Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Candidate genes for COPD: current evidence and research
Received 30 July 2015
Accepted for publication 25 September 2015
Published 19 October 2015 Volume 2015:10(1) Pages 2249—2255
DOI https://doi.org/10.2147/COPD.S80227
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Woo Jin Kim,1 Sang Do Lee2
1Department of Internal Medicine and Environmental Health Center, Kangwon National University, Chuncheon, 2Department of Pulmonary and Critical Care Medicine, Clinical Research Center for Chronic Obstructive Airway Diseases, Asan Medical Center, University of Ulsan College of Medicine, Seoul, South Korea
Abstract: COPD is a common complex disease characterized by progressive airflow limitation. Several genome-wide association studies (GWASs) have discovered genes that are associated with COPD. Recently, candidate genes for COPD identified by GWASs include CHRNA3/5 (cholinergic nicotine receptor alpha 3/5), IREB2 (iron regulatory binding protein 2), HHIP (hedgehog-interacting protein), FAM13A (family with sequence similarity 13, member A), and AGER (advanced glycosylation end product–specific receptor). Their association with COPD susceptibility has been replicated in multiple populations. Since these candidate genes have not been considered in COPD, their pathological roles are still largely unknown. Herein, we review some evidences that they can be effective drug targets or serve as biomarkers for diagnosis or subtyping. However, more study is required to understand the functional roles of these candidate genes. Future research is needed to characterize the effect of genetic variants, validate gene function in humans and model systems, and elucidate the genes’ transcriptional and posttranscriptional regulatory mechanisms.
Keywords: chronic obstructive pulmonary disease, genetics, genome-wide association study
Introduction
COPD is a common complex disease characterized by progressive airflow limitation; the condition is not fully reversible or curable.1 The global burden of this disease is high, especially in countries with older populations, and this burden is expected to increase as the populace continues aging.2 Although environmental factors, including cigarette smoking and biofuel smoke exposure, are major risk factors, genetic risk factors are also important.3 In addition, an interaction between genetics and environment is believed to drive the development of COPD.
Pathways that play a role in COPD pathogenesis include the response to oxidative stress, the protease–antiprotease imbalance, cell death, and inflammation.4–7 Genetic studies have been performed to identify genetic risk factors and to understand the pathogenesis of COPD. Family-based studies and candidate gene association studies have found associations to many genes and loci. However, α-1 antitrypsin deficiency caused by mutations in SERPINA1 is the only established genetically driven cause of COPD that has a potential intervention so far.8
Although whole-genome and exome sequencing may be the next tools used for the genetic study of COPD, genome-wide association study (GWAS) is currently the most widely used method for the discovery of candidate genes. Several GWASs have discovered novel genes and pathways that are associated with COPD susceptibility. Even more genes have been found to be significantly associated with lung function in the general population. Some of these lung function genes are also associated with COPD susceptibility. The genetic basis of different COPD-related phenotypes, including emphysema and chronic bronchitis, also overlaps with that of COPD susceptibility. After being implicated in disease pathogenesis, these genes can be used as potential drug targets or as biomarkers that can influence diagnosis and personalized treatment.
Currently, the most well-known candidate genes for COPD are CHRNA3/5 (cholinergic nicotine receptor alpha 3/5), IREB2 (iron regulatory binding protein 2), HHIP (hedgehog-interacting protein), FAM13A (family with sequence similarity 13, member A), and AGER (advanced glycosylation end product–specific receptor). They have been replicated in multiple populations. None of them are targeted by treatments for COPD yet, and the mechanisms by which they alter COPD risk are still largely unknown. There is some emerging evidence that they may be good targets for treatments or useful as biomarkers. However, more study is required to understand the functional roles of these candidate genes.
Future research is needed to characterize the effect of genetic variants, validate gene function in humans and model systems, and elucidate the genes’ transcriptional and posttranscriptional regulatory mechanisms.9 In this review, we will cover the genetic and genomic association studies of COPD that identified the candidate genes and review what is currently known about those genes’ functions.
CHRNA3, CHRNA5, and IREB2
There are several genes at chromosome 15q25 that have been identified by GWAS for affecting COPD risk, including CHRNA3, CHRNA5, and IREB2.10–12 The COPD cohorts investigated were the Norway case-control cohort (GenKOLS), the family-based ICGN cohort, the NETT (National Emphysema Treatment Trial)/NAS (normative aging study) cohorts, the Boston Early-Onset COPD cohort, and the COPDGene study cohort. The association between CHRNA3/5 and COPD has been replicated in multiple ethnic populations by direct genotyping.13–15 The CHRNA3/5 locus is associated with emphysema and smoking intensity in COPD.16,17 The CHRNA3/5 region is also associated with lung cancer and nicotine addiction. It has been debated whether this common susceptibility region is the result of a common pathogenic pathway for lung cancer and COPD, or if it is simply associated with nicotine addiction, a risk factor for both diseases. In addition, the causal variant within the CHRNA3/5 locus may be different in lung cancer than in COPD. There is some evidence that this locus has independent roles in the pathogenesis of COPD and smoking behavior.18
CHRNA3/5 and CHRNB4 are subunits of the nicotine cholinergic receptor, and the cholinergic system is active not only in cholinergic neuronal cells, but also in bronchial epithelial cells and airway inflammatory cells. The proteins are responsive to nicotine and are upregulated during chronic tobacco exposure. A recent study integrating GWAS results with expression quantitative trait loci (eQTL) study results found that single nucleotide polymorphisms (SNPs) in the 15q25 region were associated with the expression of IREB2 and CHRNA3 in blood and sputum samples.19 CHRNA3/5 and IREB2 may play different roles in the pathogenesis of COPD.
IREB2 was first identified by characterizing the differential gene expression in lung tissue between COPD patients and controls, and genotyping the SNPs within the candidate regions.20 IREB2 is a protein that binds iron-responsive elements (IREs), maintains cellular iron metabolism, and is regulated in response to oxygen and iron supply. IREB2 expression is higher in the lung tissue of COPD cases. The Ireb2-knockout mouse has abnormal iron metabolism in the brain, which causes cellular dysfunction.21 However, the role of IREB2 in COPD pathogenesis is still not known. A GWAS of the pulmonary artery measurement obtained by computed tomography (CT) in cohorts from the COPDGene study and the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) study found a genome-wide significant association to IREB2.22 This suggests a role for IREB2 in the pathogenesis of pulmonary hypertension in COPD, particularly in the vascular subtype.
HHIP
Many recently identified COPD-associated variants are located at chromosome 4q31, upstream of HHIP. This intergenic region has been associated with COPD susceptibility in several GWASs10,11,23 and has consistently been replicated in multiple ethnicities.24–27 This region is also associated with lung function in the general population.28–30 The HHIP locus is associated with fat-free mass and exacerbations in COPD subjects.16 HHIP is associated with various CT phenotypes in COPD including distinct patterns of emphysema17 and the severity of emphysema.31 HHIP is more associated with emphysema measurements than with airway phenotypes and has a more significant association in emphysema subgroups.32 This difference may reflect a different pathogenic process driven by HHIP, or may be driven by correlations between COPD status and imaging measurements. HHIP is also associated with adult height in the general population.33 Considering that the FEV1 (forced expiratory volume in 1 second) prediction is determined by height, there may be genetic factors that control both phenotypes. Using the candidate gene strategy, it was found that the HHIP gene interacted with environmental tobacco smoke in utero, suggesting that this gene is involved in the lung response to smoke exposure in early life.34
HHIP encodes a membrane glycoprotein that is an endogenous antagonist for the Hedgehog pathway. Hedgehog signaling is important for the morphogenesis of the lung and other organs.35 Although the role of HHIP in COPD is not fully understood, several studies have validated the function of this gene in COPD pathogenesis. The associated SNPs are located upstream of HHIP, suggesting they may affect promoter activity. A lung eQTL study revealed that SNPs associated with COPD affect the expression of HHIP, and the risk allele of rs1828591 decreases its expression.36 Zhou et al37 reported that HHIP expression is reduced in COPD lung tissues and the genomic region upstream of HHIP interacts with the HHIP promoter. The risk allele of a variant in the HHIP enhancer region reduces promoter activity via a differential binding affinity to transcription factors.
These studies suggest that the genetic variation of the HHIP region affects the risk of COPD by affecting HHIP expression in lung tissues. HHIP silencing in an airway epithelial cell line leads to a change in gene expression, and these differentially expressed genes are enriched in pathways related to the extracellular matrix and cell growth, which are processes relevant to COPD pathogenesis.38 Recently, Lao et al39 found that Hhip-haploinsufficient mice have increased airspace size after cigarette smoke exposure, increased lung compliance, and increased numbers of lymphoid aggregates. The functions of the genes with altered expression in Hhip+/− mice exposed to cigarette smoke were enriched in the pathway of lymphocyte activation. They used haploinsufficient mice because Hhip−/− mice die shortly after birth due to lung branching morphogenesis failure.
HHIP was also found to be associated with lung cancer by a candidate gene study.40 The Hedgehog pathway is a critical mediator of cigarette-smoke-induced lung cancer, and it may act as a common pathway for the development of COPD and lung cancer.41
FAM13A
A GWAS using three COPD cohorts, GenKOLS, NETT/NAS, and the ECLIPSE study, identified variants at chromosome 4q22 in the gene FAM13A.42 These are some of the most highly associated SNPs in COPD and are located in an intron. These associations have been replicated in a subset of the patients in the COPDGene study and the cohort of the International COPD Genetics Network. They have also been replicated in Asian populations, assayed using the candidate gene strategy.43,44 FAM13A was first found to associate with lung function in a GWAS using the general population,28 and it is associated with lung function in asthmatic subjects.45 GWASs using COPD cases with chronic bronchitis in the COPDGene study, GenKOLS, and ECLIPSE cohorts identified a significant association with FAM13A,46 whereas several GWASs for emphysema did not identify a genome-wide association. The odds ratios of FAM13A SNPs for COPD with chronic bronchitis were significantly higher than those for nonchronic bronchitis COPD, suggesting that FAM13A is more related to the pathogenesis of the chronic bronchitis subtype. Of note, FAM13A is also associated with idiopathic pulmonary disease,47 but the expression of FAM13A in lung tissues does not differ by case/control status or by genotype.
FAM13A was originally identified in cattle near a quantitative trait locus affecting milk production, and is expressed in the kidney, pancreas, lung, and thymus.48 Although the function of FAM13A has not been extensively studied, its RhoGAP domain may be related to COPD. Rho GTPases are key regulators of cytoskeletal dynamics, are involved in the pulmonary endothelial barrier, and are dysregulated in several lung diseases.49 A lung eQTL study suggested that the expression of FAM13A may be associated with particular SNPs.36 In the case of COPD, the FAM13A risk allele is associated with increased FAM13A expression in the lung, although expression does not differ in lung tissues between COPD cases and controls.43 A recent study by Jin et al50 found that FAM13A activates Wnt signaling by increasing the stability of β-catenin. Although depletion of FAM13A in a lung cancer cell line reduces Wnt signaling activity, FAM13A- knockout mice are viable and FAM13A-mutant lungs are morphologically indistinguishable from wild-type lungs, and Wnt signaling remains normal in Fam13a-knockout lungs. They also found that Akt regulates the phosphorylation of FAM13A, which can lead to cytoplasmic sequestration of FAM13A. Considering that Akt has a role in the pathogenesis of COPD,51 FAM13A may contribute to lung disease through aberrant Akt signaling. Further work is needed to validate the functional role of FAM13A in the pathogenesis of COPD.
AGER
GWASs of lung function in the general population have found that chromosome 6p21 is associated to FEV1/FVC (forced vital capacity) and FEV1, which are important physiologic parameters of COPD.30,28,52 This association was investigated in COPD patients identified from the population cohort using spirometry criteria, and the study found a suggestive association between COPD risk and AGER, although it was not statistically significant.53 A candidate gene study in NETT/NAS, GenKOLS, ECLIPSE, and a subset of the COPDGene study cohort found that it was associated with COPD susceptibility, although a subsequent GWAS did not find a significant association.54 On the other hand, an association has been found in multiple ethnic populations.55
The 6p21 region that showed significant association with COPD includes many genes, including TNXB, PPT2, AGER, and NOTCH4. However, AGER has a potential functional variant, rs2070600, and it has been studied the most in the pathogenesis of COPD. A GWAS of percent emphysema determined by CT using the Multi-Ethnic Study of Atherosclerosis cohort identified a significant association with the AGER/PPT region.56 This region did associate with emphysema severity and gas trapping in a GWAS using cohorts from the COPDGene, ECLIPSE, NETT, and GenKOLS studies.31
The protein product of AGER, the receptor for advanced glycan end products (RAGE), is a multiligand receptor of the immunoglobulin superfamily and interacts with molecules implicated in homeostatic function, inflammation, and development.57 RAGE levels are increased in the airway and alveolar walls of COPD lungs.58 RAGE expression in mice increases after cigarette smoke exposure, and cigarette-smoking-induced inflammatory responses by alveolar macrophages are diminished in RAGE-knockout mice.59 Transgenic mice with upregulated RAGE have impaired alveolar morphogenesis during lung development, distal airspace enlargement, and increased alveolar cell apoptosis.60 Another study using RAGE transgenic mice found incremental dilation of alveolar spaces, as well as pronounced inflammation in the peripheral lung and alveolar destabilization.61 A promoter variant of AGER in cystic fibrosis patients is associated with poor lung function, and it increases expression in airway epithelial cell lines, suggesting it is a modifier of lung disease severity.62
The soluble isoform of RAGE (sRAGE) contains the RAGE extracellular domain and can bind to circulating proinflammatory ligands, preventing RAGE activation. Mice that are exposed to chronic hypoxia have downregulated pulmonary RAGE protein and increased levels of sRAGE, which might be adaptive to and protective against chronic hypoxia.63 Circulatory levels of sRAGE are reduced in COPD patients.64 Reduced sRAGE levels are associated with increased emphysema in two COPD cohorts.65 Decreased plasma sRAGE levels are also associated with the progression of airflow limitation over time.66 In patients of the Treatment of Emphysema with a Selective Retinoid Agonist (TESRA) and ECLIPSE studies, sRAGE is associated with diffusing capacity, emphysema, and COPD disease status, and the variant rs2070600 is associated with circulating sRAGE levels.67 The significant association between rs2070600 and plasma sRAGE levels was also found in Dutch diabetes mellitus and control subjects.68 RAGE has been studied in metabolic diseases, and decreased levels of sRAGE are linked to vascular complications. RAGE contributes to the pathogenesis of COPD in the lung probably via the regulation of inflammation and apoptosis, and further study of the functions of this gene may lead to it being identified as a potential therapeutic target.
Other candidate genes
There have been several more regions identified in GWASs of COPD. A GWAS using subjects from the ECLIPSE, NETT/NAS, GenKOLS, and COPDGene studies identified chromosome 19q13 as being associated with COPD, along with the previously identified HHIP, FAM13A, and 15q25 regions.11 Chromosome 19q13 contains CYP2A6, RAB4B, MIA, and EGLN, which could potentially be involved in COPD pathogenesis, and EGLN2 was found to be dysregulated in the airway epithelium of smokers.69 A GWAS using the full COPDGene cohort identified additional associations with TGFB2, MMP12, and RIN3.23 TGFB2 and MMP12 have been previously studied in COPD or related phenotypes,70,71 whereas RIN3 has not been studied in COPD and needs to be investigated further. SERPINE2 was identified using a linkage analysis of gene expression changes in lung tissue.72 A recent GWAS of airway thickness identified rs734556 on chromosome 2q, which is associated with SERPINE2 expression.73
An association study of SNP in GSTDS on chromosome 4q24, which was originally shown to be associated with lung function in general population, was performed in COPD case and control subjects. Variant in GSTDS was significantly associated with COPD susceptibility.53 SNPs in this region were also tested in GenKOLS, NETT, ECLIPSE, and part of COPDGene, and association with COPD susceptibility was significant.54 GSTCD and INTS12 genes in 4q24 are expressed in human lung tissues and airway cells and expression of these genes was altered by TGFβ1 in human airway smooth muscle cells.74 Top SNPs associated with airflow obstruction defined by FEV1 and FEV1/FVC less than lower limits of normal were tested in the COPD meta-analysis. In this analysis, ADAM19 on chromosome 5q33 was nominally associated with COPD.12 This gene was also associated with FEV1/FVC in the CHARGE consortium.28 This region was nominally associated with COPD susceptibility.54 A GWAS of emphysema in the three COPD cohorts including the Norway cohort, ECLIPSE, and NETT revealed that BICD1 was the most associated with the presence or absence of emphysema.75 The emphysema-predominant mild COPD of COPDGene study was nominally associated with BICD1.
These associations require more replications and further fine-mapping studies are needed to find the causal variants of COPD, as well as studies to functionally validate the identified genes.
Future directions
Subtyping or classification, drug development, and risk prediction are potential clinical applications of the associated genes. Phenotype heterogeneity can be investigated using association studies on various COPD-related phenotypes. Although the associations merely reflect the correlations between COPD status and phenotypes, there may be subtypes with different genetic alterations. Considering that treatment with PDE4 inhibitor is effective only for the chronic bronchitis subtype, there may be a mechanism that is unique to this subtype. Candidate genes can be used to determine personalized treatment because they may help identify a subtype-unique pathogenesis, variation in a drug-action site, or variable drug metabolism.76
More regions have been identified in GWASs on FEV1 and FEV1/FVC in the general population, probably because of the larger sample sizes. GWASs using a greater sample size of COPD subjects may find more candidate genes.77 Another approach for finding more candidate genes is to identify rare variation using exome sequencing or arrays.
Functional studies for COPD candidate genes are summarized in Table 1. Although there are a few potential drug targets that are implicated in the novel COPD pathogenesis, it will take many years to apply them in the treatment.
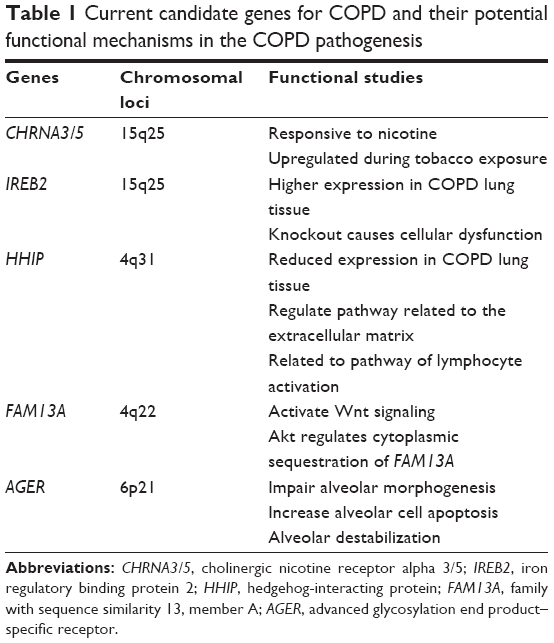 | Table 1 Current candidate genes for COPD and their potential functional mechanisms in the COPD pathogenesis |
Conclusion
Recently, several candidate genes associated with COPD risk have been identified using GWAS. Replication and functional validation studies may lead to clinical applications for these genes such as novel therapeutics, subtyping, and risk prediction for COPD.
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;187:347–365. | ||
Prince MJ, Wu F, Guo Y, et al. The burden of disease in older people and implications for health policy and practice. Lancet. 2015;385:549–562. | ||
Postma DS, Bush A, van den Berge M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet. 2015;385:899–909. | ||
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360:2445–2454. | ||
Tuder R, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122:2749–2755. | ||
Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144:266–273. | ||
Bagdonas E, Raudoniute J, Bruzauskaite I, et al. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:995–1013. | ||
Silverman EK, Sandhaus RA. Alpha1-antitrypsin deficiency. N Engl J Med. 2009;360:2749–2757. | ||
Ober C, Butte AJ, Elias JA, et al. Getting from genes to function in lung disease. Am J Respir Crit Care Med. 2010;182:732–737. | ||
Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. | ||
Cho MH, Castaldi PJ, Wan ES, et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Hum Mol Genet. 2012;21:947–957. | ||
Wilk JB, Shrine NR, Loehr LR, et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am J Respir Crit Care Med. 2012;186:622–632. | ||
Hardin M, Zielinski J, Wan ES, et al. CHRNA3/5, IREB2, and ADCY2 are associated with severe chronic obstructive pulmonary disease in Poland. Am J Respir Cell Mol Biol. 2012;47:203–208. | ||
Kim WJ, Oh YM, Kim TH, et al. CHRNA3 variant for lung cancer is associated with chronic obstructive pulmonary disease in Korea. Respiration. 2013;86:117–122. | ||
Yang L, Qiu F, Lu X, et al. Functional polymorphisms of CHRNA3 predict risks of chronic obstructive pulmonary disease and lung cancer in Chinese. PLoS One. 2012;7:e46071. | ||
Pillai SG, Kong X, Edwards LD, et al. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:1498–1505. | ||
Castaldi PJ, Cho MH, San Jose Estepar R, et al. Genome-wide association identifies regulatory Loci associated with distinct local histogram emphysema patterns. Am J Respir Crit Care Med. 2014;190:399–409. | ||
Siedlinski M, Tingley D, Lipman PJ, et al. Dissecting direct and indirect genetic effects on chronic obstructive pulmonary disease (COPD) susceptibility. Hum Genet. 2013;132:431–441. | ||
Castaldi PJ, Cho MH, Zhou X, et al. Genetic control of gene expression at novel and established chronic obstructive pulmonary disease loci. Hum Mol Genet. 2015;24:1200–1210. | ||
DeMeo DL, Mariani T, Bhattacharya S, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. 2009;85:493–502. | ||
Zumbrennen-Bullough KB, Becker L, Garrett L, et al. Abnormal brain iron metabolism in Irp2 deficient mice is associated with mild neurological and behavioral impairments. PLoS One. 2014;9:e98072. | ||
Lee JH, Cho MH, Hersh CP, et al. IREB2 and GALC are associated with pulmonary artery enlargement in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2015;52:365–376. | ||
Cho MH, McDonald ML, Zhou X, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2:214–225. | ||
Van Durme YM, Eijgelsheim M, Joos GF, et al. Hedgehog-interacting protein is a COPD susceptibility gene: the Rotterdam Study. Eur Respir J. 2010;36:89–95. | ||
Kim WJ, Oh YM, Lee JH, et al. Genetic variants in HHIP are associated with FEV1 in subjects with chronic obstructive pulmonary disease. Respirology. 2013;18:1202–1209. | ||
Xie J, Wu H, Xu Y, et al. Gene susceptibility identification in a longitudinal study confirms new loci in the development of chronic obstructive pulmonary disease and influences lung function decline. Respir Res. 2015;16:49. | ||
Wang B, Zhou H, Yang J, et al. Association of HHIP polymorphisms with COPD and COPD-related phenotypes in a Chinese Han population. Gene. 2013;531:101–105. | ||
Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42:45–52. | ||
Wilk JB, Chen TH, Gottlieb DJ, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5:e1000429. | ||
Repapi E, Sayers I, Wain LV, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42:36–44. | ||
Cho MH, Castaldi PJ, Hersh CP, et al. A genome-wide association study of emphysema and airway quantitative imaging phenotypes. Am J Respir Crit Care Med. 2015;192:559–569. | ||
Castaldi PJ, Dy J, Ross J, et al. Cluster analysis in the COPDGene study identifies subtypes of smokers with distinct patterns of airway disease and emphysema. Thorax. 2014;69:416–423. | ||
van der Valk RJP, Kreiner-Møller E, Kooijman MN, et al. A novel common variant in DCST2 is associated with length in early life and height in adulthood. Hum Mol Genet. 2015;24:1155–1168. | ||
Kerkhof M, Boezen HM, Granell R, et al. Transient early wheeze and lung function in early childhood associated with chronic obstructive pulmonary disease genes. J Allergy Clin Immunol. 2014;133:68–76.e64. | ||
Chuang PT, Kawcak T, McMahon AP. Feedback control of mammalian Hedgehog signaling by the Hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 2003;17:342–347. | ||
Lamontagne M, Couture C, Postma DS, et al. Refining susceptibility loci of chronic obstructive pulmonary disease with lung EQTLS. PLoS One. 2013;8:e70220. | ||
Zhou X, Baron RM, Hardin M, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum Mol Genet. 2012;21:1325–1335. | ||
Zhou X, Qiu W, Sathirapongsasuti JF, et al. Gene expression analysis uncovers novel hedgehog interacting protein (HHIP) effects in human bronchial epithelial cells. Genomics. 2013;101:263–272. | ||
Lao T, Glass K, Qiu W, et al. Haploinsufficiency of Hedgehog interacting protein causes increased emphysema induced by cigarette smoke through network rewiring. Genome Med. 2015;7:12. | ||
Young RP, Whittington CF, Hopkins RJ, et al. Chromosome 4q31 locus in COPD is also associated with lung cancer. Eur Respir J. 2010;36:1375–1382. | ||
Lemjabbar-Alaoui H, Dasari V, Sidhu SS, et al. Wnt and Hedgehog are critical mediators of cigarette smoke-induced lung cancer. PLoS One. 2006;1:e93. | ||
Cho MH, Boutaoui N, Klanderman BJ, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat Genet. 2010;42:200–202. | ||
Kim WJ, Lim MN, Hong Y, et al. Association of lung function genes with chronic obstructive pulmonary disease. Lung. 2014;192:473–480. | ||
Guo Y, Lin H, Gao K, et al. Genetic analysis of IREB2, FAM13A and XRCC5 variants in Chinese Han patients with chronic obstructive pulmonary disease. Biochem Biophys Res Commun. 2011;415:284–287. | ||
Li X, Hawkins GA, Ampleford EJ, et al. Genome-wide association study identifies TH1 pathway genes associated with lung function in asthmatic patients. J Allergy Clin Immunol. 2013;132:313–320.e315. | ||
Lee JH, Cho MH, Hersh CP, et al. Genetic susceptibility for chronic bronchitis in chronic obstructive pulmonary disease. Respir Res. 2014;15:113. | ||
Fingerlin TE, Murphy E, Zhang W, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–620. | ||
Cohen M, Reichenstein M, Everts-van der Wind A, et al. Cloning and characterization of FAM13A1 – a gene near a milk protein QTL on BTA6: evidence for population-wide linkage disequilibrium in Israeli Holsteins. Genomics. 2004;84:374–383. | ||
Corvol H, Hodges CA, Drumm ML, et al. Moving beyond genetics: is FAM13A a major biological contributor in lung physiology and chronic lung diseases? J Med Genet. 2014;51:646–649. | ||
Jin Z, Chung JW, Mei W, et al. Regulation of nuclear-cytoplasmic shuttling and function of Family with sequence similarity 13, member A (Fam13a), by B56-containing PP2As and Akt. Mol Biol Cell. 2015;26:1160–1173. | ||
Bozinovski S, Vlahos R, Hansen M, et al. Akt in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:31–38. | ||
Kim WJ, Lee MK, Shin C, et al. Genome-wide association studies identify locus on 6p21 influencing lung function in the Korean population. Respirology. 2014;19:360–368. | ||
Soler Artigas M, Wain LV, Repapi E, et al. Effect of five genetic variants associated with lung function on the risk of chronic obstructive lung disease, and their joint effects on lung function. Am J Respir Crit Care Med. 2011;184:786–795. | ||
Castaldi PJ, Cho MH, Litonjua AA, et al. The association of genome-wide significant spirometric loci with chronic obstructive pulmonary disease susceptibility. Am J Respir Cell Mol Biol. 2011;45:1147–1153. | ||
Li Y, Yang C, Ma G, et al. Association of polymorphisms of the receptor for advanced glycation end products gene with COPD in the Chinese population. DNA Cell Biol. 2014;33:251–258. | ||
Manichaikul A, Hoffman EA, Smolonska J, et al. Genome-wide study of percent emphysema on computed tomography in the general population. The multi-ethnic study of atherosclerosis lung/SNP health association resource study. Am J Respir Crit Care Med. 2014;189:408–418. | ||
Sorci G, Riuzzi F, Giambanco I, et al. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta. 2013;1833:101–109. | ||
Wu L, Ma L, Nicholson LFB, et al. Advanced glycation end products and its receptor (RAGE) are increased in patients with COPD. Respir Med. 2011;105:329–336. | ||
Robinson AB, Johnson KD, Bennion BG, et al. RAGE signaling by alveolar macrophages influences tobacco smoke-induced inflammation. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1192–L1199. | ||
Fineschi S, De Cunto G, Facchinetti F, et al. Receptor for advanced glycation end products contributes to postnatal pulmonary development and adult lung maintenance program in mice. Am J Respir Cell Mol Biol. 2013;48:164–171. | ||
Stogsdill MP, Stogsdill JA, Bodine BG, et al. Conditional overexpression of receptors for advanced glycation end-products in the adult murine lung causes airspace enlargement and induces inflammation. Am J Respir Cell Mol Biol. 2013;49:128–134. | ||
Beucher J, Boëlle P-Y, Busson P-F, et al. AGER-429T/C is associated with an increased lung disease severity in cystic fibrosis. PLoS One. 2012;7:e41913. | ||
Gopal P, Gosker HR, Theije CC, et al. Effect of chronic hypoxia on RAGE and its soluble forms in lungs and plasma of mice. Biochim Biophys Acta. 2015;1852:992–1000. | ||
Miniati M, Monti S, Basta G, et al. Soluble receptor for advanced glycation end products in COPD: relationship with emphysema and chronic cor pulmonale: a case-control study. Respir Res. 2011;12:37. | ||
Carolan B, Hughes G, Morrow J, et al. The association of plasma biomarkers with computed tomography-assessed emphysema phenotypes. Respir Res. 2014;15:127. | ||
Iwamoto H, Gao J, Pulkkinen V, et al. Soluble receptor for advanced glycation end-products and progression of airway disease. BMC Pulm Med. 2014;14:68. | ||
Cheng DT, Kim DK, Cockayne DA, et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:948–957. | ||
Gaens KHJ, Ferreira I, van der Kallen CJH, et al. Association of polymorphism in the receptor for advanced glycation end products (RAGE) gene with circulating RAGE levels. J Clin Endocrinol Metabol. 2009;94:5174–5180. | ||
Ryan DM, Vincent TL, Salit J, et al. Smoking dysregulates the human airway basal cell transcriptome at COPD risk locus 19q13.2. PLoS One. 2014;9:e88051. | ||
Hunninghake GM, Cho MH, Tesfaigzi Y, et al. MMP12, lung function, and COPD in high-risk populations. N Engl J Med. 2009;361:2599–2608. | ||
Artigas MS, Loth DW, Wain LV, et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011;43:1082–1090. | ||
DeMeo DL, Mariani TJ, Lange C, et al. The SERPINE2 gene is associated with chronic obstructive pulmonary disease. Am J Hum Genet. 2006;78:253–264. | ||
Dijkstra AE, Postma DS, van Ginneken B, et al. Novel genes for airway wall thickness identified with combined genome-wide association and expression analyses. Am J Respir Crit Care Med. 2014;191:547–556. | ||
Obeidat M, Miller S, Probert K, et al. GSTCD and INTS12 regulation and expression in the human lung. PLoS One. 2013;8:e74630. | ||
Kong X, Cho MH, Anderson W, et al. Genome-wide association study identifies BICD1 as a susceptibility gene for emphysema. Am J Respir Crit Care Med. 2011;183:43–49. | ||
Hersh CP. Pharmacogenetics of chronic obstructive pulmonary disease: challenges and opportunities. Pharmacogenomics. 2010;11:237–247. | ||
Silverman EK, Vestbo J, Agusti A, et al. Opportunities and challenges in the genetics of COPD 2010: an international COPD genetics conference report. COPD. 2011;8:121–135. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.