Back to Journals » OncoTargets and Therapy » Volume 10
Carcinogenicity of chromium and chemoprevention: a brief update
Authors Wang Y, Su H, Gu Y, Song X, Zhao J
Received 9 April 2017
Accepted for publication 13 June 2017
Published 16 August 2017 Volume 2017:10 Pages 4065—4079
DOI https://doi.org/10.2147/OTT.S139262
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yafei Wang,1,* Hong Su,1,* Yuanliang Gu,1 Xin Song,1 Jinshun Zhao1,2
1Department of Preventative Medicine, Zhejiang Key Laboratory of Pathophysiology, School of Medicine, Ningbo University, Ningbo, People’s Republic of China; 2Toxicology and Molecular Biology Branch, Health Effects Laboratory Division, National Institute for Occupational Safety and Health, Morgantown, WV, USA
*These authors contributed equally to this work
Abstract: Chromium has two main valence states: hexavalent chromium (Cr[VI]) and trivalent chromium (Cr[III]). Cr(VI), a well-established human carcinogen, can enter cells by way of a sulfate/phosphate anion-transport system, and then be reduced to lower-valence intermediates consisting of pentavalent chromium (Cr[V]), tetravalent chromium (Cr[IV]) or Cr(III) via cellular reductants. These intermediates may directly or indirectly result in DNA damage or DNA–protein cross-links. Although Cr(III) complexes cannot pass easily through cell membranes, they have the ability to accumulate around cells to induce cell-surface morphological alteration and result in cell-membrane lipid injuries via disruption of cellular functions and integrity, and finally to cause DNA damage. In recent years, more research, including in vitro, in vivo, and epidemiological studies, has been conducted to evaluate the genotoxicity/carcinogenicity induced by Cr(VI) and/or Cr(III) compounds. At the same time, various therapeutic agents, especially antioxidants, have been explored through in vitro and in vivo studies for preventing chromium-induced genotoxicity/carcinogenesis. This review aims to provide a brief update on the carcinogenicity of Cr(VI) and Cr(III) and chemoprevention with different antioxidants.
Keywords: hexavalent chromium, Cr(VI), trivalent chromium, Cr(III), genotoxicity, carcinogenicity, chemoprevention, antioxidant
Introduction
Chromium is a naturally occurring heavy metal that is commonly used in industrial processes and can cause severe health effects in humans.1 In the environment, chromium primarily occurs in two valence states: hexavalent chromium (Cr[VI]) and trivalent chromium (Cr[III]).2,3 Cr(VI) compounds are extensively used in diverse industries, and Cr(III) salts can be used as micronutrients and dietary supplements. Cr(III) compounds are considered to be approximately 100 times less toxic than Cr(VI).4
Although chromium can be released through natural forces, its major environmental pollution is from industrial sources. In recent years, the use of chromium has expanded around the globe as modern industries grow rapidly in developing countries, such as China. As a result, more anthropogenic chromium is being released into the environment.5 Industries with the largest contribution to chromium levels include chromate or chromium production and plating, commercial and residential fuel combustion (natural gas, coal, and oil), leather-tanning industries, textile manufacturing (effluents), pigment manufacturing, stainless steel production, welding, polishing, and grinding.6,7 Chromium is also a global marine pollutant. Through seawater pollution, chromium contaminates marine plants and eventually marine fauna that are consumed as seafood. Chromium can exist in air, water, soil, and food. The primary route of exposure is inhalation through contaminated air, while other additional potential routes include oral ingestion of contaminated water and food or direct dermal contact with products containing chromium.8 Chromates have been known to be potent inducers of cancer in exposed workers.9 All soluble Cr(VI) compounds and the chromates of potassium, sodium, calcium, and strontium are classified as carcinogen to animals.10 Although both Cr(VI) and Cr(III) are considered genotoxic, they are different in terms of the molecular mechanisms involved in toxicity.
Cr(VI) compounds can enter the cell via the sulfate-anion channel and then be subjected to reduction by a wide variety of cellular reductants, such as glutathione (GSH) and ascorbate, with the formation of highly reactive Cr(V/IV) intermediates and finally Cr(III) products.11–13 During this process, molecular oxygen is activated and reduced to superoxide anion (·O2−), which is further converted to hydrogen peroxide (H2O2) via a dismutation process. The resultant intermediates further react with H2O2 to generate a spectrum of reactive oxygen species (ROS) containing hydroxyl radicals, singlet oxygen, superoxide, and hydrogen peroxide via the Fenton pathway.12–15 Consequently, excessive ROS interaction with these intermediates may give rise to oxidative stress and DNA damage, including chromium–DNA adducts, DNA strand breaks, and DNA–protein cross-links (DPCs), all of which are unstable factors of mutagenic effects.16,17 Therefore, the International Agency for Research on Cancer (IARC) has classified Cr(VI) as a group I carcinogen.18,19 Moreover, exposure to Cr(VI) tremendously increases the risk of respiratory tract cancer in occupational settings.18,20 Meanwhile, Cr(VI) may also exert carcinogenic effects in other internal organs, such as the gastrointestinal tract.21
Cr(III) salts have a long history as a nutritional supplement. Cr(III) is an active component in the glucose tolerance factor (GTF), and plays a significant role in regulating blood-glucose levels, involving the processes of carbohydrate and lipid metabolism.22,23 Furthermore, Cr(III) complexes were previously used as insulin enhancers for the treatment of type 2 diabetes.24 However, recently studies have revealed that Cr(III) compounds can lead to DNA damage.25 Although Cr(III) complexes cannot easily pass through cell membranes, they have the ability to accumulate around cells, which could bring about surface morphological alteration.26 Therefore, Cr(III) complexes can then enter cells and react with cellular biological reductants and finally be oxidized to Cr(V).25 The process of reduction induces oxidative stress, and hydroxyl radicals, which may attack proteins, cause DNA damage, and lead to membrane lipid injuries via disruption of cellular functions and integrity.6,22 Therefore, Cr(III) is considered a potential genotoxic agent.
In order to prevent the carcinogenicity induced by chromium and its compounds, a growing number of in vitro and in vivo experiments have been conducted to explore therapeutic agents in recent years. Accordingly, this article provides a brief update on the carcinogenicity induced by chromium, including both Cr(VI) and Cr(III), and potential chemoprevention agents.
Carcinogenicity of chromium
In vitro studies
Both Cr(VI) and Cr(III) can induce oxidative stress damage in culture cells, but through different molecular mechanisms. The propidium iodide fluorescence assay indicates that the percentage of propidium iodide-permeable cells treated with Cr(III) is almost five times greater than those treated with Cr(VI) at the same concentration, whereas the lethal rate indicates that Cr(VI) is more toxic than Cr(III) in yeast cells. In addition, the GSH level in cells treated with Cr(VI) significantly declines; nevertheless, there is little change in GSH content after Cr(III) exposure, even at a very high concentration, which suggests that the toxicity of Cr(VI) is significantly higher than that of Cr(III).27,28
Cr(VI) is a well-known carcinogen, but the difference in terms of water-solubility of Cr(VI) complexes may lead to different carcinogenic potencies. Cr(VI) displays no ability to damage DNA directly, and requires reductive activation inside cells driven by such agents as ascorbic acid, GSH, and cysteine,16,29 the principal biological reducers of Cr(VI) for its genotoxic activity.13,30,31 Large amounts of reactive intermediates, such as Cr(V) and Cr(IV), are generated through the activities of Cr(VI) reduction, and then these intermediates are further reduced to a more stable and kinetically inert Cr(III)–biomolecule complex.32 This multitude of reactive intermediates is known to induce a diverse array of DNA lesions, including single-strand breaks, alkali-labile sites, DPCs, DNA–amino acid cross-links, and chromium–DNA adducts, as well as the formation of protein–Cr(III)–DNA cross-links, the most likely cause of mutations in Cr(VI)-treated cells.33–35 In addition, intracellular Cr(VI) binding to lysine- and arginine-rich peptide sequences of histones or nuclear proteins in the cytoplasm can be transported to the cell nucleus via the active protein-transport mechanism. These bindings are reversible and can be released into the vicinity of DNA, resulting in the formation of reactive Cr(V/IV) intermediates and finally leading to highly genotoxic Cr(III)–DNA adducts in the cell nucleus. This is considered a crucial pathway in Cr(VI)-induced DNA damage in vitro.36 Improper DNA repair may bring about chromosomal instability or mutation, which could evolve into tumorigenesis.37,38
Zinc chromate, a water-insoluble Cr(VI) compound, seems to be the most potential carcinogen.39 It causes DNA double-strand breaks (DSBs) and chromosome aberrations in WTHBF6 cells, a human bronchial fibroblast, in a dose-dependent manner.40 DSBs and incorrect DSB repair are most likely to induce genomic and chromosomal instability, which are hallmarks of Cr(VI)-induced tumors.37 Other studies indicate that long-term exposure to zinc chromate can cause increases in aneuploidy, resulting in centrosome amplification, spindle-assembly checkpoint bypass, and G2 arrest, finally giving rise to chromosome instability, followed by tumor formation in human lung fibroblasts.38,39
Lead chromate, one kind of particulate form of Cr(VI) compounds, can be partially dissolved in water, releasing Cr6+ and Pb2+, which can enter cells. Xie et al carried out an experiment with WTHBF6 cells, and demonstrated that lead chromate particles induced DSBs. After chronic exposure to lead–chromate complexes, the ATM protein was autoactivated and transmitted signals to SMC1. Then, γ-H2AX foci formation responded to such breaks, with a consequent impediment of cell division and S-phase arrest. Unrepaired or misrepaired DSBs might be a risk that results in chromosomal lesions, thus creating cell chromosomal instability or gene mutations and further progressing to tumorigenesis.37 γ−H2AX foci formation is a pharmacodynamic marker of DNA damage produced by DNA cross-linking agents.41 Another study conducted by Holmes et al proved that prolonged exposure to lead chromium particulates may initiate an increase of aneuploid metaphases and aberrant centrosomes with the occurrence of mitotic catastrophe and disorganized anaphase, which is potentially the mechanism of lead chromium-induced carcinogenesis.42 Wise et al evaluated the cytotoxicity and genotoxicity of lead chromate in hawksbill sea turtle cells and found lead chromate was both cytotoxic and genotoxic to the cells. Concentrations of 0, 0.1, 0.5, 1, and 5 μg/cm2 lead chromate induced damage in 4%, 10%, 15%, 26%, and 36% of cells and caused four, eleven, 17, 30, and 56 chromosome aberrations in 100 metaphases, respectively.43
MicroRNAs are a family of small, endogenous, noncoding RNA molecules that control the expression of target genes by interacting with the 3′-untranslated region of target mRNAs, with a consequence of translation repression or mRNA degradation, which exerts prominent impact on physiological and pathological processes.44,45 Among them, miR21 is a predominant onco-miR involved in the process of developing cancer.46 Pratheeshkumar et al observed that chronic exposure to Cr(VI) in human bronchial epithelial (BEAS2B) cells could lead to the rising generation of miR21, as well as a drop in expression of the tumor suppressor PDCD4, which might be a new molecular marker for understanding the potential mechanism of Cr(VI)-induced oncogenesis.47,48
Compared to the properties of Cr(VI), Cr(III) has lower water-solubility and can enter the cells through phagocytosis.49 Initially, Cr(III) was considered relatively nontoxic and an essential component of glucose-tolerance factors. However, in recent years research findings have suggested that Cr(III) may also be genotoxic.50 Novotnik et al tested the cytotoxic and genotoxic potential of Cr(VI) and Cr(III) nitrate in human hepatoma (HepG2) cells. They found that Cr(VI) at ≥0.2 μg/mL and Cr(III) at ≥1 μg/mL induced DNA damage, suggesting Cr(VI) is five times more toxic than Cr(III) in genotoxicity.51 Similarly, Figgitt et al compared the genotoxicity of physiological concentrations of Cr(III) and Cr(VI) in an in vitro study of human fibroblasts. They found that both Cr(III) and Cr(VI) at the lowest physiological dose (2 ppb) caused a significant increase in total aberrations. Cr(VI) was much more effective than Cr(III) in causing chromosome fragmentation, which were only induced at the highest doses (40 ppb).52 However, Fang et al obtained conflicting results in a study involving yeast and Jurkat cells.22 They found that both Cr(VI) and Cr(III) induced genetic mutations in yeast cells and caused DNA damage in both yeast and Jurkat cells. Furthermore, they found that the effect of Cr(III) was greater than Cr(VI). They concluded that Cr(III) and Cr(VI) caused DNA damage through different mechanisms: Cr(VI) intercalates DNA, while Cr(III) interferes with base-pair stacking. The in vitro results indicate that both Cr(VI) and Cr(III) induce chromosome or DNA damage in culture cells, while different mechanisms may be involved.
In vivo studies
Based on the genotoxicity observed in in vitro studies, a wide range of investigations in animals have been launched to study chromium carcinogenesis. Potassium dichromate (Cr[VI]) is a widely used material in industry. After oral administration to Swiss mice for 1 and 5 days at 25, 50, or 100 mg/kg body weight per day, Wang et al found that potassium dichromate induced dose- and time-dependent effects on DNA damage, hepatic oxidative stress, and hepatocyte apoptosis.53
Lead chromate (Cr[VI]) is a yellow, orange, or red crystalline inorganic compound that emits toxic chromium fumes upon heating. Though we intensively searched PubMed, we did not find any in vivo studies conducted on lead chromate in the past 20 years. As early as 1976, Furst et al investigated the carcinogenic potential of lead chromate in both Fischer 344 rats and Swiss albino mice via intramuscular and oral administration. Both male and female rats developed fibrosarcomas and rhabdomyosarcomas at the injection sites, and three male rats also generated renal tumors in the intramuscular group. Furst et al concluded that lead chromate accounts for high carcinogenic activity in rats. Swiss albino female mice could not tolerate the same high dosage as the rats. However, at the lower dose administered to the mice, no tumors were detected.54
Sodium dichromate dihydrate (Cr[VI]) is ideal for use in many circumstances, including high-temperature applications, such as in ceramic glazes and colored glass. Chronic exposure to high concentrations of Cr(VI) in drinking water has caused small intestinal tumors in mice.55 In a 2-year lifetime study, Stout et al applied a low-dose concentration of Cr(VI), similar to the human exposure dosage present in contaminated water, to male and female F344/N rats and B6C3F1 mice via oral administration. After chronic exposure to Cr(VI), both male and female F344/N rats developed increasing squamous-cell carcinoma and squamous-cell papilloma or carcinoma (combined) of the oral mucosa at 516 mg/L, some of which invaded the tongue, Harderian gland, and soft tissues surrounding the nose. Excess incidence of small-intestine adenoma or carcinoma (combined) occurred in both male and female B6C3F1 mice at all sites, including the duodenum, jejunum, and ileum, with a dose–response relationship.55 Another study showed that exposure to Cr(VI) (K2CrO4) in the drinking water increased the number of ultraviolet-induced skin cancer lesions in hairless mice in a dose-dependent manner, supporting the concern about ingesting Cr(VI) because of its carcinogenic properties.56
Cr(III) has been proposed to be an essential element. Humans ingest Cr(III) as both a natural dietary constituent and dietary supplements taken for weight loss and antidiabetic effects.57 In order to examine the potential for chromium picolinate (CP; Cr[III]) to induce carcinogenicity, the National Toxicology Program conducted studies of CP monohydrate (CPM; Cr[C6H4NO2]3) in F344/N rats and B6C3F1 mice exposed to concentrations of 0, 2,000, 10,000, or 50,000 ppm for 2 years as a dietary supplement in feed. Despite exposure to very high concentrations relative to human exposure, there was very little evidence of any adverse effect following dietary exposure of rats and mice to CPM for 2 years. In male rats, there was equivocal evidence of carcinogenic activity based on increased preputial gland adenomas. There was no evidence of carcinogenic activity in female rats or in male or female mice. In addition, CPM exposure did not induce toxicity or increase nonneoplastic lesion incidence.48 To assess genotoxicity of a Cr(III) propionate complex, a study was performed on female Wistar rats exposed through a diet supplement, and the comet assay was used to evaluate DNA damage in peripheral blood lymphocytes (PBLs). High doses (equivalent to 100 mg Cr/kg body mass/day for 4 weeks) of supplementary Cr(III) did not affect body mass, feeding-efficiency ratio, or internal organ mass. Treatment of rats with the Cr(III)–propionate complex did not significantly affect the comet-assay results in lymphocytes, which suggests that the compound does not exert genotoxic effects in rats. Most in vivo experiments have demonstrated that Cr(VI) is carcinogenic, but the carcinogenicity of Cr(III) in animals is uncertain.
Epidemiological studies
The epidemiological evidence for increased risk of cancers of the lungs and sinonasal cavity is limited to conditions of high exposure, as encountered in the chromate-production, chromate-pigment production, and chromium-plating industries.58 Occupational exposure to high concentrations of Cr(VI) compounds is a well-documented cause of respiratory cancers.32 Absorption of Cr(VI) compounds by human lung cells can bring about bulky DNA adducts and oxidative DNA damage in the p53 gene, and both adenine and guanine mutations have also been seen in the p53 gene in Cr exposure-related lung cancer.59
Davies compared lung cancer mortality among workers in three English chromate pigment factories. She found that lung cancer mortality of workers exposed to both lead and zinc chromate with medium- or high-grade exposure as operators lasting more than 1 year was greater than that of workers with low-dose exposure lasting less than a year. However, mortality of workers in a factory exposed only to lead chromate was normal (observed:expected death ratio 7:6.45), indicating that exposure to lead chromate might not contribute to lung cancer, while zinc chromate is carcinogenic.60
Langård and Vigander conducted a follow-up study on the incidence of lung cancer in 133 workers producing zinc chromate pigments. A total of 24 of 133 workers employed in pigment production for more than 3 years before January 1973 were included. Six cases of lung cancer in this subcohort were detected prior to December 1980, three of which were highly differentiated epithelial carcinoma. Based on national figures, the expected number of cases of lung cancer in this subcohort was calculated to be 0.135, while the observed number of cases was six. The observed:expected ratio was 44:1 in this subcohort. These findings proved that zinc chromate was a potent carcinogen, and indicated that exposure to Cr(VI) compounds could lead to lung cancer.38
Welling et al used relative risk (RR) to estimate the association between stomach cancer and Cr(VI) exposure in 56 cohort and case–control studies. When all studies were combined, the summary RR was 1.27 (95% CI 1.18–1.38). When analysis was limited to studies identifying increased risks of lung cancer, the summary RR for stomach cancer was higher (1.41, 95% CI 1.18–1.69). According to these results, they suggested that Cr(VI) is also a stomach carcinogen in humans, which is consistent with the tumor results reported in in vivo animal studies.61
Occupational exposure to Cr(VI) has long been a public health concern, despite improvements in working conditions. However, many people are exposed to Cr(VI) compounds and particles not through occupational exposure, but via chronic, low-level inhalation from ambient air pollution or ingestion from contaminated water.62 Exposure through drinking water may cause greater toxicity than occupational exposure, due to its long-term exposure. However, due to multiple heavy metals existing in water, it is difficult to evaluate how much Cr(VI) contributes to the development of human cancers.
Information about the carcinogenicity of Cr(III) is limited. As early as 1990, Langård evaluated the carcinogenesis of Cr(VI) and Cr(III) through a review of epidemiological evidence and selected case reports. He concluded that all Cr(VI) compounds should be considered carcinogenic among exposed populations, and that no evidence has been presented indicating that human exposure to Cr(III) is associated with increased cancer risk.63 Nurminen reviewed evaluations and studies on the carcinogenicity in humans of metallic chromium and Cr(III). He concluded that evaluations of the potential carcinogenicity of metallic chromium and Cr(III) by international and national organizations and individual scientists were in agreement that the evidence on carcinogenicity was inadequate in humans.64 Chromium compounds may contribute to lung cancers. Recent epidemiological evidence shows that Cr(VI) may also be a stomach carcinogen in humans. The carcinogenicity of Cr(VI) and Cr(III) is summarized in Table 1 and Figure 1.
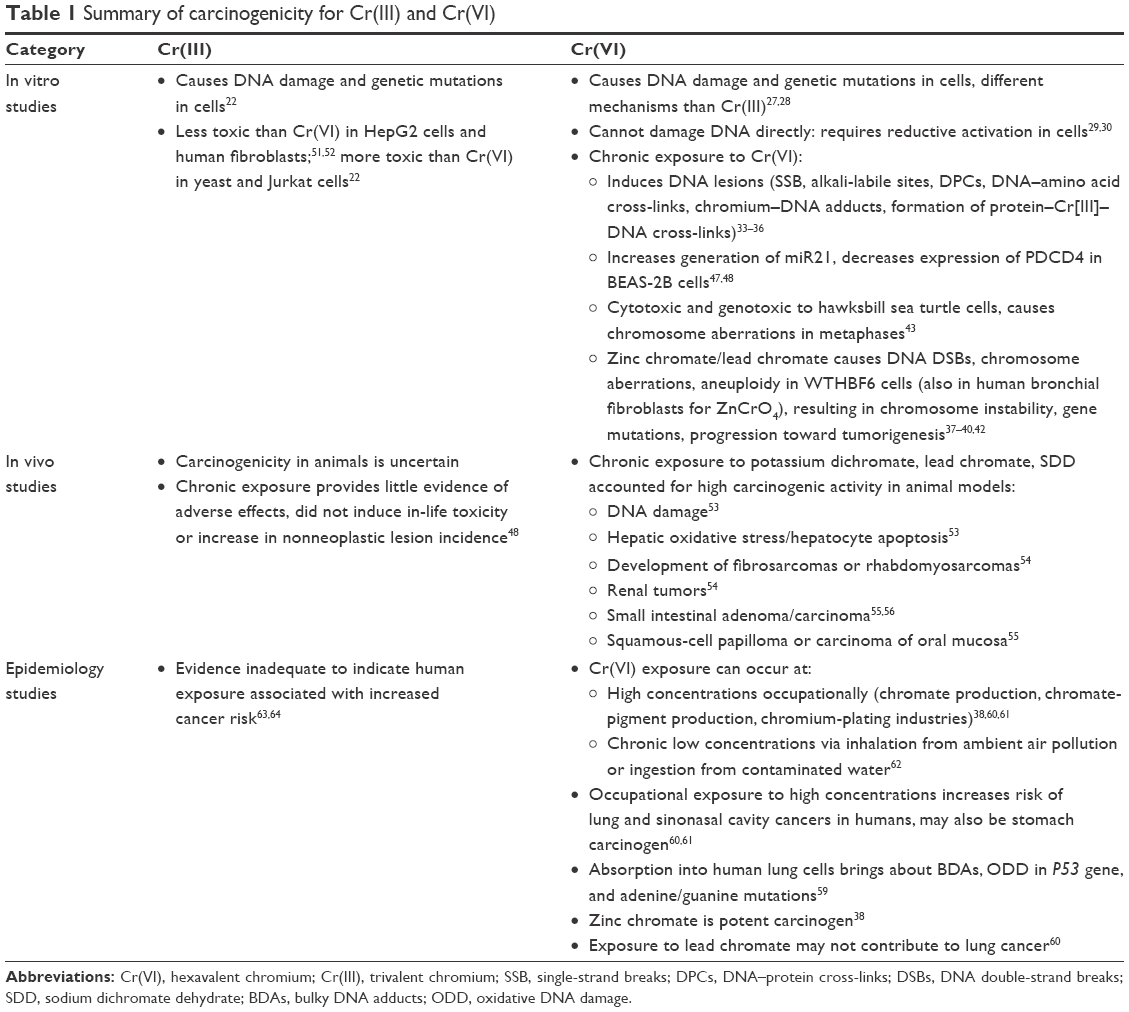 | Table 1 Summary of carcinogenicity for Cr(III) and Cr(VI) |
 | Figure 1 Possible mechanisms of Cr(VI)- and Cr(III)-induced carcinogenicity and chemoprevention of antioxidants. |
Chromium-carcinogenicity chemoprevention
In vitro chemoprevention
Intracellular Cr(VI) might be reduced to intermediate products, such as Cr(V), Cr(IV), and Cr(III). During this process, ROS are generated and then react with chromium intermediates, which cause a spectrum of cytotoxicity, genotoxicity, chromosomal aberrations, and DNA lesions. These products would then further lead to abnormal apoptosis and gene instability, or eventually carcinogenesis.
ROS-mediated oxidative stress is believed to play an important role in chromium-induced carcinogenesis.65 Exposure to Cr(VI) can cause several types of DNA lesions and gene mutations. In recent years, various antioxidants have been tested for prevention of the carcinogenesis caused by chromium compounds. It is of great importance to adopt therapeutic agents that enhance intra- and extracellular antioxidant levels and block Cr(VI)-mediated generation of ROS to prevent or attenuate Cr(VI)-induced genotoxicity.
Vitamin C plays an important role in controlling oxidative stress as a strong free-radical scavenger and protects DNA against damage induced by ROS.66,67 GSH plays a major role in protecting cells against stress caused by oxidants and heavy metals. When chromium compounds enter the human body orally, a series of organism-relevant reactions take place that attenuate their cytotoxicity and carcinogenicity.
Shi and Jiang checked the chemopreventive effects of apple juice against Cr(VI)-induced oxidative stress in human lung epithelial A549 cells. Results showed that apple juice efficiently scavenged hydroxyl radicals generated by Cr(VI) reduction catalyzed by GSH reductase/NADPH, and reduced Cr(VI) induced lipid peroxidation (LP), DNA damage, cell apoptosis, and NFκB activation in A549 cells.68 These results indicated that apple juice can prevent Cr(VI)-induced cellular injury and may help to reduce the carcinogenic potential of Cr(VI) through antioxidant properties.
Quercetin is one of the most abundant dietary flavonoids, with potent antioxidant and anticancer properties.69 MiR21 is a key onco-miR significantly elevated in the majority of human cancers that exerts its oncogenic activity by targeting the tumor-suppressor gene PDCD4.47 Pratheeshkumar et al examined the effect of quercetin on the inhibition of Cr(VI)-induced malignant cell transformation and the role of miR21–PDCD4 signaling involved in BEAS2B cells. They found that quercetin decreased ROS generation induced by Cr(VI) exposure in BEAS2B cells. Chronic Cr(VI) exposure induced malignant cell transformation, increased miR21 expression, and caused inhibition of PDCD4, which was significantly inhibited by quercetin treatment in a dose dependent manner.46 Pratheeshkumar et al examined the inhibition of luteolin on Cr(VI)-induced malignant cell transformation in BEAS2B cells. They concluded that luteolin also protected BEAS2B cells from Cr(VI)-induced carcinogenesis by scavenging ROS and modulating multiple cell-signaling mechanisms that are linked to ROS.70
Eugenia dysenterica extract (EDE; leaf, hydroalcoholic) is a phytochemical constituent associated with antioxidant properties. Pretreatment of AMJ2C11 cells with EDE protected cells against the cytotoxicity and oxidative stress induced by Cr(VI).71 N-acetylcysteine (NAC) is widely used for the assessment of the role of ROS in various biological processes.72 Luczak and Zhitkovich checked chemoprotection by the antioxidant NAC against Cr(VI) in human lung H460 cells. They found that NAC displayed significant chemoprotective activity against Cr(VI)-induced cell suppression of cytotoxicity, apoptosis, p53 activation, and HSP72 and HIF1α upregulation. However, cytoprotection by NAC seems to be independent of cellular GSH. A loss of Cr(VI) accumulation by cells was caused by NAC-mediated extracellular reduction of chromate to membrane-impermeable Cr(III). Therefore, the chemoprotective activity of NAC on Cr(VI)-induced oxidative stress damages might be completely through a ROS-independent pathway.73 However, a similar study conducted by Lee et al concluded that HaCaT cells pretreated with NAC prevented ROS-mediated cell death and cytokine expression induced by Cr(VI).74 Chen et al explored the antagonistic effect of NAC on Cr(VI)-induced apoptosis in L02 hepatocytes. They found that Cr(VI) significantly induced cell apoptosis at a dose of 20 μM/L for 6 hours. However, NAC significantly decreased the rate of cell apoptosis and alleviated the damage to mitochondria and DNA caused by Cr(VI) in L02 hepatocytes.75
Xiao et al tested the chemoprevention of vitamin C against Cr(VI)-induced genotoxicity in cultured PBLs from Sprague Dawley rats. They found that vitamin C cotreatment had a significant protective effect against the Cr(VI)-induced increase in DPCs. The mechanistic investigation revealed that cellular ROS levels were positively correlated with Cr(VI)-induced mitochondrial damage and that p53 expression was correlated with Cr(VI)-induced increase in DPCs.76 Little et al screened several drugs for their potential to protect two cell types against the cytotoxic effects of Cr(VI), specifically the reducing agents vitamin C, cysteine, and GSH and two Cr(VI) cellular uptake inhibitors. All five drugs showed dose-dependent protection against Cr(VI)-induced cytotoxicity, but only vitamin C offered complete protection.77 Capellmann et al found that vitamin C reacted much faster with Cr(VI) at physiological pH than GSH did, suggesting an influence of the reaction velocity of the redox reaction between Cr(VI) and the reducing compounds on the toxification of Cr(VI) and formation of DPCs.78
Research has shown that Lawsonia inermis fruit extracts can protect the liver from oxidative stress induced by carbon tetrachloride (CCl4).79 Guha et al explored antioxidant activity of L. inermis extracts for preventing Cr(VI)-induced DNA damage. They found that the extracts showed significant potential in scavenging free radicals (DPPH• and ABTS•+) and Fe3+ and in inhibiting LP. DNA damage caused by exposure of pBR322 plasmid DNA to Cr(VI) was markedly inhibited by the extracts. A distinct decline in Cr(VI)-induced cytotoxicity was noticed in MDAMB435S (human breast carcinoma) cells with an increase in the dosage of extracts.80 This study indicated L. inermis extracts might also be a potent therapeutic drug against Cr(VI)-induced oxidative cell damage. Guha et al also tested the ability of Phyllanthus amarus aqueous extract to inhibit Cr(VI)-induced toxicity in MDAMB435S cells, and found that the extract had high antioxidant potential (by virtue of its phenolic constituents) to inhibit Cr(VI)-induced oxidative toxicity in these cells.81 Lewinska et al found that Tempol, a nitroxide antioxidant, had inhibitory effects on Cr(VI)-induced cyto- and genotoxicity in human lymphocytes in vitro.82
Epigallocatechin-3-gallate (EGCG), one major kind of polyphenol extracted from green tea, has been identified to have protective properties against oxidative stress, inflammatory diseases, and neoplasms.83–86 It has also been proven to be a scavenger for ROS. Wu et al observed that EGCG protects BEAS-2B cells from Cr(VI)-induced genotoxicity, presumably by scavenging ROS and modulating a subset of genes.7 Shi et al used electron spin-resonance spin trapping to investigate scavenging effects on hydroxyl radicals and superoxide radicals by EGCG. Their results showed that EGCG achieved a protective effect against DNA damage, as well as inhibiting activation of the nuclear transcription factor NFκB induced by Cr(IV).87 These studies suggest an anticarcinogenic property of EGCG on Cr(VI).
Melatonin is an antioxidant that can easily cross cell membranes and the blood–brain barrier. Susa et al investigated the potent protective effect of melatonin on Cr(VI)-induced DNA single-strand breaks, cytotoxicity, and LP in primary cultures of rat hepatocytes. They concluded that melatonin protects cells from Cr(VI)-induced DNA-strand breaks, cytotoxicity, and LP, possibly through its ability to increase cellular levels of vitamins E and C and catalase activity and/or to scavenge toxic hydroxyl radicals directly in cells.88 Deb et al tested preventive effects of methanolic extract of methanolic Carum copticum extract (MCE; seeds) against Cr(VI) induced cytotoxicity, genotoxicity, apoptosis, and oxidative stress on BEAS2B and isolated human PBLs. Treatment of BEAS2B and PBLs with MCE prior to Cr(VI) treatment exhibited an increase in cell viability and decrease in DNA damage compared to Cr(VI) treatment alone. Further, MCE administration 1 hour prior to graded doses of Cr(VI) significantly decreased ROS levels. These results indicate that MCE may be beneficial in preventing Cr(VI)-induced toxicity in BEAS2B and PBL cells.89
Compared to Cr(VI), there are fewer Cr(III) chemoprevention studies. As previously cited, Lewinska et al found that the nitroxide Tempol has not only inhibitive effects on Cr(VI) but also on Cr(III)-induced cyto- and genotoxicity in human lymphocytes in vitro.82 Sankaramanivel et al investigated whether zinc pretreatment can protect human PBLs from apoptosis induced by Cr(III). Results from cytotoxicity studies showed that pretreatment of cells with zinc inhibited Cr(III)-induced cell apoptosis. The amount of ROS produced by Cr(III) was reduced with zinc pretreatment. Cell-signaling events, such as mitochondrial membrane potential, ATP levels, and activation of caspase 3 induced by Cr(III), were also reversed by pretreatment with zinc.90
In summary, more and more therapeutic agents have been explored to prevent chromium-induced genotoxicity/carcinogenesis, especially by Cr(VI) in culture cells. Evidence indicates that many antioxidants have potential chemopreventive effects against chromium-induced genotoxicity/carcinogenesis. These antioxidants include apple juice, quercetin, luteolin, EDE, NAC, vitamin C, zinc, L. inermis extracts, P. amarus aqueous extract, Tempol, EGCG, melatonin, and MCE.
In vivo chemoprevention
In recent years, many in vivo experiments have also been conducted to search for therapeutics for chemoprevention of chromium genotoxicity/carcinogenesis. As previously cited, Pratheeshkumar et al examined the effect of quercetin on the inhibition of Cr(VI)-induced malignant cell transformation and the role of miR21–PDCD4 signaling involved in BEAS2B cells. In this study, there was reduced incidence of tumors in nude mice injected with BEAS2B cells chronically exposed to Cr(VI) in the presence of quercetin compared to cells treated with Cr(VI) alone. Cr(VI) exposure increased malignant transformation and tumorigenesis in BEAS2B cells, but was suppressed with a knockdown of miR21 and an increase in PDCD4 or catalase. These results suggest that quercetin may target the miR21–PDCD4 pathway to decrease Cr(VI)-induced carcinogenesis in BEAS2B cells.46
In an in vivo study, de Avila et al found that the survival rate of mice exposed to Cr(VI) could be extended if they were treated with EDE for 10 days prior to exposure. EDE prevented pathological liver and kidney damages, corresponding to a reduction in chromium levels found in these organs and plasma. The antioxidant capability of EDE might be due to the presence of polyphenols, especially flavonoids and tannins, as confirmed by high-performance liquid chromatography photodiode array.71
Lee et al observed that dermal injection of Cr(VI) activated phospho-Akt, phospho-p65, phospho-IκBα, phospho-ERK, phospho-p38, and phospho-JNK in the epidermis of albino Hartley strain guinea pigs. They found that 5 weeks of NAC administration through gavage or intraperitoneal injection significantly inhibited activation of these signal proteins.74
Kart et al evaluated the effects of intraperitoneal administration of Cr(VI) and GSH on total oxidant status, total antioxidant capacity, oxidative stress index, and oxidative DNA damage by evaluating the level of 8-hydroxy-2′-deoxyguanosine (8-OHdG) in Swiss albino mice. They concluded that Cr(VI) given intraperitoneally caused increased oxidative stress and oxidative DNA damage in the blood of Swiss albino mice. It is possible that intraperitoneal administration of GSH acts as protection from oxidative stress and DNA damage.91
García-Rodríguez et al investigated the relationship between modulation of genotoxic damage and apoptotic activity in Hsd:ICR male mice treated with EGCG intravenously and Cr(VI) intraperitoneally. They concluded that EGCG protected against genotoxic damage induced by Cr(VI), which might contribute to elimination of DNA-damaged cells when EGCG is administered prior to Cr(VI).92 In another study, García-Rodríguez et al examined the modulating effects of the flavonoids EGCG, quercetin, and rutin on the genotoxic damage induced by Cr(VI) in polychromatic erythrocytes (PCEs) of CD1 mice. Treatments were administered intraperitoneally, and genotoxic damage was evaluated based on the number of micronucleated PCEs (MN-PCEs) obtained from the caudal vein at 0, 24, 48, and 72 hours after treatment. They found that if cells were treated with flavonoids prior to Cr(VI) exposure, there was a decrease in MN-PCE induction compared with Cr(VI) only. Effects of EGCG, quercetin, and rutin against genotoxic damage induced by Cr(VI) were observed. The magnitude of the potency of flavonoids was in the following order: rutin (82%) > quercetin (64%) > quercetin–rutin (59%) and EGCG (44%).93
Curcumin is a well-known direct and indirect antioxidant.94 Molina-Jijón et al examined the role of mitochondria on the protective effects of curcumin against renal oxidant damage induced by Cr(VI) in rats given by gavage. They found that both direct and indirect antioxidant effects were involved in the protective effects of curcumin. Preservation of mitochondrial function plays a very important role in the protective effects of curcumin pretreatment against Cr(VI)-induced renal oxidant damage.95
As an antioxidant, selenium (Se) has been shown to prevent cancer in numerous animal models.96 Soudani et al investigated the protective effect of Se on oxidative damage induced by Cr(VI) in rat erythrocytes. They found that rats exposed to Cr(VI) showed an increase in malondialdehyde and protein carbonyl levels and a decrease in sulfhydryl content, GSH, nonprotein thiol, and vitamin C levels, as well decreased enzyme, eg, catalase, GSH peroxidase, and superoxide dismutase activities. These parameters were restored to normal or near-normal values when Se was given with Cr(VI). These results suggest that Se might be useful in preventing Cr(VI)-induced erythrocyte damage.97
Kumar and Gangwar divided Wistar strain rats into five categories and administered a predetermined dose of Cr(VI), as well as antioxidants, including GSH, Se, and α-tocopherol, respectively. They studied how these three nonenzymatic antioxidants protected against LP and oxidative stress. Increased malondialdehyde and a significant depletion in GSH status were observed in the liver and kidneys of the Cr(VI)-fed rats. However, administration with antioxidants inhibited LP and oxidative stress and restored the GSH cycle in liver and kidneys of Cr(VI) treated rats.4
Krim et al evaluated the protective effect of ginger against toxicity induced by Cr(VI) in rats. Their results indicated that chromate (Cr[VI]) decreased reduced GSH levels in different tissues. Moreover, administration of ginger revealed a reduction of the intensity of oxidative stress induced by Cr(VI), demonstrating that ginger has potent antioxidant activity toward Cr(VI)-induced damage.98
Vitamin E is a fat-soluble vitamin that plays a role as an antioxidant in the body. Chandra et al explored vitamin E supplementation to protect Cr(VI)-induced spermatogenic and steroidogenic disorders in testicular tissues of rats. Simultaneous oral supplementation of vitamin E (50 mg/kg body weight/day) in Cr(VI)-exposed rats showed less oxidative damage and restored altered testicular activities and epididymal sperm numbers, indicating vitamin E might be a protective agent against Cr(VI)-induced spermatogenic and steroidogenic alteration.99
Acharya et al compared the potential role of vitamins C and E in Cr(VI)-induced spermatogenesis in Swiss mice. The possible antioxidative role of both vitamins were studied for a significant decrease in LP associated with marked elevation in sperm count and significant decrease in the percentage of abnormal sperm formation in Cr(VI)-treated mice. Of the two vitamins tested in this study, vitamin C was found to be more effective in ameliorating germ cells from degeneration and from mutation to abnormal sperm.100
Hamida et al checked the protective effect of propylthiouracil against hepatotoxicity induced by Cr(VI) in adult mice. Animals received Cr(VI) via drinking water ad libitum and propylthiouracil by intramuscular injection at a dose of 2.5 mg/100 g body weight. Exposure of mice to Cr(VI) promoted oxidative stress, with an increase in malondialdehyde, protein carbonyl, and advanced oxidation protein-product levels. They found coadministration of propylthiouracil with Cr(VI) restored these parameters to near-normal values. Histological findings confirmed these biochemical results.101
Consumption of green tea, due to its antioxidant properties, has been associated with beneficial health effects. García-Rodríguez et al investigated green tea and chemoprevention in vivo on genotoxic damage induced by carcinogenic Cr(VI). The green tea was administrated via an intragastric tube every 12 hours over 2 days (four doses of 0.25 mL infusions of 1.6 g/7.5 mL) and ad libitum (5.6 mL/day for 10 days, infusions of 3.2 g/100 mL), while Cr(VI) was administrated intraperitoneally (20 mg/kg). Blood samples were obtained from the caudal vein, and the number of micronuclei in PCEs was assessed at 0, 24, 48, and 72 hours after treatment. Their results suggested that green tea had a chemopreventive effect on Cr(VI)-induced genotoxic damage. The fact that the largest decrease in the frequency of micronuclei was observed at 72 hours and ad libitum treatment suggested that the protective effect depends on the bioavailability, pharmacodynamics, and pharmacokinetics of the active ingredient in green tea. Therefore, the administration of green tea for a long time before the exposure to Cr(VI) might have a more consistent preventive effect.102 García-Rodríguez et al found that polyphenol extracted from green tea played a pronounced role against Cr(VI)-induced genetic damage by reducing the amount of MN-PCEs in CD1 mice.103
Susa et al found that thiol compounds containing L-cysteine ethyl ester, 2,3-dimercaptosuccinic acid, or 2,3-dimercapto-1-propanesulfonic acid ameliorated lethality in Cr(VI)-treated male ddY mice under the condition of injection of Cr(VI) and thiol admixtures simultaneously. In addition, thiol compounds boosted urinary chromium excretion, which showed protective effects against chromium toxicity.104
α-Lipoic acid (α-LA), a disulfide derivative, as well as an intramolecular disulfide bond in its reduced state, dihydrolipoic acid (DHLA), serve as biological antioxidants in both hydrophilic and lipophilic environments.105,106 Both LA and DHLA directly scavenge a variety of ROS. In addition, LA–DHLA coupling has the capability of altering the redox status of cells and interacting with thiols and other antioxidants. Evidence has shown that both play an effective role in attenuating chromium-induced DPCs by counteracting oxidative stress. Budhwar and Kumar demonstrated that intraperitoneal treatment with α-LA through pre-, co-, and post-Cr(VI) exposure cut down the generation of DPCs under the circumstance of oxidative stress formation in the PBLs of Swiss albino mice.105
Very few in vivo investigations related to chemoprevention on Cr(III)-induced carcinogenicity can be found. Schrauzer examined the interactive effects of Cr(III) and Se on the appearance of mammary tumors by exposing female virgin C3H mice infected with the murine mammary tumor virus to subtoxic levels of Cr(III) and Se in the supply water. Results showed that by decreasing the inhibitory effect of Se on tumor development in a dose-dependent manner, Cr(III) increased tumor-growth rates and shortened tumor latency. Se levels in the liver and kidneys of the mice were also altered after exposure to Cr(III), indicating that Cr(III) interacted with Se and affected organ distribution. They suggested that Cr(III) must be added to the list of Se-antagonistic elements that weaken or abolish the antitumorigenic effects of Se.107
In summary, antioxidants with potential chemopreventive effects against Cr(VI)-induced genotoxicity/carcinogenicity evaluated by in vivo studies include EDE, NAC, GSH, EGCG, quercetin, rutin, curcumin, Se, α-tocopherol, ginger, vitamin C, vitamin E, propylthiouracil, green-tea polyphenol extracts, and thiol compounds, including LA. Se was the only agent investigated for prevention of Cr(III)-induced genotoxicity/carcinogenesis. Chemoprotective activity of NAC on Cr(VI)-induced oxidative stress damage was considered probably to be through ROS-independent cellular reduction of chromate to membrane impermeability Cr(III). EDE was found to prevent Cr(VI)-induced liver and kidney pathological damage, in parallel with reduction in chromium levels in these organs and plasma. In addition, thiol compounds can boost urinary chromium excretion in animals. Chemoprevention studies for Cr(III) and Cr(VI) carcinogenicity, including in vitro and in vivo experiments, are summarized in Table 2 and Figure 1.
 | Table 2 Summary of chemoprevention for Cr(III) and Cr(VI) carcinogenicity |
Conclusion
Chromium occurs mostly in Cr(VI) and Cr(III) states in the environment. Cr(VI) compounds are used extensively in industry, while Cr(III) salts are used as micronutrients and dietary supplements. Cr(VI) species are considered much more toxic than Cr(III) species. In vitro studies indicate that both Cr(VI) and Cr(III) induce genotoxicity in culture cells, although mechanisms between them may be different. Cells exposed to chromium complexes, especially Cr(VI) species, can generate a diverse array of DNA lesions, including SSB, alkali-labile sites, DPCs, DNA–amino acid cross-links and chromium–DNA adducts, as well as the formation of protein–Cr(III)–DNA cross-links, the most likely cause of mutations in Cr(VI)-treated cells. In vivo experiments have demonstrated that Cr(VI) is carcinogenic, but the carcinogenicity of Cr(III) is uncertain. Epidemiology has identified inhalation of Cr(VI) species as a great risk for lung cancer. Oral ingestion of Cr(VI) species through contaminated water and food may result in gastrointestinal tumors, such as stomach cancer. However, evidence of carcinogenicity of Cr(III) species in humans is inadequate.
In recent years, a growing number of therapeutic agents have been tested for prevention of chromium harm, especially Cr(VI)-induced genotoxicity/carcinogenesis through in vitro/in vivo studies. In vitro and in vivo experiments show that therapeutic agents with potential chemopreventive effects against chromium-induced genotoxicity/carcinogenesis are mostly antioxidant-related chemicals or compounds, such as apple juice, quercetin, luteolin, EDE, NAC, vitamin C, vitamin E, zinc, L. inermis extracts, P. amarus aqueous extract, Tempol, EGCG, melatonin, MCE, GSH, rutin, curcumin, Se, α-tocopherol, ginger, propylthiouracil, green-tea polyphenol extracts, and thiol compounds, such as LA. Furthermore, most of these agents have been explored for chemoprevention against the genotoxicity/carcinogenesis induced by Cr(VI) instead of Cr(III).
It is worthy of note that chromium is hard to be degraded or reduce in the human body. Chemoprevention effects of antioxidants are mainly through direct or indirect inhibition of oxidative stress induced by chromium. Only the chemoprotective activity of NAC on Cr(VI)-induced oxidative stress damages was found probably to be through cellular reduction of chromate to membrane-impermeable Cr(III), an ROS-independent pathway. EDE was found to prevent Cr(VI)-induced pathological liver and kidney damage, in parallel with reduction in chromium levels in these organs and plasma. In addition to having antioxidative effects, thiol compounds were found to boost urinary chromium excretion in animals. Antioxidative activities are mostly a temporary action, instead of a permanent cure for chromium species’ toxicities. New therapeutic agents with additional abilities, such as increasing the human body’s ability to clear chromium through chelating, metabolizing, or degradation, should be explored in future research. Furthermore, any in vitro or in vivo studies in future, while evaluating preventive effects on toxicity and genotoxicity/carcinogenicity, should also include examination of chromium levels in cells, organs, and tissues at the same time.
Acknowledgments
The authors would like to thank Dr Min Ding, Ms Linda Bowman, and Ms Joni Aldinger (from Toxicology and Molecular Biology Branch, Health Effects Laboratory Division, National Institute for Occupational Safety and Health, Morgantown, WV, USA) for their assistance in the preparation of this article. This work was partly supported by the National Nature Science Foundation of China (grant 81273111), Scientific Projects of Zhejiang Province (2015C33148 and 2015C37117), Ningbo Scientific Innovation Team for Environmental Hazardous Factor Control and Prevention (2016C51001), Zhejiang Key Laboratory of Pathophysiology (201703), and the KC Wong Magna Fund of Ningbo University.
Disclosure
The authors report no conflicts of interest in this work.
References
Shrivastava R, Upreti RK, Seth PK, Chaturvedi UC. Effects of chromium on the immune system. FEMS Immunol Med Microbiol. 2002;34(1):1–7. | ||
Zhitkovich A. Chromium in drinking water: sources, metabolism, and cancer risks. Chem Res Toxicol. 2011;24(10):1617–1629. | ||
Blasiak J, Kowalik J. A comparison of the in vitro genotoxicity of tri- and hexavalent chromium. Mutat Res. 2000;469(1):135–145. | ||
Kumar D, Gangwar SP. Role of antioxidants in detoxification of Cr(VI) toxicity in laboratory rats. J Environ Sci Eng. 2012;54(3):441–446. | ||
Zhao R, Wang B, Cai QT, et al. Bioremediation of hexavalent chromium pollution by Sporosarcina saromensis M52 isolated from offshore sediments in Xiamen, China. Biomed Environ Sci. 2016;29(2):127–136. | ||
Khan FH, Ambreen K, Fatima G, Kumar S. Assessment of health risks with reference to oxidative stress and DNA damage in chromium exposed population. Sci Total Environ. 2012;430:68–74. | ||
Wu F, Sun H, Kluz T, Clancy HA, Kiok K, Costa M. Epigallocatechin-3-gallate (EGCG) protects against chromate-induced toxicity in vitro. Toxicol Appl Pharmacol. 2012;258(2):166–175. | ||
Nickens KP, Patierno SR, Ceryak S. Chromium genotoxicity: a double-edged sword. Chem Biol Interact. 2010;188(2):276–288. | ||
Thompson CM, Kirman CR, Proctor DM, et al. A chronic oral reference dose for hexavalent chromium-induced intestinal cancer. J Appl Toxicol. 2014;34(5):525–536. | ||
Seidler A, Jähnichen S, Hegewald J, et al. Systematic review and quantification of respiratory cancer risk for occupational exposure to hexavalent chromium. Int Arch Occup Environ Health. 2013;86(8):943–955. | ||
Manning FC, Blankenship LJ, Wise JP, Xu J, Bridgewater LC, Patierno SR. Induction of internucleosomal DNA fragmentation by carcinogenic chromate: relationship to DNA damage, genotoxicity, and inhibition of macromolecular synthesis. Environ Health Perspect. 1994;102 (Suppl 3):159–167. | ||
Zhang XH, Zhang X, Wang XC, et al. Chronic occupational exposure to hexavalent chromium causes DNA damage in electroplating workers. BMC Public Health. 2011;11:224. | ||
Kortenkamp A, Casadevall M, Faux SP, et al. A role for molecular oxygen in the formation of DNA damage during the reduction of the carcinogen chromium(VI) by glutathione. Arch Biochem Biophys. 1996;329(2):199–207. | ||
Cheng L, Liu S, Dixon K. Analysis of repair and mutagenesis of chromium-induced DNA damage in yeast, mammalian cells, and transgenic mice. Environ Health Perspect. 1998;106 (Suppl 4):1027–1032. | ||
Cerveira JF, Sanchez-Arago M, Urbano AM, Cuezva JM. Short-term exposure of nontumorigenic human bronchial epithelial cells to carcinogenic chromium(VI) compromises their respiratory capacity and alters their bioenergetic signature. FEBS Open Bio. 2014;4:594–601. | ||
O’Brien TJ, Ceryak S, Patierno SR. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res. 2003;533(1–2):3–36. | ||
Wise JP Sr, Wise SS, Little JE. The cytotoxicity and genotoxicity of particulate and soluble hexavalent chromium in human lung cells. Mutat Res. 2002;517(1–2):221–229. | ||
Proctor DM, Suh M, Campleman SL, Thompson CM. Assessment of the mode of action for hexavalent chromium-induced lung cancer following inhalation exposures. Toxicology. 2014;325:160–179. | ||
Straif K, Benbrahim-Tallaa L, Baan R, et al. A review of human carcinogens – part C: metals, arsenic, dusts, and fibres. Lancet Oncol. 2009;10(5):453–454. | ||
Beaver LM, Stemmy EJ, Schwartz AM, et al. Lung inflammation, injury, and proliferative response after repetitive particulate hexavalent chromium exposure. Environ Health Perspect. 2009;117(12):1896–1902. | ||
Cullen JM, Ward JM, Thompson CM. Reevaluation and classification of duodenal lesions in B6C3F1 mice and F344 rats from 4 studies of hexavalent chromium in drinking water. Toxicol Pathol. 2016;44(2):279–289. | ||
Fang Z, Zhao M, Zhen H, Chen L, Shi P, Huang Z. Genotoxicity of tri- and hexavalent chromium compounds in vivo and their modes of action on DNA damage in vitro. PloS One. 2014;9(8):e103194. | ||
Liu Y, Zhao X, Zhang X, Zhao X, Liu Y, Liu J. Effects of oral administration of CrCl3 on the contents of Ca, Mg, Mn, Fe, Cu, and Zn in the liver, kidney, and heart of chicken. Biol Trace Elem Res. 2016;171(2):459–467. | ||
Levina A, Lay PA. Chemical properties and toxicity of chromium(III) nutritional supplements. Chem Res Toxicol. 2008;21(3):563–571. | ||
Shrivastava HY, Ravikumar T, Shanmugasundaram N, Babu M, Unni Nair B. Cytotoxicity studies of chromium(III) complexes on human dermal fibroblasts. Free Radic Biol Med. 2005;38(1):58–69. | ||
Parker DL, Borer P, Bernier-Latmani R. The response of Shewanella oneidensis MR-1 to Cr(III) toxicity differs from that to Cr(VI). Front Microbiol. 2011;2:223. | ||
Elshazly MO, El-Rahman SS, Morgan AM, Ali ME. The remedial efficacy of Spirulina platensis versus chromium-induced nephrotoxicity in male Sprague-Dawley rats. PloS One. 2015;10(6):e0126780. | ||
Huang Z, Kuang X, Chen Z, Fang Z, Wang S, Shi P. Comparative studies of tri- and hexavalent chromium cytotoxicity and their effects on oxidative state of Saccharomyces cerevisiae cells. Curr Microbiol. 2014;68(4):448–456. | ||
Aiyar J, Berkovits HJ, Floyd RA, Wetterhahn KE. Reaction of chromium(VI) with glutathione or with hydrogen peroxide: identification of reactive intermediates and their role in chromium(VI)-induced DNA damage. Environ Health Perspect. 1991;92:53–62. | ||
Suzuki Y, Fukuda K. Reduction of hexavalent chromium by ascorbic acid and glutathione with special reference to the rat lung. Arch Toxicol. 1990;64(3):169–176. | ||
Standeven AM, Wetterhahn KE. Ascorbate is the principal reductant of chromium(VI) in rat liver and kidney ultrafiltrates. Carcinogenesis. 1991;12(9):1733–1737. | ||
Wise JP, Wise SS, Kraus S, et al. Hexavalent chromium is cytotoxic and genotoxic to the North Atlantic right whale (Eubalaena glacialis) lung and testes fibroblasts. Mutat Res. 2008;650(1):30–38. | ||
O’Brien TJ, Jiang G, Chun G, et al. Incision of trivalent chromium [Cr(III)]-induced DNA damage by Bacillus caldotenax UvrABC endonuclease. Mutat Res. 2006;610(1–2):85–92. | ||
Casadevall M, da Cruz Fresco P, Kortenkamp A. Chromium(VI)-mediated DNA damage: oxidative pathways resulting in the formation of DNA breaks and abasic sites. Chem Biol Interact. 1999;123(2):117–132. | ||
Macfie A, Hagan E, Zhitkovich A. Mechanism of DNA-protein cross-linking by chromium. Chem Res Toxicol. 2010;23(2):341–347. | ||
Levina A, Harris HH, Lay PA. Binding of chromium(VI) to histones: implications for chromium(VI)-induced genotoxicity. J Biol Inorg Chem. 2006;11(2):225–234. | ||
Xie H, Wise SS, Holmes AL, et al. Carcinogenic lead chromate induces DNA double-strand breaks in human lung cells. Mutat Res. 2005;586(2):160–172. | ||
Langård S, Vigander T. Occurrence of lung cancer in workers producing chromium pigments. Br J Ind Med. 1983;40(1):71–74. | ||
Holmes AL, Wise SS, Pelsue SC, et al. Chronic exposure to zinc chromate induces centrosome amplification and spindle assembly checkpoint bypass in human lung fibroblasts. Chem Res Toxicol. 2010;23(2):386–395. | ||
Xie H, Holmes AL, Young JL, et al. Zinc chromate induces chromosome instability and DNA double strand breaks in human lung cells. Toxicol Appl Pharmacol. 2009;234(3):293–299. | ||
Wu J, Clingen PH, Spanswick VJ, et al. γ-H2AX foci formation as a pharmacodynamic marker of DNA damage produced by DNA cross-linking agents: results from 2 phase I clinical trials of SJG-136 (SG2000). Clin Cancer Res. 2013;19(3):721–730. | ||
Holmes AL, Wise SS, Sandwick SJ, et al. Chronic exposure to lead chromate causes centrosome abnormalities and aneuploidy in human lung cells. Cancer Res. 2006;66(8):4041–4048. | ||
Wise SS, Xie H, Fukuda T, Thompson WD, Wise JP. Hexavalent chromium is cytotoxic and genotoxic to hawksbill sea turtle cells. Toxicol Appl Pharmacol. 2014;279(2):113–118. | ||
Takeuchi-Yorimoto A, Yamaura Y, Kanki M, et al. MicroRNA-21 is associated with fibrosis in a rat model of nonalcoholic steatohepatitis and serves as a plasma biomarker for fibrotic liver disease. Toxicol Lett. 2016;258:159–167. | ||
Zhang H, Liu S, Dong T, et al. Profiling of differentially expressed microRNAs in arrhythmogenic right ventricular cardiomyopathy. Sci Rep. 2016;6:28101. | ||
Pratheeshkumar P, Son YO, Divya SP, et al. Quercetin inhibits Cr(VI)-induced malignant cell transformation by targeting miR-21-PDCD4 signaling pathway. Oncotarget. Epub 2016 Jun 17. | ||
Pratheeshkumar P, Son YO, Divya SP, et al. Hexavalent chromium induces malignant transformation of human lung bronchial epithelial cells via ROS-dependent activation of miR-21-PDCD4 signaling. Oncotarget. 2016;7(32):51193–51210. | ||
National Toxicology Program. NTP toxicology and carcinogenesis studies of chromium picolinate monohydrate (CAS no. 27882-76-4) in F344/N rats and B6C3F1 mice (feed studies). Natl Toxicol Program Tech Rep Ser. 2010;(556):1–194. | ||
Fernando VA, Weerasena J, Lakraj GP, et al. Lethal and sub-lethal effects on the Asian common toad Duttaphrynus melanostictus from exposure to hexavalent chromium. Aquat Toxicol. 2016;177:98–105. | ||
Vaidyanathan VG, Weyhermuller T, Nair BU, Subramanian J. DNA damage induced by a chromium(III) Schiff base complex is reversible under physiological condition. J Inorg Biochem. 2005;99(11):2248–2255. | ||
Novotnik B, Sčančar J, Milačič R, Filipič M, Zegura B. Cytotoxic and genotoxic potential of Cr(VI), Cr(III)-nitrate and Cr(III)-EDTA complex in human hepatoma (HepG2) cells. Chemosphere. 2016;154:124–131. | ||
Figgitt M, Newson R, Leslie IJ, Fisher J, Ingham E, Case CP. The genotoxicity of physiological concentrations of chromium (Cr(III) and Cr(VI)) and cobalt (Co(II)): an in vitro study. Mutat Res. 2010;688(1–2):53–61. | ||
Wang XF, Xing ML, Shen Y, Zhu X, Xu LH. Oral administration of Cr(VI) induced oxidative stress, DNA damage and apoptotic cell death in mice. Toxicology. 2006;228(1):16–23. | ||
Furst A, Schlauder M, Sasmore DP. Tumorigenic activity of lead chromate. Cancer Res. 1976;36(5):1779–1783. | ||
Stout MD, Herbert RA, Kissling GE, et al. Hexavalent chromium is carcinogenic to F344/N rats and B6C3F1 mice after chronic oral exposure. Environ Health Perspect. 2009;117(5):716–722. | ||
Davidson T, Kluz T, Burns F, et al. Exposure to chromium(VI) in the drinking water increases susceptibility to UV-induced skin tumors in hairless mice. Toxicol Appl Pharmacol. 2004;196(3):431–437. | ||
Stout MD, Nyska A, Collins BJ, et al. Chronic toxicity and carcinogenicity studies of chromium picolinate monohydrate administered in feed to F344/N rats and B6C3F1 mice for 2 years. Food Chem Toxicol. 2009;47(4):729–733. | ||
De Flora S, Camoirano A, Bagnasco M, Bennicelli C, Corbett GE, Kerger BD. Estimates of the chromium(VI) reducing capacity in human body compartments as a mechanism for attenuating its potential toxicity and carcinogenicity. Carcinogenesis. 1997;18(3):531–537. | ||
Arakawa H, Weng MW, Chen WC, Tang MS. Chromium(VI) induces both bulky DNA adducts and oxidative DNA damage at adenines and guanines in the P53 gene of human lung cells. Carcinogenesis. 2012;33(10):1993–2000. | ||
Davies JM. Lung cancer mortality among workers making lead chromate and zinc chromate pigments at three English factories. Br J Ind Med. 1984;41(2):158–169. | ||
Welling R, Beaumont JJ, Petersen SJ, Alexeeff GV, Steinmaus C. Chromium VI and stomach cancer: a meta-analysis of the current epidemiological evidence. Occup Environ Med. 2015;72(2):151–159. | ||
Cavallo D, Ursini CL, Fresegna AM, et al. Direct-oxidative DNA damage and apoptosis induction in different human respiratory cells exposed to low concentrations of sodium chromate. J Appl Toxicol. 2010;30(3):218–225. | ||
Langård S. One hundred years of chromium and cancer: a review of epidemiological evidence and selected case reports. Am J Ind Med. 1990;17(2):189–215. | ||
Nurminen M. Overview of the human carcinogenicity risk assessment of metallic chromium and trivalent chromium. Internet J Epidemiol. 2005;2(1):1–14. | ||
Sun L, Wang X, Yao H, et al. Reactive oxygen species mediate Cr(VI)-induced S phase arrest through p53 in human colon cancer cells. J Environ Pathol Toxicol Oncol. 2012;31(2):95–107. | ||
Parveen N, Ahmad S, Shadab GG. Iron induced genotoxicity: attenuation by vitamin C and its optimization. Interdiscip Toxicol. 2014;7(3):154–158. | ||
Sram RJ, Binkova B, Rossner P Jr. Vitamin C for DNA damage prevention. Mutat Res. 2012;733(1–2):39–49. | ||
Shi D, Jiang BH. Antioxidant properties of apple juice and its protection against Cr(VI)-induced cellular injury. J Environ Pathol Toxicol Oncol. 2002;21(3):233–242. | ||
Ding M, Zhao J, Bowman L, Lu Y, Shi X. Inhibition of AP-1 and MAPK signaling and activation of Nrf2/ARE pathway by quercitrin. Int J Oncol. 2010;36(1):59–67. | ||
Pratheeshkumar P, Son YO, Divya SP, et al. Luteolin inhibits Cr(VI)-induced malignant cell transformation of human lung epithelial cells by targeting ROS mediated multiple cell signaling pathways. Toxicol Appl Pharmacol. 2014;281(2):230–241. | ||
de Avila RI, Alvarenga CB, de Avila PH, et al. Eugenia dysenterica DC. (Myrtaceae) exerts chemopreventive effects against hexavalent chromium-induced damage in vitro and in vivo. Pharm Biol. 2016;54(11):2652–2663. | ||
Zecevic A, Hagan E, Reynolds M, Poage G, Johnston T, Zhitkovich A. XPA impacts formation but not proteasome-sensitive repair of DNA-protein cross-links induced by chromate. Mutagenesis. 2010;25(4):381–388. | ||
Luczak MW, Zhitkovich A. Role of direct reactivity with metals in chemoprotection by N-acetylcysteine against chromium(VI), cadmium(II), and cobalt(II). Free Radic Biol Med. 2013;65:262–269. | ||
Lee YH, Su SB, Huang CC, et al. N-acetylcysteine attenuates hexavalent chromium-induced hypersensitivity through inhibition of cell death, ROS-related signaling and cytokine expression. PloS One. 2014;9(9):e108317. | ||
Chen J, Zhong C, Zeng M, Liu X, Deng Y, Xiao F. [Antagonistic effect of N-acetylcysteine on apoptosis of L-02 hepatocyte induced by Cr(VI) with or without caspase inhibitor]. Wei Sheng Yan Jiu. 2010;39(6):678–681. Chinese. | ||
Xiao F, Chen D, Luo L, et al. Time-order effects of vitamin C on hexavalent chromium-induced mitochondrial damage and DNA protein crosslinks in cultured rat peripheral blood lymphocytes. Mol Med Rep. 2013;8(1):53–60. | ||
Little MC, Gawkrodger DJ, MacNeil S. Chromium- and nickel-induced cytotoxicity in normal and transformed human keratinocytes: an investigation of pharmacological approaches to the prevention of Cr(VI)-induced cytotoxicity. Br J Dermatol. 1996;134(2):199–207. | ||
Capellmann M, Mikalsen A, Hindrum M, Alexander J. Influence of reducing compounds on the formation of DNA-protein cross-links in HL-60 cells induced by hexavalent chromium. Carcinogenesis. 1995;16(5):1135–1139. | ||
Hsouna AB, Mongi S, Culioli G, et al. Protective effects of ethyl acetate fraction of Lawsonia inermis fruits extract against carbon tetrachloride-induced oxidative damage in rat liver. Toxicol Ind Health. 2016;32(4):694–706. | ||
Guha G, Rajkumar V, Kumar RA, Mathew L. Antioxidant activity of Lawsonia inermis extracts inhibits chromium(VI)-induced cellular and DNA toxicity. Evid Based Complement Alternat Med. 2011;2011:576456. | ||
Guha G, Rajkumar V, Kumar RA, Mathew L. Aqueous extract of Phyllanthus amarus inhibits chromium(VI)-induced toxicity in MDA-MB-435S cells. Food Chem Toxicol. 2010;48(1):396–401. | ||
Lewinska A, Wnuk M, Slota E, Bartosz G. The nitroxide antioxidant Tempol affects metal-induced cyto- and genotoxicity in human lymphocytes in vitro. Mutat Res. 2008;649(1–2):7–14. | ||
Okabe S, Suganuma M, Hayashi M, Sueoka E, Komori A, Fujiki H. Mechanisms of growth inhibition of human lung cancer cell line, PC-9, by tea polyphenols. Jpn J Cancer Res. 1997;88(7):639–643. | ||
Ortiz-Lopez L, Marquez-Valadez B, Gomez-Sanchez A, et al. Green tea compound epigallo-catechin-3-gallate (EGCG) increases neuronal survival in adult hippocampal neurogenesis in vivo and in vitro. Neuroscience. 2016;322:208–220. | ||
Wang Y, Li M, Xu X, Song M, Tao H, Bai Y. Green tea epigallocatechin-3-gallate (EGCG) promotes neural progenitor cell proliferation and sonic hedgehog pathway activation during adult hippocampal neurogenesis. Mol Nutr Food Res. 2012;56(8):1292–1303. | ||
Suganuma M, Okabe S, Oniyama M, Tada Y, Ito H, Fujiki H. Wide distribution of [3H](−)-epigallocatechin gallate, a cancer preventive tea polyphenol, in mouse tissue. Carcinogenesis. 1998;19(10):1771–1776. | ||
Shi X, Ye J, Leonard SS, et al. Antioxidant properties of (−)-epicatechin-3-gallate and its inhibition of Cr(VI)-induced DNA damage and Cr(IV)- or TPA-stimulated NF-κB activation. Mol Cell Biochem. 2000;206(1–2):125–132. | ||
Susa N, Ueno S, Furukawa Y, Ueda J, Sugiyama M. Potent protective effect of melatonin on chromium(VI)-induced DNA single-strand breaks, cytotoxicity, and lipid peroxidation in primary cultures of rat hepatocytes. Toxicol Appl Pharmacol. 1997;144(2):377–384. | ||
Deb DD, Parimala G, Devi SS, Chakrabarti T. Role of Carum copticum seeds in modulating chromium-induced toxicity on human bronchial epithelial cells and human peripheral blood lymphocytes. Exp Toxicol Pathol. 2012;64(7–8):889–897. | ||
Sankaramanivel S, Rajaram A, Rajaram R. Zinc protects human peripheral blood lymphocytes from Cr(III)(phenanthroline)3-induced apoptosis. Toxicol Appl Pharmacol. 2010;243(3):405–419. | ||
Kart A, Koc E, Dalginli KY, Gulmez C, Sertcelik M, Atakisi O. The therapeutic role of glutathione in oxidative stress and oxidative DNA damage caused by hexavalent chromium. Biol Trace Elem Res. 2016;174(2):387–391. | ||
García-Rodríguez MC, Montano-Rodriguez AR, Altamirano-Lozano MA. Modulation of hexavalent chromium-induced genotoxic damage in peripheral blood of mice by epigallocatechin-3-gallate (EGCG) and its relationship to the apoptotic activity. J Toxicol Environ Health. 2016;79(1):28–38. | ||
García-Rodríguez MC, Nicolas-Mendez T, Montano-Rodriguez AR, Altamirano-Lozano MA. Antigenotoxic effects of (−)-epigallocatechin-3-gallate (EGCG), quercetin, and rutin on chromium trioxide-induced micronuclei in the polychromatic erythrocytes of mouse peripheral blood. J Toxicol Environ Health. 2014;77(6):324–336. | ||
Trujillo J, Chirino YI, Molina-Jijón E, Andérica-Romero AC, Tapia E, Pedraza-Chaverrí J. Renoprotective effect of the antioxidant curcumin: recent findings. Redox Biol. 2013;1:448–456. | ||
Molina-Jijón E, Tapia E, Zazueta C, et al. Curcumin prevents Cr(VI)-induced renal oxidant damage by a mitochondrial pathway. Free Radic Biol Med. 2011;51(8):1543–1557. | ||
Ganther HE. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: complexities with thioredoxin reductase. Carcinogenesis. 1999;20(9):1657–1666. | ||
Soudani N, Amara IB, Troudi A, et al. Oxidative damage induced by chromium (VI) in rat erythrocytes: protective effect of selenium. J Physiol Biochem. 2011;67(4):577–588. | ||
Krim M, Messaadia A, Maidi I, Aouacheri O, Saka S. Protective effect of ginger against toxicity induced by chromate in rats. Ann Biol Clin (Paris). 2013;71(2):165–173. | ||
Chandra AK, Chatterjee A, Ghosh R, Sarkar M. Vitamin E-supplementation protect chromium (VI)-induced spermatogenic and steroidogenic disorders in testicular tissues of rats. Food Chem Toxicol. 2010;48(3):972–979. | ||
Acharya UR, Mishra M, Mishra I, Tripathy RR. Potential role of vitamins in chromium induced spermatogenesis in Swiss mice. Environ Toxicol Pharmacol. 2004;15(2–3):53–59. | ||
Hamida FB, Troudi A, Sefi M, Boudawara T, Zeghal N. The protective effect of propylthiouracil against hepatotoxicity induced by chromium in adult mice. Toxicol Ind Health. 2016;32(2):235–245. | ||
García-Rodríguez MC, Vilches-Larrea RE, Nicolas-Mendez T, Altamirano-Lozano MA. El te verde en la quimioprevencion in vivo del dano genotoxico inducido por metales cancerigenos (cromo [VI]). [Green tea and its role on chemoprevention in vivo of genotoxic damage induced by carcinogenic metals (chromium [VI])]. Nutr Hosp. 2012;27(4):1204–1212. Spanish. | ||
García-Rodríguez MC, Carvente-Juarez MM, Altamirano-Lozano MA. Antigenotoxic and apoptotic activity of green tea polyphenol extracts on hexavalent chromium-induced DNA damage in peripheral blood of CD-1 mice: analysis with differential acridine orange/ethidium bromide staining. Oxid Med Cell Longev. 2013;2013:486419. | ||
Susa N, Ueno S, Furukawa Y. Protective effects of thiol compounds on chromate-induced toxicity in vitro and in vivo. Environ Health Perspect. 1994;102 Suppl 3:247–250. | ||
Budhwar R, Kumar S. Substantial inhibition of chromate induced DNA-protein crosslink formation in vivo by α-lipoic acid. Environ Toxicol Pharmacol. 2005;20(1):246–249. | ||
Moini H, Packer L, Saris NE. Antioxidant and prooxidant activities of α-lipoic acid and dihydrolipoic acid. Toxicol Appl Pharmacol. 2002;182(1):84–90. | ||
Schrauzer GN. Interactive effects of selenium and chromium on mammary tumor development and growth in MMTV-infected female mice and their relevance to human cancer. Biol Trace Elem Res. 2006;109(3):281–292. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.