Back to Journals » OncoTargets and Therapy » Volume 12
Cancer Immunotherapies Targeting Tumor-Associated Regulatory T Cells
Authors Ge X, Zhao Y ![]() , Chen C
, Chen C ![]() , Wang J, Sun L
, Wang J, Sun L
Received 14 September 2019
Accepted for publication 2 December 2019
Published 13 December 2019 Volume 2019:12 Pages 11033—11044
DOI https://doi.org/10.2147/OTT.S231052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Leo Jen-Liang Su
Xiaoxu Ge,1–4,* Yamei Zhao,1–4,* Chao Chen,1–4 Jian Wang,1–4 Lifeng Sun1–4
1Department of Colorectal Surgery, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 2Cancer Institute, Key Laboratory of Cancer Prevention and Intervention, China National Ministry of Education, Hangzhou, People’s Republic of China; 3Key Laboratory of Molecular Biology in Medical Sciences, Hangzhou, People’s Republic of China; 4The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lifeng Sun
Department of Colorectal Surgery, The Second Affiliated Hospital of Zhejiang University School of Medicine, Jiefang Road 88, Hangzhou, Zhejiang 310009, People’s Republic of China
Tel +86-571-87783586
Email [email protected]
Abstract: Tumor-associated regulatory T cells (Tregs) are important effectors in the tumor microenvironment (TME), acting as accomplices in the promotion of tumor progression. Currently, the importance of removing the immunosuppressive activity in the TME has received its due attention, and Tregs have been focused on. The cytokine-receptor axes are among the essential signaling pathways in immunocytes, and tumor-associated Tregs are no exception. Therefore, manipulating cytokine-receptor pathways may be a promising effective strategy for treating various malignancies. Here, we summarize the classification, immunosuppressive mechanisms, existing immunotherapies, and potential biomarkers related to tumor-infiltrating Tregs to guide the development of effective cancer immunotherapies.
Keywords: Tregs, immune suppression, chemokine receptors, biomarkers, cancer immunotherapies
Introduction
The tumor microenvironment (TME) is the microenvironment around a tumor, consisting of the surrounding blood vessels, immunocytes, fibroblasts and extracellular matrix. The tumor cells and the TME are closely related and interact constantly. Development and progression of tumor cells involves complex genetic and epigenetic changes within the cells themselves, which also influence the TME by releasing extracellular signals. In turn, the immunocytes in the TME can affect the growth and evolution of cancer cells.1,2 Effective immunotherapies that promote the tumor-killing effect mediated by effector T cells (Teff) requires Teff activation and removal of the immunosuppressive activity in the TME, especially regarding the effects of immunosuppression-related immunocytes.
Regulatory T cells (Tregs) are a specialized subpopulation of CD4+ T cells. Tregs express transcription factor forkhead box P3 (FoxP3) and the surface molecule CD25. They have been widely regarded as critical effectors in the maintenance of healthy immune homeostasis and also play pivotal roles in preventing autoimmune diseases. Systemic depletion of Tregs can cause severe inflammation, autoimmune diseases, and allergies in both mice and humans.3,4 The increased number of Tregs in various cancer types, such as gastric, breast, cervical, hepatocellular, renal, melanoma, pancreatic and non-small cell lung cancer, is highly associated with poor prognosis and tumor grade.5–8 However, in some particular cancer types such as colorectal, bladder, and head and neck cancers, high infiltration of Tregs is positively associated with better prognosis.9,10
Inhibitory immune checkpoints such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1) are known targets in cancer immunotherapies. These conventional immunotherapeutic strategies seem to have a better therapeutic effect in patients with higher tumor-specific antigen (TSA) levels. However, TSA is rarely detected in most patients, and only 20–30% of treated patients benefit from conventional immunotherapy. What is worse, a subset of treated patients develop severe adverse reactions, including immune-associated inflammation.11–13 Additionally, CTLA-4 and PD-1 are highly expressed in Tregs, so blockage of CTLA-4 or PD-1 can simultaneously disable the systemic Tregs. Given that Tregs play an essential role in maintaining healthy immune homeostasis, this may partly explain why drugs targeting CTLA-4 or PD-1 can lead to immune-associated inflammation.14,15
Cancer vaccines can be classified as whole-cell tumor vaccines, tumor protein (or peptide) vaccines, genetically engineered (tumor DNA or RNA) vaccines and monoclonal antibody tumor vaccines. Since the US Food and Drug Administration approved the first therapeutic cancer vaccine, Provenge (which treats advanced prostate cancer) on 29 April in 2010,16 therapeutic cancer vaccines have been used to treat cancer. Whole-cell tumor vaccines lack major histocompatibility complex (MHC) dependence and TSA dependence. Whole tumor cells express an array of TSA that are both identified and unidentified. In addition, whole tumor cells contain abundant epitopes of both CD8+ and CD4+ Teff. These features can allow whole-cell tumor vaccines to activate CD4+ and CD8+ Teff more efficiently. Therefore, whole-cell tumor vaccines have better therapeutic effects than other types of cancer vaccines, and they have been regarded as the most developed and promising therapeutic cancer vaccines. However, when used alone, whole-cell tumor vaccines cannot maintain long-term anticancer effects.17 In contrast, combined use of a whole-cell tumor vaccine with a Treg scavenger results in better anticancer immune responses.12
The existing evidence indicates that enhanced tumor cytotoxicity combined with a reduction of tumor-associated Tregs can evoke more effective anticancer immune responses. Additionally, the degree of depletion of tumor-associated Tregs should be taken into account to maximally reduce side effects. Thus, identifying specific biomarkers for tumor-associated Tregs is critical. Here, we summarize the classification, immunosuppressive mechanisms, existing immunotherapies, and potential biomarkers related to tumor-infiltrating Tregs to guide the development of effective cancer immunotherapies.
Treg Classification
Double-positive (DP) CD4+CD8+ T cells undergo positive selection in the thymus. Only DP T cells that can recognize either major histocompatibility complex I (MHCI) or major histocompatibility complex II (MHCII) are allowed to undergo negative selection. During negative selection, transiently activated single-positive (SP) CD4+ T cells show high affinity for antigen-MHCII complexes and can differentiate into regulatory CD4+ T cells. However, persistently activated SP T cells show high affinity for antigen-MHCI/II complexes and lead to apoptosis.13 Tregs formed in the thymus are referred to as natural Tregs (nTregs), which possess high efficiency at limiting overactive immune responses as they can be activated by a lower antigen-MHC complex concentration compared with Teff.18 On the other hand, mature naïve CD4+ T cells can differentiate into Tregs in the presence of transforming growth factor beta (TGF-β) and all-trans retinoic acid (ATRA; a metabolic product of vitamin A), and this type of Treg that form in the periphery are referred to as inducible Tregs (iTregs)19 (Figure 1A).
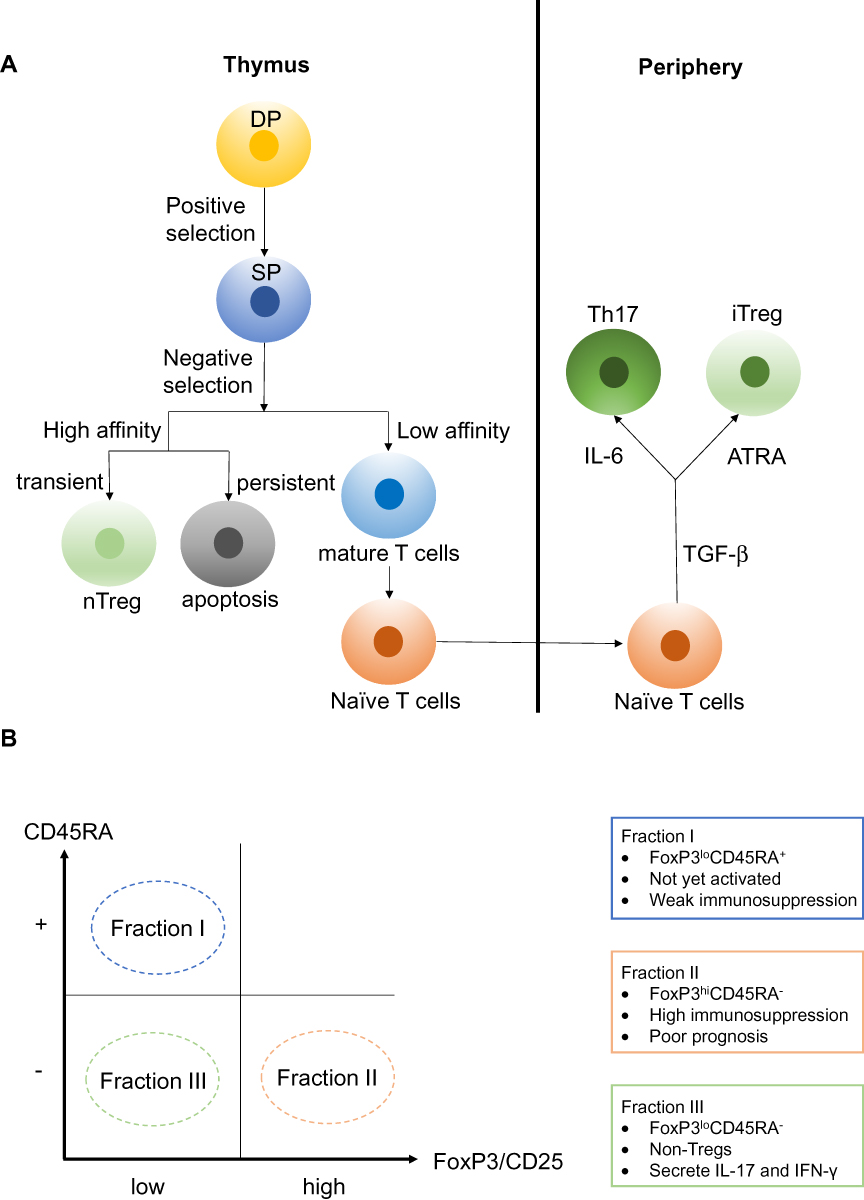 |
Figure 1 (A) Double-positive (DP) CD4+CD8+ T cells can recognize either MHCI or MHCII and are allowed to undergo negative selection. During negative selection, transiently activated single-positive (SP) CD4+ T cells show high affinity for antigen-MHCII complexes and can differentiate into regulatory CD4+ T cells. However, persistently activated SP T cells show high affinity for antigen-MHC complexes and lead to apoptosis. Tregs formed in the thymus are referred to as natural Tregs (nTregs). On the other hand, mature naïve CD4+ T cells can differentiate into Tregs in the presence of transforming growth factor beta (TGF-β) and all-trans retinoic acid (ATRA), and this type of Tregs formed in the periphery are referred to as inducible Tregs (iTregs). (B) Tregs can divide into three fractions based on the differential expression levels of CD45RA and FoxP3. Fraction I is the FoxP3loCD45RA+ subpopulation, also referred to as naïve Tregs (nTregs). This set of Tregs is not yet activated and thus possess weak immunosuppressive activity. Fraction II is the FoxP3hiCD45RA− subpopulation, referred to as effector Tregs (eTregs), and they possess high immunosuppressive activity. Fraction III is the FoxP3loCD45RA− subpopulation, referred to as non-Tregs, and they lack immunosuppressive activity. However, FoxP3loCD45RA− non-Tregs can secrete pro-inflammatory cytokines such as IL-17 and IFN-γ. |
nTregs and iTregs are classified based on the site of differentiation. A second classification explains why a high level of tumor-infiltrating Tregs can indicate different prognoses even within the same malignancy type. In this classification, Tregs can be divided into three fractions based on the differential expression levels of CD45RA and FoxP3 (or CD25 can replace the indicator FoxP3). Fraction I is the FoxP3loCD45RA+ subpopulation, also referred to as naïve Tregs (nTregs). This set of Tregs is not yet activated and thus possesses weak immunosuppressive activity. In addition, Fraction I can differentiate into Fraction II under antigenic stimulation. Fraction II is the FoxP3hiCD45RA− subpopulation, referred to as effector Tregs (eTregs), which possess high immunosuppressive activity. Colorectal cancer (CRC) patients with tumor-infiltrating Tregs dominated by Fraction II tend to have a poor prognosis.10 Fraction III is the FoxP3loCD45RA− subpopulation, referred to as non-Tregs. This subpopulation lacks immunosuppressive activity. However, FoxP3loCD45RA− non-Tregs can secrete pro-inflammatory cytokines such as interleukin (IL)-17 and interferon (IFN)-γ. CRC patients with tumor-infiltrating FoxP3+ T cells dominated by this Fraction III tend to have a better prognosis10,20 (Figure 1B).
Immunosuppressive Mechanisms of Tregs
The immunoregulatory mechanisms of Tregs are complicated. There are several known mechanisms (Figure 2): (1) Direct intercellular contact may be the primary suppressive mechanism of Tregs.21 (2) Tregs secrete immunosuppressive cytokines, such as IL-10, IL-35 and TGF-β. Additionally, they induce adjacent immune cells such as dendritic cells (DC) to secrete IL-10.22 IL-10 is often considered as a significant factor controlling the immunosuppressive TME. IL-10 can stimulate the expression of the E3 ubiquitin ligase March-I in activated macrophages. Additionally, IL-10 can inhibit DC self-activation, thereby downregulating MHC-II and antigen presentation to CD4 T cells.23 However, it has been reported that IL-10 can activate CD4+ T cells and CD8+ Teff under certain in vitro and in vivo conditions,24 and it has also been reported that the IL10-dependent signaling pathway may not be the critical immunosuppressive mechanism of Tregs.25 Therefore, the paradoxical effects of IL-10 on tumors need further clarification. (3) Tregs produce granzyme B, which directly leads to Teff’s death.26 (4) Direct contact of Tregs with DCs can induce DCs to produce and secrete indoleamine 2, 3-dioxygenase (IDO). IDO selectively impairs the function of Teff by producing the toxic catabolic product kynurenine.27,28 (5) Tregs secrete the extracellular enzymes CD39 (ectonucleoside triphosphate diphosphohydrolase) and CD73. CD39 can degrade ATP or ADP into AMP, and then AMP is degraded into adenosine by CD73. Adenosine binds to the adenosine receptors (or P1 receptors), which are expressed by activated Teff, thus inducing an immunosuppressive effect.29 (6) Immune checkpoints such as TIGIT, LAG-3 and CTLA-4 are highly expressed on the surface of tumor-infiltrating eTregs. TIGIT can induce adjacent DCs to secrete IL-10 and LAG-3 binds to ligands such as MHC-II, which is expressed by antigen presentation cells (APCs), to induce APCs’ death.30,31 The human monoclonal antibodies BMS-986207 and MK-4280 (which target TIGIT and LAG-3, respectively) plus nivolumab or pembrolizumab (anti-PD-1 antibodies) are currently undergoing Phase 1 clinical trials in patients with advanced solid tumors. The results show that no autoimmune disease has appeared so far.32,33 Moreover, autoimmunity did not occur in TIGIT-deficient or LAG-3- deficient mice.32,33 This evidence indicates that TIGIT and LAG-3 are mainly expressed on tumor-associated Tregs and may play a dispensable role in maintaining homeostasis. (7) Tregs inhibit Teff proliferation due to Tregs highly constitutively expressing CD25 (also known as IL-2RA, a high-affinity receptor for IL-2). IL-2 is produced mainly by conventional T cells (Tconv), and it is hardly secreted by Tregs because FoxP3 binds to and attenuates two transcription factors that are required for the production of IL-2. IL-2 is an essential cytokine for the proliferation of both T and B lymphocytes. Therefore, CD25 highly constitutively expressed on Tregs gives Tregs an advantage regarding competitively binding to IL-2 to inhibit Teff proliferation.34,35 (8) The antigen concentration required for stimulating CD25+CD4+ T cells to exert suppression is much lower than that required for stimulating CD25-CD4+ T cells to proliferate.18 (9) The programmed cell death 1 (PD-1)/PD-ligand (PD-L) pathway can promote the development and enhance the function of Tregs. At sites where TGF-β is present, PD-L1 can promote the de novo generation of CD4+FoxP3+ iTregs from naïve CD4+ T cells. PD-L1 can also enhance and maintain the suppressive function of established iTregs.36 In mechanistic studies, PD‐L1 induces the production of iTregs from naïve T cells by attenuating Akt‐mTOR signaling and concomitantly upregulating PTEN.36 More specifically, the development of PD-L1 iTregs is mediated through the downregulation of phospho-Akt, mTOR, S6, ERK2 and the concomitant upregulation of PTEN.14,36
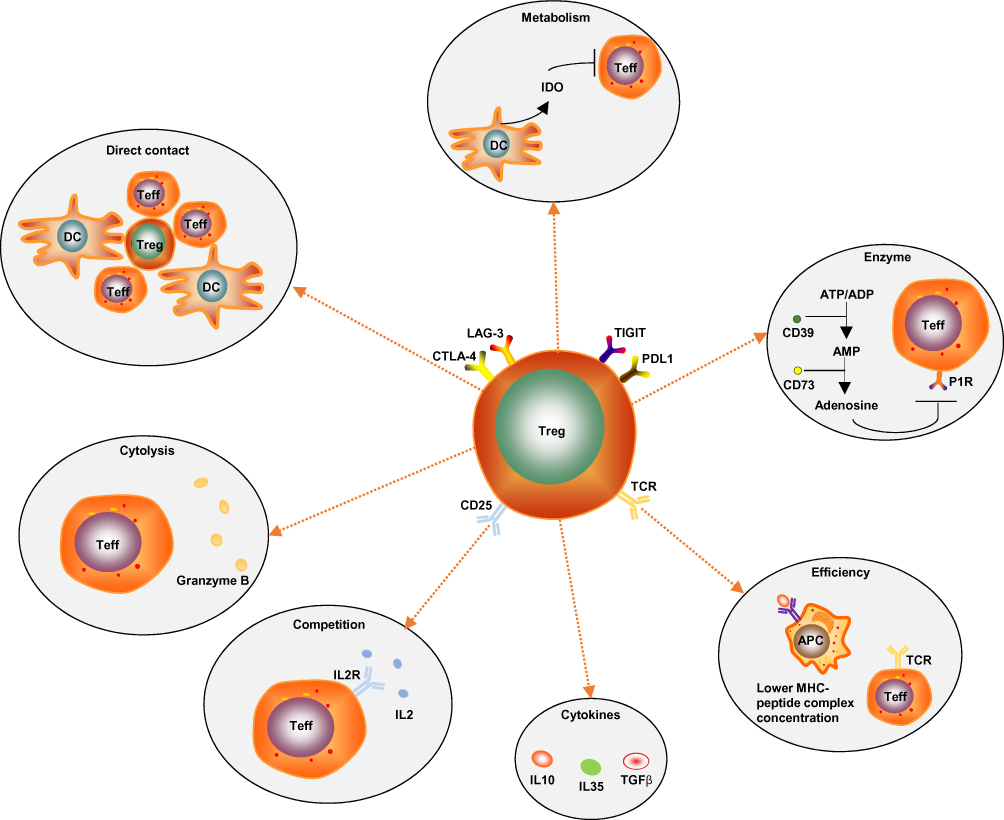 |
Figure 2 Known immunoregulatory mechanisms of Tregs: (1) Direct intercellular contact may be the primary suppressive mechanism of Tregs. (2) Tregs produce granzyme B, which directly leads to Teff’s death. (3) CD25 is highly constitutively expressed on Tregs, which gives Tregs an advantage regarding competitively combining with IL-2 to inhibit Teff proliferation. (4) Tregs secrete immunosuppressive cytokines, such as IL-10, IL-35 and TGF-β. (5) The antigen concentration required for stimulating CD25+CD4+ T cells to exert suppression is much lower than that required for stimulating CD25-CD4+ T cells to proliferate. (6) Tregs secrete the extracellular enzymes CD39 and CD73. CD39 can degrade ATP or ADP into AMP, and then AMP is degraded into adenosine by CD73. Adenosine binds to the adenosine receptors (P1 receptors), which are expressed by activated Teff, thus inducing an immunosuppressive effect. (7) Direct contact of Tregs with DCs can induce DCs to produce and secrete indoleamine 2, 3-dioxygenase (IDO). IDO selectively impairs Teff function by producing the toxic catabolic product kynurenine. (8) Immune checkpoints such as TIGIT, LAG-3, CTLA-4 and PD-L1 are highly expressed on the surface of tumor-infiltrating eTregs. |
Existing Treg Immunotherapies
The known mechanisms of tumor immune escape involve altering tumor antigens so that they are expressed less by immunogenic tumor cells, and causing the MHC allele in leukocytes to be lost to reduce their ability to present neoantigens.37–39 Furthermore, high infiltration of eTregs in the TME is positively associated with a poor prognosis for many cancer types. Developing therapies to combat these mechanisms will expand the therapeutic anticancer strategies. Here, we summarize the existing Treg immunotherapies to treat cancers.
Anti-CTLA-4 Antibodies
CD28 is the natural ligand of B7 family members (including CD80 and CD86), and the inhibitory immune checkpoint CTLA-4 is the congener of CD28. B7 family members are expressed on activated APCs, and the B7-CD28 signaling pathway plays an essential role in activating the second co-stimulatory signals of T cells. Physiologically, when the second signals are not activated, even though the first signals have been initiated by the binding of the antigen-MHC complexes to TCRs, the T cells cannot achieve complete activation. The affinity between CTLA-4 and B7 family members is a hundred times greater than the affinity between CD28 and B7 family members. CTLA-4 is highly expressed in both Tregs and Teff in patients with tumors.40 Due to these characteristics of CTLA-4, it can outcompete CD28 regarding binding to B7 family members and inhibit the second signals of Teff.15 Additionally, CTLA-4+ Tregs can reduce the expression level of B7 family members on APCs, thereby promoting tumor immune escape.41
The anti-CTLA-4 antibodies ipilimumab and tremelimumab were initially used with the aim of restoring the tumor cytotoxicity of Teff. However, the major antitumor effect of anti-CTLA-4 antibodies is now considered to be a result of their cytotoxicity against Tregs.42 Moreover, Fc receptors (FcR), which are mostly expressed on innate immunocytes, can recognize and combine with the Fc fragment of antibodies, thus inducing innate immunocytes to mediate antibody-dependent cell-mediated cytotoxicity (ADCC).43 The effect of anti-CTLA-4 antibodies partly depends on the binding affinity of the human Fc fragment of IgG receptors (FcγRs) and highly immunogenic tumors such as advanced melanoma. Preclinical trial results44 showed that antibodies with isotypes equivalent to anti-CTLA-4 antibodies mediate tumor-associated Treg depletion in vivo. Antibodies with improved FcγR binding affinity have superior antitumor responses and survival outcomes and, in particular,45 the IgG1 isotype confers higher relative binding affinity than IgG2.
A pooled meta-analysis of the long-term outcomes of patients with melanoma treated with ipilimumab showed that in Phase II and III clinical trials, some patients have a long survival period, sometimes exceeding 10 years. However, only 20–30% of the participants had long-lasting antitumor immune responses, and these participants frequently experienced severe autoimmune disease.11 In a phase II clinical trial for patients with mesothelioma treated with tremelimumab plus durvalumab (an anti-PD-L1 antibody), 63% of patients experienced disease control, but 75% of patients developed treatment-related adverse effects.46
Anti-CD25 Antibodies
CD25 is highly constitutively expressed on Tregs, which gives Tregs an advantage regarding competitively binding with IL-2 to inhibit the proliferation of Teff. This characteristic of Tregs is considered as an immunotherapeutic target. In a preclinical trial, the anti-CD25 antibody daclizumab resulted in selective downregulation of FoxP3 among Fraction II Tregs; moreover, Fraction II Tregs in the daclizumab group could be converted into Fraction III Tregs and obtain the ability to produce IFN-γ.47 In a clinical trial for patients with metastatic breast cancer treated with daclizumab plus an anticancer vaccine, the daclizumab group had a significant and prolonged decrease in Tregs.47 In contrast, a preclinical trial showed that anti-CD25 antibody depletes peripheral Tregs, but not tumor-infiltrating Tregs.48 In Phase I and II clinical trials for patients with metastatic melanoma pretreated with daclizumab before DC vaccine treatment, the daclizumab pretreatment group had all CD25high immune cells depleted from their circulation, but there was no significant effect on the progression-free survival compared with the control group, as daclizumab pretreatment could not maintain a durable depletion of CD25high immune cells.49
Cyclophosphamide (CTX)
CTX is an alkylating nitrogen mustard antineoplastic agent that undergoes biotransformation in the liver to produce the active form aldophosphamide. The immunosuppressive mechanisms of CTX involve inducing cross-linkages between DNA strands, inhibiting nucleic acid replication and inducing polarization of Th1 cells (a subpopulation of T helper cells). Its mechanism also involves transiently increasing the levels of interferon regulatory factor-1 (IRF-1). Downstream effectors like caspase-1 and IL-1β are subsequently increased in a direct IRF-1-dependent manner, while IL-6 and CXCL10 are decreased in an indirect IRF-1-dependent manner.50
The application of high-dose CTX severely affects all T cell types, whereas low-dose CTX with an extended treatment cycle selectively reduces the high proliferation of Tregs by decreasing the expression of FoxP3.51 In phase I and II clinical trials for patients with metastatic CRC treated with 2-week-long courses of low-dose CTX, there was significant Teff activation with an absolute reduction of Tregs.52 Similar therapeutic effects were shown in a phase I clinical trial for patients with metastasized breast cancer; depletion of Tregs was mirrored by a significant boost in tumor-reactive T cells.53 However, CTX often causes adverse reactions such as myelosuppression, excessive immunosuppression and opportunistic infections.
The existing immunotherapies (summarized in Table 1) have similar drawbacks in that they are not specific enough to target tumor-associated eTregs. Given that peripheral Tregs are essential in maintaining host homeostasis, this may partly explain why these immunotherapies often lead to severe autoimmune disease. The following strategy may provide an idea for how to solve this problem:54 based on identifying new biomarkers that allow accurate identification of tumor-associated eTregs, targeted medicines may preserve peripheral Tregs and prevent autoimmune diseases. Therefore, it is imperative to identify potential biomarkers specific to tumor-associated eTregs.
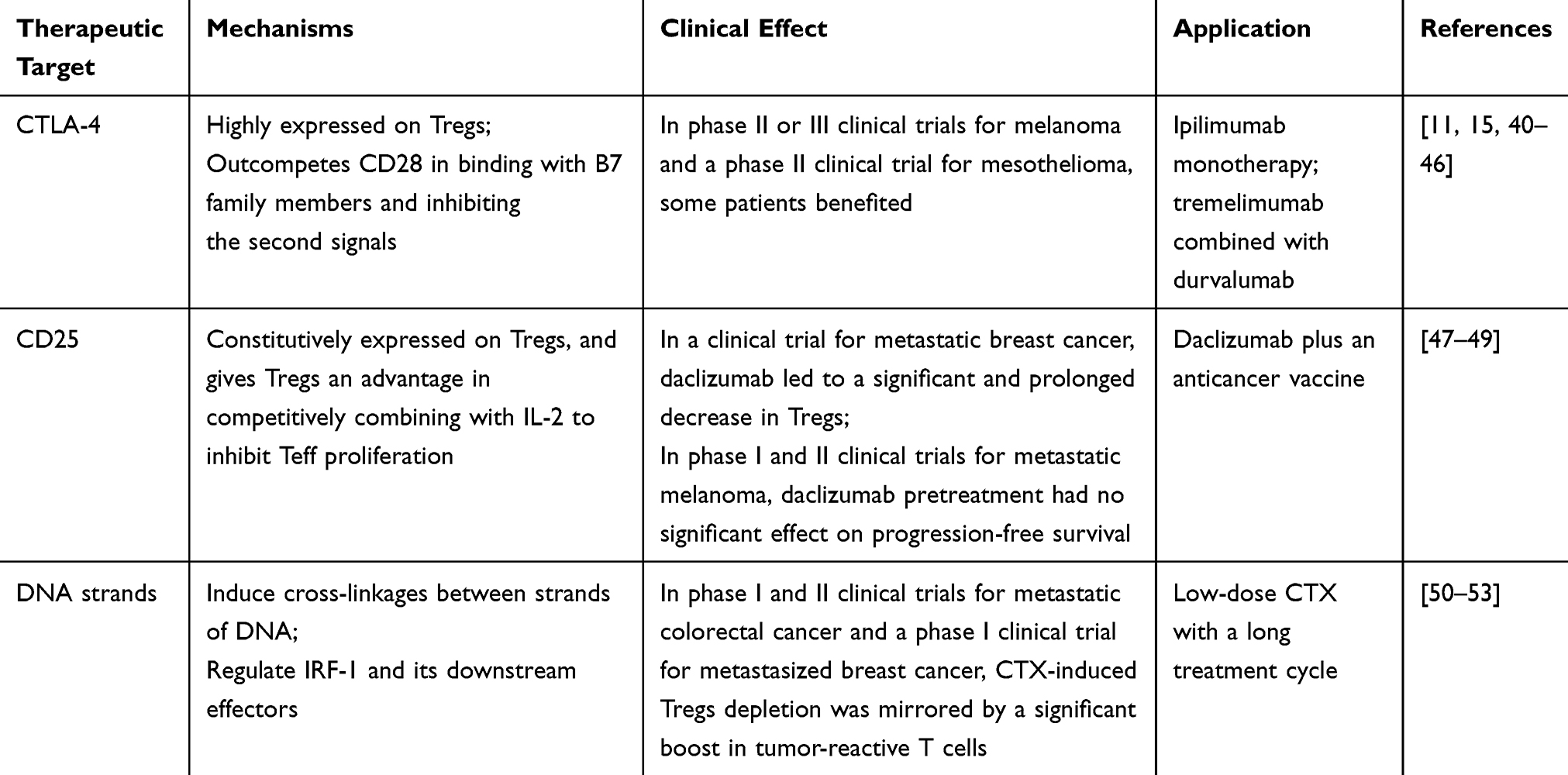 |
Table 1 Existing Treg Immunotherapies |
Potential Treg Biomarkers
Cancer immunotherapies can be classified into two types: those that can recover the tumor-killing effect of Teff and those that remove the immunosuppression of the TME. Chimeric antigen receptor effector T cells and TCR-engineered T cells belong to the former type, while the existing immunotherapies mentioned in this review belong to the latter type. Given that tumor-associated Tregs are important effectors in the TME, the intention of the following section is to summarize and describe more specific targets regarding tumor-associated Tregs in order to attain better therapeutic effects and minimize off-target adverse events.
CCR6
The number of thymic recirculating Tregs is lower in CCR6−/- mice than in wild-type controls, which suggests that CCR6 is a critical component involved in the circulation of peripheral Tregs. Additionally, high CCR6 expression in tumor-associated Tregs is positively associated with tumor progression, including in patients with laryngeal squamous cell carcinoma, hepatocellular carcinoma and breast cancer.55–58 Besides, CCL20 (a CCR6 ligand) has been detected in medullary thymic epithelial cells (mTECs)59 and diverse cancer stem cells (CSCs) including cells from pancreatic, colorectal, gastric, lung, breast and head and neck cancer.60–62 In the preclinical trial for advanced cutaneous T-cell lymphoma, knockdown of CCR6 by micro RNA-150 (miR-150) led to a distinct decrease in tumor metastasis and invasion.63 Similarly, altering the enrichment of CCR6+ Tregs in the TME can lead to a beneficial antitumor effect against breast cancer.64 The CCR6-CCL20 axis is therefore considered as an essential pathway in chemotaxis and functions in tumor-associated Tregs.
CXCR4
CXCR4+ Tregs are abundant in the bone marrow of terminal cancer patients, and CXCL12 (a CXCR4 ligand), which can recruit CXCR4+ Tregs to enter the bone marrow, is highly expressed in the bone marrow.65 Besides, CXCR4-CXCL12 is positively related to tumor advancement and metastasis in ovarian carcinoma and non-small cell lung cancer (NSCLC).6,66–68 Administration of granulocyte colony-stimulating factor (G-CSF) to deplete CXCL12 can remove Tregs in the bone marrow in both humans and mice.69 This partly explains the many kinds of late malignancies that often accompany osseous metastasis. CXCR4 is also expressed on tumor cells, and CXCL12 can directly promote tumor progression via the CXCR4-CXCL12 axis under the synergistic effect of vascular endothelial growth factor (VEGF). In preclinical trials for primary brain tumors, non-Hodgkin’s lymphoma and breast cancer, blocking CXCR4 inhibited tumor progression and prolong survival.70–72 The mechanisms involved, including blockage of CXCR4, can enhance antitumor immune responses mediated by Teff and induce Treg conversion into T helper cells. The effect of CXCR4 blockage is related to both high CXCR4 expression and chemotactic responses to CXCL12 in ovarian cancer.73,74
CCR4
CCR4 is abundantly expressed on tumor-associated eTregs in various types of cancer, including breast, bladder, colorectal, ovarian and oral squamous cancer and Hodgkin’s lymphoma. Additionally, the expression of CCR4 on Tregs is positively associated with poor prognosis in ovarian carcinoma, oral squamous cell carcinoma, Hodgkin’s lymphoma, colon adenocarcinoma, primary breast cancer and bladder cancer.6,75–79 CCR4 is essential for the migration of Tregs to non-lymphoid tissues; in contrast, Tregs that scarcely express CCR4 lack the ability to migrate.80 The mechanisms of immunosuppression of CCR4+ Tregs relates to the fact that CCR4+ Tregs possess high chemotaxis ability and can also inhibit the activation of T and NK cells via TGF-β signaling pathways.81 CCR4 blockage can selectively deplete tumor-associated eTregs and effectively increase the number of Teff in human and canine models.79,82 The anti-CCR4 antibody mogamulizumab first underwent a clinical trial in humans in 2007.83 It was approved in Japan in 2012 for the treatment of CCR4+ adult T-cell lymphoma (ATCLL), and for CCR4+ cutaneous T cell lymphoma (CTCL) in 2014.84 However, the therapeutic effect of CCR4 blockage in other types of solid tumors requires further investigations.
CCR8
CCR8 is mainly expressed in tumor-associated eTregs in NSCLC, CRC and breast cancer,40,85–88 and high expression of CCR8 in NSCLC and CRC is positively associated with poor prognosis. Furthermore, peripheral Fraction II Tregs are phenotypically the closest to tumor-associated Tregs, which implies that tumor-associated Tregs in the TME may be derived from the peripheral blood.87,88 The effects of CCR8 on Tregs involve prompting differentiation, survival, function and migration of Tregs via the STAT3 signaling pathway, but CCR8 does not influence Treg proliferation in CRC and graft-versus-host disease.25,89 The anti-CCR8 antibody prevents naïve T cells from differentiating into Tregs and inhibiting the immunosuppression of tumor-associated Tregs, but without influencing the function of peripheral Tregs in CRC.90 CCL1 is a recognized ligand of CCR8 that is secreted by activated T cells or Tregs. CCL1 upregulates the expression of CCR8 and other factors such as FoxP3, CD39, granzyme B and IL-10 in Tregs.25 The anti-CCL1 antibody can prevent de novo conversion of Tregs, which is consistent with the effect of the anti-CCR8 antibody.91 Another known ligand of CCR8 is CCL18. Tumor-associated Tregs secrete IL-10, which causes tumor-associated macrophages (TAM) to abundantly express the chemokine CCL18. This enhances the immunosuppression of tumor-associated Tregs, thus forming a vicious cycle that leads to tumor progression.92–94 Knockdown of CCL18 reduces tumor growth and invasiveness in bladder cancer and esophageal squamous cell carcinoma.93,95
Conclusion and Perspective
The existing immunotherapeutic strategies need to be improved. Take anti-CTLA4 antibodies as an example. The binding affinity of human FcγRs and their relative abundance are rarely considered, but the effect of anti-CTLA-4 antibodies partly depends on the binding affinity of human FcγRs and highly immunogenic tumors. Thus, FcγR polymorphism status and tumor mutational burden should be taken into account during the selection of patients who are likely to benefit from an anti-CTLA-4 antibody.45 In addition, to obtain the best curative effect, adjusting the therapeutic dosage, course and administration route is of prime importance. For instance, it is necessary to deplete tumor-associated eTregs before activating Teff.96
Existing immunotherapies targeting Tregs can lead to severe autoimmune diseases due to both robust on-target mechanisms as well as off-target mechanisms. On-target adverse events are dependent on drug characteristics and patient heterogeneity, which is hard to control. Therefore, the Potential Treg biomarkers section in this review aimed to summarize and describe more specific targets regarding tumor-associated Tregs in order to minimize off-target adverse events. The chemokines that we selected in this review have comparatively clear and specific ligands, and the chemokine-receptor axes mentioned are involved in Treg chemotaxis, function or differentiation and show high potential as phenotypic and functional markers of tumor-associated eTregs (as summarized in Table 2). However, evidence showing the efficiency of the potential biomarkers mainly come from animal models, and clinical data are still lacking. Thus, we hope that more researchers can focus on this research field and give pay attention to the potential biomarkers.
 |
Table 2 Potential Treg Biomarkers |
Abbreviations
eTregs, effector regulatory T cells; Tregs, regulatory T cells; NSCLC, non-small cell lung cancer; Teff, effector T cells; TSA, tumor specific antigens; TME, tumor microenvironment; DP, double positive cells; SP, single positive cells; nTregs, natural Tregs or naive Tregs; cTregs, central Tregs; CRC, colorectal cancer; DC, dendritic cells; IDO, indoleamine 2, 3-dioxygenase; APCs, antigen-presenting cells; CTX, cyclophosphamide; IRF-1, interferon regulatory factor-1; mTECs, medullary thymic epithelial cells; CSCs, cancer stem cells; miR-150, micro RNA-150; G-CSF, granulocyte colony stimulating factor; VEGF, vascular endothelial growth factor; TAM, tumor-associated macrophages; ATCLL, CCR4+ adult T-cell lymphoma; CTCL, CCR4+ cutaneous T cell lymphoma; GZMB, granzyme B; CTLA-4, Cytotoxic T-Lymphocyte Associated Protein 4; PD-1, programmed cell death 1; PD-L, PD-ligand; TGF-β, transforming growth factor beta; ATRA, all-transretinoic acid; FoxP3, forkhead transcription factor P3; MHCI, major histocompatibility complex I; MHCII, major histocompatibility complex II; Tconv, conventional T cells; TCR, T cells receptor; FcR, Fc receptors; ADCC, antibody-dependent cell-mediated cytotoxicity; FcγRs, Fc fragment of IgG receptors.
Disclosure
The authors of this review state that they have written the complete article by themselves, and they have no conflicts of interest in this work.
References
1. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80. doi:10.1126/science.aaa6204
2. Spill F, Reynolds DS, Kamm RD, Zaman MH. Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol. 2016;40:41–48. doi:10.1016/j.copbio.2016.02.007
3. Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27(1):20–21. doi:10.1038/83713
4. Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27(1):68–73. doi:10.1038/83784
5. Sasada T, Kimura M, Yoshida Y, Kanai M, Takabayashi A. CD4(+)CD25(+) regulatory T cells in patients with gastrointestinal malignancies - possible involvement of regulatory T cells in disease progression. Cancer. 2003;98(5):1089–1099. doi:10.1002/cncr.11618
6. Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi:10.1038/nm1093
7. Bates GJ, Fox SB, Han C, et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol. 2006;24(34):5373–5380. doi:10.1200/JCO.2006.05.9584
8. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. 2015;5:15179. doi:10.1038/srep15179
9. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi:10.1038/nrc3245
10. Saito T, Nishikawa H, Wada H, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. 2016;22(6):679–684. doi:10.1038/nm.4086
11. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi:10.1016/j.ccell.2015.03.001
12. Le DT, Jaffee EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res. 2012;72(14):3439–3444. doi:10.1158/0008-5472.CAN-11-3912
13. Bending D, Martin PP, Paduraru A, et al. A timer for analyzing temporally dynamic changes in transcription during differentiation in vivo. J Cell Biol. 2018;217(8):2931–2950. doi:10.1083/jcb.201711048
14. Francisco LM, Salinas VH, Brown KE, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–3029. doi:10.1084/jem.20090847
15. Walker LSK. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun. 2013;45:49–57. doi:10.1016/j.jaut.2013.06.006
16. [No authors listed]. Cancer vaccine approval could open floodgates. Nat Med. 2010;16(6):615. doi:10.1038/nm0610-615b
17. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi:10.1016/j.cell.2017.01.017
18. Takahashi T, Kuniyasu Y, Toda M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10(12):1969–1980. doi:10.1093/intimm/10.12.1969
19. Weaver C, Hatton R. Interplay between the TH17 and TReg cell lineages: a (co-)evolutionary perspective. Nat Rev Immunol. 2009;9(12):883–889. doi:10.1038/nri2660
20. Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30(6):899–911. doi:10.1016/j.immuni.2009.03.019
21. Maganto-Garcia E, Bu DX, Tarrio ML, et al. Foxp3+-inducible regulatory T cells suppress endothelial activation and leukocyte recruitment. J Immunol. 2011;187(7):3521–3529. doi:10.4049/jimmunol.1003947
22. Whitehead GS, Wilson RH, Nakano K, Burch LH, Nakano H, Cook DN. IL-35 production by inducible costimulator (ICOS)-positive regulatory T cells reverses established IL-17-dependent allergic airways disease. J Allergy Clin Immunol. 2012;129(1):
23. Mittal SK, Cho KJ, Ishido S, Roche PA. Interleukin 10 (IL-10)-mediated Immunosuppression: march-I induction regulates antigen presentation by macrophages but not dendritic cells. J Biol Chem. 2015;290(45):27158–27167. doi:10.1074/jbc.M115.682708
24. Ouyang W, O’Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. 2019;50(4):871–891. doi:10.1016/j.immuni.2019.03.020
25. Barsheshet Y, Wildbaum G, Levy E, et al. CCR8(+)FOXp3(+) Treg cells as master drivers of immune regulation. Proc Natl Acad Sci U S A. 2017;114(23):6086–6091. doi:10.1073/pnas.1621280114
26. Perrella A, D’Antonio A, Sbreglia C, et al. CD4+/CD25+ T cells suppress autologous CD4+/CD25- lymphocytes and secrete granzyme B during acute and chronic hepatitis C. Pathog Dis. 2014;72(2):124–130. doi:10.1111/2049-632X.12190
27. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–143. doi:10.1016/j.it.2012.10.001
28. Jiang T, Sun Y, Yin Z, Feng S, Sun L, Li Z. Research progress of indoleamine 2,3-dioxygenase inhibitors. Future Med Chem. 2015;7(2):185–201. doi:10.4155/fmc.14.151
29. Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204(6):1257–1265. doi:10.1084/jem.20062512
30. Joller N, Lozano E, Burkett PR, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40(4):569–581. doi:10.1016/j.immuni.2014.02.012
31. Bauche D, Joyce-Shaikh B, Jain R, et al. LAG3(+) regulatory T cells restrain interleukin-23-producing CX3CR1(+) gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity. 2018;49(2):342–352.e345. doi:10.1016/j.immuni.2018.07.007
32. Bhagwat B, Cherwinski H, Sathe M, et al. Establishment of engineered cell-based assays mediating LAG3 and PD1 immune suppression enables potency measurement of blocking antibodies and assessment of signal transduction. J Immunol Methods. 2018;456:7–14. doi:10.1016/j.jim.2018.02.003
33. Jiang H, Kozhich A, Cummings J, et al. Singlicate ligand binding assay using an automated microfluidic system: a clinical case study. AAPS J. 2017;19(5):1461–1468. doi:10.1208/s12248-017-0105-5
34. Wyss L, Stadinski BD, King CG, et al. Affinity for self antigen selects Treg cells with distinct functional properties. Nat Immunol. 2016;17(9):1093–1101. doi:10.1038/ni.3522
35. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201(5):723–735. doi:10.1084/jem.20041982
36. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi:10.1111/j.1600-065X.2010.00923.x
37. Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–404. doi:10.1038/nature10755
38. McGranahan N, Rosenthal R, Hiley CT, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171(6):1259–1271.e1211. doi:10.1016/j.cell.2017.10.001
39. Scheper W, Kelderman S, Fanchi LF, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. 2018.
40. Plitas G, Konopacki C, Wu K, et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity. 2016;45(5):1122–1134. doi:10.1016/j.immuni.2016.10.032
41. Alissafi T, Banos A, Boon L, et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J Clin Invest. 2017;127(7):2789–2804. doi:10.1172/JCI92079
42. Selby MJ, Engelhardt JJ, Quigley M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1(1):32–42. doi:10.1158/2326-6066.CIR-13-0013
43. Lee CH, Romain G, Yan W, et al. IgG Fc domains that bind C1q but not effector Fcgamma receptors delineate the importance of complement-mediated effector functions. Nat Immunol. 2017;18(8):889–898.
44. Arce Vargas F, Furness AJS, Litchfield K, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. 2018;33(4):649–663.e644. doi:10.1016/j.ccell.2018.02.010
45. Ha D, Tanaka A, Kibayashi T, et al. Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U S A. 2019;116(2):609–618. doi:10.1073/pnas.1812186116
46. Calabro L, Morra A, Giannarelli D, et al. Tremelimumab combined with durvalumab in patients with mesothelioma (NIBIT-MESO-1): an open-label, non-randomised, Phase 2 study. Lancet Respir Med. 2018;6(6):451–460. doi:10.1016/S2213-2600(18)30151-6
47. Rech AJ, Mick R, Martin S, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4(134):134ra162. doi:10.1126/scitranslmed.3003330
48. Arce Vargas F, Furness AJS, Solomon I, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. 2017;46(4):577–586. doi:10.1016/j.immuni.2017.03.013
49. Jacobs JF, Punt CJ, Lesterhuis WJ, et al. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res. 2010;16(20):5067–5078. doi:10.1158/1078-0432.CCR-10-1757
50. Buccione C, Fragale A, Polverino F, et al. Role of interferon regulatory factor 1 in governing Treg depletion, Th1 polarization, inflammasome activation and antitumor efficacy of cyclophosphamide. Int J Cancer. 2018;142(5):976–987. doi:10.1002/ijc.v142.5
51. Motoyoshi Y, Kaminoda K, Saitoh O, et al. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol Rep. 2006;16(1):141–146.
52. Scurr M, Pembroke T, Bloom A, et al. Low-dose cyclophosphamide induces antitumor T-cell responses, which associate with survival in metastatic colorectal cancer. Clin Cancer Res. 2017;23(22):6771–6780. doi:10.1158/1078-0432.CCR-17-0895
53. Ge Y, Domschke C, Stoiber N, et al. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunol Immunother. 2012;61(3):353–362. doi:10.1007/s00262-011-1106-3
54. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–118. doi:10.1038/cr.2016.151
55. Chen B, Zhang D, Zhou J, et al. High CCR6/CCR7 expression and Foxp3+ Treg cell number are positively related to the progression of laryngeal squamous cell carcinoma. Oncol Rep. 2013;30(3):1380–1390. doi:10.3892/or.2013.2603
56. Li WM, Liu HR. CCL20-CCR6 cytokine network facilitate Treg activity in advanced grades and metastatic variants of hepatocellular carcinoma. Scand J Immunol. 2016;83(1):33–37. doi:10.1111/sji.12367
57. Chen KJ, Lin SZ, Zhou L, et al. Selective recruitment of regulatory T cell through CCR6-CCL20 in hepatocellular carcinoma fosters tumor progression and predicts poor prognosis. PLoS One. 2011;6(9):e24671. doi:10.1371/journal.pone.0024671
58. Xu L, Xu W, Qiu S, Xiong S. Enrichment of CCR6+Foxp3+ regulatory T cells in the tumor mass correlates with impaired CD8+ T cell function and poor prognosis of breast cancer. Clin Immunol. 2010;135(3):466–475. doi:10.1016/j.clim.2010.01.014
59. Cowan JE, Baik S, McCarthy NI, et al. Aire controls the recirculation of murine Foxp3(+) regulatory T-cells back to the thymus. Eur J Immunol. 2018;48(5):844–854. doi:10.1002/eji.201747375
60. Marsigliante S, Vetrugno C, Muscella A. CCL20 induces migration and proliferation on breast epithelial cells. J Cell Physiol. 2013;228(9):1873–1883. doi:10.1002/jcp.24349
61. van Vlerken-ysla LE, Rios-Doria J, Moynihan J, et al. Targeting the CCL20-CCR6 axis as a novel opportunity to stimulataneously modulate cancer stem cells and the tumor-immune infiltrate by a dual anti-cancer mechanism. Cancer Res. 2017:77.
62. Kryczek I, Lin Y, Nagarsheth N, et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. 2014;40(5):772–784. doi:10.1016/j.immuni.2014.03.010
63. Ito M, Teshima K, Ikeda S, et al. MicroRNA-150 inhibits tumor invasion and metastasis by targeting the chemokine receptor CCR6, in advanced cutaneous T-cell lymphoma. Blood. 2014;123(10):1499–1511. doi:10.1182/blood-2013-09-527739
64. Hu Y, Wang C, Li Y, et al. MiR-21 controls in situ expansion of CCR6(+) regulatory T cells through PTEN/AKT pathway in breast cancer. Immunol Cell Biol. 2015;93(8):753–764. doi:10.1038/icb.2015.37
65. Zhao E, Wang L, Dai J, et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology. 2012;1(2):152–161. doi:10.4161/onci.1.2.18480
66. Kryczek I, Lange A, Mottram P, et al. CXCL12 and vascular endothelial growth factor synergistically induce neoangiogenesis in human ovarian cancers. Cancer Res. 2005;65(2):465–472.
67. Jung MJ, Rho JK, Kim YM, et al. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene. 2013;32(2):209–221. doi:10.1038/onc.2012.37
68. Cioffi M, D’Alterio C, Camerlingo R, et al. Identification of a distinct population of CD133(+)CXCR4(+) cancer stem cells in ovarian cancer. Sci Rep. 2015;5:10357. doi:10.1038/srep10357
69. Zou L, Barnett B, Safah H, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004;64(22):8451–8455. doi:10.1158/0008-5472.CAN-04-1987
70. Rubin JB, Kung AL, Klein RS, et al. A small-molecule antagonist of CXCR4 inhibits intracranial growth of primary brain tumors. Proc Natl Acad Sci U S A. 2003;100(23):13513–13518. doi:10.1073/pnas.2235846100
71. Bertolini F, Dell’Agnola C, Mancuso P, et al. CXCR4 neutralization, a novel therapeutic approach for non-Hodgkin’s lymphoma. Cancer Res. 2002;62(11):3106–3112.
72. Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005;65(3):967–971.
73. Righi E, Kashiwagi S, Yuan J, et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 2011;71(16):5522–5534. doi:10.1158/0008-5472.CAN-10-3143
74. Zeng Y, Li B, Liang Y, et al. Dual blockade of CXCL12-CXCR4 and PD-1-PD-L1 pathways prolongs survival of ovarian tumor-bearing mice by prevention of immunosuppression in the tumor microenvironment. FASEB J. 2019;33(5):6596–6608. doi:10.1096/fj.201802067RR
75. Watanabe Y, Katou F, Ohtani H, Nakayama T, Yoshie O, Hashimoto K. Tumor-infiltrating lymphocytes, particularly the balance between CD8(+) T cells and CCR4(+) regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2010;109(5):744–752. doi:10.1016/j.tripleo.2009.12.015
76. Ishida T, Ishii T, Inagaki A, et al. Specific recruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lymphoma fosters immune privilege. Cancer Res. 2006;66(11):5716–5722. doi:10.1158/0008-5472.CAN-06-0261
77. Svensson H, Olofsson V, Lundin S, et al. Accumulation of CCR4(+)CTLA-4 FOXP3(+)CD25(hi) regulatory T cells in colon adenocarcinomas correlate to reduced activation of conventional T cells. PLoS One. 2012;7(2):e30695. doi:10.1371/journal.pone.0030695
78. Gobert M, Treilleux I, Bendriss-Vermare N, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res. 2009;69(5):2000–2009. doi:10.1158/0008-5472.CAN-08-2360
79. Maeda S, Murakami K, Inoue A, Yonezawa T, Matsuki N. CCR4 blockade depletes regulatory T cells and prolongs survival in a canine model of bladder cancer. Cancer Immunol Res. 2019;7(7):1175–1187. doi:10.1158/2326-6066.CIR-18-0751
80. Sather BD, Treuting P, Perdue N, et al. Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med. 2007;204(6):1335–1347. doi:10.1084/jem.20070081
81. Liu W, Wei X, Li L, et al. CCR4 mediated chemotaxis of regulatory T cells suppress the activation of T cells and NK cells via TGF-beta pathway in human non-small cell lung cancer. Biochem Biophys Res Commun. 2017;488(1):196–203. doi:10.1016/j.bbrc.2017.05.034
82. Sugiyama D, Nishikawa H, Maeda Y, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A. 2013;110(44):17945–17950. doi:10.1073/pnas.1316796110
83. Ueda R. Clinical application of Anti-CCR4 monoclonal antibody. Oncology. 2015;89(Suppl 1):16–21. doi:10.1159/000431059
84. Yu X, Marshall MJE, Cragg MS, Crispin M. Improving antibody-based cancer therapeutics through glycan engineering. BioDrugs. 2017;31(3):151–166. doi:10.1007/s40259-017-0223-8
85. Freeman CM, Chiu BC, Stolberg VR, et al. CCR8 is expressed by antigen-elicited, IL-10-producing CD4+CD25+ T cells, which regulate Th2-mediated granuloma formation in mice. J Immunol. 2005;174(4):1962–1970. doi:10.4049/jimmunol.174.4.1962
86. Soler D, Chapman TR, Poisson LR, et al. CCR8 expression identifies CD4 memory T cells enriched for FOXP3+ regulatory and Th2 effector lymphocytes. J Immunol. 2006;177(10):6940–6951. doi:10.4049/jimmunol.177.10.6940
87. De Simone M, Arrigoni A, Rossetti G, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016;45(5):1135–1147. doi:10.1016/j.immuni.2016.10.021
88. Wang L, Simons DL, Lu X, et al. Connecting blood and intratumoral Treg cell activity in predicting future relapse in breast cancer. Nat Immunol. 2019;20:1220–1230. doi:10.1038/s41590-019-0429-7
89. Coghill JM, Fowler KA, West ML, et al. CC chemokine receptor 8 potentiates donor Treg survival and is critical for the prevention of murine graft-versus-host disease. Blood. 2013;122(5):825–836. doi:10.1182/blood-2012-06-435735
90. Villarreal DO, L’Huillier A, Armington S, et al. Targeting CCR8 induces protective antitumor immunity and enhances vaccine-induced responses in colon cancer. Cancer Res. 2018;78(18):5340–5348. doi:10.1158/0008-5472.CAN-18-1119
91. Hoelzinger DB, Smith SE, Mirza N, Dominguez AL, Manrique SZ, Lustgarten J. Blockade of CCL1 inhibits T regulatory cell suppressive function enhancing tumor immunity without affecting T effector responses. J Immunol. 2010;184(12):6833–6842. doi:10.4049/jimmunol.0904084
92. Bonecchi R, Locati M, Mantovani A. Chemokines and cancer: a fatal attraction. Cancer Cell. 2011;19(4):434–435. doi:10.1016/j.ccr.2011.03.017
93. Liu X, Xu X, Deng W, et al. CCL18 enhances migration, invasion and EMT by binding CCR8 in bladder cancer cells. Mol Med Rep. 2019;19(3):1678–1686. doi:10.3892/mmr.2018.9791
94. Melief SM, Schrama E, Brugman MH, et al. Multipotent stromal cells induce human regulatory T cells through a novel pathway involving skewing of monocytes toward anti-inflammatory macrophages. Stem Cells. 2013;31(9):1980–1991. doi:10.1002/stem.1432
95. Wang W, Wu D, He X, et al. CCL18-induced HOTAIR upregulation promotes malignant progression in esophageal squamous cell carcinoma through the miR-130a-5p-ZEB1 axis. Cancer Lett. 2019;460:18–28. doi:10.1016/j.canlet.2019.06.009
96. Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol. 2016;28(8):401–409. doi:10.1093/intimm/dxw025
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.