Back to Journals » Cancer Management and Research » Volume 11
Candidate tumor suppressor gene IRF6 is involved in human breast cancer pathogenesis via modulating PI3K-regulatory subunit PIK3R2 expression
Authors Xu HF, Huang TJ, Yang Q, Xu L ![]() , Lin F, Lang YH, Hu H
, Lin F, Lang YH, Hu H ![]() , Peng LX, Meng DF
, Peng LX, Meng DF ![]() , Xie YJ, Tan L, Qian CN, Huang BJ
, Xie YJ, Tan L, Qian CN, Huang BJ
Received 26 January 2019
Accepted for publication 13 May 2019
Published 21 June 2019 Volume 2019:11 Pages 5557—5572
DOI https://doi.org/10.2147/CMAR.S203060
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Hong-Fa Xu,1,2,* Tie-Jun Huang,3,* Qin Yang,1 Liang Xu,1 Fen Lin,1 Yan-Hong Lang,1 Hao Hu,4 Li-Xia Peng,1 Dong-Fang Meng,1 Yu-Jie Xie,1 Li Tan,5 Chao-Nan Qian,1 Bi-Jun Huang1
1Department of Experimental Research, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-Sen University Cancer Center, Guangzhou 510060, People’s Republic of China; 2Zhuhai Precision Medical Center, Zhuhai People’s Hospital Affiliated to Jinan University, Zhuhai 519000, People’s Republic of China; 3Department of Nuclear Medicine, The Second People’s Hospital of Shenzhen, Shenzhen 518035, People’s Republic of China; 4Department of Traditional Chinese Medicine, The First Affiliated Hospital of Sun Yat-Sen University, Guangzhou 510060, People’s Republic of China; 5Center of Hematology, the First Affiliated Hospital of Guangzhou Medical University, Guangzhou 510230, People’s Republic of China
*These authors contributed equally to this work
Background/Aims: The tumor-suppressive functions of interferon regulatory factor 6 (IRF6) in some tumors have been preliminarily established, but its pathogenesis and underlying molecular mechanisms in breast cancer, the most common malignancy in women, remains poorly understood.
Methods: Pairs of typical breast cancer cell lines (high- and low-aggressive) in addition to 27 breast cancer tissue samples and 31 non-cancerous breast tissues were used to investigate the expression level of IRF6 and Lentivirus-mediated gain-of-function studies, short hairpin RNA-mediated loss-of-function studies in vivo and in vitro were used to validate the role of IRF6 in breast cancer. Next, we performed RNA-Seq analysis to identify the molecular mechanisms of IRF6 involved in breast cancer progression.
Results: Our findings showed that IRF6 was downregulated in highly invasive breast cancer cell lines but upregulated in poorly aggressive ones. Functional assays revealed that elevated IRF6 expression could suppress cell proliferation and tumorigenicity, and enhanced cellular chemotherapeutic sensitivity. To identify the molecular mechanisms involved, we performed a genome-wide and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis in breast cancer cells using RNA sequencing of gene expression profiles following the overexpression of IRF6. Genome-wide and KEGG analyses showed that IRF6 might mediate the PI3K-regulatory subunit PIK3R2, which in turn modulated the PI3K/AKT pathway to control breast cancer pathogenesis.
Conclusion: We provide the first evidence of the involvement of IRF6 in breast cancer pathogenesis, which was found to modulate the PI3K/AKT pathway via mediating PIK3R2; indicating that IRF6 can be targeted as a potential therapeutic treatment of breast cancer.
Keywords: breast cancer, IRF6, proliferation, RNA sequencing, PIK3R2, PI3K/AKT
Introduction
Breast cancer is the most common malignancy affecting women, with an increasing worldwide incidence, and fatal to ~600,000 women annually.1,2 It is a highly heterogeneous disease, and based on gene expression studies, breast cancer can be divided into several clinically relevant molecular subtypes: luminal A (ER+and/or PR+, HER2-) and B (ER+ and/or PR+, HER2+), HER2+ (ER+ and/or PR+, HER2+), and basal-like (ER- and/or PR-, HER2-).3,4 Basal-like breast cancer has been defined as triple negative breast cancer (TNBC) where the epithelial-to-mesenchymal transition (EMT) can be increased compared to non-TNBC tumors,5 with a frequent exhibition of P53 mutations (80%) and a loss of retinoblastoma (RB1) and BRCA1. Patients with basal-like breast carcinomas are at an increased risk for metastasis and relapse, which is incurable and the leading cause of female mortality.6–8 Numerous studies have shown that the luminal subtypes have better differentiated tumors and the best prognosis compared to other subtypes.8–10,36
IRF6 is a developmental transcription factor and a member of the IRF family of transcription factors. There are 9 members of the IRF transcription factor family (IRF1-9), but unlike the other members, IRF6 is not involved in interferon (IFN) gene expression.11–13 The human IRF6 gene maps to chromosome 1q32.2 and encodes transcriptional factor proteins.13,14 IRF6 has been found to regulate embryonic craniofacial development and epidermal maturation, which are related to the lamination, scalding and keratinization of the epidermis during embryonic development.13,14 Further, it can initiate a switch between keratinocyte proliferation and differentiation, thereby regulating the balance between the differentiation and proliferation of epidermal stem cells.15,16 Gene mutations in IRF6 can lead to popliteal pterygium syndrome and van der Woude syndrome.17,18 The syntrophicloss of terminal differentiation is due to the hyperproliferative epidermis, which causes the fusion of soft tissue in the craniofacial region.13,17,19 In the process of embryonic facial maturation, IRF6 regulates the formation of facial morphology by promoting epidermal cell apoptosis in addition to regulating cell-cycle-dependent proliferation and differentiation.20,21 In addition, deficiencies or mutations of IRF6 cause heterauxesis of the embryo, which do not occur with the other 8 members of the IFN family. As such, IRF6 is closely involved in the differentiation and proliferation of embryonic stem cells.
The evolutionarily conserved phosphatidylinositol 3-kinase (PI3K)-signal transducer, an activator of protein kinase B (AKT) and mammalian target of rapamycin (mTOR) signaling pathway mediate the cellular responses to cytokines and growth factors. Earlier studies have shown that the PI3K/AKT pathway is crucial for intracellular signaling that are involved in growth control and homeostasis maintenance in various cells and tissues. Dysregulation in the PI3K/AKT signaling is significantly associated with tumorigenesis22–24,45 and altered responses to a variety of breast cancer therapies.23,25,26
It has been reported that p85β, which is encoded by the gene PIK3R2, is the major isoform of the regulatory subunit of PI3K,27,28 and the PI3K/AKT pathway can be activated by p85β.29,30 Despite these findings, evidence regarding the association between IRF6, p85β and PI3K/AKT in human breast cancers remains inconclusive.
The downregulation of IRF6 has also been seen in squamous cell carcinomas,11 nasopharyngeal carcinomas (NPCs),31 colon and rectal cancer32 and breast cancer.33–35 Notably, elevated IRF6 protein levels can inhibit tumor malignancy and is associated with good prognosis in NPCs, but the role of IRF6 in the oncogenesis of breast cancer has not been fully elucidated.
On the basis of existing medical literature, for this present study, we hypothesized that IRF6 would show tumor-suppressive functions in breast cancer and we thereby attempted to clarify the mechanisms altering the IRF6 expression in breast cancer pathogenesis. We utilized RNA-Seq to assess the differences in the genomic expression profiles in MDA-MB-231 cells between vector control cells and those bearing overexpression of IRF6. Our results provided a comprehensive assessment of the key genetic changes in response to IRF6 overexpression at the cellular level via transcriptomics.
The findings of this study indicate that IRF6 is downregulated in highly malignant human and mouse breast cancer cell lines. Also, the modulation of the expression of IRF6 in breast cancer cell lines demonstrated the potential to alter cell proliferation ability. To confirm these observations, various in vitro and in vivo studies were conducted. RNA-Seq identified several cellular pathways modified by the overexpression of IRF6. In all, this study provides the first comprehensive evidence of the key genetic changes in response to IRF6 overexpression which has been found to regulate the PI3K/AKT pathway via downregulating PIK3R2 in the pathogenesis of human breast cancer.
Materials and methods
Cell lines and cell culture
Human and mice breast cancer cell lines were maintained with 5% CO2 at 37°C in Dulbecco modified Eagle medium (GIBCO USA) supplemented with 10% fetal bovine serum. MCF-10A was grown in DMEM/F12 (GIBCO USA) with 5% horse serum (GIBCO, 26050070), 100 U penicillin/streptomycin (MDbio, P003-10 g, S007-25 g), 20 ng/mL EGF (SIGMA, SRP3027), 20 ng/mL insulin (Beyotime, P3376), 100 ng/mL cholera toxin (MCE, HY-P1446), and 0.5 μg/mL hydrocortisone (MCE, HY-N0583). All the cells were purchased from ATCC and the culture period did not exceed two months.
Cell proliferation assays and colony-formation assays
Colorimetric MTS assays (Promega; G3582) were performed to determine the growth and viability of the breast cancer cells, as previously reported.34,35 Briefly, 800 cells/well were treated in a 96-well culture plate (Corning) in triplicate. After various times post-seeding, then harvested the parallel culture plates, and added 20 μL MTS solution (promega, G3580) to each well. Then, the solution was incubated for 3 hrs and the optical density value was determined at 490 nm.
For the colony formation experiments, 500–800 cells/well were bred in a 6-well plate (Corning). The culture medium was changed every 3days. After 10–13 days, the resulting colonies were gently washed with PBS, fixed in paraformaldehyde for 10 mins at room temperature and stained with 1% crystal violet. Only the colonies that included more than 50 cells were considered. All experiments were independently conducted at least thrice.
Quantitative real-time PCR
Gene mRNA levels were determined by real-time PCR. Total RNA of breast cancer cells were extracted using Trizol reagent (Invitrogen, 15596018, CA, USA), and were reverse-transcribed using a RevertAid First Strand cDNA Synthesis Kit (Thermo, K1622). GAPDH was used as the normalization gene. The mRNA levels of the genes were reckoned as two power values of ΔCt (the Ct of the GAPDH minus the Ct of the target gene). The sequences of the PCR primer are listed in Table S1.
Immunoblotting analysis
Western blotting was performed according to the previously described standard method.15 The primary antibodies used were anti-IRF6 (abcam, ab132057), caspase-3 (CST, 9662), cleaved-caspase3 (CST, 9661), p85β (abcam, ab137815), mTOR (abcam, ab32028), p-mTOR (abcam, ab109268), AKT1 antibody (CST, 9272), p-AKT (CST, 4058), GAPDH (proteintech, 60004–1-Ig), anti-rabbit, anti-mouse peroxidase-conjugated secondary antibodies (proteintech).
Plasmid transfection assays
Plasmid construction was performed according to previously described standard method.36 In brief, human full-length IRF6 cDNAs were cloned and linked into pcDNA3.1. MDA-MB-231 cells were treated at 1×105 cells/well in a 6-well plate, 0.5–2 μg plasmid DNA and the empty vector plasmid were transfected using X-treme GENE HP DNA transfection reagent (Roche, 6365779001) according to the manufacturer’s instructions.
Lentiviral transduction studies
Lentivirus-expressing IRF6 shRNA were produced to structure stable IRF6 knockdown cell lines. Two more effective shRNA sequences (shRNA plasmid DNAs purchased from Obio Technology; the TRC numbers for the IRF6 sh1 and sh2 plasmids are Y3667 and Y3668, respectively) were selected for the following research. Lentiviruses were generated by 293T cells with the shRNA using the X-tremeGENE HP DNA transfection reagent (Roche). Forty-eight hours after transfection, the harvested infectious lentiviruses were filtered through a 0.45 µm filter (Millipore, GLQ025), transduced with lentiviruses IRF6 shRNA and control shRNA, and selected with 2 μg/mL puromycin (SIGMA, A1113803) for 5–7 days. The efficiency of the IRF6 knockdown was analyzed using real-time PCR and immunoblotting.
Lentiviral plasmids were co-transfected with Lenti-pac HIV mix (Genecopoeia, LT003) and EndoFection mix (Genecopoeia, LT003) into 293T cells to establish the IRF6 stable overexpression MDA-MB-231 cell line. A homologous vector carrying a Flag sequence was used as the control. Cells were transfected with lentiviruses IRF6 or vector. Stable cell lines were selected in 2 μg/mL puromycin for 5–7 days and the IRF6 overexpression efficiency was confirmed by RT-qPCR and immunoblotting.
Sphere culture
Sphere culture was performed as previously described.37 The cells were seeded on a 6-well ultra-low attachment plates (800 cells/well). Serum-free DMEM-F12 medium with epidermal growth factor (20 ng/mL) was added in addition to basic fibroblast growth factor (20 ng/mL, GIBCO, PHG0264) and 2% B27 (GIBCO, 17504044). The cells were then cultured for 7 days. The experiments were performed at least thrice.
Animal assays
Female BALB/c athymic mice, age 3–4 weeks, were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. For the tumorgenicity experiment, BALB/c nude mice were randomly separated into three groups, 5 per groups. Cells were suspended in 100 µL DMEM, in 50% Matrigel (BD Biosciences, 356230), then injected subcutaneously into the right or left axillary area of each mice. Their tumor diameter was measured twice every week after apparent tumor formation. The animal experiment was terminated after 1month. All animal experiments were conducted based on the regulation of The Research Animal Resource Center of Sun Yat-Sen University.
RNA extraction, library construction, and sequencing
Total RNA from cultured cell lines was extracted using Trizol reagent (Invitrogen). RNA samples with an OD 260/280 nm ratio more than 1.8 were chosen to use for deep sequencing.
The purity, concentration, and integrity of total RNA were verified using Nanodrop, Qubit 3.0, and Agilent 2100 Bioanalyzer. The rRNA was removed, and mRNA was enriched using mRNA Capture Beads (Thermofish, 61012). Using VAHTSTM DNA Clean Beads to select and purify the cDNA library products, the RNA sequencing analysis was performed using Illumina HiSeqXTen.
Sequencing data analysis
Clean RNA-Seq data were analyzed based on the following standard protocol.38 The clean reads were acquired by removing reads including adapter and reads including unrecognized-ploy-N and poor-quality reads from the raw data. We then calculated the duplication, GC-content and Q30 level of the clean reads. Clean reads with high-quality were mapped to the NCBI (National Center for Biotechnology Information), UCSC, Ensembl reference genome sequence using Hisat2. Only with a perfect match or just one mismatch reads were further analyzed. Known genes expression were evaluated by fragments per kilobase of transcript per million fragments mapped (FPKM) during the different samples.
Differentially expressed genes (DEG) and KEGG analysis
DESeq R package was used to analyze the DEG. The DEG cutoff values were selected as the fold changevalue ≥1.5 and the corrected p-value≤0.05. T Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment was implemented using the KOBAS server.39
Statistical analysis
In this study, all data analysis were represented as mean±SD. Statistical analyses were performed using SPSS (version 17) software under the student’s t-test (two-tailed) between any two groups of data. The Pearson correlation was used to evaluate the relationship between RNA-Seq and RT-qPCR value. p<0.05 was considered significant difference.
Results
IRF6 expression is elevated in breast cancer tissues and breast cell lines
In previous studies, MDA-MB-231 and MDA-MB-468 were shown to have higher invasive and self-renewal abilities, whereas MCF7 had poorly aggressive abilities and MCF10A was a normal breast epithelial cells.36,40 To investigate the role of IRF6 in breast cancer, the expression of IRF6 was assessed in different breast cells lines using RT-qPCR, agarose gel electrophoresis, and immunoblotting analyses. Decreased IRF6 mRNA and protein levels were observed in MDA-MB-231 cells compared with the levels seen in immortalized normal breast epithelial cells, MCF10A, and poorly aggressive cells MCF7 (Figure 1A–C). Also, IRF6 mRNA expression was detectable by RT-qPCR in 27 breast cancer and 31 non-cancerous breast tissues. The expression levels of IRF6 were relatively lower in the breast cancer tissues than in the non-cancerous breast tissues (Figure 1D). Therefore, we hypothesized that IRF6 could play an important role in breast cancer progression.
 | Figure 1 IRF6 expression is elevated in breast cancer tissues and breast cell lines. (A and B) The mRNA expression levels of IRF6 in breast cell lines were determined by RT-qPCR and agarose gel electrophoresis. (C) The protein levels of IRF6 in breast cancer cell lines and non-cancerous breast cell lines were confirmed by immunoblotting analysis; GAPDH was used as a normalized control. (D) The mRNA expression levels of IRF6 in breast cancer tissues and non-cancerous breast tissues were determined by RT-qPCR. Student’s t-test, *p<0.05. |
Regulation of IRF6 expression affects growth in breast cancer cells in vitro and in vivo
To explore the role of IRF6 in breast cancer cell proliferation, we generated transiently transfected MDA-MB-231, then IRF6 mRNA and protein expression levels were confirmed by RT-qPCR and immunoblotting, respectively (Figure 2A). We observed that the transient overexpression of IRF6 in MDA-MB-231 cells resulted in decreased cell proliferation (Figure 2B) and cell colony formation (Figure 2C).
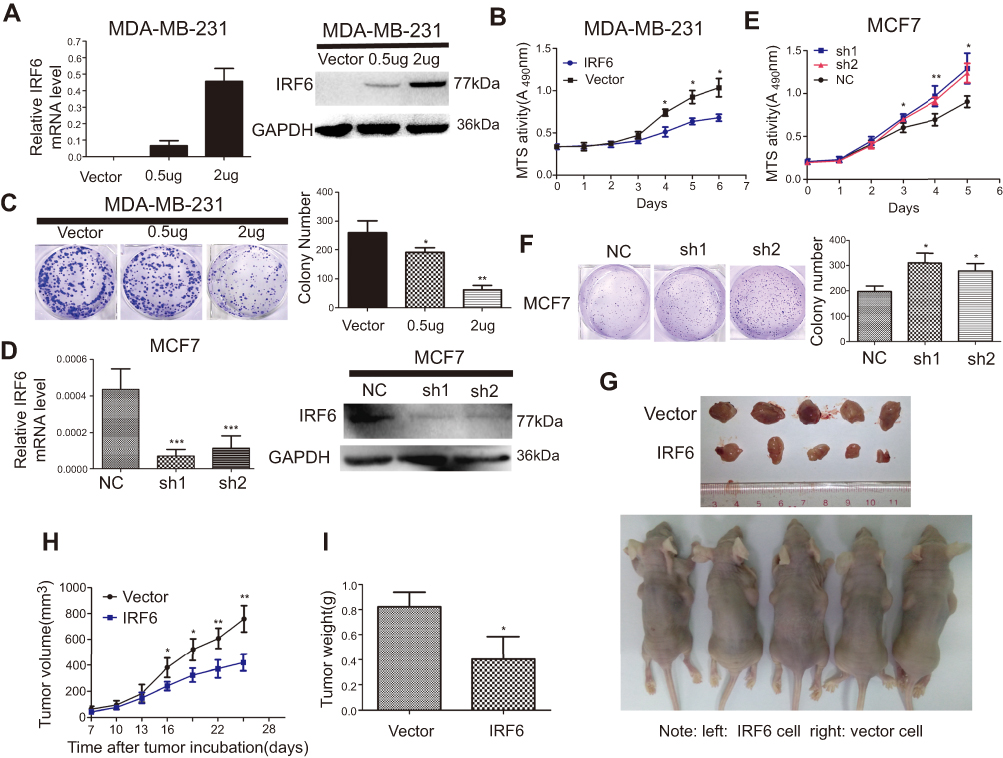 | Figure 2 Regulation of IRF6 expression affects growth in breast cancer cells in vitro and in vivo. (A and D) Transient suppression and overexpression of IRF6 in breast cancer cells were determined by RT-qPCR and immunoblotting analysis, GAPDH was used as the normalized control. (B and E) Cell proliferation was measured by the MTS assay with 1% FBS culture medium. (C and F) Colony formation assays were performed following the overexpression or suppression of IRF6; representative photos and a quantification chart of cells stained with 1% crystal violet are shown. (G) Subcutaneous injection of IRF6-overexpressing cells and vector control cells into nude mice. (H) The growth curve shows MDA-MB-231 growth suppression following IRF6 overexpression in vivo. (I) The total tumor weights were lower following injection with IRF6-overexpressing cells than injection with vector control cells. Student’s t-test, data represent the mean±SD; *p<0.05, **p<0.01, ***p<0.001. |
We then transfected MCF7 with shRNA (IRF6 sh1 and sh2) and negative control shRNA (scramble) and the IRF6 expression levels were confirmed in it using RT-qPCR and immunoblotting, respectively (Figure 2D). We observed that the IRF6 suppression resulted in increased breast cancer cell growth (Figure 2E) and cell colony formation (Figure 2F).
The tumorigenesis ability of MDA-MB-231 cells in vivo was reduced when IRF6 was overexpressed (Figure 2G). When 1×106 cells in 100 μL DMEM containing 50% Matrigel were injected into the left and right axillary areas of nude mice, the MDA-MB-231-IRF6 cells group demonstrated a slower growth rate as compared to the vector control cells (Figure 2H and I). From these data, we can conclude that in vitro and in vivo IRF6 overexpression has an inhibitory effect on breast cancer cell proliferation.
IRF6 enhances dose-dependent sensitivity to cisplatin (CDDP) and suppresses the tumorigenicity
Cisplatin is the first-line chemotherapeutic drug for breast cancer patients in clinical practice.41 When comparing the colony formation assays of MDA-MB-231 cell colonies with that of vector control cell colonies, we found that the transient overexpression of IRF6 could substantially reduce the number of cisplatin-treated (0.5, 1, 2 µg/mL) (Figure 3A). Moreover, IRF6-overexpressing cells showed lower cleaved caspase-3 levels upon cisplatin treatment but higher caspase-3 levels than the vector cells in a time-dependent manner; further confirming that IRF6 confers sensitivity to cisplatin (Figure 3B). However, opposing effects can be found in IRF6-knockdown cell lines (Figure S1A and B). These data highlight the important role of IRF6 in causing cisplatin resistance in MDA-MB-231 and MCF7 cell lines. Additionally, we found that the number of mammospheres generated by IRF6-overexpressing cell lines was significantly lower than that generated by the vector control cells (Figure 3C). To further explore the role of IRF6 in breast cancer growth, in vivo serial dilutions of MDA-MB-231 vector cells or IRF6 overexpression cells were subcutaneously injected into nude mice, all the mice were observed twice every week until the formation of palpable tumors. When 1×106 cells were injected into the nude mice, both the IRF6 overexpression cells and the vector control cells developed tumors at a similar rate (5/5). When the injected cell numbers were reduced to 5×105, 5 mice inoculated with vector cells demonstrated tumor growth while only 3 mice in the IRF6-overexpressing group exhibited tumor growth (3/5) (Figure 3D), suggesting that IRF6 expression is associated with tumorigenicity or tumor-initiating capacity in vivo.
 | Figure 3 IRF6 enhances dose-dependent sensitivity to cisplatin (CDDP) and suppresses the breast tumorigenicity. (A) The cells were seeded with different doses of cisplatin. Representative photos and the survival index of the formed colonies are presented. Student’s t-test, *p<0.05. (B) The cells treated with increasing doses of cisplatin followed by Western blotting analysis of caspase-3 and cleaved caspase-3 expression. (C) Images (left panel) and quantification of the number of oncospheres (right panel) formed in transiently transfected cell lines in DMEM/F12 medium with a 20 ng/mL concentration of FGF and EGF for 6–7 days. Student’s t-test, **p<0.01. (D) Tumorigenesis abilities: stabled IRF6-overexpressing cells in five cell concentrations were injected subcutaneously into the right and left armpits of nude mice and observed for 20 days. The tumor formation rates in different groups are shown and the right upper panel shows photographs of their respective isolated tumors. |
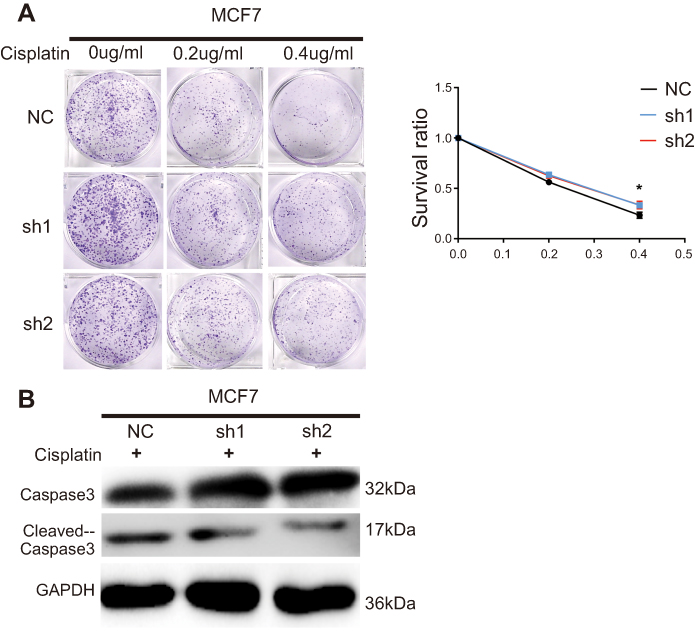 | Figure S1 Knockdown of IRF6 reduces the dose-dependent sensitivity to cisplatin in breast cancer. (A) IRF6-knockdown cells and scramble cells were seeded with different doses of cisplatin. Representative photos and the survival index of the formed colonies were presented. Student’s t-test, *p<0.05. (B) The cells were treated with 0.2 μg/mL cisplatin followed by Western blotting analysis of caspase-3 and cleaved caspase-3 expression. |
RNA-Seq and KEGG analysis reveals IRF6 downregulates the expression of the PIK3R2 gene, a PI3K regulatory subunit that is comprehensively involved in many cancer-related signaling pathways
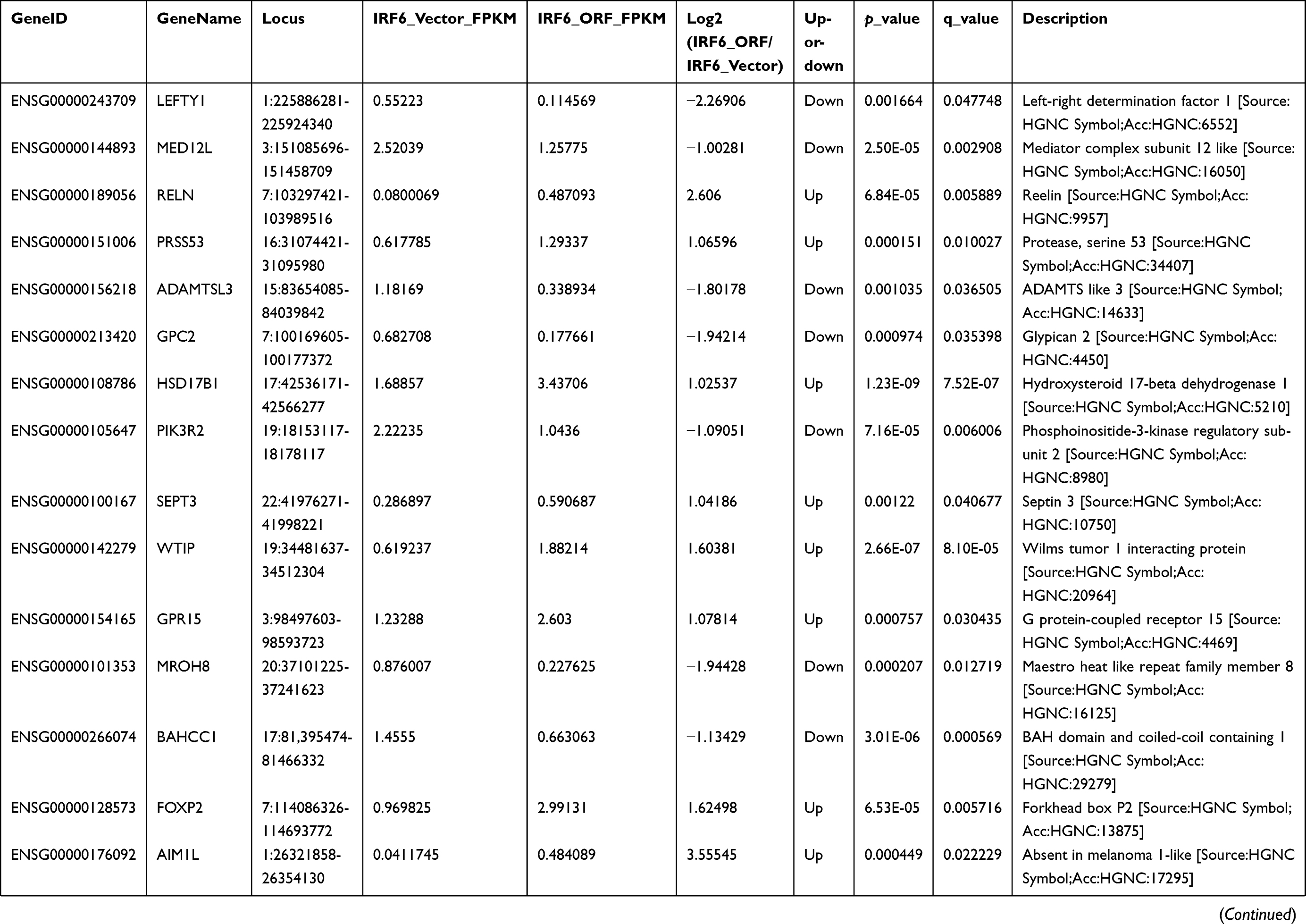
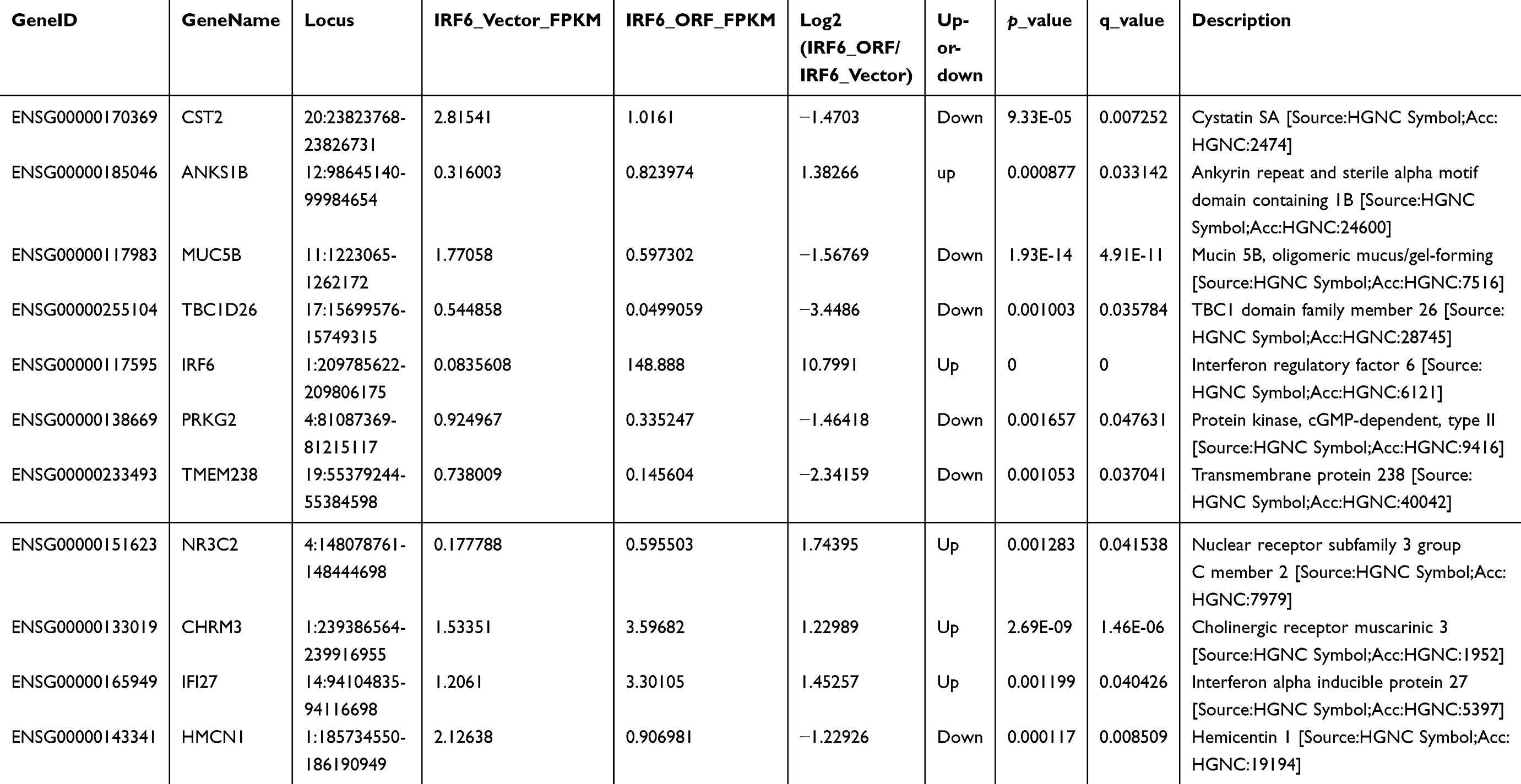
We assessed the expression and evaluated the significance of DEGs between vector control and IRF6-overexpressing cells. A total of 642 DEGs were identified between vector and IRF6-overexpressing cell line using non-supervised clustering analysis; as illustrated by the expression patterns in Figure 4A, and 26 were found to be significantly dysregulated genes with log2 ratios >1 and q-values<0.05 were identified (Figure 4B). The descriptions of these genes are shown in Table S2. Notably, the red outline indicates the gene PIK3R2 (p85β), which is a PI3K regulatory subunit. The PI3K pathway can regulate various biological activities, including cellular proliferation and survival.42 PI3K-mediated production can trigger a signaling pathway cascade that leads to the activation of AKT and several of its downstream targets. To calculate the RNA-Seq data reliability as well as further validate the DEG patterns, 5 genes were randomly selected and assessed using RT-qPCR (Figure 4C). Pearson’s correlation analysis demonstrated that the RNA-Seq data were positively correlated with RT-qPCR data (Pearson’s r2=0.9774, P=0.0015), suggesting the high reliability of the RNA-Seq analysis. At the same time, we also found that in the top 20 KEGG pathways of the DEGs that were significantly enriched, PIK3R2 participated in most of the cancer-related and biological activity signaling pathways, such as regulation of actin cytoskeleton, cholinergic synapse, toll-like receptor signaling pathway, pancreatic cancer, insulin signaling pathway, and so on. Especially the phosphatidylinositol signaling system, which also contains the PI3K/Akt signaling pathway. (Figure 4D). These results are similar to those of previous study,11 implying that PIK3R2 is a probable checkpoint for the activation of various malignancy signaling pathways. Hence, here, we mainly analyzed the potential contributions of the IRF6-mediated PIK3R2 in breast cancer.
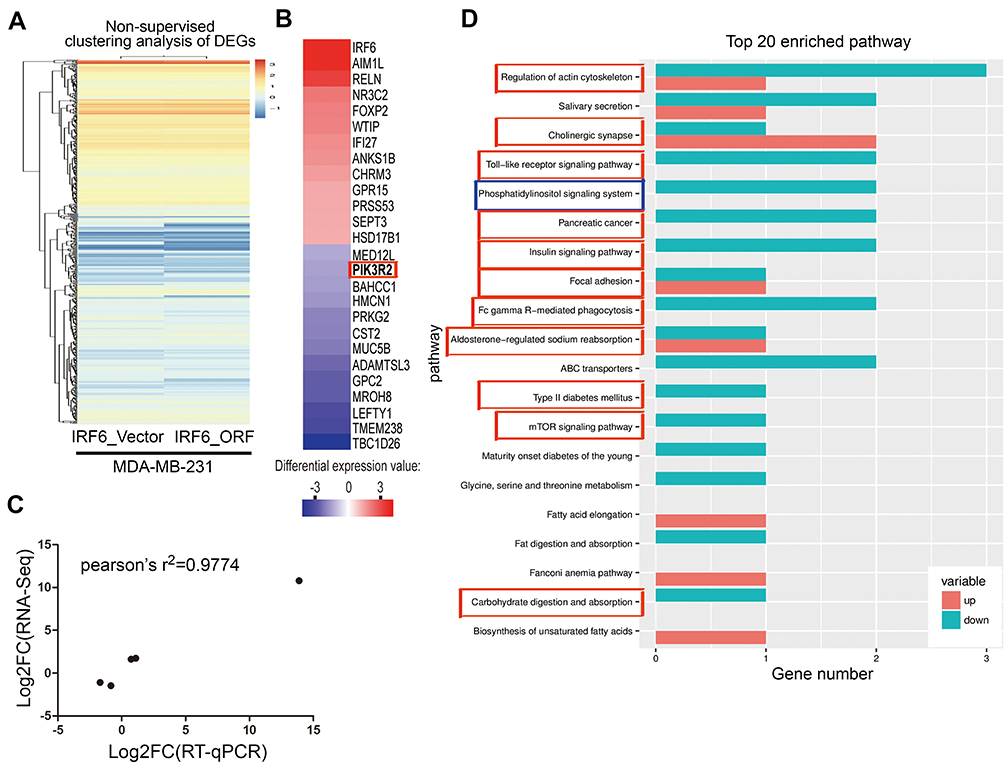 | Figure 4 RNA-Seq and KEGG analysis revealed IRF6 downregulates the expression of the PIK3R2 gene, a PI3K regulatory subunit that is comprehensively involved in many cancer-related signaling pathways. (A) Non-supervised clustering of all DEGs. The heatmap represents the mRNA K-means clustering of log10-transformed expression value differences between IRF6-overexpressing and vector cells. Red represents increased expression, and blue represents reduced expression. (B) Gene expression profiling was performed on IRF6overexpressing and vector cells lines. Here presentation of part of the distinctly upregulated and down-regulated genes in IRF6-overexpressing cell lines relative to the vector cell lines. Red, the gene has increased expression in IRF6-overexpressing cell lines compared to the vector cell lines. Blue, the gene has decreased expression. The red box indicates the gene PIK3R2 (p85β). (C) A scatter diagram including RNA-Seq and RT-qPCR data from correlation analysis including 5 genes: IRF6, PIK3R2, NR3C2, CST2, FOXP2. Pearson’s analysis, r2=−0.9774; p=0.0015. (D) Pathway analysis showed that 20 pathways were dramatically enriched; the numbers for each pathway were based on Fisher’s exact test, indicating the fold enrichment. The red outline box indicates that the gene PIK3R2 (p85β) participated in the corresponding pathway, such as regulation of actin cytoskeleton, cholinergic synapse, toll-like receptor signaling pathway, and phosphatidylinositol signaling system. The blue outline box indicates the phosphatidylinositol signaling system, which contains the PI3K/Akt signaling pathway. |
IRF6-mediated PIK3R2 expression modulates PI3K/AKT pathway activity in breast cancer pathogenesis
PIk3R2 mRNA expression was detected in 31 non-cancerous and 27 breast cancer patient tissue samples by RT-qPCR. The expression level of PIK3R2 was remarkably higher in the breast cancer tissues than in the non-cancerous breast tissue samples (Figure 5A), and there was an inverse relationship between IRF6 and PIK3R2 levels in the primary breast cancer tissues (r=−0.4228) (Figure 5B). At the same time, we also demonstrated that the MDA-MB-231 cell line had higher endogenous expression levels of p-AKT and CD44 proteins than the MCF7 cell line (Figure 5C), further showing that the AKT signaling pathway was activated and in MDA-MB-231 cell lines compared to the counterpart cell line. Subsequently, we measured the protein expression levels of p85β, p-AKT, AKT, p-mTOR, and mTOR using Western blot analysis of cell lines with the transient overexpression and suppression of IRF6 (Figure 5D). MDA-MB-231 cells transfected with IRF6 plasmids at different concentrations showed substantially decreased expression of p85β, p-AKT, and p-mTOR in a dose-dependent manner. Meanwhile, we found that the protein expression levels of p85β, p-AKT, and p-mTOR were increased in IRF6 knockdown cells. These data collectively demonstrate an inverse relationship between IRF6 and p85β, p-AKT and p-mTOR. Therefore, we hypothesized that IRF6 might mediate PIK3R2 expression to regulate the PI3K/AKT pathway, subsequently triggering a signaling cascade leading to the activation of downstream targets. Thus, we used increasing doses of an AKT inhibitor (LY294002) that is a cell-permeable, potent and a specific PI3K inhibitor to act on the ATP-binding site of the enzyme (Figure 5E). LY294002 was found to also inhibit non-homologous end-joining repair through the inhibition of DNA-PK catalytic subunits.43 MDA-MB-231 cells were treated with LY294002, and we found that the number of the mammospheres generated were lower than those of the control group (Figure 5F and G), implying that the suppression of endogenous AKT activity can block tumor self-renewal.
 | Figure 5 IRF6-mediated PIK3R2 expression modulates PI3K/AKT pathway to involve in breast cancer pathogenesis. (A) The mRNA expression levels of PIK3R2 in breast cancer and non-cancerous breast cancer tissues were determined by RT-qPCR. (B) PIK3R2mRNA levels were negatively correlated with IRF6 expression in breast cancer tissues as assessed by RT-qPCR (Pearson’s analysis, r=−0.4228; p<0.05). (C) Protein expression levels of p-AKT, AKT, and CD44 in MCF7 and MDA-MB-231 cells were assessed by immunoblotting analysis. GAPDH was used as a reference control. (D) Effect of IRF6 on the expression level of proteins involved in p85β-activated AKT signaling cascades. For the immunoblot analysis of IRF6, p85β, p-AKT, AKT, p-mTOR and mTOR in transient overexpression or suppression of IRF6 cell lines. GAPDH was used as a loading control. (E–G) MDA-MB-231 cells were supplemented with increasing doses of an AKT inhibitor (LY294002) and calculated the number of spheres generated between cells seeded with LY294002 and DMSO. (H and I) IRF6 knockdown cells (sh1) and shRNA control cells treated with DMSO vehicle control and increasing doses of an AKT inhibitor (LY294002); compared to control cells, sh1 cells showed increased sensitivity to LY294002. Data represent the mean±SD, *p<0.05,**p<0.01. |
To further confirm whether the effect of PI3K/AKT on cancer progression is through the IRF6-mediated pathway, we treated MCF7 cells with suppression of IRF6 and negative control shRNA (scramble) with increasing doses of LY294002. Specifically, we found that the percentage of spheroid formations was inhibited more obviously in the IRF6-suppression group compared with negative control counterparts (Figure 5H and I). These results further demonstrated that the activation of the PI3K/AKT signaling pathway via the upregulation of p85β by the inhibition of IRF6 expression could enhance a dose-dependent sensitivity to LY294002.
Discussion
The transcription functions of IRFs family are activated by the phosphorylation of regulatory serine/threonine residues and acts to promote protein–protein interactions involving in the IAD and CTD.44 It is well known that phosphorylation mainly occurs in serine-rich region of the C-terminus, which plays an auto-inhibitory effect in a non-phosphorylated state by blocking the protein–protein interactions.45 These phosphorylation events trigger the re-organization of the CTD such that it no longer serves as an autoinhibitory function.46 The precise study has demonstrated the IRF6 C-terminal serine-rich region can cause a similar auto-inhibitory effect, and the IRF6 IAD is both necessary and sufficient for an interaction with maspin and that the inclusion of the C-terminal serine-rich region abrogates this interaction.33 Additionally, M.Q. Kwa et al have also shown that the phosphorylation of Ser413 and Ser424 induces IRF6 activation through RIPK4.47 There was one study which reported that nonsense mutations (eg, p.Ser424X) may abolish RIPK4-mediated IRF6 activation in Van der Woude syndrome.48 Furthermore, these nonsense mutations not only promote IRF6 degradation but also prevent the activation of IRF6 by RIPK4.47 However, the transactivator functions of IRF6 and the initiating signal for IRF6 activation still need to be further investigated.
In this study, we confirmed that IRF6 is downregulated in highly invasive breast cancer cells. Conversely, IRF6 is upregulated in poorly aggressive breast cancer cells. Functional studies revealed that elevated IRF6 expression suppressed cell proliferation and the tumorigenicity, enhanced cell chemotherapeutic sensitivity. Therefore, our findings suggest that IRF6 is a viable therapeutic target in breast cancer.
The PI3K/AKT pathway has been described as having an important effect on tumor development.49 PIK3R2 is a PI3K regulatory subunit, and PIK3R2 genes encode p85β regulatory subunits. PI3K consists of p110α/p85β, and p85β mediates p110 translocation to receptors at the cell membrane;50 augmented p110α/p85β levels lead to maximal PI3K activation, showing that p85β possibly modulates p110α activation in a distinct manner.50,51 p85β expression augments PIP3 levels and activates the PI3K effector PKB1/2 in the absence of a stimulus, triggering downstream AKT signaling cascades. Nonetheless, in the absence of a stimulus, p85β alone and p110α/p85β together induce moderate PI3K activation, suggesting that p85β regulates malignancy progression.50
In the present study, the suppression of IRF6 resulted in the upregulation of PIK3R2, thus enhance the PI3K/AKT signaling activation, which was always accompanied by increased levels of mTOR. However, the results from the overexpression of IRF6 showed that these changes were reversible. More importantly, using RNA-Seq analysis, we found that PIK3R2 participated in most of the cancer-related and biological activity signaling pathways, such as the phosphatidylinositol signaling pathway and mTOR signaling pathway, strongly implying that PIK3R2 is a checkpoint in the activation of various malignancy signaling pathways. Additionally, the ability of these cells to form spheres was notably decreased when IRF6-knockdown cells were treated with the PI3K/AKT-specific inhibitor LY294002, further demonstrating that the PI3K/AKT signaling pathway plays a key role in the process of tumor self-renewal. However, until now there has been no study conducted that illuminates the mechanisms of IRF6 involved in breast cancer development.
In this study, we demonstrated for the first time that IRF6 may play an important role in breast cancer pathogenesis via the mediation of PIK3R2. The novel findings are summarized in Figure 6, which unveil a possible relationship between IRF6 and PIK3R2. PIK3R2 function as an important role in the activation of PI3K/AKT pathway and once upregulated the IRF6 expression releases its inhibition of the PIK3R2 element and consequently decreases the PIK3R2 levels, and subsequently regulate the activation of the PI3K/AKT pathway as involvement in breast cancer pathogenesis, such as tumor proliferation, chemoresistance, and so on. However, whether IRF6 directly binds to PIK3R2 remains to be elucidated, and the mechanisms of how IRF6-mediates the inverse regulation of PIK3R2 requires further investigation.
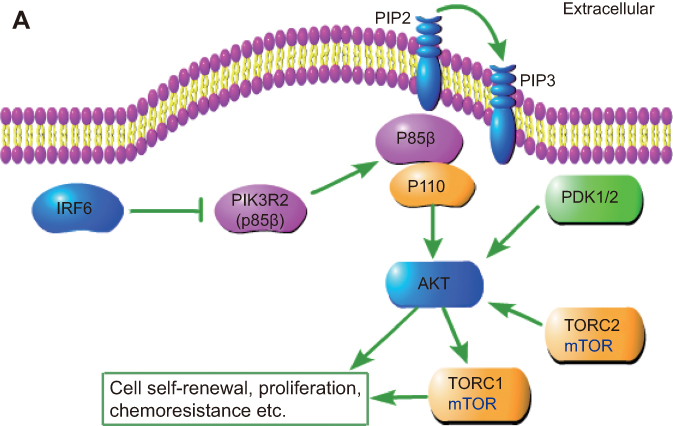 | Figure 6 Schematic depiction. Schematic representation of IRF6-mediated PIK3R2 expression modulation of the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway. |
Conclusion
We have shown that the re-expression of IRF6 can restrain PIK3R2 expression and regulate the PI3K/AKT pathway to involve breast cancer pathogenesis. Collectively, these experiments on IRF6 levels revealed an important role of IRF6 in the suppressing of the progression of breast cancer and could, therefore, serve as a potential therapeutic target for breast cancer treatment.
Ethics approval and consent to participate
All animal experiments were conducted based on the regulation of The Research Animal Resource Center of Sun Yat-Sen University. All human tissue samples were obtained with patient consent and the approval of the Institutional Clinical Ethics Review Board at Sun Yat-Sen University Cancer Center.
Availability of data and materials
For all manuscripts, especially those containing data from the Sun Yat-sen University Cancer Center it is recommended that the authors to deposit the data in the Research Data Deposit at the following website:
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (grant no. 81773162, 81572901, 31170151, 81672872, 81472386), the Science and Technology Planning Project of Guangdong Province, China (grant no. 2014A020209024, 2014B020212017, 2014B050504004, 2015B050501005), the Natural Science Foundation of Guangdong Province, China (2017A030313866, 2016A030311011), and the National Key Research and Development Program of China (No. 2016YFC0902000).
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Consent for publication
The manuscript does not contain any individual person’s data in any form.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Feng RM, Zong YN, Cao SM, Xu RH. Current cancer situation in China: good or bad news from the 2018 global cancer statistics? Cancer Commun (Lond). 2019;39(1):22. doi:10.1186/s40880-019-0368-6
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Podo F, Buydens LM, Degani H, et al. Triple negative breast cancer: present challenges and new perspectives. Mol Oncol. 2010;4:209–229. doi:10.1016/j.molonc.2010.04.006
4. Cianfrocca M, Gradishar W. New molecular classifications of breast cancer. CA Cancer J Clin. 2009;59:303–313. doi:10.3322/caac.20029
5. Karihtala P, Auvinen P, Kauppila S, Haapasaari KM, Jukkola-Vuorinen A, Soini Y. Vimentin, zeb1 and Sip1 are up-regulated in triple-negative and basal-like breast cancers: association with an aggressive tumour phenotype. Breast Cancer Res Treat. 2013;138:81–90.
6. Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann Oncol. 2012;23:2223–2234. doi:10.1093/annonc/mds067
7. Reeder-Hayes KE, Carey LA, Sikov WM. Clinical trials in triple negative breast cancer. Breast Dis. 2010;32:123–136. doi:10.3233/BD-2010-0310
8. Toft DJ, Cryns VL. Minireview. Basal-like breast cancer: from molecular profiles to targeted therapies. Mol Endocrinol. 2011;25:199–211. doi:10.1210/me.2010-0389
9. Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi:10.1038/35021093
10. Zuo T, Zeng H, Li H, et al. The influence of stage at diagnosis and molecular subtype on breast cancer patient survival: a hospital-based multi-center study. Chin J Cancer. 2017;36(1):84. doi:10.1186/s40880-017-0250-3
11. Botti E, Spallone G, Moretti F, et al. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc Natl Acad Sci USA. 2011;108:13710–13715. doi:10.1073/pnas.1110931108
12. Savitsky D, Tamura T, Yanai H, Taniguchi T. Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol Immunother. 2010;59:489–510. doi:10.1007/s00262-009-0804-6
13. Ingraham CR, Kinoshita A, Kondo S, et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat Genet. 2006;38:1335–1340. doi:10.1038/ng1903
14. Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–655. doi:10.1146/annurev.immunol.19.1.623
15. Richardson RJ, Dixon J, Malhotra S, et al. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet. 2006;38:1329–1334. doi:10.1038/ng1894
16. Richardson RJ, Hammond NL, Coulombe PA, et al. Periderm prevents pathological epithelial adhesions during embryogenesis. J Clin Invest. 2014;124:3891–3900. doi:10.1172/JCI71946
17. Kondo S, Schutte BC, Richardson RJ, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi:10.1038/ng985
18. Little HJ, Rorick NK, Su LI, et al. Missense mutations that cause Van der Woude syndrome and popliteal pterygium syndrome affect the DNA-binding and transcriptional activation functions of IRF6. Hum Mol Genet. 2009;18:535–545. doi:10.1093/hmg/ddn381
19. Richardson RJ, Dixon J, Jiang R, Dixon MJ. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum Mol Genet. 2009;18:2632–2642. doi:10.1093/hmg/ddp201
20. Bailey CM, Abbott DE, Margaryan NV, Khalkhali-Ellis Z, Hendrix MJ. Interferon regulatory factor 6 promotes cell cycle arrest and is regulated by the proteasome in a cell cycle-dependent manner. Mol Cell Biol. 2008;28:2235–2243. doi:10.1128/MCB.01866-07
21. Ferretti E, Li B, Zewdu R, et al. A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev Cell. 2011;21:627–641. doi:10.1016/j.devcel.2011.08.005
22. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–6091. doi:10.1158/0008-5472.CAN-07-6854
23. Karlsson E, Pérez-Tenorio G, Amin R, et al. The mTOR effectors 4EBP1 and S6K2 are frequently coexpressed, and associated with a poor prognosis and endocrine resistance in breast cancer: a retrospective study including patients from the randomised Stockholm tamoxifen trials. Breast Cancer Res. 2013;15:R96. doi:10.1186/bcr3557
24. Chandarlapaty S, Sakr RA, Giri D, et al. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012;18:6784–6791. doi:10.1158/1078-0432.CCR-12-1785
25. Barone I, Cui Y, Herynk MH, et al. Expression of the K303R estrogen receptor-alpha breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway. Cancer Res. 2009;69:4724–4732. doi:10.1158/0008-5472.CAN-08-3660
26. Razis E, Bobos M, Kotoula V, et al. Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Res Treat. 2011;128:447–456. doi:10.1007/s10549-011-1572-5
27. Park SW, Zhou Y, Lee J, et al. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat Med. 2010;16:429–437. doi:10.1038/nm.2099
28. Ito Y, Hart JR, Ueno L, Vogt PK. Oncogenic activity of the regulatory subunit p85beta of phosphatidylinositol 3-kinase (PI3K). Proc Natl Acad Sci USA. 2014;111:16826–16829. doi:10.1073/pnas.1420281111
29. Hale BG, Jackson D, Chen YH, Lamb RA, Randall RE. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc Natl Acad Sci USA. 2006;103:14194–14199. doi:10.1073/pnas.0606109103
30. Li W, Wang G, Zhang H, et al. Inability of NS1 protein from an H5N1 influenza virus to activate PI3K/Akt signaling pathway correlates to the enhanced virus replication upon PI3K inhibition. Vet Res. 2012;43:36. doi:10.1186/1297-9716-43-36
31. Xu L, Huang TJ, Hu H, et al. The developmental transcription factor IRF6 attenuates ABCG2 gene expression and distinctively reverses stemness phenotype in nasoparyngeal carcinoma. Cancer Lett. 2018;431:230–243. doi:10.1016/j.canlet.2017.10.016
32. Slattery ML, Lundgreen A, Bondurant KL, Wolff RK. Interferon-signaling pathway: associations with colon and rectal cancer risk and subsequent survival. Carcinogenesis. 2011;32:1660–1667. doi:10.1093/carcin/bgr189
33. Bailey CM, Khalkhali-Ellis Z, Kondo S, et al. Mammary serine protease inhibitor (Maspin) binds directly to interferon regulatory factor 6: identification of a novel serpin partnership. J Biol Chem. 2005;280:34210–34217. doi:10.1074/jbc.M503523200
34. Zengin T, Ekinci B, Kucukkose C, Yalcin-Ozuysal O. IRF6 is involved in the regulation of cell proliferation and transformation in MCF10A cells downstream of notch signaling. PLoS One. 2015;10:e0132757. doi:10.1371/journal.pone.0132757
35. Bailey CM, Margaryan NV, Abbott DE, et al. Temporal and spatial expression patterns for the tumor suppressor Maspin and its binding partner interferon regulatory factor 6 during breast development. Dev Growth Differ. 2009;51:473–481. doi:10.1111/j.1440-169X.2009.01110.x
36. Subik K, Lee JF, Baxter L, et al. The expression patterns of eR, pR, HeR2, cK5/6, eGFR, Ki-67 and AR by Immunohistochemical analysis in breast cancer cell lines. Breast Cancer. 2010;4:35–41.
37. Xie P, Yang J-P, Cao Y, et al. Promoting tumorigenesis in nasopharyngeal carcinoma, NEDD8 serves as a potential theranostic target. Cell Death Dis. 2017;8(6):e2834. doi:10.1038/cddis.2017.518
38. Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi:10.1038/nprot.2012.016
39. Xie C, Mao X, Huang J, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(Web Server issue):W316–W322. doi:10.1093/nar/gkr483
40. Croker AK, Goodale D, Chu J, et al. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. 2009;13:2236–2252. doi:10.1111/j.1582-4934.2008.00455.x
41. He K, Wang X, Guan X, et al. Vinorelbine plus gemcitabine or cisplatin as first-line treatment of HER2-negative advanced breast cancer. Anticancer Res. 2017;37:5647–5653. doi:10.21873/anticanres.12000
42. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi:10.1126/science.296.5573.1655
43. Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64:4394–4399. doi:10.1158/0008-5472.CAN-04-0343
44. Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi:10.1146/annurev.immunol.26.021607.090400
45. Eroshkin A, Mushegian A. Conserved transactivation domain shared by interferon regulatory factors and Smad morphogens. J Mol Med (Berl). 1999;77(5):403–405.
46. Qin BY, Liu C, Lam SS, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. 2003;10(11):913–921. doi:10.1038/nsb1002
47. Kwa MQ, Huynh J, Aw J, et al. Receptor-interacting protein kinase 4 and interferon regulatory factor 6 function as a signaling axis to regulate keratinocyte differentiation. J Biol Chem. 2014;289(45):31077–31087. doi:10.1074/jbc.M114.589382
48. de Lima RL, Hoper SA, Ghassibe M, et al. Prevalence and nonrandom distribution of exonic mutations in interferon regulatory factor 6 in 307 families with Van der Woude syndrome and 37 families with popliteal pterygium syndrome. Genet Med. 2009;11(4):241–247. doi:10.1097/GIM.0b013e318197a49a
49. Qian CN, Furge KA, Knol J, et al. Activation of the PI3K/AKT pathway induces urothelial carcinoma of the renal pelvis: identification in human tumors and confirmation in animal models. Cancer Res. 2009;69:8256–8264. doi:10.1158/0008-5472.CAN-09-1689
50. Cortés I, Sánchez-Ruíz J, Zuluaga S, et al. p85β phosphoinositide 3-kinase subunit regulates tumor progression. Pnas. 2012;109(28):11318–11323. doi:10.1073/pnas.1118138109
51. Alcázar I, Cortés I, Zaballos A, et al. p85β phosphoinositide 3-kinase regulates CD28 coreceptor function. Blood. 2009;113:3198–3208. doi:10.1182/blood-2008-04-152942
Supplementary materials
 |  |  |  | Table S1 The sequences of the PCR primer are listed |
| Table S2 The description of parts of DEGs |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.