Back to Journals » Drug Design, Development and Therapy » Volume 13
Cardiovascular adverse events during treatment with darunavir-based regimens in an Italian observational study
Authors Antinori A, Rusconi S ![]() , Gianotti N
, Gianotti N ![]() , Bini T, Mancusi D
, Bini T, Mancusi D ![]() , Termini R
, Termini R ![]()
Received 20 July 2018
Accepted for publication 2 April 2019
Published 14 May 2019 Volume 2019:13 Pages 1667—1685
DOI https://doi.org/10.2147/DDDT.S180981
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Cristiana Tanase
A Antinori,1 S Rusconi,2 N Gianotti,3 T Bini,4 D Mancusi,5 R Termini5
1HIV/AIDS Department, National Institute for Infectious Diseases “Lazzaro Spallanzani” IRCCS, Rome, Italy; 2Infectious Diseases Unit, DIBIC Luigi Sacco, University of Milan, Milan, Italy; 3Infectious Diseases, San Raffaele Scientific Institute, Milan, Italy; 4Clinic of Infectious Diseases, ASST “Santi Paolo e Carlo” Hospital, Milan, Italy; 5Medical Affairs Department, Infectious Diseases, Janssen-Cilag SpA, Cologno Monzese (MI), Italy
Background: The protease inhibitor (PI) darunavir (DRV) has proven to be highly effective and well tolerated for HIV treatment. The DAD (Data collection on Adverse Effects of Anti-HIV Drugs) cohort showed an increased 5-year cumulative cardiovascular (CV) risk in patients given various PIs, including DRV, whereas two other recent studies found no association between DRV and CV diseases.
Methods: We performed a post-hoc analysis of CV adverse events (CVAEs) in an Italian cohort, the TMC114-HIV4042 observational study, where 875 patients treated with ritonavir-boosted DRV-based regimens were followed for a total of 1,566 patient-years.
Results: We observed 23 CVAEs of any type, including 17 [12 (95%CI, 7–19) per 1,000 patient-years] primary; 14 [10 (95%CI, 5–17) per 1,000 patient-years] were primary Framingham-type general CVAEs, close to what expected according to the Framingham algorithm based on traditional risk factors. Age and systolic blood pressure (SBP) at the time of study enrolment were the only relevant (p<0.01) independent predictors of CVAEs in all models; patients with any CVAE were on average 10 years older and had an SBP 14 mmHg higher than patients without CVAEs. When controlling for age and SBP, the association with other traditional factors, including serum lipids, and with HIV-specific factors was not statistically significant (p>0.05). Models that also adjusted for previous ARV exposure showed no statistically significant association between any-type CVAEs and either DRV doses, 1,200 or 800 mg/daily (as also suggested by propensity score stratification), or previous DRV exposure duration.
Conclusion: We found no evidence of a relationship between DRV use and increased CV risk.
Keywords: HIV infection, darunavir, cardiovascular risk, observational study
Introduction
The prognosis of human immunodeficiency virus (HIV) infection had considerably improved over the years since the introduction of highly active antiretroviral therapy (HAART) two decades ago. With decreased mortality due to HIV-specific causes, there has been an increasing prevalence in death due to non-infectious causes, notably neoplastic and cardiovascular (CV) events.1 An analysis of the DAD (Data collection on Adverse effects of anti-HIV Drugs) cohort showed an increased 5-year cumulative risk of CV adverse events (CVAEs) in patients given regimens based on protease inhibitors (PIs), including darunavir (DRV), compared with the risk observed within populations unexposed to these drugs.2,3 On the other hand, a recent large analysis of data from pooled clinical trials, pharmacovigilance reporting and US administrative claims databases did not find any signal for an association between DRV and cardiovascular diseases (CVDs),4 and a recent case-control study showed no evidence of an increased risk of myocardial infarction (MI) following exposure to DRV or atazanavir, another PI.5
TMC114-HIV4042 was a non-interventional study aiming to collect data on the efficacy and safety of antiretroviral (ARV) regimens based on darunavir/ritonavir (DRV/r) in clinical practice; the main efficacy and overall safety results were previously published.6 The analysis reported here aims to describe the CVAEs that occurred during this observational study and to identify any predictive factor that could be associated with CV risk.
Methods
Study design and execution
The TMC114-HIV4042 study was conducted as described in Antinori et al.6 Both DRV-experienced and DRV-naïve patients were enrolled. Some DRV-experienced patients, referred to hereafter as ex-EAP, started DRV/r before marketing authorization (July 2007), as part of the DRV/r Early Access Program (EAP) (subjects included in the EAP were heavily experienced, not achieving virological suppression on their current regimen, were at risk of clinical or immunological progression, and had limited or no treatment options); the remaining DRV-experienced patients, referred to hereafter as DRV-experienced not-EAP, started DRV/r in routine clinical practice after marketing authorization. The DRV-naïve group included both ARV-experienced and ARV-naïve patients. Patients received a DRV/r-based regimen in routine practice, together with other active ARV drugs, and were observed for 12–42 months up to the end 2012 or earlier discontinuation. The study was approved by the ethical committees of the participating centers, as detailed in the
Definition of cardiovascular events and cardiovascular risk factors
Hard CVAEs were defined as cardiac death, MI and stroke, according to the ACC/AHA atherosclerotic cardiovascular disease (ASCVD) events definition,7 whereas the DAD study definition also included invasive heart surgery.3 General CVAEs were defined as the hard ACC/AHA CVAEs plus congestive heart failure, coronary insufficiency, angina, transient ischaemic attack, and clinical (ie, symptomatic) peripheral artery diseases, similarly to the Framingham general CVD definition.8 The definition of any CVAE included, in addition to the Framingham-type general CVAEs, subclinical artery diseases such as atherosclerosis and carotid artery occlusion or stenosis. CVAEs were adjudicated after examination of the original AE reports.
The baseline values for laboratory variables were defined as the value nearest to study entry from 90 days before to 1 day after, included. The body mass index (BMI) was computed as weight/height^2. The estimated glomerular filtration rate (eGFR) was obtained according to the CKD-EPI equation.9 Serum low-density lipoprotein (LDL) cholesterol levels were either directly measured (14% of samples) or estimated from total and high-density lipoprotein (HDL) cholesterol and triglyceride levels using tables from the Very Large Database of Lipids.10 Active diabetes at study entry was reviewed to include antidiabetic drug use ongoing or started at baseline or fasting serum glucose >125 mg/dL, as described in the Framingham study.8
The Framingham general CVD 10-year risk was calculated as specified.8 The Framingham risk functions derive from Cox models separate for males and females and include age, systolic blood pressure (SBP), use of antihypertensive medication (yes/no), serum total and HDL cholesterol levels, current smoking status (yes/no) and diabetes (yes/no). For the numerical values outside the range for which the index was calculated (age 30–74 years, SBP 90–200 mmHg, total cholesterol 100–405 mg/dL and HDL cholesterol 10–100 mg/dL), the limit value exceeded was used, as recommended in the spreadsheet calculator implementing this instrument. A Framingham risk function using BMI instead of cholesterol levels and previously shown to perform similarly8 was used in this study when total or HDL cholesterol levels were missing. The hard ACC/AHA CVD 10-year risk was calculated as a function of age, gender, race, SBP, antihypertensive medication, serum total and HDL cholesterol levels, smoking and diabetes, as previously reported.7 The limit value exceeded was used for the numerical values outside the validated range (age 40–79 years, SBP 90–200 mmHg, total cholesterol 130–320 mg/dL and HDL cholesterol 20–100 mg/dL).
Statistical methods
The incidence rate (IR) of CVAEs was calculated as the ratio between the number of events and the cumulative follow-up duration; the exact 95% confidence interval (CI) of the Poisson distribution was estimated using the chi-square method. Kaplan-Meier curves were obtained showing CVAE incidence over time.
The expected number of Framingham-type CVAEs was calculated as the sum of the individual probabilities with methods similar to those described for the DAD cohort.11,12 As the Framingham risk calculation is validated only for primary events, it was restricted to patients without previous CVDs. Individual probabilities were obtained from each patient’s Framingham general CVD 10-year risk8 and follow-up duration, assuming either the Weibull accelerated failure time distribution as described in former Framingham equations,13 or a constant rate. For this calculation, the missing data required for the Framingham score were imputed using surrogate information, as detailed in the
CVAE incidence by DRV dose was examined within propensity score (PS) strata. The PS for DRV 600 mg bis in die (b.i.d.), ie the probability of receiving that dose rather than 800 mg quantum die (q.d.) given certain patient characteristics, was obtained empirically by examining its relationship, essentially nonlinear, with key features on which DRV-dosage recommendations are based: previous ARV therapy, HIV-RNA load or CD4+ counts.
The association between CVAEs and several possible predictors was examined using Cox proportional-hazard models. The predictors tested included the Framingham 10-year risk score, the variables used for its computation (age, gender, smoking, SBP, antihypertensive medication, diabetes, serum total and HDL cholesterol, and BMI), other traditional factors (LDL cholesterol, triglycerides, diastolic blood pressure, eGFR, statin medication, anticoagulant medication, and family history of CVD), and HIV-specific variables (disease stage, HIV-RNA load, CD4+ count, and history of intravenous drug use). Models for Framingham-type CVAEs were studied in patients without previous CVDs, as the Framingham score was developed for primary events only. Models for any-type CVAEs were analyzed in all patients, with or without previous CVDs; CVD history was added to the characteristics examined as potential prognostic factors. Patients who were ARV-naïve at DRV start (who had no CVAE during the study) were excluded from some analyses to reduce confounding by variables that represent exposure to the disease and its therapy. The effect of these exposure variables (duration of HIV infection, duration of ARV therapies, duration and dose of DRV therapy) on any-type CVAEs was studied in models that included both ARV-experienced and ARV-naïve patients; these analyses were adjusted for age and SBP at baseline, the most relevant traditional factors. Subject exclusions from exposure models were avoided by imputing, for missing data, the most likely value based on surrogate information, as detailed in the
Laboratory values at baseline and throughout the study were summarised for patients with and without CVAEs; Student’s two-sample t-tests were used for comparisons.
The statistical analyses were carried out using SAS© 9.4.
Results
Patients and treatments
Eight hundred seventy-five patients were enrolled: 235 (DRV-experienced ex-EAP) were already receiving DRV/r since July 2007 or earlier, 407 (DRV-experienced not EAP) were already receiving DRV/r since August 2007 or later, before the inclusion in this study, and 233 (DRV-naïve) started DRV/r at entry in this study. Of the DRV-experienced not EAP patients, 75 had received no ARV therapy before DRV/r, like 117 patients enrolled in this study as DRV-naïve; all other patients were ARV-experienced at DRV initiation.
The baseline characteristics are shown in Table 1. The mean age was 46 years; 77.8% of the patients were men, 40.7% were in clinical stage C according to the Center for Disease Control and Prevention (CDC) criteria, 49.1% were active smokers, 15.7% were on antihypertensive therapy and 5.8% had diabetes; the mean serum total cholesterol was 189 mg/dL in the 748 patients with an available baseline measure. Previous CVDs, either atherosclerotic in nature or potentially related to atherosclerotic risk, were reported in 9.8% of the patients overall, including 25 (2.9%) with MI and 10 (1.1%) with stroke or cerebral haemorrhage, as detailed in the
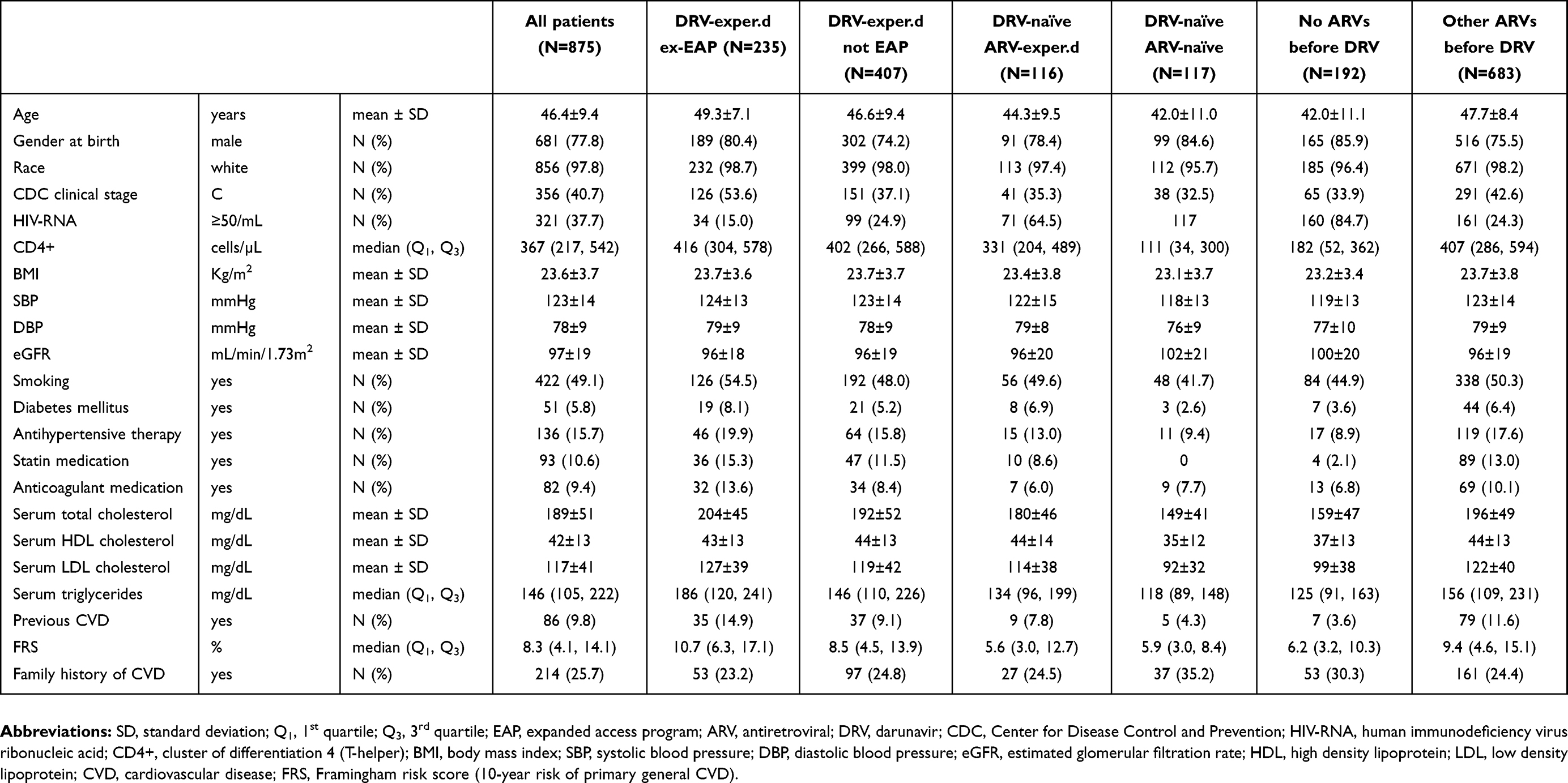 | Table 1 Patients’ characteristics at entry into the study |
ARV therapy before and during the study is reported in Table 2, and a list of previously used ARV drugs is provided in the
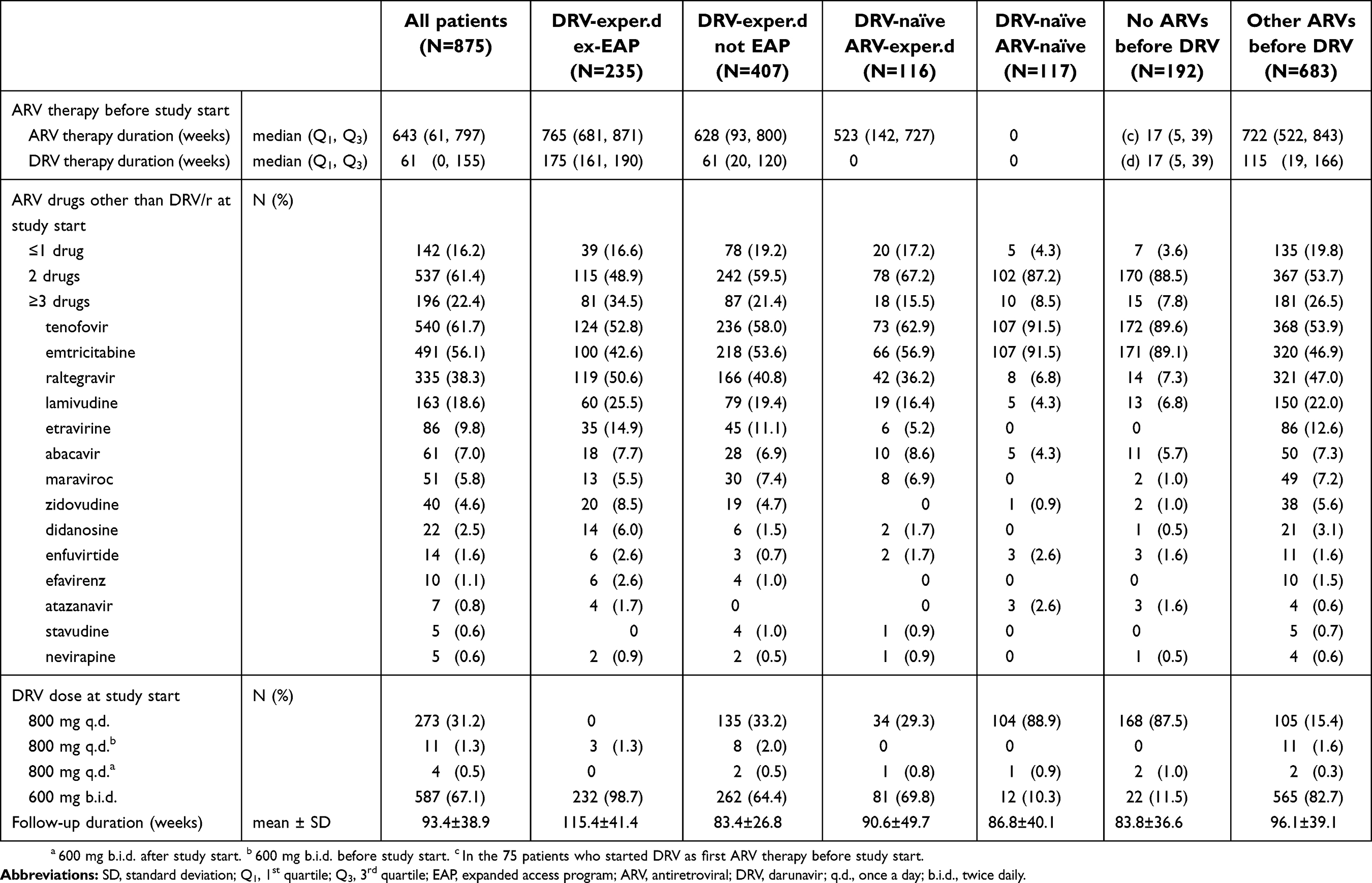 | Table 2 ARV therapy history, concomitant ARV treatments at study start, DRV doses and follow-up duration |
Incidence rates of cardiovascular events
A summary of the CVAEs reported in this study by previous CVD (yes or no), patient group and event type is shown in Table 3. An individual description of all CVAEs is provided in the
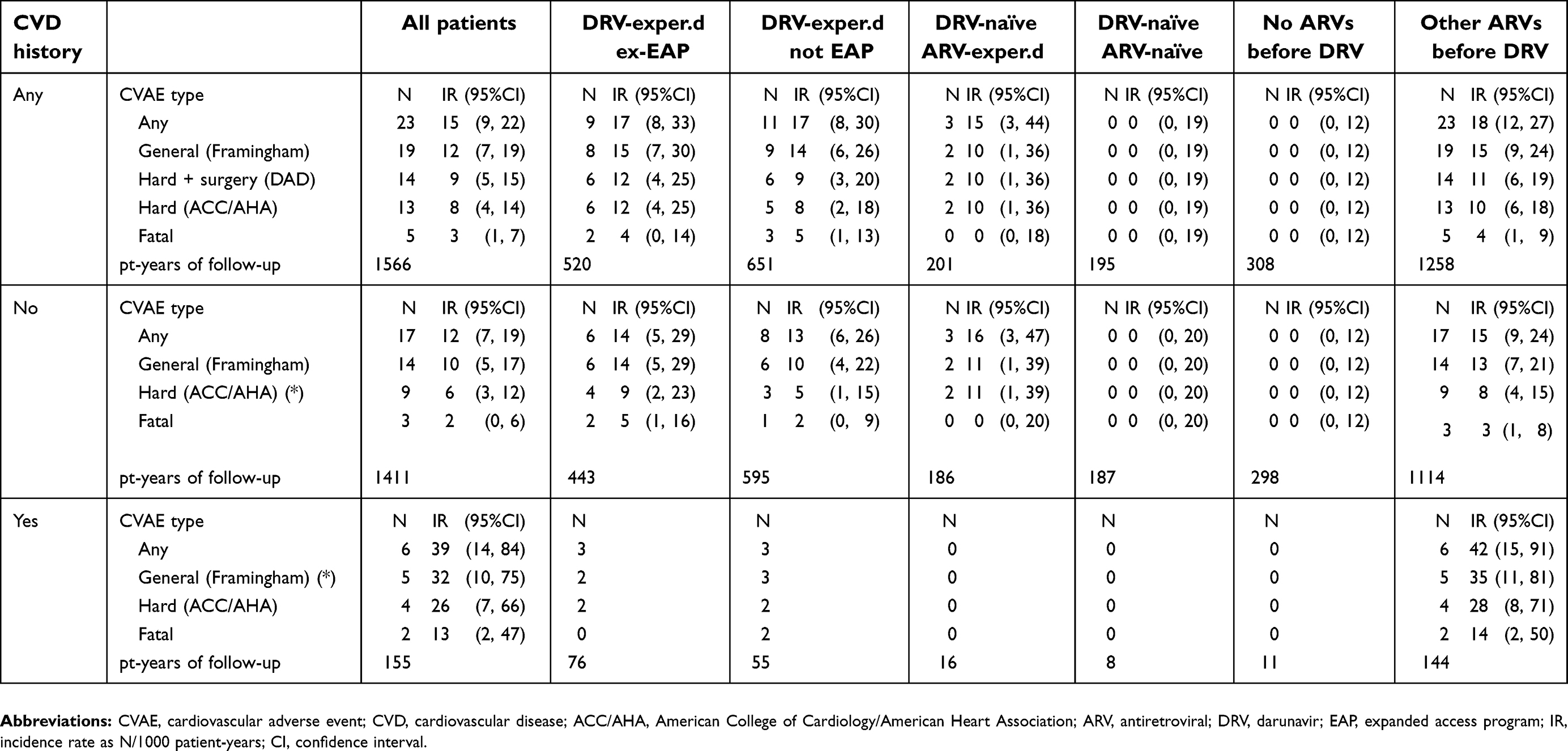 | Table 3 CVAEs emerging during the study |
The IRs remained relatively constant in all groups throughout the study (Figure 1A and B).
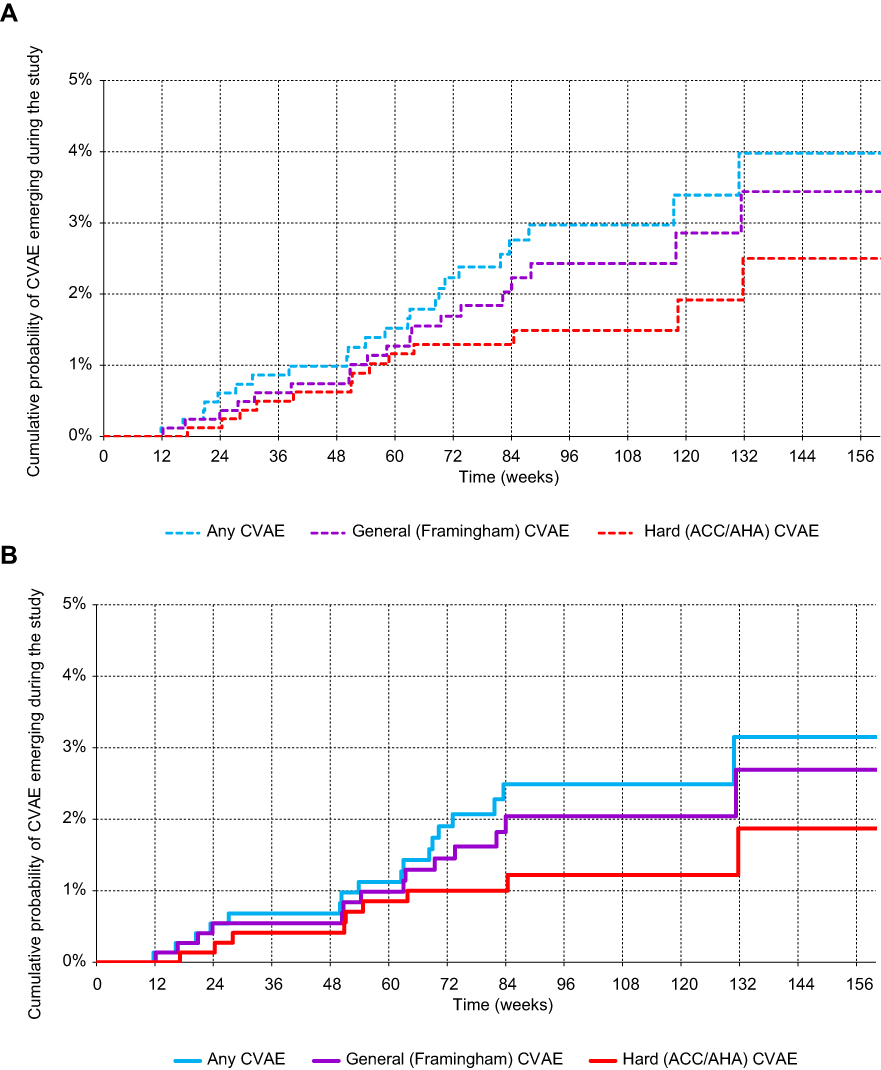 | Figure 1 Kaplan-Meier curves of time to cardiovascular adverse event (CVAE). (A) All patients (N=875). (B) Patients without previous CVD (N=789). |
Darunavir dose, background therapy and cardiovascular events
The PS for DRV 600 mg b.i.d., ie, the probability of receiving that dose rather than 800 mg q.d., is reported in Table 4 as a function of previous ARV therapy, HIV-RNA load or CD4+ counts. Almost all ex-EAP patients were still receiving DRV 600 mg b.i.d at entry into this study, as well as all non-EAP patients who were on DRV for more than 3 years. Conversely, 89% of patients who were ARV-naïve when DRV was started, received DRV 800 mg q.d. In the remaining DRV-experienced non-EAP patient group, the PS for DRV 600 mg b.i.d. was increased due to a longer history of ARV and DRV therapy, whereas the preference for DRV 600 mg b.i.d in the DRV-naïve ARV-experienced cohort was mainly determined by higher HIV-RNA load and lower CD4+ counts at study entry. The crude incidence of any CVAEs in the patient subsets with different PS for DRV 600 mg b.i.d. is shown in Table 4. Patients with no ARV therapy before DRV start, whose PS for DRV 600 mg b.i.d. was <0.15, had no CVAEs; CVAEs occurred only in patients with previous ARV therapy, whose PS for DRV 600 mg b.i.d. ranged from 0.5 to 1. Therefore, although the crude rate of CVAEs was greater in patients receiving DRV 600 mg b.i.d., this was largely due to a confounding by the previous ARV therapy, and it was no longer observed when comparing IRs within dose PS strata (Table 5). In the group having PS for 600 mg b.i.d. of 0.50–0.65, where both DRV doses were similarly represented, the IR for any CVAE was slightly higher with 600 mg b.i.d., but this was not true for general and hard CVAEs; the IRs in the other strata are hardly comparable due to the small number of patients given one of the alternative doses (Table 5 and Supplementary materials,
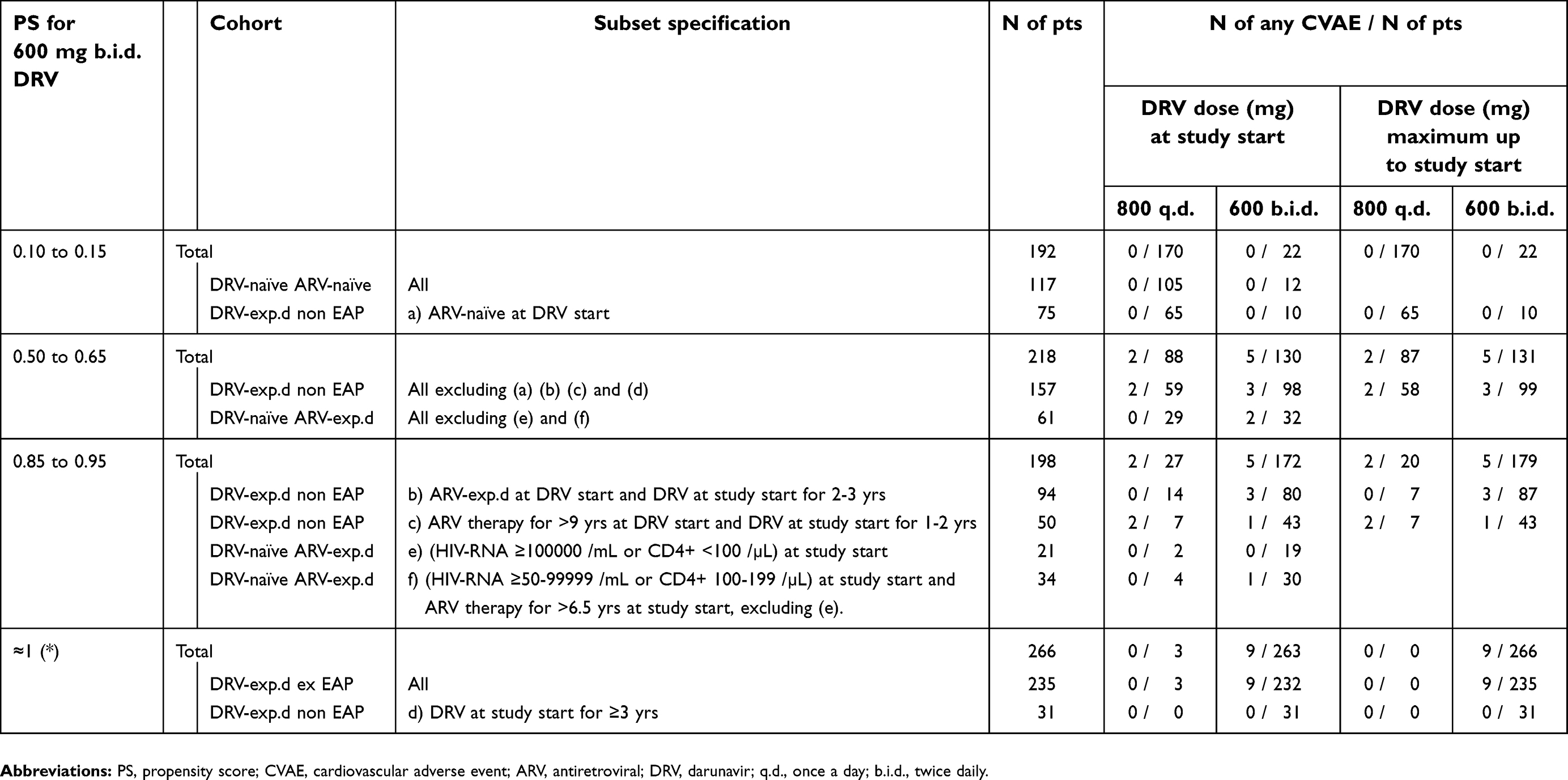 | Table 4 Size and CVAE counts in patient subsets defined by PS for DRV doses |
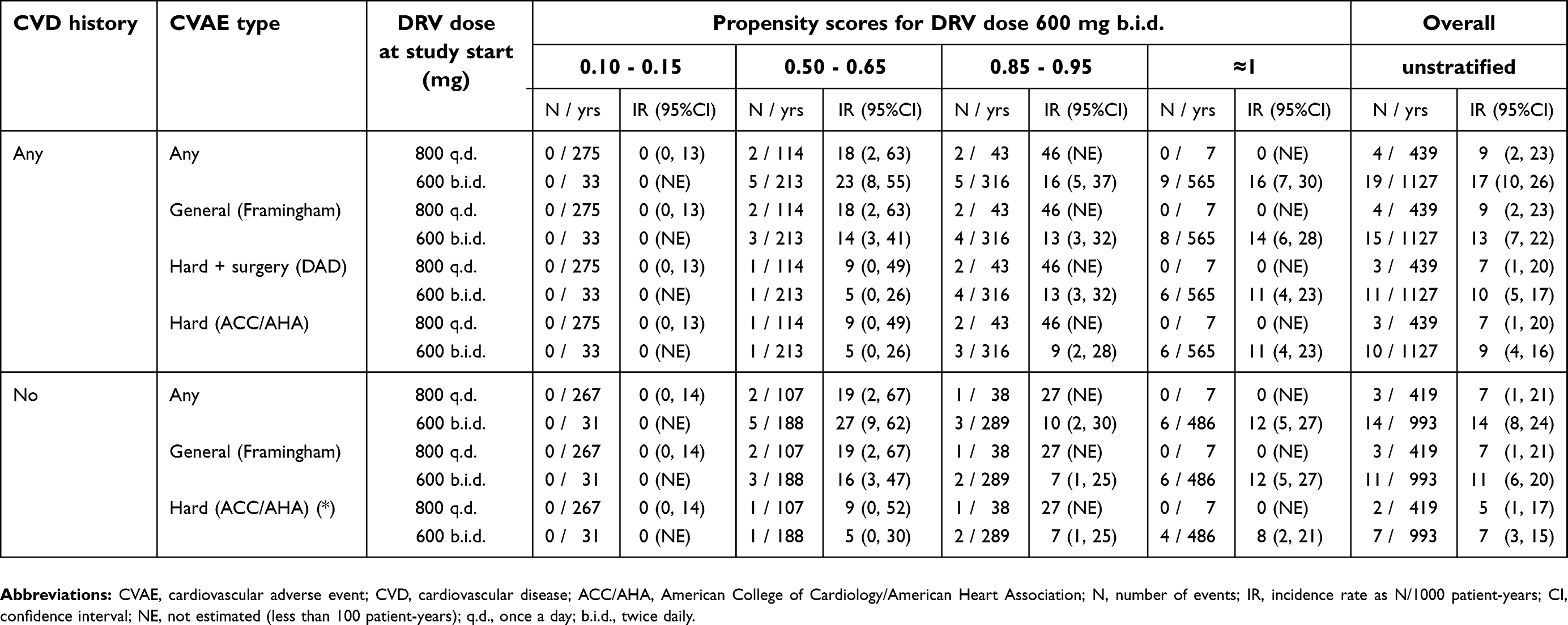 | Table 5 Incidence rates of CVAEs emerging during the study according to DRV dose at study start |
For the patients who had CVAEs, exposure to the two different doses of DRV and to background ARV drugs before and during the study is listed in the
Cardiovascular risk scores and cardiovascular events
The Framingham general CVD 10-year risk score at study entry was calculated in 626 (79.3%) of all 789 patients without previous CVDs including 485/604 (80.3%) ARV-experienced patients at DRV start. The median score was 8.3% (1st quartile 4.1%, 3rd quartile 14.1%) overall, as reported in Table 1; the median score was 26.1% (15.2, 45.4) in 13 patients who actually experienced Framingham-type CVAEs during the study, and 8.1% (4.1, 13.8) in patients without such events. Notably, the median score was 9.2% (4.5, 14.9) in patients given ARV therapies before DRV start and 6.2% (3.2, 10.3) in ARV-naïve patients at DRV start.
In these 626 patients with 13 primary Framingham-type CVAEs actually observed, 10 such events were expected during the study, given their Framingham score, by the accelerated risk model described in the Methods section, while 13 were expected assuming that the risk was uniformly distributed over 10 years. Considering all 789 patients without previous CVDs (including 163 with incomplete data), with an actual observation of 14 Framingham-type CVAEs, the number of expected events of this type was 12 in the accelerated-risk model and 16 in the constant-risk hypothesis, when imputing the most likely values for missing data; instead, when imputing the optimal values, the expected events were 11 and 15, respectively. As a reference, four (assuming an accelerated IR), or six (assuming a constant IR) Framingham-type CVAEs would be expected in an age- and gender-matched population with all optimal values of modifiable risk factors.
The ACC/AHA hard ASCVD 10-year risk could be calculated in 451 (57.2%) of all 789 patients without previous CVDs. The median score was 4.0% (1st quartile 1.8%, 3rd quartile 7.8%) overall, 8.2% (3.8, 24.8) in seven patients who experienced these events during the study and 4.0% (1.8, 7.8) in those who did not.
Cardiovascular events by risk factors at baseline
The main results of the Cox models examining the association between the incidence of CVAEs and patient characteristics are reported in Table 6. Details of all analysis models are provided in the Supplementary materials (
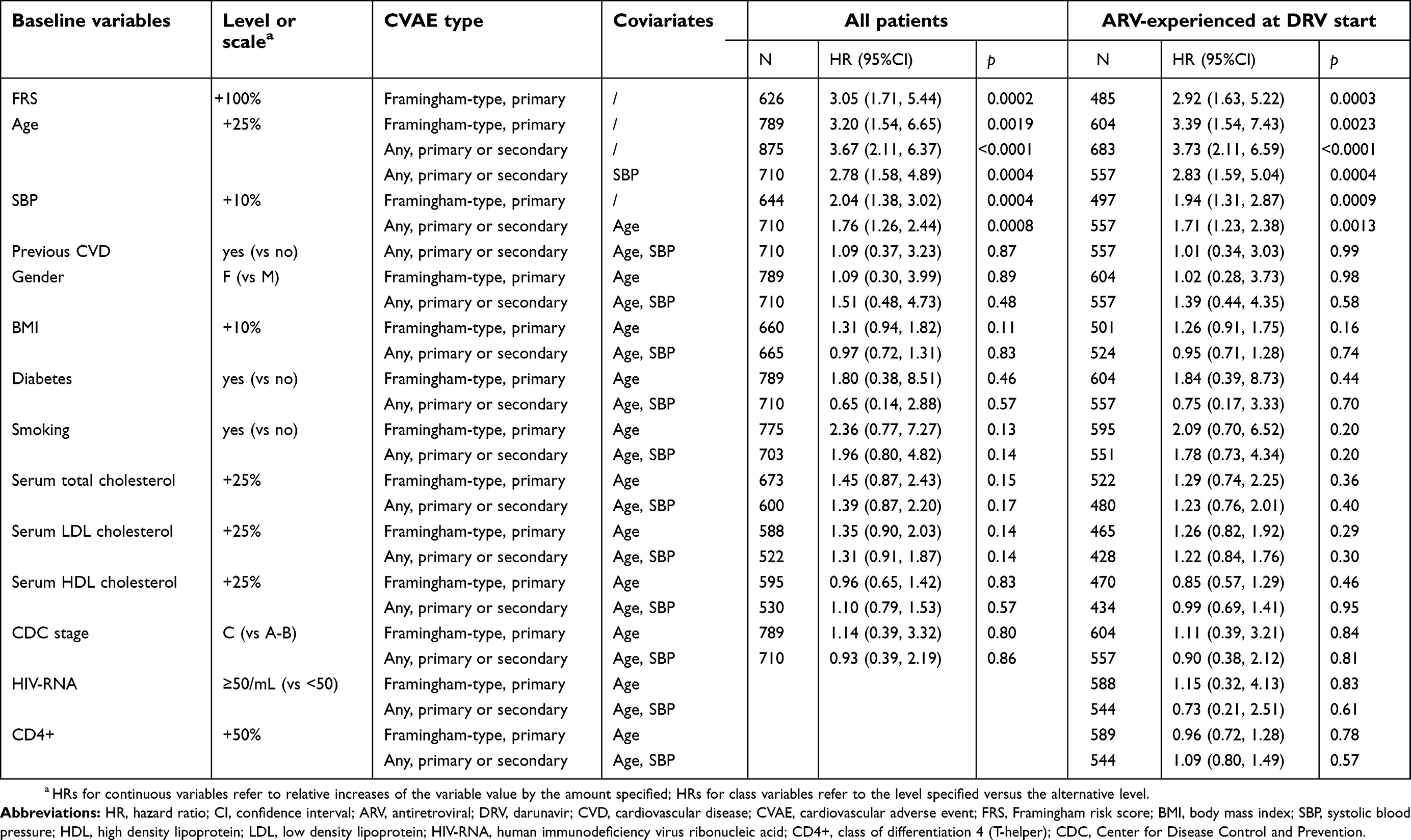 | Table 6 Main results of Cox models for primary Framingham-type general CVAEs (14 events) and for any CVAEs (23 events) |
The Framingham primary CVD 10-year risk index was strongly (p<0.001) associated with Framingham-type general CVAEs, both in the analysis of all patients and in the analysis of patients who were ARV-experienced at DRV start (thereby excluding the 192 patients ARV-naïve at DRV start, who had no CVAE). The predictive model based on the Framingham risk score was more efficient (according to Akaike’s information criterion) than the models containing any combination of other variables, either included in the score calculation or not; other variables not included in the Framingham risk score had no significant effect after adjusting for it. The Framingham risk index, however, was not available in approximately 20% of the patients, and it was not used in the models for any primary or secondary CVAEs, as it was developed for primary events only. Age was the single variable without missing data that was most significantly (p<0.001) associated with both Framingham-type general CVAEs and all CVAEs in univariate analyses. Multivariable analyses showed that age and SBP at enrolment were the most important (p<0.01) independent predictors of both Framingham-type CVAEs and of any CVAEs, consistently across various models (Table 6 and Supplementary materials,
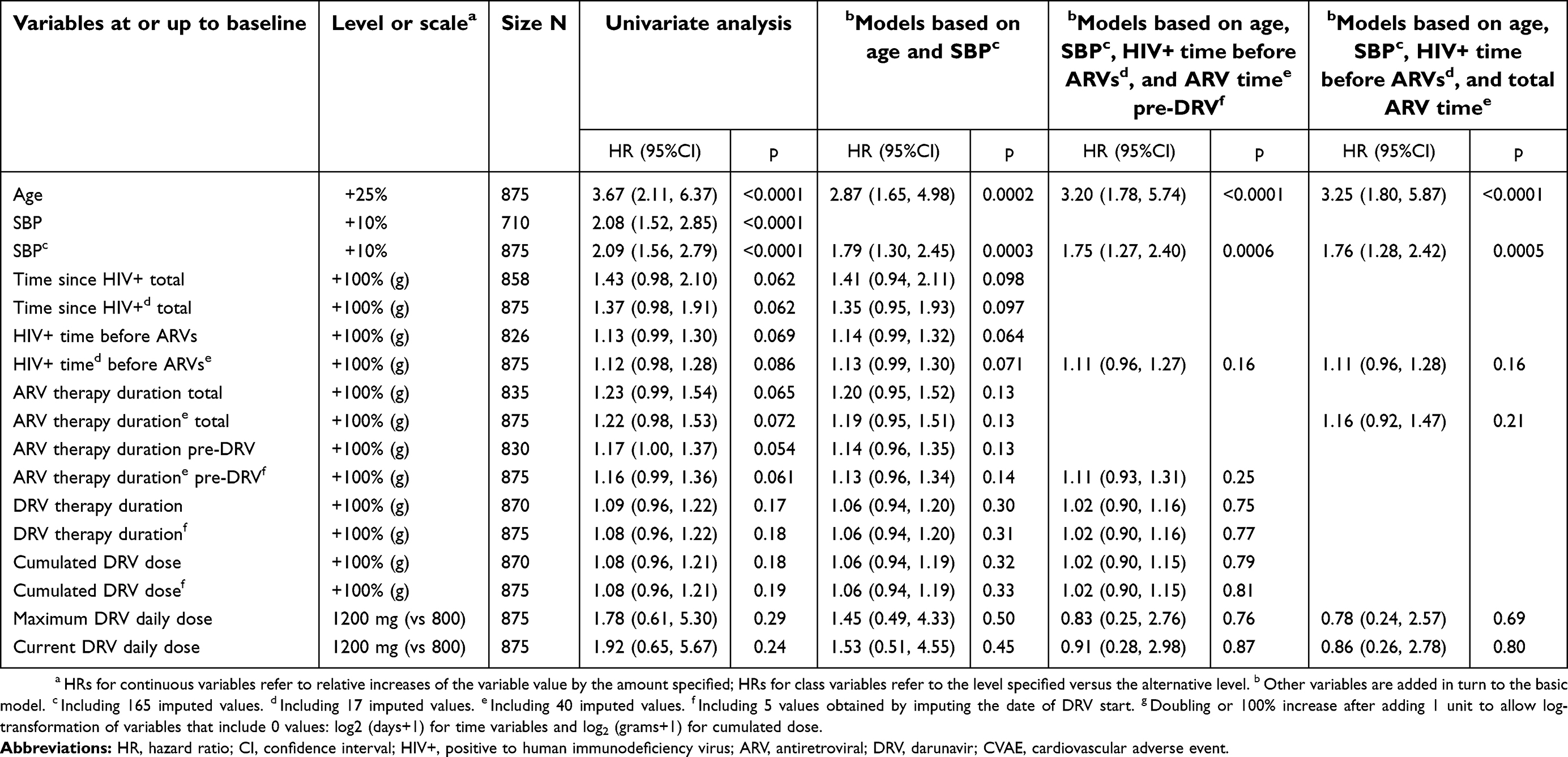 | Table 7 Cox models for any primary or secondary CVAEs (23 events) in all patients (N=875) |
Other traditional risk factors, including gender, diabetes, BMI, serum lipids, and eGFR, were at most weakly and not significantly (p>0.10) associated with CVAEs when controlling for age (in primary Framingham-type CVAE models) or for age and SBP (in all-type CVAE models) (Table 6 and Supplementary materials,
Cardiovascular events by HIV infection duration and previous exposure to therapy
The effect of HIV infection length and exposure to ARV therapy, including DRV, on the risk of any CVAE was further examined in all patients by the Cox models reported in Table 7. Time since HIV+ diagnosis is reported both as the total time up to study entry and as the time from diagnosis to ARV therapy start, and ARV therapy duration is reported both as total time up to study entry and as the partial time up to DRV start. When both durations were included in the same model, overlaps between the exposure times were avoided. The variables used for adjustment included imputed values to avoid patient exclusion due to missing data; the HRs for these variables with and without imputation were always close enough to justify this step. In the univariate analysis, all variables that represent the length of HIV infection and exposure to ARV therapy were associated with CVAE incidence with p-values of 0.05–0.09, while the variables that represent exposure to DRV showed a weaker association with non-significant p-values. Adjusting for age and SBP, the risk factors highlighted the most relevant in the previous analyses, somewhat attenuated all these associations. When additionally adjusting for time from HIV+ diagnosis to ARV therapy start and for ARV therapy duration before DRV start, the association of previous DRV therapy duration and cumulated DRV dose with CVAE incidence was further reduced, with HRs around 1. This model (or a similar one with ARV therapy duration up to study entry rather than up to DRV start) overrides the unadjusted differences between DRV daily doses, yielding adjusted HRs of approximately 0.9 for 600 mg b.i.d. versus 800 mg q.d. at study start. Such result is explained by the above consideration that the PS for DRV 600 mg b.i.d. was increased by longer histories of previous ARV therapy, which are in turn associated with greater CVAE incidence, and that patients with no previous ARV therapy, who had no CVAE, were mostly given DRV 800 mg q.d.
Cardiovascular events and biochemical parameters during the study
The values of several biochemical parameters around the CVAEs (within three months before or shortly after) were compared with before-CVAE means (from baseline included to CVAE excluded) in the same patients and with average measures in patients without CVAEs. Complete figures are provided in the
The mean intrapatient change in lipid values registered at the CVAE compared with the values before the CVAE was +6 mg/dL for total cholesterol and +2 mg/dL for LDL cholesterol. Compared to the average measures in patients without CVAEs, values in patients who developed CVAEs were 18 mg/dL higher at baseline for total cholesterol and 14 mg/dL higher for LDL cholesterol, while the means throughout the study (before the CVAE) were higher by 11 and 10 mg/dL, respectively. These differences were not statistically significant (p>0.10), and were further attenuated by excluding patients who were ARV-naive at DRV start (with no CVAEs). Essentially no increase was found for HDL cholesterol, triglycerides, and blood glucose. Of 19 CVAEs with available data, 2 were preceded by hypercholesterolemia of DAIDS grade 3 (total cholesterol ≥300 mg/dL or LDL cholesterol ≥190 mg/dL).
CD4+ counts at baseline and throughout the study were similar in patients with and without CVAEs. Two CVAEs, of 20 with available data, were preceded by a CD4+ count <200 cells/µL, including one with confirmed HIV-RNA >50 copies/mL.
Neutrophil counts around CVAEs were on average 20% higher than the values previously measured in the same patients and 32% higher than those measured in CVAE-free patients. The neutrophil/lymphocyte ratio also increased at the time of CVAEs (+28% within patients and +40% between patients), as well as the total WBC count (+11% within patients and +17% between patients). These results, however, were driven by the remarkably high neutrophil counts that preceded three CVAEs (7.9, 10.3 and 10.6 103/µL), while the other 15 CVAEs with available data were preceded by normal neutrophil counts. Essentially no increase was found for monocyte and platelet counts.
Discussion and conclusions
CV morbidity and mortality in HIV-infected subjects has improved in the last few years.14,15 However, the risk of incident CVDs is still greater in HIV-infected than in uninfected subjects, even after adjustment for demographic characteristics and traditional risk factors.16–19 The causes of this increased risk are only partly understood; they may be related to HIV infection itself or to therapy with ARV drugs, especially boosted PIs, via increased serum levels of LDL cholesterol or through metabolic changes such as lipodystrophy and chronic inflammation.18,20,21 In this paper we provide a description of all CVAEs that occurred during the TMC114-HIV4042 study; the main efficacy and overall safety results were previously published.6
In this study we followed 875 HIV-infected subjects for a total of 1,566 patient-years. The incidence of primary CVAEs, expressed as number/1000 patient-year, was as follows: any, 12; Framingham-type general, 10; DAD-type and ACC/AHA-type hard, 6; and fatal, 2. All CVAEs occurred in the 683 patients exposed to ARV therapy before DRV start.
These results are better interpreted with respect to underlying CVD risk. CVD risk scores, ie equations that calculate the probability of CVDs in a given time frame based on known risk factors, are available for the outcomes reported above: Framingham,8 ACC/AHA7 and DAD.22 However, only the Framingham risk could be calculated in a sufficiently high proportion of our patients, ie approximately 80%, as allowed by the existence of an alternative Framingham function that uses BMI instead of cholesterol values, which were frequently missing in our study. Although the Framingham prediction functions were developed in the general population, they have been shown to correctly estimate the absolute CVD risk also in HIV-infected patients.23,24 The ACC/AHA risk score could be calculated in less than 60% of our sample, mainly because of missing values of SBP and cholesterol. DAD scores were not calculated because they require, in addition to the variables above, a three-level classification of smoking (current, ex, no) whereas a two-level classification (yes/no) was used in our study. Moreover, DAD equations were developed for time-updated rather than baseline values of quantitative variables, whereas post-baseline blood pressure values were generally not available in our study.
CVD incidence is expected to increase during follow-up as age, the main risk factor, also increases. Therefore, to calculate the absolute risk over time periods shorter than 10 years, a correction derived from other Framingham equations that account for an accelerated event time13 was used.11,12 On the other hand, empirical Kaplan-Meier curves often show constant or nearly constant IRs over 10 years.25,26 Using Framingham equations, the number of primary Framingham-type general CVDs expected in our study was 12 assuming the accelerated event-time model and 16 assuming constant IRs, compared to the 14 actually observed. This result should be viewed cautiously due to the small number of events and the short follow-up time compared to the 10 years for which the Framingham functions were developed. However, it suggests that the CVD risk in our patients, in spite of a long previous exposure to HIV infection and ARV therapy for most of them, was not increased compared to what expected in an HIV-negative population matched for the traditional risk factors.
We examined the role of possible risk factors in several multivariable Cox models, both for primary Framingham-type general CVAEs and for any primary or secondary CVAEs. In predicting primary Framingham-type CVAEs, the model based on the Framingham risk score was more efficient than the models containing any combination of other covariates, including the variables used to calculate the score. In patients who had all data required for calculating the scores, the median Framingham general CVD risk at 10 years was 26.1% in patients who actually experienced these events during the study and 8.1% in patients who did not. Age and SBP at entry were by far the most important independent predictors of Framingham-type general CVAEs, aside from the Framingham risk score to which both contribute, and of any CVAEs. The association of age and SBP with the incidence of CVAEs was highly significant (p<0.01) across all models. Patients who had any CVAE during the study were on average 10 years older and had an SBP 14 mmHg higher than patients without CVAEs. When controlling for these two factors, no association with other factors, including smoking, BMI, diabetes, serum lipids and eGFR, was statistically significant (p>0.05), but it should be noted that the models had sufficient power to highlight only very strong associations as significant. This was especially true for binary variables, such as smoking and family CVD history, which had p-values >0.10 across all models in spite of HRs of almost 2. The risk of any CVAE in patients with previous CVDs was approximately three times that of patients without CVD; however, this unadjusted difference was entirely explained by older age and higher SBP values.
As previously reported,6,27 mean serum lipid concentrations during this study were stable in DRV-experienced patients while in DRV-naïve patients, especially if ARV-naïve, they started from lower levels and increased over 3–6 months to reach values similar to DRV-experienced patients. The increase of non-HDL cholesterol levels has been supposed to represent a connection between ARV therapy and increased CVD risk, although this may not necessarily be the only one.28,29 In our study, mean LDL and total cholesterol in patients who developed CVAEs were 14 and 18 mg/dL higher at baseline, respectively, compared to patients who did not, and 10–11 times higher throughout the study; this may have contributed to increased CVD risk. Multivariable models showed that this contribution was smaller than that of age and SBP and did not achieve statistical significance. Moreover, mean LDL and total cholesterol values were fairly stable up to the time of CVAEs and around it. No relevant change in blood glucose levels occurred during the study6,27 nor was it associated with CVAEs in the analysis reported here. HIV infection itself and the related opportunistic infections have been suggested to contribute to increase CVD risk through immunosuppression and increased inflammation.18,20,21,30 Low CD4+ counts and detectable viremia were independently associated with increased CVD risk in several studies,31–35 (although not all),36 as was the neutrophil/lymphocyte ratio26 among other markers of systemic inflammation. In this study, we found no evidence of an association of CD4+ counts or HIV-RNA load with CVAEs, while the neutrophil/lymphocyte ratio was markedly increased around the time of three CVAEs out of 18 with available data. A recent systematic review of the literature showed that other inflammatory markers (interleukin-6, high-sensitivity C-reactive protein, lipoprotein-associated phospholipase A2 and LDL subfraction Apo A-I) did not increase during treatment with DRV/r and that lipid profile changes were overall similar compared to ritonavir-boosted atazanavir.37
Prolonged exposure to ARV drugs, especially PIs (or at least some of them), has often been found to increase CVD risk,28,29,38,39 although not all studies agree on this point.31,40 The effects of previous exposure to HIV infection and to ARV therapy on CVD risk can hardly be disentangled because one is contained in the other. In our study, both were moderately associated with the incidence of CVAEs in the univariate analysis or in the multivariable analysis adjusting for traditional factors. Previous exposure to DRV, reported either as time or as cumulated dose, showed a weaker association that was further reduced after adjustment for age, SBP and the duration of HIV infection until the start of ARV therapy and of ARV therapy until DRV start. The possible relationship between daily doses of DRV and CVD risk cannot be examined without considering the confounding effect of the characteristics of the patients for whom the alternative doses, 800 mg q.d. or 600 mg b.i.d., are indicated. The propensity to give DRV 600 mg b.i.d. was expectedly increased by longer histories of previous ARV therapy, which were in turn associated with greater CVAEs incidence; patients with no ARV therapy before DRV, who had no CVAE, mostly received the 800 mg q.d. dose. Therefore, although the crude rate of CVAEs in patients receiving DRV 600 mg b.i.d. was almost twice that of patients receiving DRV 800 mg q.d.; this apparent effect was mostly due to confounding by ARV treatment history, and was no longer observed in stratified analysis and in models that adjust for previous ARV history in addition to traditional factors. These analyses, although insufficiently powered, show the inadequacy of comparing doses without accounting for the different characteristics of the patients. A dosage of 600 mg b.i.d. may be considered a proxy of more advanced disease, with more comorbidities and exposure to more risk factors. Much larger studies that, unlike the DAD, provide information on DRV dosing are clearly required to clarify whether the dosing schedule is related to CVD risk or simply reflect a bias due to the preferred use of 600 mg b.i.d. in highly experienced populations. Even large studies, however, may fail to clarify this issue if confounding by indication is complete or almost complete or is not adequately adjusted for in the statistical analysis.
In addition to the relatively small size of the population studied, the analysis presented here has other limitations. As it was not planned when the study was conducted, relevant information such as blood pressure and serum lipids was not as systematically collected as were virological and immunological data. Indeed, blood pressure is not routinely recorded in hospital charts except in patients having known hypertension. Missing baseline data further reduced the analysis sample size, and moreover, blood pressure, a key predictor of CVAEs, was not measured again during the study. Lack or incompleteness of relevant information relating to traditional CV risk factors has been a problem also in previous studies on the use of ARV treatments in HIV-infected patients. Future prospective studies should include the systematic collection of these data, given the importance of CV risk assessment in this population. The possible effect of individual ARV drugs that have been reported to increase CV risk, such as abacavir and older PIs, could not be accounted for in the multivariable analyses because of confounding with overall ARV therapy duration, and the limited sample size did not allow the inclusion of both variables in the same model. Furthemore, while the length of overall ARV exposure was available, that of individual ARV drugs was not. Another concerning issue is a possible selection bias. Upon the inclusion in this study, 87% of the patients were ARV-experienced, many of them for several years, and 73% were DRV-experienced; to be enrolled, they had obviously survived fatal events, including fatal CVDs, were not lost to observation and had not discontinued DRV for any reason. Whether this selection affected the results of our analysis is uncertain.
In conclusion, we did not find an increased CVD risk in our patients, despite most of them had long previous exposure to HIV infection and ARV therapy, compared to what was expected in an HIV-negative population matched for traditional risk factors. This is consistent with the results of a recent review of data from pooled clinical studies and pharmacovigilance reporting, which did not find any signal suggesting that CVD could be connected with DRV use.4 Indeed, in our analysis two traditional factors, age and blood pressure, were by far the most significant predictors of CVD risk in HIV patients treated with DRV/r. Serum lipids, a traditional factor possibly related to ARV therapy, and previous exposure to HIV infection or ARV therapy were of lesser importance, while CD4+ count and previous length of exposure to DRV or DRV dose were not associated with CVD risk. A lack of evidence of such associations is not proof that they do not exist; they may have been not fully captured in this study due to the small sample size or the other limitations pointed at above. Our analysis, however, confirms that traditional factors were overwhelmingly important compared to HIV-specific factors in the development of CVDs.
Acknowledgments
We acknowledge the contribution of Giuseppe Airoldi (Studio Associato Airoldi Cicogna and Ghirri, Milan) who performed the statistical analysis and provided support for writing the manuscript. This study was funded by Janssen-Cilag Spa – Italy.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
A preliminary analysis of these data was accepted as oral presentation at 9th Italian Conference of AIDS and Antiviral Research (ICAR) 2017. The slides are available at the congress repository:
References
1. Eyawo O, Franco-Villalobos C, Hull MW, et al;
2. Ryom L, Lundgren JD, El-Sadr W, et al. Cardiovascular disease and use of contemporary protease inhibitors: the D:A:D international prospective multicohort study. Lancet HIV. 2018. pii: S2352-3018(18)30043-2. Epub ahead of print. doi:10.1016/S2352-3018(18)30043-2
3. Friis-Møller N, Thiébaut R, Reiss P, et al. Predicting the risk of cardiovascular disease in HIV-infected patients: the Data collection on Adverse effects of anti-HIV Drugs Study. Eur J Cardiovasc Prev Rehabil. 2010;17:491–501. doi:10.1097/HJR.0b013e328336a150
4. Opsomer M, Dimitrova D, Verspeelt J, et al. Evaluation of cardiovascular disease risk in HIV-1–infected patients treated with darunavir. Drugs R D. 2018;18:199–210. doi:10.1007/s40268-018-0238-8.
5. Costagliola D, Potard V, Lang S, et al. Is the risk of myocardial infarction in PLHIV associated with atazanavir or darunavir exposure?
6. Antinori A, Meraviglia P, d’Arminio Monforte A, et al. Effectiveness, durability and safety of darunavir/ritonavir in HIV-1-infected patients in routine clinical practice in Italy: a postauthorization non-interventional study. Drug Des Dev Ther. 2016;10:1589–1603.
7. Goff DC
8. D’Agostino RB, Ramachandran SV, Pencina MJ, et al.
9. Levey AS, Stevens LA, Schmid CH, et al.
10. Martin SS, Blaha MJ, Elshazly MB, et al. Comparison of a novel method vs the Friedewald equation for estimating low-density lipoprotein cholesterol levels from the standard lipid profile. JAMA. 2013;310:2061–2068. doi:10.1001/jama.2013.280532
11. Law M, Friis-Møller N, Weber R, et al. Modelling the 3-year risk of myocardial infarction among participants in the Data collection on Adverse events of anti-HIV Drugs (DAD) study. HIV Med. 2003;4:1–10.
12. Law MG, Friis-Møller N, El-Sadr WM, et al. The use of the Framingham equation to predict myocardial infarctions in HIV-infected patients: comparison with observed events in the D:A:D Study. HIV Med. 2006;7:218–230. doi:10.1111/j.1468-1293.2006.00362.x
13. Anderson KM, Odell PM, Wilson PW, Kannel WB. Cardiovascular disease risk profiles. Am Heart J. 1991;121(1 Pt 2):293–298.
14. Smith CJ, Ryom L, Weber R, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet. 2014;384:241–248. doi:10.1016/S0140-6736(14)60604-8
15. Klein DB, Leyden WA, Xu L, et al. Declining relative risk for myocardial infarction among HIV-positive compared with HIV-negative individuals with access to care. Clin Infect Dis. 2015;60:1278–1280. doi:10.1093/cid/civ014
16. Triant VA, Lee H, Hadigan C, Grinspoon SK. Increased acute myocardial infarction rates and cardiovascular risk factors among patients with human immunodeficiency virus disease. J Clin Endocrinol Metab. 2007;92:2506–2512. doi:10.1210/jc.2006-2190
17. Freiberg MS, Chang CCH, Kuller LH, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med. 2013;173(8):614–622. doi:10.1001/jamainternmed.2013.3728
18. Hemkens LG, Bucher HC. HIV infection and cardiovascular disease. Eur Heart J. 2014;35:1373–1381. doi:10.1093/eurheartj/eht528
19. Quiros-Roldan E, Raffetti E, Focà E, et al. Incidence of cardiovascular events in HIV-positive patients compared to general population over the last decade: a population-based study from 2000 to 2012. AIDS Care. 2016;28(12):1551–1558. doi:10.1080/09540121.2016.1198750
20. Chastain DB, Henderson H, Stover KR. Epidemiology and management of antiretroviral-associated cardiovascular disease. Open AIDS J. 2015;9:23–37. doi:10.2174/1874613601509010023
21. Martin-Iguacel R, Llibre JM, Friis-Møller N. Risk of cardiovascular disease in an aging HIV population: where are we now? Curr HIV/AIDS Rep. 2015;12(4):375–387. doi:10.1007/s11904-015-0284-6
22. Friis-Møller N, Ryom L, Smith C, et al. An updated prediction model of the global risk of cardiovascular disease in HIV-positive persons: the Data-collection on Adverse effects of anti-HIV Drugs (D:A:D) study. Eur J Prev Cardiol. 2016;23:214–223. doi:10.1177/2047487315579291
23. Thompson-Paul AM, Lichtenstein KA, Armon C, et al. Cardiovascular disease risk prediction in the HIV outpatient study. Clin Infect Dis. 2016;63:1508–1516. doi:10.1093/cid/ciw615
24. De Socio GV, Pucci G, Baldelli F, Schillaci G. Observed versus predicted cardiovascular events and all-cause death in HIV infection: a longitudinal cohort study. BMC Infect Dis. 2017;17:414. doi:10.1186/s12879-017-2510-x
25. Assmann G, Cullen P, Schulte H. Simple scoring scheme for calculating the risk of acute coronary events based on the 10-year follow-up of the Prospective Cardiovascular Munster (PROCAM) study. Circulation. 2002;105:310–315.
26. Quiros-Roldan E, Raffetti E, Donato F, et al. Neutrophil to lymphocyte ratio and cardiovascular disease incidence in HIV-infected patients: a population-based cohort study. PLoS One. 2016;11(5):e0154900. doi:10.1371/journal.pone.0154900
27. Antinori A, Di Perri G, Meraviglia P, et al. P141 Metabolic safety during treatment with darunavir-based regimens in an Italian observational study.
28. Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med. 2007;356:1723–1735. doi:10.1056/NEJMoa062744
29. Lundgren J, Mocroft A, Ryom L. Contemporary protease inhibitors and cardiovascular risk. Curr Opin Infect Dis. 2018;31(1):8–13. doi:10.1097/QCO.0000000000000425
30. Lambert CT, Sandesara PB, Hirsh B, et al. HIV, highly active antiretroviral therapy and the heart: a cellular to epidemiological review. HIV Med. 2016;17(6):411–424. doi:10.1111/hiv.12346
31. Lichtenstein KA, Armon C, Buchacz K, et al. Low CD4+ T cell count is a risk factor for cardiovascular disease events in the HIV Outpatient Study. Clin Infect Dis. 2010;51:435–447. doi:10.1086/655144
32. Triant VA, Regan S, Lee H, et al. Association of immunologic and virologic factors with myocardial infarction rates in a U.S. health care system. J Acquir Immune Defic Syndr. 2010;55:615–619. doi:10.1097/QAI.0b013e3181f4b752
33. Lang S, Mary-Krause M, Simon A, et al. HIV replication and immune status are independent predictors of the risk of myocardial infarction in HIV-infected individuals. Clin Infect Dis. 2012;55(4):600–607. doi:10.1093/cid/cis489
34. Helleberg M, Kronborg G, Larsen CS, et al. CD4 decline is associated with increased risk of cardiovascular disease, cancer, and death in virally suppressed patients with HIV. Clin Infect Dis. 2013;57:314–321. doi:10.1093/cid/cit232
35. Silverberg MJ, Leyden WA, Xu L, et al. Immunodeficiency and risk of myocardial infarction among HIV-positive individuals with access to care. J Acquir Immune Defic Syndr. 2014;65:160–166. doi:10.1097/QAI.0000000000000009
36. Sabin CA, Ryom L, De Wit S, et al. Associations between immune depression and cardiovascular events in HIV infection. AIDS. 2013;27:2735–2748. doi:10.1097/01.aids.0000432457.91228.f3
37. Menshawy A, Ismail A, Abushouk AI, et al. Efficacy and safety of atazanavir/ritonavir-based antiretroviral therapy for HIV-1 infected subjects: a systematic review and meta-analysis. Arch Virol. 2017;162:2181–2190. doi:10.1007/s00705-017-3346-9.
38. May M, Sterne JAC, Shipley M, et al. A coronary heart disease risk model for predicting the effect of potent antiretroviral therapy in HIV-1 infected men. Int J Epidemiol. 2007;36:1309–1318. doi:10.1093/ije/dym135
39. Lang S, Mary-Krause M, Cotte L, et al. Impact of individual antiretroviral drugs on the risk of myocardial infarction in human immunodeficiency virus–infected patients. Arch Intern Med. 2010;170(14):1228–1238. doi:10.1001/archinternmed.2010.197
40. Bozzette SA, Ake CF, Tam HK, et al. Cardiovascular and cerebrovascular events in patients treated for human immunodeficiency virus infection. N Engl J Med. 2003;348:702–710. doi:10.1056/NEJMoa022048
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.