Back to Journals » Breast Cancer: Targets and Therapy » Volume 6
Candidate prognostic markers in breast cancer: focus on extracellular proteases and their inhibitors
Received 7 April 2014
Accepted for publication 12 May 2014
Published 3 July 2014 Volume 2014:6 Pages 81—91
DOI https://doi.org/10.2147/BCTT.S46020
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
David M Roy,1 Logan A Walsh2
1Weill Cornell/Rockefeller/Sloan-Kettering Tri-Institutional MD-PhD Program, New York, NY, USA; 2Human Oncology and Pathogenesis Program, Memorial Sloan-Kettering Cancer Center, New York, NY, USA
Abstract: The extracellular matrix (ECM) is the complex network of proteins that surrounds cells in multicellular organisms. Due to its diverse nature and composition, the ECM has a multifaceted role in both normal tissue homeostasis and pathophysiology. It provides structural support, segregates tissues from one another, and regulates intercellular communication. Furthermore, the ECM sequesters a wide range of growth factors and cytokines that may be released upon specific and well-coordinated cues. Regulation of the ECM is performed by the extracellular proteases, which are tasked with cleaving and remodeling this intricate and diverse protein matrix. Accordingly, extracellular proteases are differentially expressed in various tissue types and in many diseases such as cancer. In fact, metastatic dissemination of tumor cells requires degradation of extracellular matrices by several families of proteases, including metalloproteinases and serine proteases, among others. Extracellular proteases are emerging as strong candidate cancer biomarkers for aiding and predicting patient outcome. Not surprisingly, inhibition of these protumorigenic enzymes in animal models of metastasis has shown impressive therapeutic effects. As such, many of these proteolytic inhibitors are currently in various phases of clinical investigation. In addition to direct approaches, aberrant expression of extracellular proteases in disease states may also facilitate the selective delivery of other therapeutic or imaging agents. Herein, we outline extracellular proteases that are either bona fide or probable prognostic markers in breast cancer. Furthermore, using existing patient data and multiple robust statistical analyses, we highlight several extracellular proteases and associated inhibitors (eg, uPA, ADAMs, MMPs, TIMPs, RECK) that hold the greatest potential as clinical biomarkers. With the recent advances in high-throughput technology and targeted therapies, the incorporation of extracellular protease status in breast cancer patient management may have a profound effect on improving outcomes in this deadly disease.
Keywords: uPA, RECK, ADAMs, MMPs, TIMPs, ECM
Introduction
Extracellular proteases are complex and heterogeneous enzymes that play a key role in many pathophysiologic processes. Included in this group are metalloproteinases such as matrix metalloproteinases (MMPs) and serine proteases such as plasmin, among others.1 These proteins have the capacity to completely remodel the extracellular matrix (ECM) and can therefore alter a variety of biologic processes, including angiogenesis, growth factor bioavailability, cytokine modulation, receptor shedding, cell migration, proliferation, invasion, and apoptosis.2,3 Not surprisingly, these proteases and their protein inhibitors have been implicated in many diseases, including cancer. Specifically, several extracellular proteases have been shown to alter tumor aggressiveness and patient response to therapy.4–6 Herein, we summarize the most recent and relevant literature detailing the role of extracellular proteases and their inhibitors as prognostic indicators and putative therapeutic targets in breast cancer.
Materials and methods
Tumors analyzed in this study were from a previously published dataset of over 2,000 woman diagnosed with breast cancer.7 Expression and clinical data were downloaded from Oncomine™ (http://www.oncomine.org). Nonparametric analyses were performed since expression values were not normally distributed. Mann–Whitney U test was used to compare gene expression values with clinicopathological features (P<0.05= significant). For patient outcomes, data were analyzed in two ways. First, gene expression values were divided into “high” and “low” expression groups based on median values in all samples and Kaplan–Meier tests were used to determine significance (log-rank). Second, gene expression values were treated as a continuous variable and subjected to a univariate Cox regression analysis and Wald test (P<0.01= significant). All statistics were carried out using SPSS software (v 20; IBM Corporation, Armonk, NY, USA).
The extracellular matrix (ECM)
The ECM is the complex network of proteins that surrounds and supports cells in multicellular organisms. It is composed of three main types of proteins with distinct roles: structural proteins (eg, collagen, elastin), specialized glycoproteins (eg, fibronectin), and proteoglycans (eg, syndecans).8 Initially, it was believed that the sole function of the ECM was to provide tissues with structural support.9 More recently, however, it has been shown that the ECM plays a more active – and critical – role in many fundamental cellular processes such as cell growth, proliferation, migration, and differentiation.10 In fact, it is the intrinsic diversity of the ECM that underlies its pleiotropic role as a structural scaffold, cytokine reservoir, and regulator of developmental and physiologic signaling.
Although the exact protein composition can vary considerably due to unique tissue architecture and function, the major protein component of the ECM is collagen. In fact, collagen is the most abundant protein across the animal kingdom, serving to provide tissues with strength and resilience.11 Accordingly, there are many diseases that stem directly from defects in collagen production and homeostasis, either from underlying genetic alterations and/or abnormal collagen processing (eg, osteogenesis imperfecta, Alport syndrome, Ehlers–Danlos syndrome).12 In addition, the integrity of collagen in the ECM plays a key role in cancer – the active degradation of type IV collagen by extracellular proteases facilitates tumor cell invasion through the basement membrane.13 In fact, this hallmark histopathologic feature of epithelial cancers is what defines the transition from carcinoma in situ to invasive carcinoma, carrying significant prognostic significance in breast cancer.14
Specialized glycoproteins are important for proper cell–ECM adhesion. For example, cells can bind to fibronectin via integrin receptors to form focal adhesions, which facilitate cellular migration.15 As with other ECM components, improper remodeling of glycoproteins is associated with pathological processes including tumor growth and metastasis.16
In addition to direct structural and cell adhesion roles, the ECM also sequesters and moderates the passage of many cytokines and growth factors between cells. This process is governed by proteoglycans, of which syndecans are a major constituent. Syndecans are cell surface proteoglycans that are ubiquitously expressed and contain a heparan sulfate side chain, which allows interactions with heparan-binding proteins, including cell surface receptors.17 Cleavage of these proteoglycans and the subsequent release of cytokines can stimulate cellular signaling cascades.18 In fact, one particularly overlooked function of the ECM is its role in facilitating intercellular communication. The extensive array of cytokines and growth factors sequestered by the ECM can powerfully regulate cell behavior through activation of signaling cascades. Accordingly, the importance of extracellular proteases, the enzymes tasked with maintaining homeostasis within the ECM microenvironment, cannot be overstated.19 Considering the many structural and functional roles played by the ECM, it is evident that its cleavage and remodeling must be highly regulated. Not surprisingly, altered extracellular protease activity in cancer is common and is increasingly linked to significant changes in patient outcomes.20
The urokinase plasminogen activator (uPA) system
Several biomarkers for breast cancer have been currently validated at the highest level of evidence (LOE-1) and are already utilized in the clinical setting to guide management strategy.21 Patient prognosis and treatment efficacy are strongly correlated to these biomarkers, which include estrogen receptor (ER), progesterone receptor (PR), and receptor tyrosine-protein kinase erbB-2 (HER2).22 In fact, testing the status of these proteins via tissue-based assay is now the standard of care in all newly diagnosed breast cancer patients. Despite this, there exists a continued need for new biomarkers in breast cancer in order to better stratify patients for therapeutic regimens and improve outcomes.23,24
Two components of the uPA system hold exciting potential as biomarkers in breast cancer: the serine protease uPA and plasminogen activator inhibitor-1 (PAI-1), a serine protease inhibitor (serpin).25 Although they are not yet in widespread clinical use, they have already been validated as biomarkers for clinical breast cancer management at the highest level;26,27 therefore, they could be an important addition to existing clinical prognostic tools used in breast cancer (Table 1).28,29
 | Table 1 Clinically utilized biomarkers in breast cancer |
The uPA system consists of the serine protease uPA, its membrane-anchored receptor uPAR, and the serpins PAI-1 and PAI-2.30 The uPA system is causally involved in multiple steps of breast cancer progression, including ECM remodeling, increased cell proliferation and migration, and modulating cell adhesion.31 Therefore, it is not surprising that uPA in primary breast cancer is independently associated with adverse outcome.32 Interestingly, and somewhat paradoxically, high levels of PAI-1 (an inhibitor of plasminogen activation) also correlate with poor prognosis in breast cancer patients.33 Nevertheless, the prognostic value of uPA/PAI-1 in axillary node-negative breast cancer patients has been validated in two independent level 1 evidence studies – a prospective randomized trial and a pooled analysis of primary data.34
Less is known about the predictive clinical value of uPA system components in regard to systemic therapy for recurrent breast cancer. In a recent study by Harbeck et al, uPA/PAI-1 levels in primary breast tumors were predictive for response to adjuvant systemic chemotherapy.33 The benefits of chemotherapy as opposed to endocrine therapy were greatly enhanced in patients with high uPA/PAI-1 levels. These data support earlier findings by Jänicke et al, who studied 556 patients with lymph node-negative breast cancer and found that uPA and PAI-1 status was sufficient to classify a subset of patients as low risk (50%), for whom adjuvant chemotherapy may be avoided.26
To date, steroid hormone receptor status is the prevailing parameter assessed in deciding whether to treat with endocrine therapy. Interestingly, both ER and PR status are inversely correlated with uPA (P=10−6 and P=0.05, respectively) or uPAR (P=10−6 and P=0.0005, respectively) expression in breast invasive carcinoma (Memorial Sloan-Kettering Cancer Center cBio Cancer Genomics Portal).35,36 Selective ER modulators such as tamoxifen and raloxifene, GnRH (gonadotropin-releasing hormone) agonists, and aromatase inhibitors are the most widely used endocrine therapies.37 The nonsteroidal estrogen antagonist tamoxifen is a type II competitive inhibitor of estradiol at its receptor and is the most popular endocrine treatment in premenopausal women. Endocrine treatment is a mainstay therapy for patients with ER-positive cancers and non-life-threatening advanced disease. Nonetheless, despite the fact that endocrine therapy has a relatively low morbidity, only one-half of patients treated with tamoxifen will receive clinical benefit.38 Therefore, there is a strong need for more sensitive and predictive biomarkers for response to endocrine therapy. In a recent study by Meijer-van Gelder et al, uPA, uPAR, and PAI-1 were all predictive for improved efficacy of tamoxifen therapy in patients treated for recurrent breast cancer.39 Therefore, the status of uPA system proteins in patient tumor specimens may be useful in developing individualized therapy protocols.
Matrix metalloproteinases
MMPs are members of a large multigene family of zinc-dependent endopeptidases. There are more than 24 MMPs known to play a vital role in remodeling the ECM.40 In addition to ECM proteins, MMPs target and cleave a wide range of substrates such as other proteases, growth factors, cell adhesion molecules, clotting factors, and cell surface receptors. Thus, MMPs are essential in regulating many cellular interactions under conditions that promote tissue turnover.41 Not surprisingly, MMPs are primarily active during development, when the majority of ECM remodeling occurs. In adults, the majority of remaining MMP activity is isolated to remodeling processes such as wound repair and angiogenesis. However, in human disease, aberrant MMP hyperactivity has been observed in many pathological conditions, such as osteoarthritis, multiple sclerosis, and cancer.2,42
MMPs are translated as inactive pro-enzymes, secreted, and then activated through the catalytic removal of their pro-domain by other proteases in the ECM. Membrane-type MMPs (MT-MMPs), however, are a family of six proteins that contain a furin cleavage site motif that is recognized for intracellular activation prior to secretion.43 Though most MMPs cleave a number of specific substrates, including several ECM components, MT-MMPs have an additional role in pro-MMP activation. It is important to note that MMPs have considerable structural and functional similarity. Likewise, this relative redundancy may account for the fact that most MMP knock-out mice are viable.44 However, despite high levels of homology, MMPs still retain some substrate specificity and tissue-specific expression patterns. As a result, there are a number of MMPs that have been shown to possess great prognostic potential for clinical outcome in breast cancer.45–47
Currently, there exist many studies and review articles that detail the potential prognostic utility of individual MMPs in specific cancers.48–53 However, most of the published original data are derived solely from retrospective analyses performed on patient cohorts of limited size. Recently, McGowan and Duffy analyzed a well-defined published database of 295 breast cancer patients in the most comprehensive study of MMPs in breast cancer to date.54 Of the 17 MMPs investigated in their study, five (MMP-1, -9, -12, -14, and -15) were significantly associated with poor outcome. Additionally, MMP-14 was found to be an independent predictor of outcome, irrespective of tumor size, grade, lymph node status, and ER status. Despite these MMPs being linked to adverse outcome previously, there were several novel findings worth noting. First, MMP-1 expression was significantly higher in basal-like breast cancer compared to other subtypes. Also, although low MMP-9 expression was shown to be significantly associated with poor prognosis in the total population of breast cancer patients, it was not associated with outcome in a systemically untreated cohort.54 This suggests that MMPs may be only predictive of outcome depending on the therapeutic regimen.
Here, we perform our own analysis of MMP status in breast cancer using a previously published dataset that contains microarray data in over 1,500 breast primary breast tumors, the largest dataset currently available.7 Details of this dataset are outlined in Table 2. In our investigation, we chose to analyze patient survival by two distinct methods. First, we stratified tumor samples into “high” and “low” MMP expression groups based on the median MMP expression for each gene and performed Kaplan–Meier analysis for disease-specific survival. For our second analysis, we treated MMP expression in each patient as a continuous variable and performed a Cox regression analysis and Wald test for any significance between MMP expression and disease-specific survival (Table 3). Of the 19 MMPs tested, only MMP-9, -11, and -15 were significantly associated with worse survival in both analyses (P<0.01). Interestingly, MMP-11 was not previously identified by McGowan and Duffy as holding any prognostic value in breast cancer.54 However, Cheng et al examined MMP-11 expression in paired tumor and adjacent normal tissue and discovered that increased MMP-11 expression correlated with more aggressive clinical features.55 Furthermore, Tan et al found that MMP-11 has a necessary paracrine function during mammary gland development that may later be co-opted to promote breast cancer progression.56 Additionally, another putative role of MMP-11 in breast cancer was described by Takeuchi et al, who demonstrated that MMP-11 overexpression significantly increased resistance to anoikis, favoring anchorage-independent growth over programmed cell death in the setting of reduced cell–ECM contact.57 To identify any additional associations, we explored whether MMP-9, -11, and -15 expression was linked with several common clinical and/or pathological features of breast cancer (Table 4). Both MMP-9 and MMP-15 were significantly associated with higher tumor grade, ER-negative status, and PAM50 basal subtype. This result is consistent with a wide body of literature implicating MMP-9 in breast cancer progression in both clinical and preclinical investigations.58 It is important to note that our analysis is limited to gene expression. MMPs are also regulated at the posttranslational level, which may account for discrepancies between our analysis and other studies investigating the expression of MMPs in breast cancer at the protein level. For example, multiple studies have shown that MMP-2 is associated with poor outcome in breast cancer by assessing MMP-2 levels via immunohistochemistry.59,60
 | Table 2 Characteristics of 1,545 breast cancer samples (Curtis dataset) |
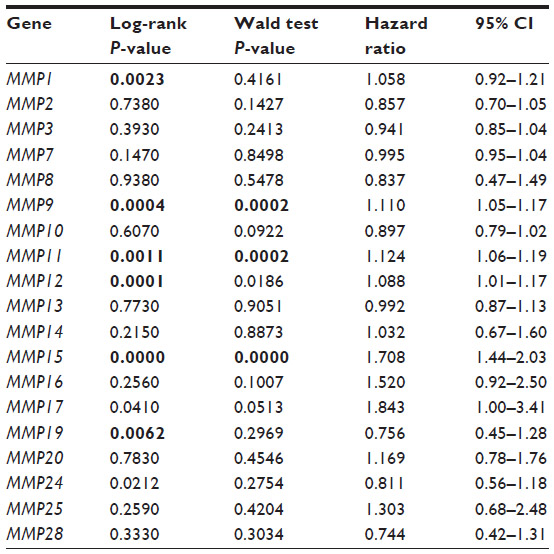 | Table 3 Relationship between MMP expression and disease-specific survival in 1,545 breast cancer patients |
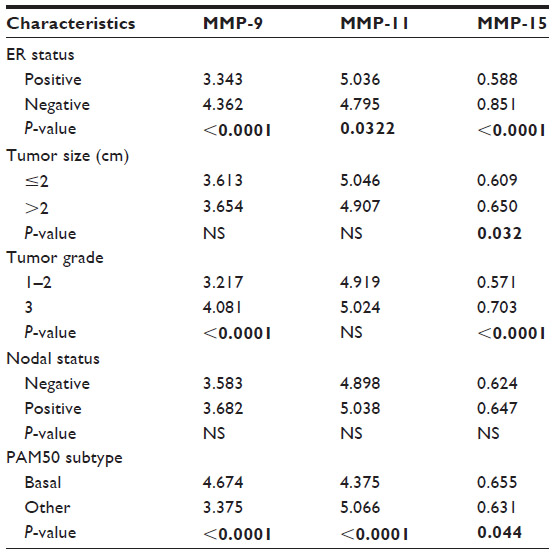 | Table 4 Relationship between MMP-9, -11, and -15 expression and tumor characteristics |
A disintegrin and metalloproteases (ADAMs)
ADAM proteins are zinc-dependent transmembrane metalloproteases responsible for proteolytic cleavage of extracellular domains of membrane-bound growth factors, cytokines, and receptors.61 Accordingly, they are known to have myriad diverse effects on cellular behavior. In total, there are 21 ADAMs that are believed to be functional in humans. Together they cleave a diverse array of ECM constituents and alter extracellular protein localization and bioavailability.62 Altered ADAM expression has been associated with several pathologies, though none as convincingly and well-demonstrated as in cancer.63,64 There are several excellent reviews that highlight the mechanism by which ADAMs are involved in tumorigenesis, but none have focused exclusively on ADAMs in breast cancer.65
For years, both clinical and preclinical studies have detailed the important role that individual ADAMs play in breast cancer. The most extensively characterized of all ADAMs is ADAM-17.66 ADAM-17 has been demonstrated to act via several oncogenic mechanisms, though one of the most important and relevant may be that it actively releases HER2/EGFR ligands through proteolytic cleavage, thereby increasing proliferation, invasion, and angiogenesis.67 In fact, forced overexpression of ADAM-17 in breast cancer cells increases invasion and proliferation in vitro, and the opposite effect is seen following ADAM-17 inhibition and knockdown.68 At both mRNA and protein levels, ADAM-17 expression is significantly upregulated in tumor samples compared with normal breast tissue.69,70
The prognostic potential of ADAMs in breast cancer has been highlighted on a case-by-case basis. Collectively, ADAM-9, ADAM-12, ADAM-15, ADAM-17, ADAM-22, and ADAM-28 have been implicated in the occurrence of breast cancer.71 Here, we analyze the prognostic potential of 15 ADAMs genes in the Curtis breast cancer dataset (Table 1). The results are summarized in Table 5. Only ADAM-8 and ADAM-17 were significantly associated with worse disease-specific survival in both of our analyses. ADAM-8 was the focus of a recent study that found that ADAM-8 was abundantly expressed in breast tumors and metastases compared to normal tissue, especially in triple-negative breast cancers.72 Further, elevated ADAM-8 expression predicted poor patient outcome. Additionally, in their breast cancer xenograft mouse model, treatment of tumors with an anti-ADAM-8 antibody reduced primary tumor burden and the number of metastases.72 Together, our analysis and these data demonstrate that ADAM-8 is a promising novel biomarker and a therapeutic candidate in breast cancer.
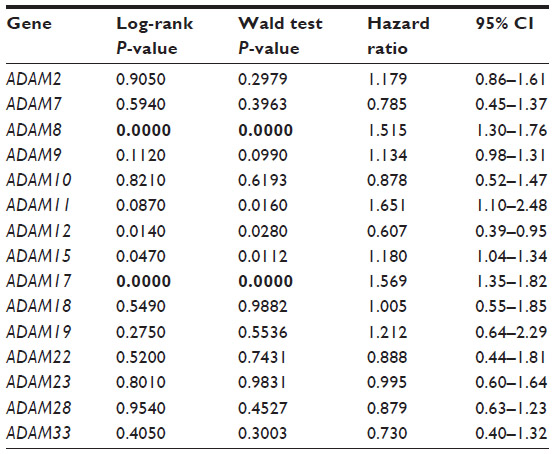 | Table 5 Relationship between ADAMs expression and disease-specific survival in 1,545 breast cancer patients |
We also analyzed the association between ADAM-8 and ADAM-17 and common clinical and pathologic features of breast cancer (Table 6). ADAM-17 is most significantly associated with the presence of lymph node metastases. Both ADAM-17 and ADAM-8 expression are significantly associated with ER-negative status, increased tumor grade, and basal tumor subtype (Table 6). Therefore, these data reveal that both ADAM-17 and ADAM-8 are putative biomarkers in breast cancer and promising candidates for new targeted therapies.
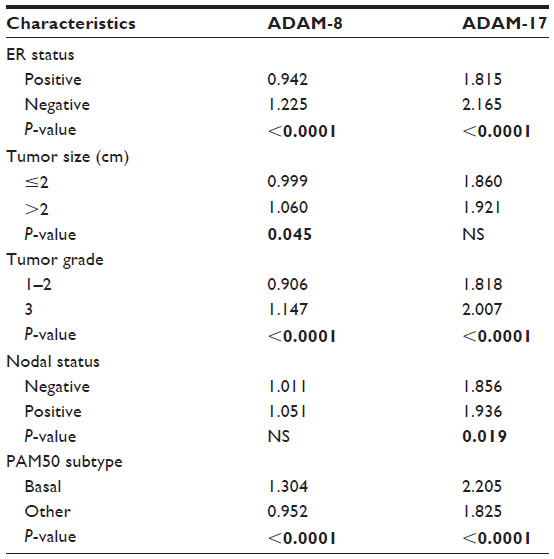 | Table 6 Relationship between ADAM-8 and -17 expression and tumor characteristics |
Tissue inhibitor of metalloproteinases (TIMPs)
The TIMPs comprise a highly conserved and homologous set of proteins. To date, four TIMPs have been identified (TIMP1–4). TIMPs are small, secreted proteins consisting of structurally and functionally distinct N- and C-terminal domains.73 Originally, TIMPs were characterized based on their ability to inhibit MMP activity, though, more recently, TIMPs have been shown to possess a number of MMP-independent functions.74 For example, TIMPs can bind directly to cell surface receptors to stimulate cell-signaling pathways, thereby leading to changes in cell growth, proliferation, and apoptosis. In fact, this activity requires the C-terminus of TIMP proteins and is therefore independent of their MMP-inhibitory activity, which occurs exclusively at the N-terminus.75 Notably, TIMPs play a fundamental role in controlling cell–ECM interactions, specifically under conditions that promote tissue turnover. Considering the profound effects that ECM remodeling can have on pathophysiology, it is not surprising that TIMPs play an important role in tumor behavior.
A number of promising prognostic candidates exist among TIMPs. In fact, TIMP-1 holds potential utility in breast cancer, as a number of studies have demonstrated an association between both high serum and tumor levels of TIMP-1 and lower overall survival.76,77 These findings are somewhat paradoxical as one might predict that elevated levels of TIMPs would result in decreased MMP proteolytic activity and therefore suppression of breast cancer invasion and metastasis. However, these data are more consistent with the MMP-independent role of TIMPs in breast cancer progression, thereby reconciling this apparent conundrum while simultaneously highlighting the dual and opposing roles that TIMPs may have. Recently, CD63 and integrin β1 have been identified as cell surface binding partners for TIMP-1 which may modulate its antiapoptotic signaling activity, though the specific details of this pathway still remain poorly understood.78 Similarly, TIMP-2 has been shown to bind to α3β1 integrin. This association modulates the MMP regulator reversion-inducing cysteine-rich protein with kazal motifs (RECK).79 RECK is another MMP inhibitor and important metastasis suppressor, and is discussed in the next section of this review. However, again somewhat paradoxically, elevated TIMP-2 levels have been correlated with poor prognosis in breast cancer.80 Conversely, low levels of TIMP-3 have been associated with poor prognosis.81 Additionally, TIMP-3 has been associated as a biomarker for successful endocrine therapy.82
A delicate balance exists between MMPs and TIMPs in the ECM. This dynamic relationship is tightly and coordinately regulated, and any alterations may carry profound effects on breast cancer tumorigenesis and progression. As a result, we considered it would be of interest to determine which TIMPs were predictive of survival in the Curtis dataset. Interestingly, the only TIMP that was significantly associated with prognosis was TIMP-4 (Table 7). More specifically, decreased levels of TIMP-4 are significantly associated with poor disease-specific survival, ER-negative status, tumor size >2 cm, higher grade, and basal PAM50 subtype (all: P<0.0001). Decreased TIMP-4 is also associated with the presence of positive lymph node metastases (P=0.011). Of note, TIMP-4 is the least studied of all the TIMPs, particularly in breast cancer. Nonetheless, in an early study by Wang et al, forced overexpression of TIMP-4 was shown to significantly inhibit breast cancer growth both in vitro and in vivo.83 In addition, Liss et al investigated TIMP-4 as a potential breast cancer biomarker via immunohistochemical staining of TIMP-4 in 314 tumors from patients with early-stage disease (defined as tumors smaller than 2 cm and no positive lymph nodes).84 They found that tumors with elevated TIMP-4 were correlated with a reduced probability of long-term disease-free survival, especially in patients with ER-negative tumors.
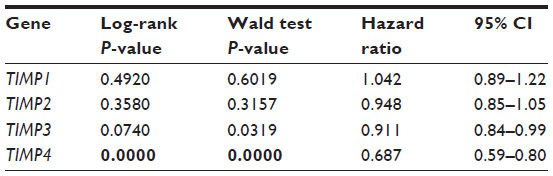 | Table 7 Relationship between TIMP expression and disease-specific survival in 1,545 breast cancer patients |
Despite the fact that our analysis only identified TIMP-4 as a prognostic marker for breast cancer outcome, we again must highlight that our analysis is restricted to gene expression. Many of the aforementioned studies clearly identify a strong relationship between other TIMPs and breast cancer. TIMP secretion and localization is vital for TIMP-mediated effects on cancer cell behavior, which cannot be assessed by gene expression analysis alone. Nonetheless, TIMP-4 represents an intriguing new prognostic candidate.
Reversion-inducing cysteine-rich protein with Kazal Motifs
RECK is a membrane-anchored glycoprotein that negatively regulates MMPs and potently inhibits tumor angiogenesis.85 RECK is suppressed across many cancer types and transformed cell line models. Functional studies in non-transformed cells have shown that RECK serves as a negative regulator of MMP-9 and is a target for repression itself by the oncomiR miR-21.86,87 Over a decade ago, Span et al assessed the prognostic value of RECK expression in tumor tissue specimens from 278 breast carcinoma patients via quantitative reverse transcription polymerase chain reaction.88 Multivariate Cox regression analysis showed that RECK expression held significant independent prognostic value for improved recurrence-free survival. More recently, Zhang et al performed a retrospective analysis of 119 patients with invasive breast cancer and analyzed RECK expression by immunohistochemical staining of tumor specimens.89 They found a significant positive correlation between RECK and 5-year overall survival. Additional multivariate analyses confirmed that reduced RECK expression was an independent and significant factor in predicting a poor prognosis. In an interesting study, Hill et al profiled DNA methylation patterns genome-wide in sporadic breast tumors using the HumanMethylation27 BeadChip® (Illumina, San Diego, CA, USA) to assess relationships between epigenetic regulation and tumor features.90 They identified several individually methylated genes, including RECK, and discovered a significant inverse correlation between promoter hypermethylation and relapse-free survival.
Likewise, we recently demonstrated a RECK-associated disease-specific survival advantage in several independent breast cancer datasets.91 Our group also performed a comprehensive functional characterization of RECK using multiple in vitro and in vivo model systems. In this study, we demonstrated that RECK is a bona fide metastasis suppressor gene in breast cancer. In addition to known mechanisms, we revealed that RECK can also regulate metastasis and neoangiogenesis via suppression of uPA, VEGF, and STAT3 signaling.91 These data all support RECK as a strong biomarker for breast cancer prognosis and a putative target for future therapy.
Discussion
Breast cancer is the most common form of cancer among women and one of the deadliest, second only to lung cancer. Each year, over 230,000 new cases of breast cancer are diagnosed and nearly 40,000 deaths occur in the United States.92 Despite increasing incidence of breast cancer, mortality rates have been declining in recent decades, largely due to increased screening and early detection.93 Classically, clinicopathologic features (eg, tumor size, histological grade) helped guide decision-making in the clinic, enabling physicians to tailor treatment regimens according to well-established probabilities of risk versus response. However, these prognostic markers alone have not been able to completely predict treatment efficacy and survival outcomes. Fortunately, several biomarkers have been validated and are now in routine clinical use, such as ER and HER2 status, allowing further treatment stratification following diagnosis. In addition, new technologies have given rise to entire gene panel sets as prognostic tools (eg, Oncotype DX®, Genomic Health Inc., Redwood City, CA, USA; MammaPrint™, Agendia Inc., Irvine, CA, USA). It goes without question that a clinician’s ever-growing repertoire of prognostic tools has improved therapeutic management and patient outcomes in breast cancer. Regardless, there are many potential biomarkers that remain undiscovered or not validated and therefore warrant further investigation.
There has been renewed interest in recent years in the use of extracellular proteases as biomarkers in breast cancer. For example, both uPA and PAI-1 have now been validated at the highest level of evidence, though their use in the clinic is not yet widespread. In fact, studies have shown that uPA, uPAR, and PAI-1 status can be used to predict efficacy of tamoxifen therapy or when patients may avoid chemotherapy altogether. Other extracellular proteases have been significantly associated with patient outcomes, notably MMP-9, -11, and -15 and ADAM-8 and -17. Interestingly, several inhibitors of extracellular proteases have also been identified as potential breast cancer biomarkers. In particular, TIMP-4 may represent an understudied and potentially powerful new biomarker. Perhaps most interesting is the identification of RECK – a negative regulator of MMPs – as a robust biomarker for improved prognosis (overall survival and relapse-free survival) in breast cancer. Due to the plethora of evidence implicating altered ECM remodeling in cancer progression and metastasis, it is not surprising that alterations in several of these proteases correlate to patient outcomes.
The greatest unknown about the utility of extracellular protease status in breast cancer may be whether this knowledge can extend beyond tailoring existing treatment regimens. Targeting proteases, such as MMPs, has long been the subject of preclinical and clinical trials, though the early data were mostly disappointing.94,95 Several reasons for these failures have been proposed, including the focus on treating metastases, lack of specificity among inhibitors, and broad action of MMPs on both pro- and antiangiogenic proteins (eg, ADAMTSs).94,95 Since MMP activation occurs early in tumor progression, it is reasonable that trials on early-stage cancers using novel, narrow-specificity protease inhibitors may attain greater success. In this vein, several exciting inhibitors of extracellular proteases are in various stages of development for use in cancer (Table 8).
 | Table 8 Select candidate inhibitors of extracellular proteases in cancer |
Conclusion
Exploiting proteolytic activity for the selective delivery of other therapeutics is a promising new avenue and may depend on the localization and specificity of extracellular proteases.96 Regardless, although both direct and indirect therapeutic approaches represent a more optimistic and challenging goal, the use of extracellular proteases as biomarkers in breast cancer presents an exciting new frontier in cancer management.
Acknowledgment
DMR was supported by NIH MSTP grant T32GM007739. LAW was supported by the Canadian Institutes of Health Research PDF Award MFE-127325.
Disclosure
The authors report no conflicts of interest in this work.
References
Overall CM, Blobel CP. In search of partners: linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007;8(3):245–257. | |
Yadav L, Puri N, Rastogi V, Satpute P, Ahmad R, Kaur G. Matrix metalloproteinases and cancer – roles in threat and therapy. Asian Pac J Cancer Prev. 2014;15(3):1085–1091. | |
Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291(2):113–135. | |
Wang X, Wilson MJ, Slaton JW, Sinha AA, Ewing SL, Pei D. Increased aggressiveness of human prostate PC-3 tumor cells expressing cell surface localized membrane type-1 matrix metalloproteinase (MT1-MMP). J Androl. 2009;30(3):259–274. | |
Chimal-Ramírez GK, Espinoza-Sánchez NA, Utrera-Barillas D, et al. MMP1, MMP9, and COX2 expressions in promonocytes are induced by breast cancer cells and correlate with collagen degradation, transformation-like morphological changes in MCF-10A acini, and tumor aggressiveness. Biomed Res Int. 2013;2013:279505. | |
Sun J. Matrix metalloproteinases and tissue inhibitor of metalloproteinases are essential for the inflammatory response in cancer cells. J Signal Transduct. 2010;2010:985132. | |
Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. | |
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(Pt 24):4195–4200. | |
Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol. 2011;209(2):139–151. | |
Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. J Pathol. 2003;200(4):423–428. | |
Di Lullo GA, Sweeney SM, Korkko J, Ala-Kokko L, San Antonio JD. Mapping the ligand-binding sites and disease-associated mutations on the most abundant protein in the human, type I collagen. J Biol Chem. 2002;277(6):4223–4231. | |
Salam A, Simpson MA, Stone KL, et al. Next generation diagnostics of heritable connective tissue disorders. Matrix Biol. 2014;33:35–40. | |
Tanjore H, Kalluri R. The role of type IV collagen and basement membranes in cancer progression and metastasis. Am J Pathol. 2006;168(3):715–717. | |
Polyak K. Molecular markers for the diagnosis and management of ductal carcinoma in situ. J Natl Cancer Inst Monogr. 2010;2010(41):210–213. | |
Schmidt S, Friedl P. Interstitial cell migration: integrin-dependent and alternative adhesion mechanisms. Cell Tissue Res. 2010;339(1):83–92. | |
Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4(2):165–178. | |
Schaefer L, Schaefer RM. Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 2010;339(1):237–246. | |
Leonova EI, Galzitskaya OV. Structure and functions of syndecans in vertebrates. Biochemistry (Mosc). 2013;78(10):1071–1085. | |
Barbouri D, Afratis N, Gialeli C, Vynios DH, Theocharis AD, Karamanos NK. Syndecans as modulators and potential pharmacological targets in cancer progression. Front Oncol. 2014;4:4. | |
Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196(4):395–406. | |
Lamy PJ, Martin PM, Romieu G, Jacot W. Re: clinical validity/utility, change in practice patterns, and economic implications of risk stratifiers to predict outcomes for early-stage breast cancer: a systematic review. J Natl Cancer Inst. 2013;105(2):149. | |
Tessari A, Palmieri D, Di Cosimo S. Overview of diagnostic/targeted treatment combinations in personalized medicine for breast cancer patients. Pharmgenomics Pers Med. 2013;7:1–19. | |
Turner N, Pestrin M, Galardi F, De Luca F, Malorni L, Di Leo A. Can biomarker assessment on circulating tumor cells help direct therapy in metastatic breast cancer? Cancers (Basel). 2014;6(2):684–707. | |
Mulrane L, Klinger R, McGee SF, Gallagher WM, O’Connor DP. microRNAs: a new class of breast cancer biomarkers. Expert Rev Mol Diagn. 2014;14(3):347–363. | |
Andres SA, Edwards AB, Wittliff JL. Expression of urokinase-type plasminogen activator (uPA), its receptor (uPAR), and inhibitor (PAI-1) in human breast carcinomas and their clinical relevance. J Clin Lab Anal. 2012;26(2):93–103. | |
Jänicke F, Prechtl A, Thomssen C, et al; German N0 Study Group. Randomized adjuvant chemotherapy trial in high-risk, lymph node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. J Natl Cancer Inst. 2001;93(12):913–920. | |
Look MP, van Putten WL, Duffy MJ, et al. Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J Natl Cancer Inst. 2002;94(2):116–128. | |
Tang L, Han X. The urokinase plasminogen activator system in breast cancer invasion and metastasis. Biomed Pharmacother. 2013;67(2):179–182. | |
Harbeck N, Sotlar K, Wuerstlein R, Doisneau-Sixou S. Molecular and protein markers for clinical decision making in breast cancer: today and tomorrow. Cancer Treat Rev. 2014;40(3):434–444. | |
McMahon B, Kwaan HC. The plasminogen activator system and cancer. Pathophysiol Haemost Thromb. 2008;36(3–4):184–194. | |
Mekkawy AH, Pourgholami MH, Morris DL. Involvement of urokinase-type plasminogen activator system in cancer: an overview. Med Res Rev. Epub February 18, 2014. | |
Urban P, Vuaroqueaux V, Labuhn M, et al. Increased expression of urokinase-type plasminogen activator mRNA determines adverse prognosis in ErbB2-positive primary breast cancer. J Clin Oncol. 2006;24(26):4245–4253. | |
Binder BR, Mihaly J. The plasminogen activator inhibitor “paradox” in cancer. Immunol Lett. 2008;118(2):116–124. | |
Duffy MJ. The urokinase plasminogen activator system: role in malignancy. Curr Pharm Des. 2004;10(1):39–49. | |
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. | |
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. | |
Lumachi F, Luisetto G, Basso SM, Basso U, Brunello A, Camozzi V. Endocrine therapy of breast cancer. Curr Med Chem. 2011;18(4):513–522. | |
Schiavon G, Smith IE. Endocrine therapy for advanced/metastatic breast cancer. Hematol Oncol Clin North Am. 2013;27(4):715–736, viii. | |
Meijer-van Gelder ME, Look MP, Peters HA, et al. Urokinase-type plasminogen activator system in breast cancer: association with tamoxifen therapy in recurrent disease. Cancer Res. 2004;64(13):4563–4568. | |
Woessner JF Jr. MMPs and TIMPs – an historical perspective. Mol Biotechnol. 2002;22(1):33–49. | |
Gibson DJ, Schultz GS. Molecular wound assessments: matrix metalloproteinases. Adv Wound Care (New Rochelle). 2013;2(1):18–23. | |
Javaid MA, Abdallah MN, Ahmed AS, Sheikh Z. Matrix metalloproteinases and their pathological upregulation in multiple sclerosis: an overview. Acta Neurol Belg. 2013;113(4):381–390. | |
Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92(8):827–839. | |
Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011;41(2):271–290. | |
Kulić A, Dedić Plavetić N, Vrbanec J, Sirotković-Skerlev M. Low serum MMP-1 in breast cancer: a negative prognostic factor? Biomarkers. 2012;17(5):416–421. | |
Puzovic V, Brcic I, Ranogajec I, Jakic-Razumovic J. Prognostic values of ETS-1, MMP-2 and MMP-9 expression and co-expression in breast cancer patients. Neoplasma. Epub March 20, 2014. | |
Fernandez-Garcia B, Eiró N, Marín L, et al. Expression and prognostic significance of fibronectin and matrix metalloproteases in breast cancer metastasis. Histopathology. 2014;64(4):512–522. | |
van der Jagt MF, Wobbes T, Strobbe LJ, Sweep FC, Span PN. Metalloproteinases and their regulators in colorectal cancer. J Surg Oncol. 2010;101(3):259–269. | |
de la Peña S, Sampieri CL, León-Córdoba K. [Matrix metalloproteases as molecular markers in gastric cancer]. Med Clin (Barc). 2010;134(3):123–126. Spanish. | |
Rydlova M, Holubec L Jr, Ludvikova M Jr, et al. Biological activity and clinical implications of the matrix metalloproteinases. Anticancer Res. 2008;28(2B):1389–1397. | |
Łukaszewicz M, Mroczko B, Szmitkowski M. [The role of metalloproteinases and their inhibitors in pancreatic cancer]. Postepy Hig Med Dosw (Online). 2008;62:141–147. | |
Libra M, Scalisi A, Vella N, et al. Uterine cervical carcinoma: role of matrix metalloproteinases (review). Int J Oncol. 2009;34(4):897–903. | |
Lim SC. Expression of c-erbB receptors, MMPs and VEGF in head and neck squamous cell carcinoma. Biomed Pharmacother. 2005;59 Suppl 2:S366–S369. | |
McGowan PM, Duffy MJ. Matrix metalloproteinase expression and outcome in patients with breast cancer: analysis of a published database. Ann Oncol. 2008;19(9):1566–1572. | |
Cheng CW, Yu JC, Wang HW, et al. The clinical implications of MMP-11 and CK-20 expression in human breast cancer. Clin Chim Acta. 2010;411(3–4):234–241. | |
Tan J, Buache E, Alpy F, et al. Stromal matrix metalloproteinase-11 is involved in the mammary gland postnatal development. Oncogene. Epub October 21, 2013. | |
Takeuchi T, Adachi Y, Nagayama T, Furihata M. Matrix metalloproteinase-11 overexpressed in lobular carcinoma cells of the breast promotes anoikis resistance. Virchows Arch. 2011;459(3):291–297. | |
Song J, Su H, Zhou YY, Guo LL. Prognostic value of matrix metalloproteinase 9 expression in breast cancer patients: a meta-analysis. Asian Pac J Cancer Prev. 2013;14(3):1615–1621. | |
Talvensaari-Mattila A, Pääkkö P, Turpeenniemi-Hujanen T. Matrix metalloproteinase-2 (MMP-2) is associated with survival in breast carcinoma. Br J Cancer. 2003;89(7):1270–1275. | |
Jezierska A, Motyl T. Matrix metalloproteinase-2 involvement in breast cancer progression: a mini-review. Med Sci Monit. 2009;15(2):RA32–RA40. | |
Tortorella MD, Malfait F, Barve RA, Shieh HS, Malfait AM. A review of the ADAMTS family, pharmaceutical targets of the future. Curr Pharm Des. 2009;15(20):2359–2374. | |
Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139(20):3693–3709. | |
Mochizuki S, Okada Y. ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007;98(5):621–628. | |
Pollheimer J, Fock V, Knöfler M. Review: the ADAM metalloproteinases – novel regulators of trophoblast invasion? Placenta. 2014; 35 Suppl:S57–S63. | |
Flannery CR. MMPs and ADAMTSs: functional studies. Front Biosci. 2006;11:544–569. | |
Duffy MJ, Mullooly M, O’Donovan N, et al. The ADAMs family of proteases: new biomarkers and therapeutic targets for cancer? Clin Proteomics. 2011;8(1):9. | |
Gooz M. ADAM-17: the enzyme that does it all. Crit Rev Biochem Mol Biol. 2010;45(2):146–169. | |
McGowan PM, Ryan BM, Hill AD, McDermott E, O’Higgins N, Duffy MJ. ADAM-17 expression in breast cancer correlates with variables of tumor progression. Clin Cancer Res. 2007;13(8):2335–2343. | |
Kenny PA, Bissell MJ. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J Clin Invest. 2007;117(2):337–345. | |
McGowan PM, McKiernan E, Bolster F, et al. ADAM-17 predicts adverse outcome in patients with breast cancer. Ann Oncol. 2008;19(6):1075–1081. | |
Bolger JC, Young LS. ADAM22 as a prognostic and therapeutic drug target in the treatment of endocrine-resistant breast cancer. Vitam Horm. 2013;93:307–321. | |
Romagnoli M, Mineva ND, Polmear M, et al. ADAM8 expression in invasive breast cancer promotes tumor dissemination and metastasis. EMBO Mol Med. 2014;6(2):278–294. | |
Murphy G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011;12(11):233. | |
Stetler-Stevenson WG. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal. 2008;1(27):re6. | |
Seo DW, Li H, Guedez L, et al. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003;114(2):171–180. | |
Wu ZS, Wu Q, Yang JH, et al. Prognostic significance of MMP-9 and TIMP-1 serum and tissue expression in breast cancer. Int J Cancer. 2008;122(9):2050–2056. | |
Schrohl AS, Holten-Andersen MN, Peters HA, et al. Tumor tissue levels of tissue inhibitor of metalloproteinase-1 as a prognostic marker in primary breast cancer. Clin Cancer Res. 2004;10(7):2289–2298. | |
Toricelli M, Melo FH, Peres GB, Silva DC, Jasiulionis MG. Timp1 interacts with beta-1 integrin and CD63 along melanoma genesis and confers anoikis resistance by activating PI3-K signaling pathway independently of Akt phosphorylation. Mol Cancer. 2013;12:22. | |
Oh J, Seo DW, Diaz T, et al. Tissue inhibitors of metalloproteinase 2 inhibits endothelial cell migration through increased expression of RECK. Cancer Res. 2004;64(24):9062–9069. | |
Remacle A, McCarthy K, Noël A, et al. High levels of TIMP-2 correlate with adverse prognosis in breast cancer. Int J Cancer. 2000;89(2):118–121. | |
Mylona E, Magkou C, Giannopoulou I, et al. Expression of tissue inhibitor of matrix metalloproteinases (TIMP)-3 protein in invasive breast carcinoma: relation to tumor phenotype and clinical outcome. Breast Cancer Res. 2006;8(5):R57. | |
Span PN, Lindberg RL, Manders P, et al. Tissue inhibitors of metalloproteinase expression in human breast cancer: TIMP-3 is associated with adjuvant endocrine therapy success. J Pathol. 2004;202(4):395–402. | |
Wang M, Liu YE, Greene J, et al. Inhibition of tumor growth and metastasis of human breast cancer cells transfected with tissue inhibitor of metalloproteinase 4. Oncogene. 1997;14(23):2767–2774. | |
Liss M, Sreedhar N, Keshgegian A, et al. Tissue inhibitor of metalloproteinase-4 is elevated in early-stage breast cancers with accelerated progression and poor clinical course. Am J Pathol. 2009;175(3):940–946. | |
Oh J, Takahashi R, Kondo S, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107(6):789–800. | |
Reis ST, Pontes-Junior J, Antunes AA, et al. miR-21 may acts as an oncomir by targeting RECK, a matrix metalloproteinase regulator, in prostate cancer. BMC Urol. 2012;12:14. | |
Chang CK, Hung WC, Chang HC. The Kazal motifs of RECK protein inhibit MMP-9 secretion and activity and reduce metastasis of lung cancer cells in vitro and in vivo. J Cell Mol Med. 2008;12(6B):2781–2789. | |
Span PN, Sweep CG, Manders P, Beex LV, Leppert D, Lindberg RL. Matrix metalloproteinase inhibitor reversion-inducing cysteine-rich protein with Kazal motifs: a prognostic marker for good clinical outcome in human breast carcinoma. Cancer. 2003;97(11):2710–2715. | |
Zhang Y, Cheng S, Zhang G, et al. Low expression of RECK indicates a shorter survival for patients with invasive breast cancer. Cancer Sci. 2012;103(6):1084–1089. | |
Hill VK, Ricketts C, Bieche I, et al. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011;71(8):2988–2999. | |
Walsh LA, Roy DM, Reyngold M, et al. RECK controls breast cancer metastasis by modulating a convergent, STAT3-dependent neoangiogenic switch. Oncogene. Epub 2014 Jun 16. | |
American Cancer Society. Cancer Facts and Figures. Atlanta, GA: The Society:v. | |
Weigel MT, Dowsett M. Current and emerging biomarkers in breast cancer: prognosis and prediction. Endocr Relat Cancer. 2010;17(4):R245–R262. | |
Overall CM, López-Otín C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2(9):657–672. | |
Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002; 295(5564):2387–2392. | |
Vartak DG, Gemeinhart RA. Matrix metalloproteases: underutilized targets for drug delivery. J Drug Target. 2007;15(1):1–20. | |
Leighl NB, Paz-Ares L, Douillard JY, et al. Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR.18. J Clin Oncol. 2005;23(12):2831–2839. | |
Bernardo MM, Brown S, Li ZH, Fridman R, Mobashery S. Design, synthesis, and characterization of potent, slow-binding inhibitors that are selective for gelatinases. J Biol Chem. 2002;277(13):11201–11207. | |
Levitt NC, Eskens FA, O’Byrne KJ, et al. Phase I and pharmacological study of the oral matrix metalloproteinase inhibitor, MMI270 (CGS27023A), in patients with advanced solid cancer. Clin Cancer Res. 2001;7(7):1912–1922. | |
Griffin MO, Fricovsky E, Ceballos G, Villarreal F. Tetracyclines: a pleitropic family of compounds with promising therapeutic properties. Review of the literature. Am J Physiol Cell Physiol. 2010;299(3):C539–C548. | |
Hirte H, Vergote IB, Jeffrey JR, et al. A phase III randomized trial of BAY 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: a National Cancer Institute of Canada Clinical Trials Group Study. Gynecol Oncol. 2006;102(2):300–308. | |
Macaulay VM, O’Byrne KJ, Saunders MP, et al. Phase I study of intrapleural batimastat (BB-94), a matrix metalloproteinase inhibitor, in the treatment of malignant pleural effusions. Clin Cancer Res. 1999;5(3):513–520. | |
Lu C, Lee JJ, Komaki R, et al. Chemoradiotherapy with or without AE-941 in stage III non-small cell lung cancer: a randomized phase III trial. J Natl Cancer Inst. 2010;102(12):859–865. | |
Chu QS, Forouzesh B, Syed S, et al. A phase II and pharmacological study of the matrix metalloproteinase inhibitor (MMPI) COL-3 in patients with advanced soft tissue sarcomas. Invest New Drugs. 2007; 25(4):359–367. | |
Bissett D, O’Byrne KJ, von Pawel J, et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J Clin Oncol. 2005;23(4):842–849. | |
Khan SA, Chatterton RT, Michel N, et al. Soy isoflavone supplementation for breast cancer risk reduction: a randomized phase II trial. Cancer Prev Res (Phila). 2012;5(2):309–319. | |
Miller KD, Gradishar W, Schuchter L, et al. A randomized phase II pilot trial of adjuvant marimastat in patients with early-stage breast cancer. Ann Oncol. 2002;13(8):1220–1224. | |
Hundhausen C, Misztela D, Berkhout TA, et al. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102(4):1186–1195. | |
McGowan PM, Mullooly M, Caiazza F, et al. ADAM-17: a novel therapeutic target for triple negative breast cancer. Ann Oncol. 2013;24(2):362–369. | |
Joh T, Kataoka H, Tanida S, et al. Helicobacter pylori-stimulated interleukin-8 (IL-8) promotes cell proliferation through transactivation of epidermal growth factor receptor (EGFR) by disintegrin and metalloproteinase (ADAM) activation. Dig Dis Sci. 2005;50(11):2081–2089. | |
Zhou BB, Peyton M, He B, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10(1):39–50. | |
Witters L, Scherle P, Friedman S, et al. Synergistic inhibition with a dual epidermal growth factor receptor/HER-2/neu tyrosine kinase inhibitor and a disintegrin and metalloprotease inhibitor. Cancer Res. 2008;68(17):7083–7089. | |
Heinemann V, Ebert MP, Laubender RP, Bevan P, Mala C, Boeck S. Phase II randomised proof-of-concept study of the urokinase inhibitor upamostat (WX-671) in combination with gemcitabine compared with gemcitabine alone in patients with non-resectable, locally advanced pancreatic cancer. Br J Cancer. 2013;108(4):766–770. | |
Stillfried GE, Saunders DN, Ranson M. Plasminogen binding and activation at the breast cancer cell surface: the integral role of urokinase activity. Breast Cancer Res. 2007;9(1):R14. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.