μg RNA was subjected to reverse transcription reaction to obtain cDNAs (Reverse Transcription System, promega, USA). Specific quantitative real-time PCR experiments were performed using GoScriptTM RT System (Promega, USA) following the manufacturer’s protocol (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal control. The primers used in this research are listed in Table S1 (Supplementary information).
Western Blotting
Total protein was extracted from cells with RIPA buffer (Beyotime, China). Nuclear protein and cytoplasmic protein were extracted with a nuclear cytoplasmic protein extraction kit (Beyotime, China). All protein concentrations were measured by BCA protein assay kit (Beyotime, China). Approximately 40 μg protein extract per sample was separated using a SDS polyacrylamide gel and transferred onto the PVDF membrane (Millipore, USA), and 5% bovine serum albumin was used to block membrane. The membranes were incubated with rabbit anti-GFP (1:1000, Beyotime, China), anti-β-catenin (1:1000, Cell Signaling, USA), anti-Survivin (1:1000, Abcam, USA), anti-cyclin D (1:1000, Cell Signaling, USA), anti-c-myc (1:1000, Abcam, USA), anti-β-catenin Ser45 phosphorylation (1:1000, Cell Signaling, USA), and mouse antibody against β-actin (1:10,000, Sigma, USA), anti-Histone 3 (1:1000, Sigma, USA) overnight at 4°C, followed by incubation for 1hr with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000). After extensive washing in TBST, the expression levels of the protein were detected by Quantity-one software (Bio-Rad Laboratories, USA) using the ECL kit.
Xenograft Tumor Growth
The 6-week-old male athymic BALB/c nude mice were purchased from Hunan SLAC Jingda Experimental Animal Company (Changsha, China). Nude mice were maintained in laminar flow cabinets under conditions of specific pathogenic free. Five mice in each group were subcutaneously injected with 5.0 × 106 stable HCT-116 corrected cells or control cells in 0.2 mL PBS with 50% matrigel. Five weeks after the injection, the nude mice were sacrificed, and the dorsal tumors were excised and weighed after they had been photographed and measured. Sections from the tumors were stained with hematoxylin and eosin (H&E) for general histology, or immunohistochemistry (IHC) stained with specific antibodies (anti-Ki67 for proliferation).All animal experiments were approved by the Ethical Committee of the First People’s Hospital of Chenzhou City, university of South China. We followed the guideline, Laboratory Animal-guideline for Ethical Review of Animal Welfare of P.R. China, for the welfare of animals.
Statistics
Quantitative data are expressed as mean ± SD. SPSS.20 was used for data analysis. The Student’s t-test or one-way ANOVA was applied to determine significant differences in multiple comparisons. In these analyses, p values were two-sided, and p values <0.05 were considered statistically significant.
Results
Correction of GFP Silent Mutation in MCF-7 Cells Using CRISPR/Cas9-sgRNA and ssODN
To verify whether using CRISPR/Cas9-sgRNA and ssODN can correct gene mutations in mammalian cells, we constructed a MCF-7-GFP-mut cell line containing GFP silent mutation by lentivirus-mediated transduction (Fig. S1). The vector of Cas9-sgRNA specific targeting GFP mutation sequence was used to transfect into MCF-7-GFP-mut cells combined with GFP wild-type ssODN (sense or antisense ssODN) (Fig. S1, Table S1). Compared to no Cas9-sgRNA transfection groups, the GFP-positive cells using CRISPR/Cas9-sgRNA and ssODN to correct were obviously increased by FACS assay (Figure 1A and B). The efficiency of using antisense ssODN as templates is higher than that using sense ssODN in our test, which is consistent with the previous reports.11,18 These results were further confirmed by fluorescence microscopy, DNA sequencing and Western blot showing the correction of GFP silent mutation in MCF-7-GFP-mut cells using CRISPR/Cas9-sgRNA and ssODN (Figure 1C–E).
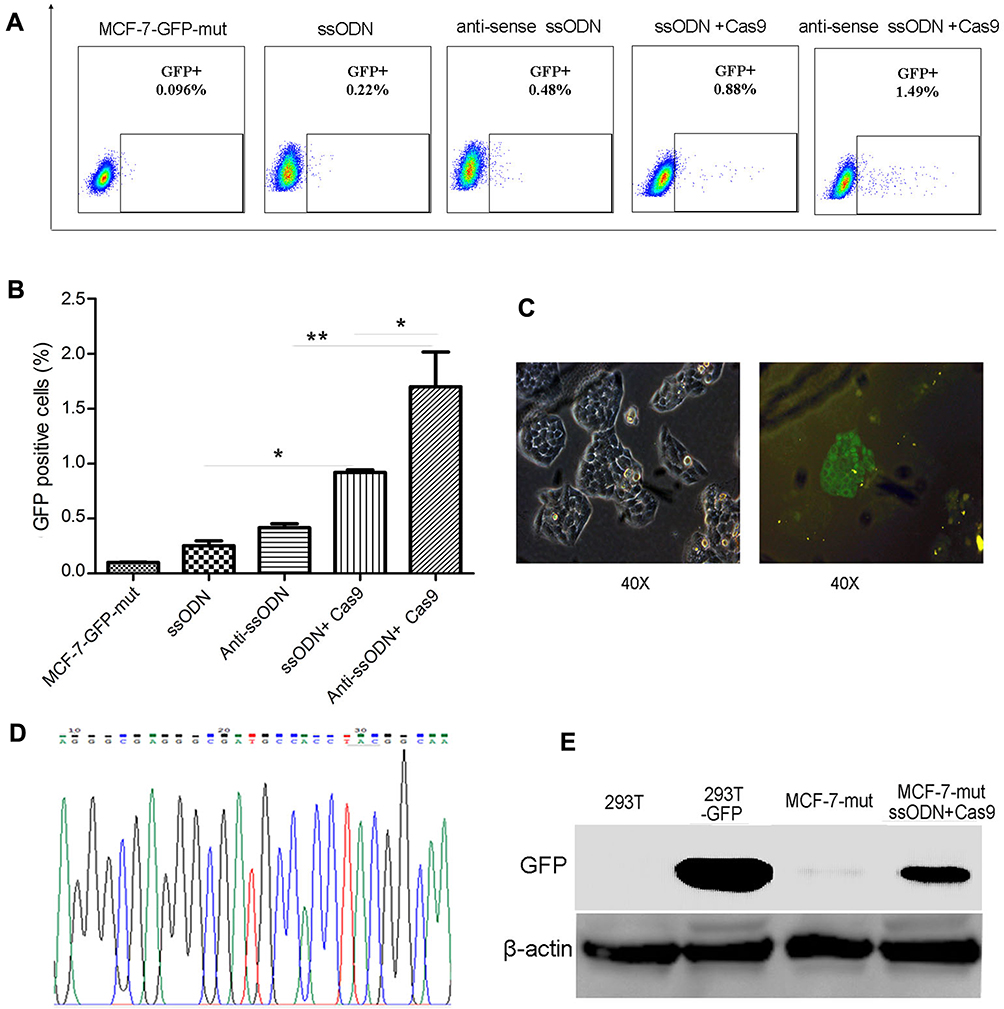 |
Figure 1 Correction of GFP silent mutation in MCF-7 cells using CRISPR/Cas9-sgRNA and ssODN. The FACS assay results of using CRISPR/Cas9-sgRNA and ssODN to correct GFP silent mutation in MCF-7-GFP-mut cells. Represented images (A), and the statistics results of the GFP-positive cell rate (*p<0.05, **p<0.01) (B). (C–E) Further verified results after using CRISPR/Cas9-sgRNA and ssODN to correct GFP silent mutation in MCF-7-GFP-mut cells. (C) The results of fluorescence microscopy assays. (D) The results of DNA sequencing. (E) The results of Western blot. MCF-7-GFP-mut/MCF-7-mut: MCF-7 cells with GFP silent mutation; ssODN: GFP-sense-ssODN-90bp; anti-sense ssODN: GFP-antisense-ssODN-90bp; Cas9: CRISPR/Cas9-sgRNA vector targeting to GFP mutation sequence;293T-GFP: GFP transfected 293T cells; MCF-7-mut ssODN + Cas9: MCF-7-GFP-mut cells transfected using the vector of CRISPR/Cas9-sgRNA and GFP ssODN (sense-ssODN or antisense-ssODN). |
Correction of β-catenin ΔTCT Ser45 Deletion Mutation in HCT-116 Cells Using CRISPR/Cas9-sgRNA-GFP Vector and ssODN
The human colorectal cell line HCT-116 was chosen for this study as it has a heterozygous deletion mutation (ΔTCT) of β-catenin genes, which is responsible for encoding the regulatory 45 serine (Ser45) at the N-terminal region of the protein.19 We confirmed the location of ΔTCT Ser45 deletion mutation on the exon 3 of β-catenin gene and the CRISPR/Cas9 PAM site by gene sequencing (Figure 2A).
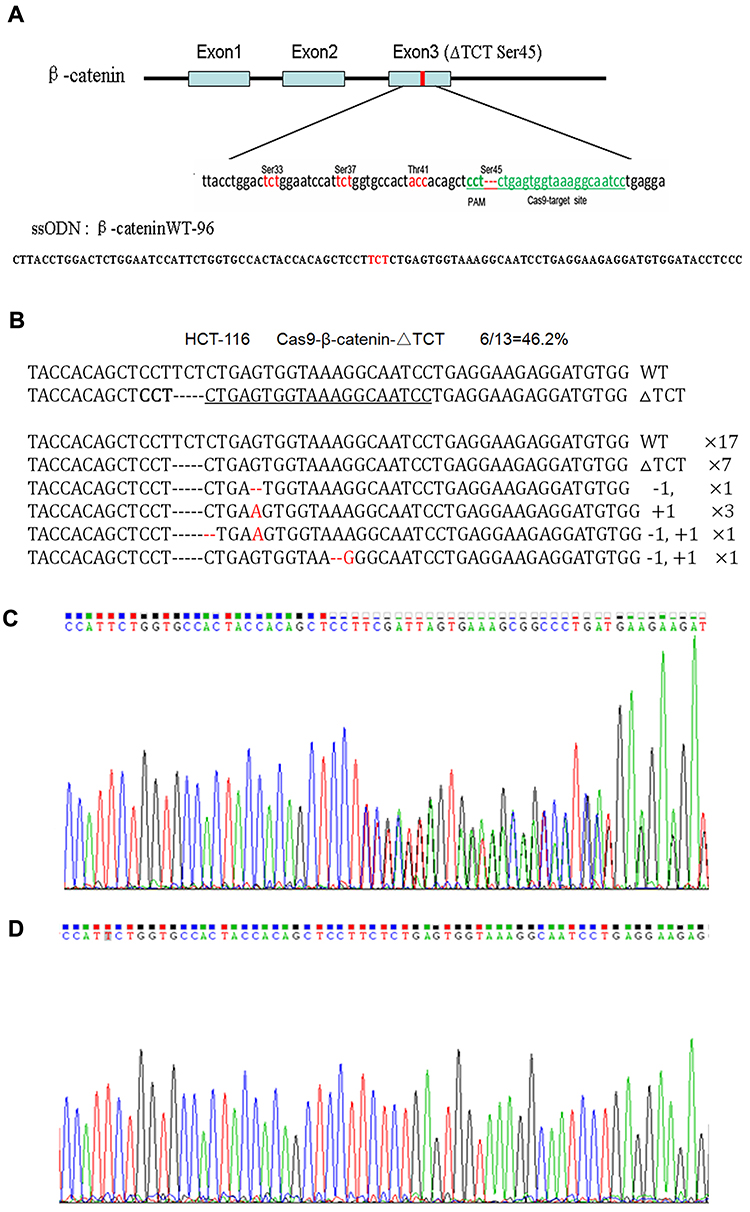 |
Figure 2 CRISPR/Cas9-directed mutation correction of β-catenin ΔTCT Ser45 mutation in HCT-116 cells. (A) Depiction of the CRISPR/Cas9 targeted mutation site (ΔTCT Ser45) in green letters and surrounding sequences in exon 3 of β-catenin gene and the sequence of ssODN. The genetic codes of serine and threonine phosphorylated amino acids around the target site are marked in red-color letters. (B) The disruption rate induced by transfecting the Cas9-GFP/sgRNA (ΔTCT Ser45) co-expression vector into HCT-116 cells. (C, D) The representative results of using PCR products directed sequencing of the β-catenin gene for HCT-116 wild-type cells (C), and mutation-corrected cell clones (D). |
With sgRNAs targeting to β-catenin ΔTCT Ser45 deletion sequences obtained by annealing two synthesized partially complementary oligonucleotides, we constructed a Cas9-GFP/sgRNA co-expression vector. A 96-nt ssODN containing the wild-type β-catenin gene sequence as a template for HDR (Figure 2A). After HCT-116 cells were co-transfected with the vector and ssODN, GFP-positive cells were sorted by FACS and expanded, and the mutation correction efficiency was calculated by TA cloning and sequencing. The disruption rate of specific targeting β-catenin ΔTCT Ser45 deletion sequence was 46.2% by this method (Figure 2B).
Due to a deletion mutation in one allele of the gene, the sequencing map of β-catenin in HCT-116 cells showed overlapped peaks starting from the mutation locus (Figure 2C). After mutation was corrected, we found the presence of a TCT sequence in the mutation locus and the overlapped peaks starting from the mutation locus was disappeared accordingly (Figure 2D). These data indicated that co-transfected with the Cas9-GFP/sgRNA co-expression vector and ssODN could correct the β-catenin ΔTCT Ser45 deletion mutation in HCT-116 cells.
Restoration of β-catenin Phosphorylation at Ser45
Ser45 of the β-catenin is an important phosphorylation site.20 Deletion mutation of the codon ΔTCT will result in a significant decrease of β-catenin Ser45 phosphorylation in HCT-116 cells. Therefore, to study whether β-catenin ΔTCT deletion mutation correction resulted in restoration of the expression of Ser45 phosphorylated β-catenin, we tested the expression of Ser45 phosphorylated β-catenin and total β-catenin by Western blotting. As expected, uncorrected HCT-116 cells had very weak detectable Ser45 phosphorylated β-catenin expression, compared to the DLD1 colorectal cancer cells without β-catenin ΔTCT deletion mutations (Figure 3A). Importantly, the level of Ser45 phosphorylated β-catenin protein was significantly increased in the mutation-corrected cells (Figure 3A). There is no difference in the expression of total β-catenin among the DLD1 cells, HCT-116 cells and mutation-corrected HCT-116 cells. In summary, our data showed that β-catenin ΔTCT deletion mutation correction resulted in the restoration of β-catenin Ser 45 phosphorylation.
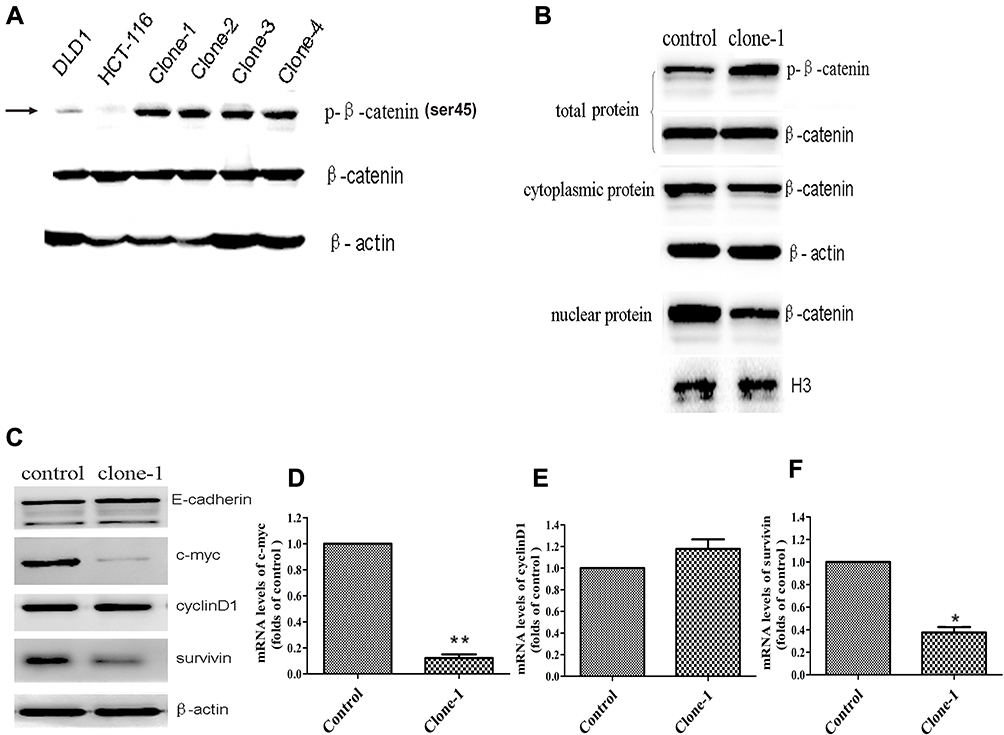 |
Figure 3 Functional effects of CRISPR/Cas9-mediated β-catenin ΔTCT Ser45 mutation correction. (A) Western blotting analysis of phospho-β-catenin (Serine 45) and total β-catenin levels in DLD1, the control HCT-116 cells and multiple mutation-corrected HCT-116 cell clones (clone1-4). β-actin was used as an internal control. (B) Western blotting (representative of one clone) shows total, nuclear and cytoplasmic β-catenin levels in the control and one mutation-corrected HCT-116 cell clone. The H3 histone and β-actin were used as loading controls. (C) Western blotting shows the expression of E-cadherin, c-myc, cyclinD1 and survivin of the control cells and one mutation-corrected HCT-116 cell clone. (D–F) Quantitative RT-PCR analysis the expression levels of c-myc, cyclinD1 and survivin genes in one corrected clones compared with the control cells. Control: uncorrected cells with transfection of Cas9 control vector. Clone-1: one of clones with β-catenin mutation correction. *p<0.05, **p<0.01. |
Effects on β-catenin Nuclei Transfer
β-Catenin is a key conductance regulator in the canonical Wnt signaling pathway. The genetic mutation of the site encoding phosphorylation residue of the β-catenin prevents it from being phosphorylated and degraded, results in its accumulation in the cytoplasm and transportation into the nucleus. In order to investigate whether the expression level of β-catenin changes in the nucleus, we detected the cytoplasmic and nuclear β-catenin by Western blotting. As expected, compared with the uncorrected HCT-116 cells, the level of cytoplasmic β-catenin had no significant change but the level of nuclear β-catenin was dramatically decreased in the corrected cells (Figure 3B). These findings indicate that corrected β-catenin mutation restored phosphorylation and degradation of β-catenin and reduced the transportation of β-catenin in the nucleus.
Effects on Expression of Downstream Target Genes
β-Catenin, the transcription modulator in classical Wnt/β-catenin pathway, interacts with TCF/LEF and leads the expression of related downstream proteins, such as c-myc and cyclinD1.21–24 In this study, we detected the expression of c-myc, cyclinD1 and survivin in the control cells and mutation-corrected cells by Western blotting and quantitative real-time PCR. The results showed the correction of β-catenin genetic mutation restored phosphorylation and attenuated both mRNA and protein expression of c-myc and survivin (Figure 3C–F), though the expression of cyclinD1 was not significantly changed. Therefore, our data indicate that the correction of β-catenin genetic mutation regulated the activity of the Wnt/β-catenin signaling pathway.
Effects of Mutation Correction on Cell Growth in vitro
The increased expression of β-catenin in the nucleus can regulate cell proliferation and growth. After correcting the genetic mutation of β-catenin in HCT-116 cells, its effect on cell growth was evaluated by CCK8, EDU, and colony formation assays. The results of CCK8 assay showed that β-catenin correction significantly inhibited the proliferation of HCT-116 cells compared with control cells as observed at the time points of 24, 48, and 72 hrs (Figure 4E). The inhibited growth of HCT-116 cells with β-catenin correction was also documented by EDU dyeing assay (Figure 4A and B). The results of colonic formation assay were consistent with the CCK8 and EDU assays (Figure 4C and D). In summary, these results indicate that β-catenin mutation correction disrupted the abnormal cell proliferation in HCT-116 cells.
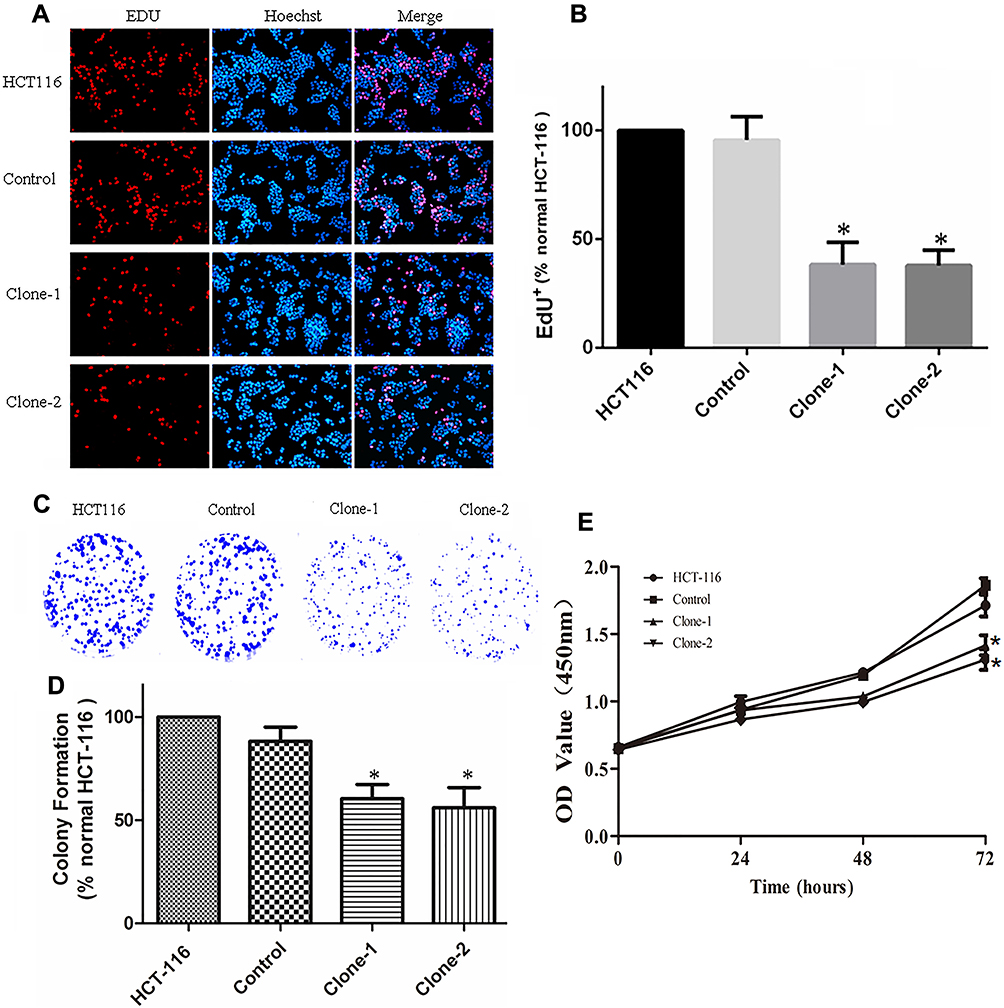 |
Figure 4 CRISPR/Cas9-mediated β-catenin ΔTCT Ser45 mutation correction inhibit cell growth and colony formation. (A) Representative images of EDU dyeing assay from the control and mutation-corrected HCT-116 cell clones. Red, EDU+ cells; Blue, Hoechst; Pink, EDU+ cells in the merged image. (B) Quantification of percent EDU+ cells. The proportion of EDU+ nuclei is decreased in mutation-corrected HCT-116 cell clones. (C) Colony formation assay showed that correction of β-catenin Ser45 mutation reduced the colony formation of HCT-116 cells. (D) Normalized fold changes of colony formation in the control and mutation-corrected HCT-116 cell clones. (E) CCK8 assay showed that the proliferation rate of mutation-corrected HCT-116 cells was reduced. *p< 0.05. |
Effects on Tumor Growth in Mouse Xenograft
To determine the effects of β-catenin mutation correction in vivo, we xenografted uncorrected control cells and mutation-corrected cell clones into nude mice. Compared to controls, mice with mutation-corrected cells demonstrated significantly smaller size and less weight of lumps, indicating decreased proliferative ability of β-catenin mutation-corrected HCT-116 cells in vivo (Figure 5A–C). IHC results showed that the expression of Ki67 in β-catenin mutation-corrected cells was significantly decreased compared with the uncorrected cells (Figure 5D and E). These data suggest that correction of β-catenin mutation in HCT-116 cells inhibits proliferation in vivo.
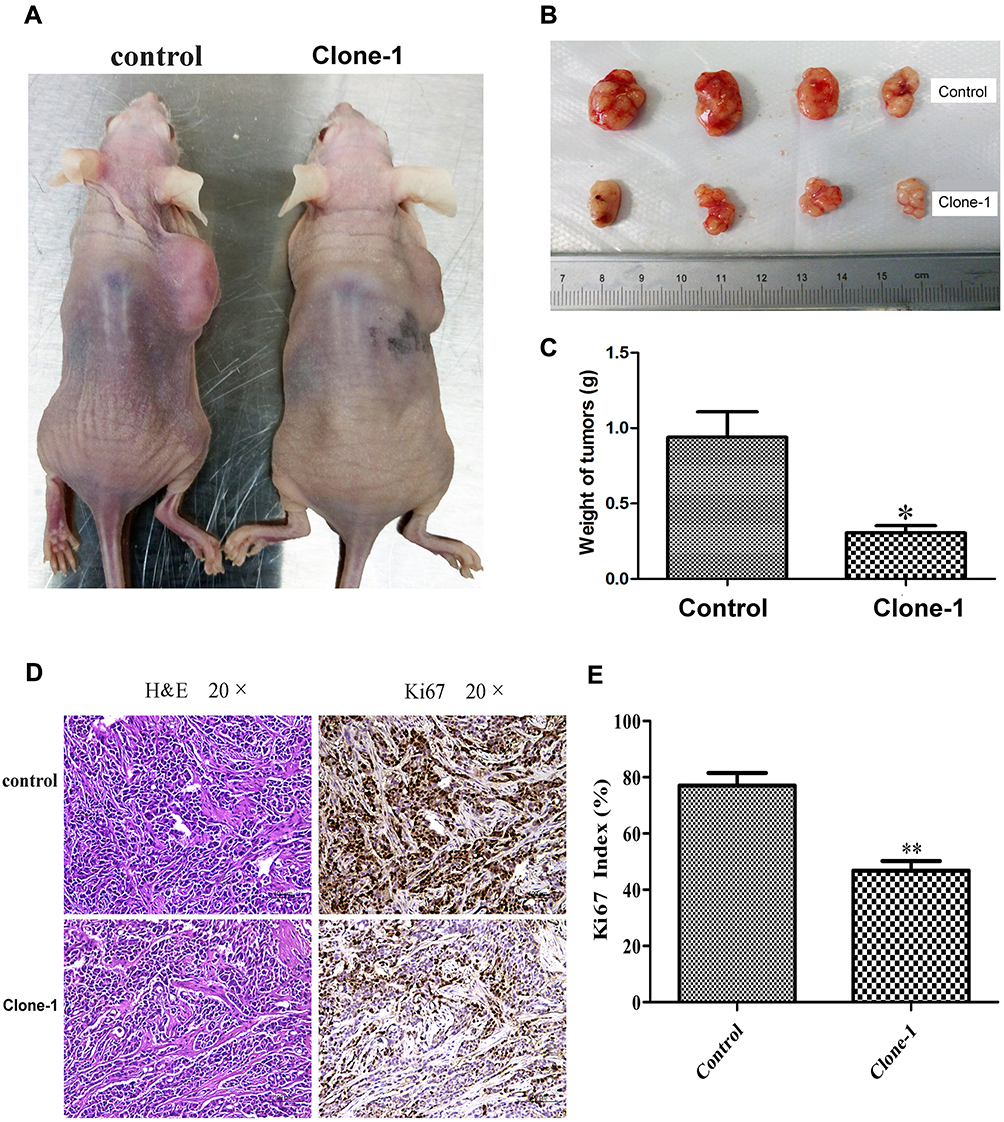 |
Figure 5 In vivo effects of CRISPR/Cas9-mediated correction of β-catenin Ser45 mutation in mouse xenografts. (A) Mutation-corrected HCT-116 cells inhibited the formation of subcutaneous tumor in nude mice. Cells were inoculated subcutaneously on the back of nude mice. (B) The tumor size in the control group and mutation-corrected cell clone group. (C) Normalized fold changes of tumor weight in the control and mutation-corrected HCT-116 cell clone. (D) The tumor sections were subjected to H&E and IHC staining using antibodies against Ki-67. (E) Normalized fold changes of Ki-67 index in the control and mutation-corrected HCT-116 cell clone. The Ki-67 index was calculated as the number of Ki-67 positive cells divided by the number of total cells × 100%. The data is the mean ± SD of 4 independent experiments. *p<0.05, **p<0.01, compared to the control. |
Discussion
Colorectal cancer is the third most common cancer. CRC has a strong association with deregulation of Wnt/β-catenin signaling pathway.25–28 Wnt/β-catenin is the canonical β-catenin dependent Wnt signaling of the Wnt signaling pathway, which controls numerous developmental processes and leads to tumor formation when aberrantly activated.29–32 The key step of Wnt/β-catenin pathway involves the balance between stabilization and degradation of β-catenin. Normally, β-catenin is phosphorylated at Ser45 by casein kinase 1 (CK1), and consecutively at Ser33, Ser37 and Thr41 by glycogen synthase kinase-3β (GSK-3β), which trigger the subsequent ubiquitination and proteasomal degradation.20,33–35 When Wnt protein bind, β-catenin is stabilized by a supercomplex Wnt-Frz-LRP5/6-DVL-AXIN, which lead β-catenin accumulation in the nucleus and regulate transcription of Wnt target genes, such as c-myc, survivin and cyclinD1.21,36–40 Regulation of β-catenin fails when β-catenin itself contains mutations.41 A high frequency of β-catenin gene mutations is found in 6% CRC and hotspot mutation region is observed in exon 3 of β-catenin gene, which is responsible for encoding the regulatory sequence DS33GIHS37GAVT41QAPS45 in the N-terminus of the protein.3,42 Mutations of these Ser/Thr residues alters functionally significant phosphorylation sites, inhibits the β-catenin phosphorylation-degradation cascade and results in constitutive activation of Wnt/β-catenin signaling pathway. Then, correcting β-catenin genetic mutation by gene-editing technology may develop a next-generation therapeutic approach for colon cancer.43–45
The CRISPR/Cas9 is a prokaryotic nucleic acid-based adaptive immune system to confer resistance to foreign genetic elements such as viral DNA or other foreign DNA.46,47 Due to the guidance of sgRNA complementary to the target DNA sequence, Cas9 protein binds to a specific genomic locus and creates a site-specific DSB.48 DSB initiates inaccurate but dominant NHEJ or low efficient but more precise HDR.49 Providing wild-type homologous DNA repair templates can achieve precise gene editing. Previous studies demonstrated that ssODN could be used as templates to generate point mutations and short sequence insertions in human cells and animal models.11–13,50,51 Our previous study also suggested that the use of ssODN as repaired templates facilitates the CRISPR/Cas9-directed HDR.16 In this study, we sought to explore this strategy of combination of CRISPR/Cas9 and ssODN to correct a specific ΔTCT Ser45 heterozygous mutation of β-catenin, a gene frequently mutated in colon cancer.
In the present study, the plasmid integrated CRISPR/Cas9, eGFP and sgRNA targeting the β-catenin deletion mutation sequence was constructed and co-transfected with a β-catenin wild-type ssODN into HCT-116 cells. Mutation-corrected single-cell clones were screened out by GFP-positive flow cytometry and Sanger’s sequencing of PCR products. We observed that β-catenin Ser45 serine phosphorylation level was restored completely compared with control HCT-116 cells. Cell proliferation and the growth rate of the mutant-corrected cells were much slower than control HCT-116 cells documented by CCK8, cell-plated clones assay and EDU dyeing analysis. Furthermore, in nude mice subcutaneous xenograft experiments, the tumor weight of mutation-corrected cells was obviously lighter than that of the control cells, indicating that the tumorigenic ability of the corrected cells was weakened. These phenotypes of mutant-corrected cells might be resulted from the reducing translocation of β-catenin from cytoplasm to nucleus leading to the decreasing expression of downstream genes such as c-myc and survivin. Our study also showed that mutant-corrected cells expressed lesser c-myc and survivin proteins as compared with control HCT-116 cells. The expression of cyclinD1 in mutation-corrected cells has no difference compared to that in control cells either in protein or in mRNA, indicating this gene is not fully regulated by β-catenin in HCT-116 cells.
Due to the high mutation frequency of β-catenin in patients with CRC, the experimental results from the present study provide the basis for further development of gene therapy protocols for patients with β-catenin gene mutations. However, there are still many obstacles in the clinical application of tumor gene therapy that need to be studied and overcome. For example, multiple genes’ mutations in the same cancer drive tumor progression. Therefore, several genes’ mutations need to be corrected simultaneously. Furthermore, the mutations at various stages of the tumor are also varied. More importantly, for an effective target gene therapy for patients with tumor, the current situation of gene mutations should be determined by accurate genetic detection. This study, to our knowledge, is the first report of the β-catenin genetic correction in colon cancer cells. The data of this study suggest that a combination of gene-editing Cas9/sgRNA and ssODN could be a useful method to correct targeted gene mutation of the genome in situ at cancer cells, which have potential applications in targeted gene therapy to genetic diseases.
Abbreviations
CRC, the colorectal cancer; CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats; ZFN, zinc-finger nuclease; TALEN, transcription activator-like effector nuclease; DSB, double-strand break; ssODN, single-strand oligonucleotide; bp, base pairs; FACS, fluorescence activating cell sorter; GFP, green fluorescent protein; HDR, homology-directed repair; NHEJ, non-homologous end-joining; PAM, proto-spacers adjacent motif; sgRNA, single-guide RNA; EDU, 5-ethynyl-2ʹ-deoxyuridine; TBST, mixture of Tris-buffered saline (TBS) and Tween 20; IHC, immunohistochemistry.
Ethics Approval and Consent to Participate
All experiments were carried out according to the guidelines set by the institutional Animal Ethics Committee of the First People’s Hospital of Chenzhou, Hunan, P.R. China.
Acknowledgments
This work was funded by the China Postdoctoral Science Foundation (No.2016T90765), the Key R&D Program of Hunan Province (2017SK2172), the Natural Science Foundation of Hunan Province (2017JJ2004), the Health Department Project of Hunan Province (B20180378, C20180166, C2019003), the Science and Technique Foundation of Chenzhou (jsyf2017021, zdyf201838), the Key Project of the First People’s Hospital of Chenzhou (N2019-001). We appreciate Jia Li, Weihao Luo, Lili Duan, Rongzhang He and Jian zhang, the colleagues from Translational Medicine Institute of the First People’s Hospital of Chenzhou for their kind help in technical support, data analysis, statistics, and revised suggestions to this manuscript. We are also very grateful to Dr. Kai Li from Genetalks Bio-tech Limited Liability Company, Hunan, China, and Dr. Xinlan Xu from Taizhou People’s Hospital, pharmaceutical high-tech zone, Taizhou, Jiangsu, China for their critical editing of grammar and spelling.
Author Contributions
ZH and DXL conceived the study and designed the experiments. ZH, YLL, XNL conducted the experiments. DXL and ZH analyzed the data. JYQ and ZH wrote the paper. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi:10.1002/ijc.25516
2. Pawa N, Arulampalam T, Norton JD. Screening for colorectal cancer: established and emerging modalities. Nat Rev Gastroenterol Hepatol. 2011;8(12):711–722. doi:10.1038/nrgastro.2011.205
3. Johnson V, Volikos E, Halford SE, et al. Exon 3 beta-catenin mutations are specifically associated with colorectal carcinomas in hereditary non-polyposis colorectal cancer syndrome. Gut. 2005;54(2):264–267. doi:10.1136/gut.2004.048132
4. Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787–1790. doi:10.1126/science.275.5307.1787
5. Christie M, Jorissen RN, Mouradov D, et al. Different APC genotypes in proximal and distal sporadic colorectal cancers suggest distinct WNT/beta-catenin signalling thresholds for tumourigenesis. Oncogene. 2013;32(39):4675–4682. doi:10.1038/onc.2012.486
6. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi:10.1038/nature11252
7. Zhang X, Wang L, Liu M, Li D. CRISPR/Cas9 system: a powerful technology for in vivo and ex vivo gene therapy. SCI CHINA LIFE SCI. 2017;60(5):468–475. doi:10.1007/s11427-017-9057-2
8. Men K, Duan X, He Z, Yang Y, Yao S, Wei Y. CRISPR/Cas9-mediated correction of human genetic disease. Sci China Life Sci. 2017;60(5):447–457. doi:10.1007/s11427-017-9032-4
9. Lone BA, Karna SKL, Ahmad F, Shahi N, Pokharel YR. CRISPR/Cas9 system: a bacterial tailor for genomic engineering. Genet Res Int. 2018;2018:3797214. doi:10.1155/2018/3797214
10. Hefferin ML, Tomkinson AE. Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair (Amst). 2005;4(6):639–648. doi:10.1016/j.dnarep.2004.12.005
11. Bialk P, Rivera-Torres N, Strouse B, Kmiec EB. Regulation of gene editing activity directed by single-stranded oligonucleotides and CRISPR/Cas9 systems. PLoS One. 2015;10(6):e0129308. doi:10.1371/journal.pone.0129308
12. Chen F, Pruett-Miller SM, Davis GD. Gene editing using ssODNs with engineered endonucleases. Methods Mol Biol. 2015;1239:251–265.
13. Kan Y, Ruis B, Takasugi T, Hendrickson EA. Mechanisms of precise genome editing using oligonucleotide donors. Genome Res. 2017;27(7):1099–1111. doi:10.1101/gr.214775.116
14. Li J, Guo Y, Duan L, et al. AKR1B10 promotes breast cancer cell migration and invasion via activation of ERK signaling. Oncotarget. 2017;8(20):33694–33703. doi:10.18632/oncotarget.16624
15. Wang Q, He R, Tan T, et al. A novel long non-coding RNA-KAT7 is low expressed in colorectal cancer and acts as a tumor suppressor. Cancer Cell Int. 2019;19:40.
16. Hu Z, Shi Z, Guo X, et al. Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR-Cas9 and ssODN in human cancer cells. Cell Biosci. 2018;8:12. doi:10.1186/s13578-018-0200-z
17. Hu Z, Wang L, Shi ZY, et al. Customized one-step preparation of sgRNA transcription templates via overlapping PCR using short primers and its application in vitro and in vivo gene editing. Cell Biosci. 2019;9:87. doi:10.1186/s13578-019-0350-7
18. Pierce EA, Liu Q, Igoucheva O, et al. Oligonucleotide-directed single-base DNA alterations in mouse embryonic stem cells. Gene Ther. 2003;10(1):24–33. doi:10.1038/sj.gt.3301857
19. Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci U S A. 1997;94(19):10330–10334. doi:10.1073/pnas.94.19.10330
20. Amit S, Hatzubai A, Birman Y, et al. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002;16(9):1066–1076. doi:10.1101/gad.230302
21. Rennoll S, Yochum G. Regulation of MYC gene expression by aberrant Wnt/beta-catenin signaling in colorectal cancer. World J Biol Chem. 2015;6(4):290–300. doi:10.4331/wjbc.v6.i4.290
22. Yochum GS, Sherrick CM, Macpartlin M, Goodman RH. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5ʹ and 3ʹ Wnt responsive enhancers. Proc Natl Acad Sci U S A. 2010;107(1):145–150. doi:10.1073/pnas.0912294107
23. Yochum GS. Multiple Wnt/ss-catenin responsive enhancers align with the MYC promoter through long-range chromatin loops. PLoS One. 2011;6(4):e18966. doi:10.1371/journal.pone.0018966
24. Yochum GS, Cleland R, Goodman RH. A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol Cell Biol. 2008;28(24):7368–7379. doi:10.1128/MCB.00744-08
25. Kolligs FT, Bommer G, Goke B. Wnt/beta-catenin/tcf signaling: a critical pathway in gastrointestinal tumorigenesis. Digestion. 2002;66(3):131–144. doi:10.1159/000066755
26. Buchert M, Rohde F, Eissmann M, et al. A hypermorphic epithelial beta-catenin mutation facilitates intestinal tumorigenesis in mice in response to compounding WNT-pathway mutations. Dis Model Mech. 2015;8(11):1361–1373. doi:10.1242/dmm.019844
27. Burgess AW, Faux MC, Layton MJ, Ramsay RG. Wnt signaling and colon tumorigenesis–a view from the periphery. Exp Cell Res. 2011;317(19):2748–2758. doi:10.1016/j.yexcr.2011.08.010
28. Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer. A review in the theme: cell signaling: proteins, pathways and mechanisms. Am J Physiol Cell Physiol. 2015;309(8):C511–C521. doi:10.1152/ajpcell.00117.2015
29. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi:10.1146/annurev.cellbio.20.010403.113126
30. Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4:5. doi:10.1101/cshperspect.a008052
31. Duchartre Y, Kim Y-M, Kahn M. The Wnt signaling pathway in cancer. Crit Rev Oncol Hematol. 2016;99:141–149. doi:10.1016/j.critrevonc.2015.12.005
32. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8(5):387–398. doi:10.1038/nrc2389
33. Liu C, Li Y, Semenov M, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108(6):837–847. doi:10.1016/S0092-8674(02)00685-2
34. Stamos JL, Weis WI. The beta-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5(1):a007898. doi:10.1101/cshperspect.a007898
35. Nakamura T, Hamada F, Ishidate T, et al. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells. 1998;3(6):395–403. doi:10.1046/j.1365-2443.1998.00198.x
36. Mosimann C, Hausmann G, Basler K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol. 2009;10(4):276–286. doi:10.1038/nrm2654
37. Moon RT. Wnt/beta-catenin pathway. Sci STKE. 2005;2005(271):cm1.
38. Kafka A, Basic-Kinda S, Pecina-Slaus N. The cellular story of dishevelleds. Croat Med J. 2014;55(5):459–467. doi:10.3325/cmj.2014.55.459
39. Tauriello DV, Jordens I, Kirchner K, et al. Wnt/beta-catenin signaling requires interaction of the dishevelled DEP domain and C terminus with a discontinuous motif in Frizzled. Proc Natl Acad Sci U S A. 2012;109(14):E812–E820. doi:10.1073/pnas.1114802109
40. Zeng X, Huang H, Tamai K, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008;135(2):367–375. doi:10.1242/dev.013540
41. Kishida M, Koyama S, Kishida S, et al. Axin prevents Wnt-3a-induced accumulation of beta-catenin. Oncogene. 1999;18(4):979–985. doi:10.1038/sj.onc.1202388
42. Kim S, Jeong S. Mutation hotspots in the beta-catenin gene: lessons from the human cancer genome databases. Mol Cells. 2019;42(1):8–16. doi:10.14348/molcells.2018.0436
43. Sebio A, Kahn M, Lenz HJ. The potential of targeting Wnt/beta-catenin in colon cancer. Expert Opin Ther Targets. 2014;18(6):611–615. doi:10.1517/14728222.2014.906580
44. Cheng X, Xu X, Chen D, Zhao F, Wang W. Therapeutic potential of targeting the Wnt/beta-catenin signaling pathway in colorectal cancer. Biomed Pharmacother. 2019;110:473–481. doi:10.1016/j.biopha.2018.11.082
45. Gupta SK, Shukla P. Gene editing for cell engineering: trends and applications. Crit Rev Biotechnol. 2017;37(5):672–684. doi:10.1080/07388551.2016.1214557
46. Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–1712. doi:10.1126/science.1138140
47. Santiago-Fernandez O, Osorio FG, Quesada V, et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat Med. 2019;25(3):423–426. doi:10.1038/s41591-018-0338-6
48. Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi:10.1126/science.1232033
49. Salsman J, Dellaire G. Precision genome editing in the CRISPR era. Biochem Cell Biol. 2017;95(2):187–201. doi:10.1139/bcb-2016-0137
50. Bialk P, Sansbury B, Rivera-Torres N, Bloh K, Man D, Kmiec EB. Analyses of point mutation repair and allelic heterogeneity generated by CRISPR/Cas9 and single-stranded DNA oligonucleotides. Sci Rep. 2016;6:32681. doi:10.1038/srep32681
51. Rivera-Torres N, Banas K, Bialk P, Bloh KM, Kmiec EB. Insertional mutagenesis by CRISPR/Cas9 ribonucleoprotein gene editing in cells targeted for point mutation repair directed by short single-stranded DNA oligonucleotides. PLoS One. 2017;12(1):e0169350. doi:10.1371/journal.pone.0169350
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.