Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Can resistive breathing injure the lung? Implications for COPD exacerbations
Authors Vassilakopoulos T, Toumpanakis D
Received 30 May 2016
Accepted for publication 1 August 2016
Published 26 September 2016 Volume 2016:11(1) Pages 2377—2384
DOI https://doi.org/10.2147/COPD.S113877
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Theodoros Vassilakopoulos, Dimitrios Toumpanakis
Pulmonary and Critical Care Medicine, Medical School, National and Kapodistrian University of Athens, Greece
Abstract: In obstructive lung diseases, airway inflammation leads to bronchospasm and thus resistive breathing, especially during exacerbations. This commentary discusses experimental evidence that resistive breathing per se (the mechanical stimulus) in the absence of underlying airway inflammation leads to lung injury and inflammation (mechanotransduction). The potential implications of resistive breathing-induced mechanotrasduction in COPD exacerbations are presented along with the available clinical evidence.
Keywords: resistive breathing, COPD, mechanotransduction, bronchoconstriction, inflammation
Introduction
Resistive breathing (breathing through increased airway resistance) is the hallmark of the most prevalent pulmonary diseases, COPD, and asthma. During severe exacerbations, these diseases often culminate in respiratory failure with increased morbidity and mortality. In COPD, inflammation of the airways results in variable airway narrowing through a variety of mechanisms such as mucus hypersecretion, goblet cell hyperplasia, mucosal edema, cytokine upregulation1 and inflammation, and alveoli destruction (emphysema), leading to a loss of elastic distending traction on the distal airways and bronchospasm.2,3 During COPD exacerbations, the inciting factor (eg, mainly infections and/or environmental stress, such as air pollution4) augments inflammation, which worsens airway mucosal edema and through release of mediators, which can cause smooth muscle contraction, leads to bronchoconstriction/bronchospasm, further increasing airway resistance. This sequence of events often culminates in both hypercapnic and/or hypoxemic respiratory failure through largely unexplored mechanisms.
During resistive breathing, the inspiratory muscles contract strenuously to inflate the lung, resulting in large negative intrathoracic pressures during inspiration. Both the strenuous muscle contractions and the large negative intrathoracic pressures lead to mechanical stress on cells residing in the diaphragm and the lungs, respectively. This mechanical stress may lead to injury of both the diaphragm and the lung through either direct “tearing” forces5 or through mechanotransduction (ie, transformation of mechanical distortion into intracellular cascade activation,6 including the formation of oxidative stress).7
Thus, it is plausible that bronchoconstriction can injure both the lung and the diaphragm even in the absence of underlying airway inflammation (ie, in the previous healthy lung). The clinical paradigm is acute upper airway obstruction in a previously healthy human can culminate in pulmonary edema.
It is also tempting to speculate that in the presence of underlying airway inflammation (like in COPD), the augmentation of bronchoconstriction would by itself cause inflammation and injury in the lung periphery and the diaphragm. The clinical paradigm would be a severe COPD exacerbation requiring hospitalization.
Effect of inspiratory resistive breathing on the lung
We have long been interested in studying the “isolated” effects of resistive breathing/(acute) airway obstruction on the lung and the diaphragm. Over the last 15 years, we have developed an experimental model of resistive breathing in healthy animals and humans.8–12 The model consists of external resistors of appropriate magnitude in the inspiratory line of a two-way valve, with the expiratory line having no resistance. Our aim was to decipher the effects of mechanical stressor (increased inspiratory airway resistance) from the effects of the underlying airway inflammation which is observed in COPD. A series of studies have been performed by adding an inspiratory resistance, so that the tidal pressure during inspiration is set at 50% of maximum (Pi/Pi,max=0.5).9,10 The major findings of these studies are that in a healthy rat lung, inspiratory resistive breathing increases the permeability of the alveolar-capillary membrane of the lung, deranges the respiratory system mechanics, shifting the pressure–volume curve to the right and downward (ie, making the lung less distensible), induces lung inflammation, activates matrix metalloproteinases, and results in acute lung injury and respiratory failure (Figure 1).9,10 These findings raise the intriguing possibility that during severe COPD exacerbations, bronchoconstriction leads to distal lung inflammation and acute lung injury, thus contributing to the development of hypoxemic respiratory failure. The resulting less distensible lung contributes to the development of dyspnea. Furthermore, bronchoconstriction-resistive breathing leads to the activation of matrix metalloproteinases (MMPs, mainly MMP-9 and MMP-12) in the lung tissue (Figure 1G), which in turn leads to the destruction of lung tissue (ie, loss of functional lung units [emphysema]).10 This finding may explain the observation that patients with frequent COPD exacerbations experience more rapid loss of lung function over time (ie, at each exacerbation, bronchoconstriction would cause MMP activation and thus alveolar destruction and loss of lung function).13,14 Although the exact signaling pathways that mediate inspiratory resistive breathing-induced lung injury are largely unknown, preliminary results from our ongoing research have shown that early after the initiation of resistive breathing, sensors of mechanical stress, such as Src kinase,15 are activated in the lung tissue,16 which is followed by ERK activation.9 Interestingly, inhibition of Src kinase activation partially prevented inspiratory resistive breathing-induced lung injury.16
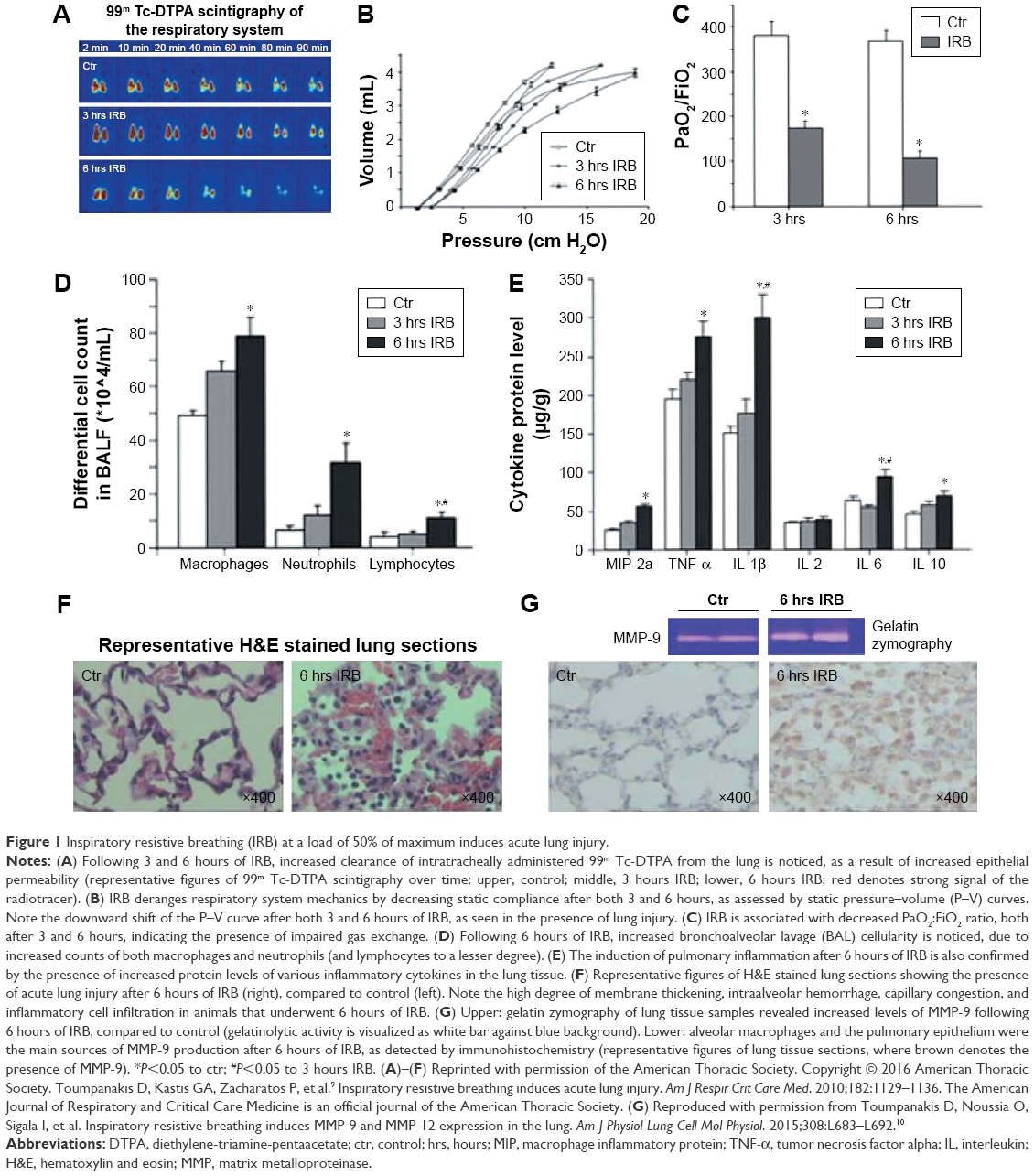 | Figure 1 Inspiratory resistive breathing (IRB) at a load of 50% of maximum induces acute lung injury. |
The load used in our studies was chosen based on extrapolation from data on COPD exacerbations. The actual load faced by the respiratory muscles of patients with a severe COPD exacerbation is not known, because it is extremely difficult to perform pathophysiological studies in the emergency department. Thus, only indirect estimates can be made. In patients with stable severe COPD (forced expiratory volume in 1 second [FEV1], 0.79±0.35 L) with hypercapnia (PCO2, 62.4±5.5 mmHg), the mean inspiratory/maximum inspiratory pressure was 0.27±0.15.17 Similarly, a group of Italian investigators found mean pleural/maximum pleural pressure (Ppleural/Ppleural,max) values amounting to 0.35 in patients with stable severe COPD (stage IV: FEV1, 0.57±0.06 L [20.6%±2.12% of predicted]) and with less severe hypercapnia (PCO2, 45.8±1.65 mmHg).18 We can speculate that these values would be expected to be even higher during an exacerbation because of worsening lung mechanics (increased bronchoconstriction delaying lung emptying) and the accompanying dynamic hyperinflation, which decreases the capacity of the respiratory muscles to generate pressure.
A relatively good model of COPD exacerbations, which mimics its pathophysiological characteristics, is patients with COPD requiring mechanical ventilation or in the weaning phase who have similar respiratory muscle loads to severely exacerbated COPD patients. In this situation physiological measurements are allowed because the patient is in the controlled environment of the intensive care unit. In these exacerbated COPD patients, the load on their respiratory muscles is quite similar to the one selected for the animal studies we conducted. Accordingly, our group has shown that in COPD patients requiring invasive mechanical ventilation, the peak/maximum inspiratory pressure value was 0.62±0.15 (the peak inspiratory pressure is the sum of the elastic and resistive pressure required for the given tidal volume).19 In COPD patients who fail to wean from mechanical ventilation, the Ppleural/Ppleural,max was ~0.6 (range, 0.38–0.95) and the transdiaphragmatic pressure to maximal transdiaphragmatic pressure (Pdi/Pdi,max) ratio was 0.62 (range, 0.42–0.85).20 Similarly, our group has shown that COPD patients who fail to wean from mechanical ventilation have mean inspiratory/maximum inspiratory ratios amounting to 0.490±0.09.21 Vitacca et al22 found Pdi/Pdi,max values of 0.58 at the end of a failing weaning trial. Thus, the choice of loads in our animal model with inspiratory pressures amounting to 50% of maximum inspiratory pressures is within the range of severe COPD exacerbation. It is currently unknown whether inspiratory resistances leading to loads smaller than 50% of maximum inspiratory pressures can cause lung injury and inflammation and which is the smallest injurious load.
Combined inspiratory and expiratory resistive breathing
It could be claimed that isolated inspiratory resistive breathing cannot exist in real life, since irrespective of where in the airways is the area of bronchoconstriction, both the inspiratory and the expiratory resistances are elevated. If bronchoconstriction is intrathoracic, the expiratory resistance is greater than the inspiratory resistance, due to the positive intrathoracic pressure during expiration, which narrows the airways, and the negative intrathoracic pressure during inspiration, which dilates the airways. This pattern is usually observed in COPD and asthma patients. If bronchoconstriction is extrathoracic, the reverse is true, inspiration reducing and expiration increasing the airway surface area. This pattern is observed in patients with upper airway obstruction.
To have a model of concurrent inspiratory and expiratory resistances, thus better mimicking COPD exacerbations, we developed a mouse model of resistive breathing through tracheal banding.23 Resistive breathing in a healthy mouse increases the permeability of the alveolar-capillary membrane of the lung, deranges respiratory system mechanics, induces lung inflammation, and results in acute lung injury, similarly to the rat inspiratory resistive breathing model (Figure 2A–C).23 Willms and Shure24 suggested that the presence of expiratory resistance with its accompanying hyperinflation (increased end expiratory volume) and intrinsic positive end expiratory pressure opposes during expiration the large negative swings in intrathoracic pressure that are developed during inspiration and produce pulmonary edema. In our mouse model, the presence of expiratory resistance in the face of inspiratory resistance did not prevent lung injury23 (supposedly induced by inspiratory resistance based on the rat inspiratory resistance model).9,10
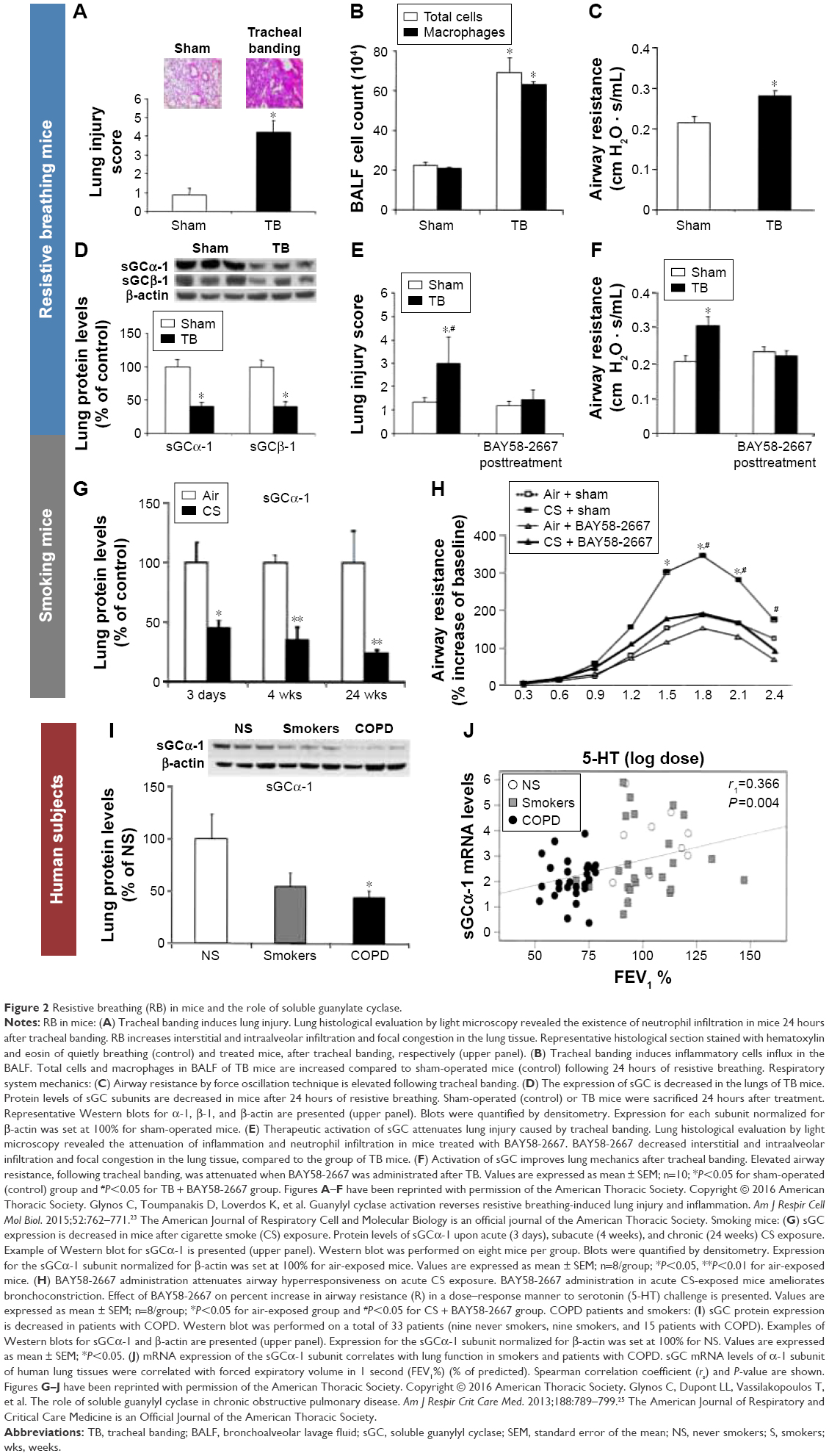 | Figure 2 Resistive breathing (RB) in mice and the role of soluble guanylate cyclase. |
Interestingly, resistive breathing in mice downregulated the expression of soluble guanylate cyclase in the lung (Figure 2D).23 Activation of soluble guanylate cyclase even after injury, which was well established, significantly reversed the lung injury (Figure 2E and F).23 This finding might suggest a new therapeutic category in obstructive airway diseases. In fact, we have shown that soluble guanylate cyclase is downregulated secondary to chronic smoking exposure in a mice model of COPD (Figure 2G and H) and in the lungs of COPD patients (Figure 2I–J).25
However, even this mouse model of tracheal banding does not accurately mimic COPD exacerbations, as tracheal banding is an extrathoracic bronchoconstriction, whereas COPD exacerbations result in intrathoracic obstruction. To date, we have had no data with regard to the effects of expiratory resistances on a healthy lung. Hyperinflation could generate an inflammatory process through excessive deformation of the lung cells.26 However, this hypothesis has never been tested experimentally. Furthermore, the effects of increased expiratory resistance on healthy lung (greater than those caused by inspiratory resistance, which is the actual clinical situation) has not been established.
In a recent article published in the International Journal of Chronic Obstructive Pulmonary Disease, Loverdos et al27 added the missing information. They applied a series of pure inspiratory resistances of various magnitudes and determined that injury starts developing at inspiratory loads as low as 30% of maximum inspiratory pressure. As presented before, such inspiratory loads can be present even in stable severe COPD patients without being exacerbated.17,18 It could also be observed in less severe COPD patients when an exacerbation has led to bronchoconstriction.
Loverdos et al27 showed that pure expiratory resistance (up to 70% of maximum expiratory pressure) can also cause inflammation with load (ie, resistance)-dependent influx of neutrophils and cytokine upregulation. Interestingly, macrophages were downregulated, which is a behavior different from the one observed in the other models (inspiratory resistive breathing and tracheal banding).27 Lung mechanics were not affected (in contrast to the other models) and increased protein levels in the bronchoalveolar lavage fluid (suggesting the presence of increased permeability pulmonary edema) occurred only at the highest studied load (70%).27
Combined inspiratory and expiratory resistive breathing, with the expiratory load being higher than the inspiratory load and thus mimicking intrathoracic obstruction similar to that occurs during COPD exacerbations, resulted in a dose-dependent injury and inflammation of the lung.27 Thus, similar to our mouse model of resistive breathing through tracheal banding, the presence of expiratory resistance did not prevent lung injury and inflammation induced by resistive breathing, and may have even augmented it.
All the aforementioned results were obtained in animals with healthy lungs.9,10,23,27 It has to be acknowledged that no data are available on the effects of resistive breathing-bronchoconstriction on the lung that has preexisting underlying inflammation (as occurs in COPD). Yet, we can speculate that a similar or even augmented response would be exhibited by the lung with COPD, which would partly explain why severe COPD exacerbations culminate in respiratory failure, often requiring noninvasive and even invasive mechanical ventilation.
Evidence from human studies
Do we have any human data? In healthy humans, inspiratory resistive breathing leads to plasma elevation of tumor necrosis factor alpha (TNF-α), interleukin 1β (IL-1β), and IL-6,12 and stimulates the hypothalamic pituitary adrenal axis resulting (through cleavage of pro-opiomelanocortin) in adrenocorticotropic hormone and β-endorphin release.11 β-Endorphins are endogenous opioids that affect the control of breathing rendering breathing shallow.28 Thus, this could contribute to the development of hypercapnic respiratory failure frequently observed during COPD exacerbations.29 Preliminary results from our laboratory suggest that inspiratoryresistive breathing leads to elevation of IL-6 in the induced sputum of healthy volunteers (Figure 3).
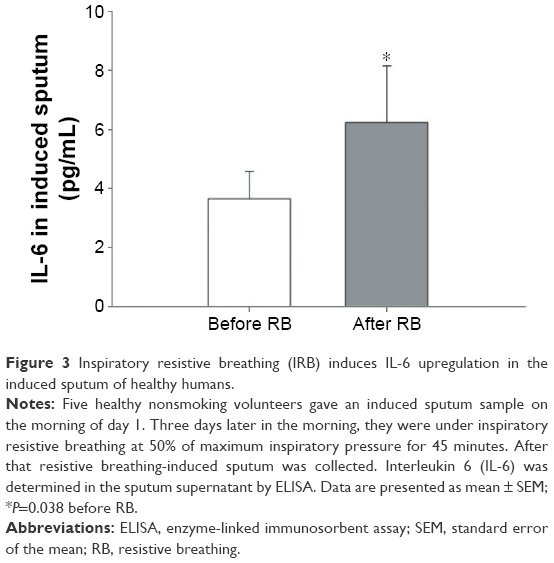 | Figure 3 Inspiratory resistive breathing (IRB) induces IL-6 upregulation in the induced sputum of healthy humans. |
Plasma cytokines observed in humans after resistive breathing do not originate from monocytes.12 One clear source of these plasma cytokines observed after resistive breathing in humans is the lung.9,10,23,27 Yet another source is the strenuously contracting respiratory muscles. In the rat diaphragm, inspiratory resistive breathing results in MAPK and NF-κB activation and cytokine upregulation (Figure 4).8,30 These cytokines are produced by resident myocytes and are under the control of oxidative stress and nitric oxide (Figure 4).8,30,31 Indeed, although low levels of reactive oxygen species (ROS) are present in the skeletal muscles, including the diaphragm, under normal function, the levels of ROS are significantly augmented under stress, including strenuous contraction, hypoxia, and heat stress.32 Do we have any human data? In stable COPD patients, Casadevall et al33 have shown that the intercostal muscles express cytokines. Preliminary results from our study show that cytokines are upregulated in the diaphragms of COPD GOLD III and IV patients.
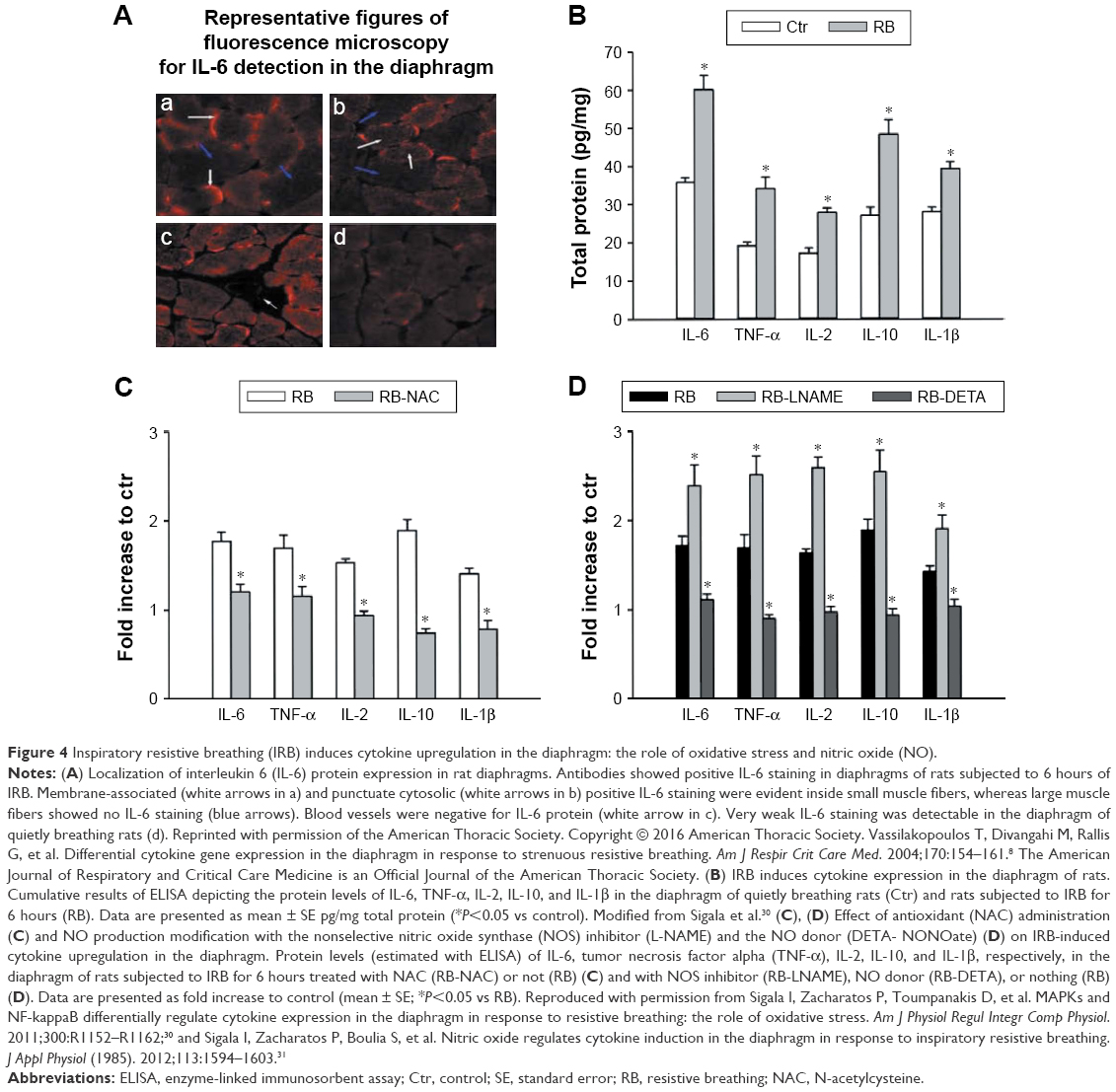 | Figure 4 Inspiratory resistive breathing (IRB) induces cytokine upregulation in the diaphragm: the role of oxidative stress and nitric oxide (NO). |
Regarding COPD patients, there is still little data to support the possible injurious effect of resistive breathing on the lung. Our group recently tested the hypothesis that the respiratory muscles and/or the mechanically stressed “inflammatory” lung contribute to the elevated plasma cytokine levels in patients with severe acute exacerbation of COPD. Hillas et al34 studied 30 patients with severe acute exacerbation of COPD, which led to endotracheal intubation and mechanical ventilation (intervention group); patients who had acute exacerbation of COPD received the same drugs as the intubated patients but were not intubated (control group 1), and patients with COPD at steady state were intubated and mechanically ventilated for elective surgery (control group 2). Application of mechanical ventilation in patients with acute exacerbation of COPD, which resulted in endotracheal intubation, led to decreased plasma levels of circulating IL-6, IL-10, eotaxin, and epidermal growth factor within 30 minutes to 6 hours.34 These findings suggest that the respiratory muscles and especially the contracting diaphragm and/or the mechanically stressed inflammatory lung contributed to the elevated levels of these cytokines during the exacerbation. It was the relief of mechanical stress with the application of mechanical ventilation, which unloaded the diaphragm and alleviated the large negative swings in intrathoracic pressure that stressed and injured the lung, that was responsible for the decrease in the cytokine level (given that the cytokines did not decrease in acutely exacerbated COPD patients who received only drugs but not mechanical ventilation). These results might provide mechanistic insights into the beneficial effects of noninvasive mechanical ventilation during severe COPD exacerbations.
Based on the previously mentioned evidence, it is plausible that resistive breathing-bronchoconstriction per se can injure both the lung and the diaphragm even in the absence of underlying airway inflammation (ie, in the previous healthy lung). In the lung with underlying lung inflammation, this resistive breathing-bronchoconstriction may perpetuate distal lung injury leading to the development of respiratory failure.
Acknowledgment
The authors thank Drs Sigala, Glynos, Hillas, and Loverdos for carefully reading the manuscript and for providing comments.
Disclosure
The authors report no conflicts of interest in this work.
References
Zuo L, Nogueira L, Hogan MC. Effect of pulmonary TNF-α overexpression on mouse isolated skeletal muscle function. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1025–R1031. | ||
Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. | ||
Barnes PJ. Chronic obstructive pulmonary disease. N Engl J Med. 2000;343:269–280. | ||
Ni L, Chuang CC, Zuo L. Fine particulate matter in acute exacerbation of COPD. Front Physiol. 2015;6:294. | ||
West JB. Invited review: pulmonary capillary stress failure. J Appl Physiol (1985). 2000;89:2483–2489. | ||
Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10:63–73. | ||
Zuo L, Hallman AH, Yousif MK, Chien MT. Oxidative stress, respiratory muscle dysfunction, and potential therapeutics in chronic obstructive pulmonary disease. Front Biol. 2012;7:506–513. | ||
Vassilakopoulos T, Divangahi M, Rallis G, et al. Differential cytokine gene expression in the diaphragm in response to strenuous resistive breathing. Am J Respir Crit Care Med. 2004;170:154–161. | ||
Toumpanakis D, Kastis GA, Zacharatos P, et al. Inspiratory resistive breathing induces acute lung injury. Am J Respir Crit Care Med. 2010;182:1129–1136. | ||
Toumpanakis D, Noussia O, Sigala I, et al. Inspiratory resistive breathing induces MMP-9 and MMP-12 expression in the lung. Am J Physiol Lung Cell Mol Physiol. 2015;308:L683–L692. | ||
Vassilakopoulos T, Zakynthinos S, Roussos C. Strenuous resistive breathing induces proinflammatory cytokines and stimulates the HPA axis in humans. Am J Physiol. 1999;277:R1013–R1019. | ||
Vassilakopoulos T, Katsaounou P, Karatza MH, et al. Strenuous resistive breathing induces plasma cytokines: role of antioxidants and monocytes. Am J Respir Crit Care Med. 2002;166:1572–1578. | ||
Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847–852. | ||
Makris D, Moschandreas J, Damianaki A, et al. Exacerbations and lung function decline in COPD: new insights in current and ex-smokers. Respir Med. 2007;101:1305–1312. | ||
Han B, Bai XH, Lodyga M, et al. Conversion of mechanical force into biochemical signaling. J Biol Chem. 2004;279:54793–54801. | ||
Toumpanakis D, Zacharatos P, Michailidou T, Tsoukalas G, Vassilakopoulos T. The role of Src kinase in inspiratory resistive breathing-induced pulmonary inflammation. Thorax. 2015;70:A39. | ||
Begin P, Grassino A. Inspiratory muscle dysfunction and chronic hypercapnia in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1991;143:905–912. | ||
Duranti R, Misuri G, Gorini M, et al. Mechanical loading and control of breathing in patients with severe chronic obstructive pulmonary disease. Thorax. 1995;50:127–133. | ||
Zakynthinos SG, Vassilakopoulos T, Roussos C. The load of inspiratory muscles in patients needing mechanical ventilation. Am J Respir Crit Care Med. 1995;152:1248–1255. | ||
Purro A, Appendini L, De GA, et al. Physiologic determinants of ventilator dependence in long-term mechanically ventilated patients. Am J Respir Crit Care Med. 2000;161:1115–1123. | ||
Vassilakopoulos T, Zakynthinos S, Roussos C. The tension-time index and the frequency/tidal volume ratio are the major pathophysiologic determinants of weaning failure and success. Am J Respir Crit Care Med. 1998;158:378–385. | ||
Vitacca M, Ambrosino N, Clini E, et al. Physiological response to pressure support ventilation delivered before and after extubation in patients not capable of totally spontaneous autonomous breathing. Am J Respir Crit Care Med. 2001;164:638–641. | ||
Glynos C, Toumpanakis D, Loverdos K, et al. Guanylyl cyclase activation reverses resistive breathing-induced lung injury and inflammation. Am J Respir Cell Mol Biol. 2015;52:762–771. | ||
Willms D, Shure D. Pulmonary edema due to upper airway obstruction in adults. Chest. 1988;94:1090–1092. | ||
Glynos C, Dupont LL, Vassilakopoulos T, et al. The role of soluble guanylyl cyclase in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:789–799. | ||
Agusti A, Soriano JB. Dynamic hyperinflation and pulmonary inflammation: a potentially relevant relationship? Eur Respir Rev. 2006;15:68–71. | ||
Loverdos K, Toumpanakis D, Litsiou E, et al. The differential effects of inspiratory, expiratory, and combined resistive breathing on healthy lung. Int J COPD. 2016;11(1):1623–1638. | ||
Scardella AT, Santiago TV, Edelman NH. Naloxone alters the early response to an inspiratory flow-resistive load. J Appl Physiol (1985). 1989;67:1747–1753. | ||
Vassilakopoulos T, Roussos C, Zakynthinos S. The immune response to resistive breathing. Eur Respir J. 2004;24:1033–1043. | ||
Sigala I, Zacharatos P, Toumpanakis D, et al. MAPKs and NF-kappaB differentially regulate cytokine expression in the diaphragm in response to resistive breathing: the role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1152–R1162. | ||
Sigala I, Zacharatos P, Boulia S, et al. Nitric oxide regulates cytokine induction in the diaphragm in response to inspiratory resistive breathing. J Appl Physiol (1985). 2012;113:1594–1603. | ||
Zuo L, Best TM, Roberts WJ, Diaz PT, Wagner PD. Characterization of reactive oxygen species in diaphragm. Acta Physiol (Oxf). 2015;213:700–710. | ||
Casadevall C, Coronell C, Ramirez-Sarmiento AL, et al. Upregulation of pro-inflammatory cytokines in the intercostal muscles of COPD patients. Eur Respir J. 2007;30:701–707. | ||
Hillas G, Perlikos F, Toumpanakis D, et al. Controlled mechanical ventilation attenuates the systemic inflammation of severe chronic obstructive pulmonary disease exacerbations. Am J Respir Crit Care Med. 2016;193:696–698. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.