Back to Journals » OncoTargets and Therapy » Volume 12
c-Src is required for hypoxia-induced metastasis-associated functions in prostate cancer cells
Received 12 January 2019
Accepted for publication 20 March 2019
Published 8 May 2019 Volume 2019:12 Pages 3519—3529
DOI https://doi.org/10.2147/OTT.S201320
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Yao Dai, Dietmar Siemann
Department of Radiation Oncology, University of Florida, Gainesville, FL 32608, USA
Background: Metastasis is the major cause of therapeutic failure in prostate cancer patients, and hypoxia has been shown to promote metastatic functions. However, whether Src family kinases (SFKs) can be upregulated under hypoxia is unclear.
Materials and methods: In the current study, we evaluated the effects of hypoxia on cellular functions and activities of different SFK members (c-Src, Lyn, Fyn) in prostate cancer cells. Prostate cancer cell functions were determined in vitro including migration (wound-healing assay), invasion (Matrigel-based transwell assay) and clonogenic cell survival (colony formation assay). Protein expression was detected by Western blotting and gene knockdown was accomplished by siRNA transfection.
Results: SRC, but not LYN and FYN, is associated with overall survival in prostate cancer patients, while all three phosphorylated proteins are highly expressed in tumors compared to normal tissues. Short-term hypoxic exposure significantly enhances cell migration, invasion, clonogenic survival, and consistently, c-Src phosphorylation in both PC-3ML and C4-2B cells. Knockdown of SRC, but not LYN or FYN, abolished hypoxia-induced functions. Finally, small molecule Src inhibitors strongly inhibited cell behaviors and c-Src activation under hypoxic conditions.
Conclusion: Our data show that hypoxia is able to enhance metastatic-associated cell functions by activating c-Src in prostate cancer cells. Importantly, SFK inhibition by small molecule inhibitors was able to impair hypoxia-induced metastasis associated cell functions, suggesting a possible role of SFK inhibitors for prostate cancer treatment.
Keywords: hypoxia, Src, metastasis, prostate cancer
Introduction
For patients with advanced/metastatic prostate cancer, androgen deprivation therapy (ADT) remains the standard of care.1 While these patients clearly benefit from such therapy for palliating their symptoms and for improving their quality of life, this treatment very rarely results in cure, with the majority of the patients eventually developing recurrences that are not only castration-resistant but also more aggressive.2
Hypoxia, a common occurrence in most solid tumors, long known to be a detriment to cancer treatment, is now also recognized as a key contributor to metastasis.3 Hypoxia transcriptionally regulates ~1–2% of all human genes, many of which are driven by the hypoxia-inducible factor 1 (HIF-1); a heterodimeric complex consisting of α and β subunits.4 Under hypoxic conditions, HIF-1α activation leads to a more aggressive tumor cell phenotype. HIF-1α upregulation has been considered an early event in carcinogenesis5 and highly correlated with the risk of metastases in prostate cancer.6
The Src family kinases (SFKs) are a family of non-receptor tyrosine kinases that are frequently hyper-activated in human cancers7 and contribute to increased aggressiveness and adverse outcomes in patients.8 In prostate cancer, overexpressed and/or hyper-activated c-Src is associated with carcinogenesis,9 hormone insensitivity,10 metastasis11 and castration resistance.12 Although c-Src is the most often studied SFK, other non-c-Src members have also been shown to be overexpressed in prostate cancer patients.13,14 Recent studies have indicated the pro-metastatic potential of Fyn15 and Yes,16 as well as the involvement of Lyn in tumor growth17 and castration resistance18 in preclinical prostate cancer models. In addition, SFKs and androgen receptor (AR) are able to cooperate to drive prostate cancer tumorigenesis and invasion.19 For example, c-Src acts as a crucial facilitator of invasiveness in AR negative cells,20,21 implying an independent role for c-Src as a mediator of metastasis irrespective of AR status. Previous studies in our laboratory have suggested treatment potential of SFK inhibitors in prostate cancer invasion and metastasis.22–25
A few reports have proposed Src activation in response to hypoxia in different preclinical models. Pham et al have reported that Src/FAK signaling tends to be associated with chronic hypoxia in pancreatic cancer xenografts.26 In a melanoma model, HIF-1α was shown to drive melanoma cell invasion and invadopodia formation through direct activation of Src.27 In prostate cancer, interactions between c-Src and HIF-1 also have been reported.28,29 In all these cases it is remains unclear however, whether such hypoxia activation occurs generally in SFKs or just in a specific family member. and studies on the direct impact of hypoxia on SFK activity remain very limited.
In the current study, we examined the effect of hypoxia on the activation of three key Src family members and consequent functional behaviors in prostate cancer cells. Furthermore, we evaluated the treatment effects of small molecule Src inhibitors in prostate cancer cells under both aerobic and hypoxic conditions.
Materials and methods
Reagents
Saracatinib was a gift from AstraZeneca (Macclesfield, U.K.). Dasatinib was a gift from Bristol-Meyer Squibb (Piscataway, NJ). The powder was dissolved in dimethyl sulfoxide (DMSO) and stored as aliquots (10 mM) at −20 °C. Additional chemicals were purchased from Sigma (St. Louis, MO) unless otherwise indicated.
Patient tumors
Frozen patient tissue samples were acquired from Biorepository Core of Clinical-Translational Science Institute (CTSI) at University of Florida. All samples were deidentified with approved IRB protocol (IRB201400467). The tumor tissues were from prostate cancer patients with the protocol approved by CTSI.
Cell culture and hypoxia
Human prostate cancer PC-3ML cells, a highly invasive subline of PC-3 cells, were kindly provided by Dr. A. Fatatis (Drexel University, Philadelphia, PA), while C4-2B cells were purchased from ATCC. PC-3ML cells were authenticated by Idexx BioResearch (Westbrook, ME), and were maintained in Ham’s F-12K supplemented with 10% fetal bovine serum (FBS), while C4-2B cells were maintained in T-medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were cultured in a 5% CO2 humidified incubator at 37 °C. For hypoxic culture conditions, cells were incubated in a hypoxia incubator chamber (Oxygen Sensors) flushed with a gas mixture containing 1% O2 balanced with 5% CO2 and N2 at 37 °C. Cells were used for all experiments in 20 passages.
Cell migration
Cell migration was evaluated using a “wound-healing” assay. Cells were seeded in a 6-well plate and allowed to grow for 48 h. Wounds were generated on the confluent cell monolayer by making a scratch using a sterilized 1 ml pipette tip. Cells were then exposed to aerobic or 1% O2 for desired times and the cells that had migrated into the denuded area were photographed. To quantify migrated cells, 8 fields (original magnification ×100) were randomly selected in each sample, and cells that moved into the defined area were scored.
Cell invasion
Invasion was examined using a commercial transwell insert (8 μm pore membrane) pre-loaded with Matrigel (BD Biosciences, San Diego, CA). Inserts were incubated with serum-free medium at 37 °C for 2 h to allow rehydration of Matrigel. After exposure to hypoxia, cells suspended in serum-free medium were loaded onto the top chamber (5×103/insert). Complete medium (containing 10% FBS) was used in the lower chamber as a chemo-attractant. After incubation for 24 h, the Matrigel was removed and the inserts were stained with crystal violet. Invaded cells on the underside of the filter were counted.
Colony formation
Cells were seeded into 6-well plates in triplicate (200/well), after exposed to 1% O2 or aerobic conditions with or without drug treatment. Cells were incubated for 2~3 weeks and plates were stained with crystal violet. Cell colonies (≥50 cells) were counted macroscopically.
SiRNA transfection
The siRNA specific for human SRC, LYN and FYN, and control siRNA were purchased from IDT Technologies (Des Moines, IA). siRNA fragments (22 pmol) were transfected into prostate cancer cells using jetPRIME (Polyplus, New York, NY), following the manufacturer’s instructions. Twenty-four hours after transfection, cells were processed for functional assays. The siRNA knockdown effects were confirmed by Western blotting.
Western blotting
Cells were harvested and disrupted in a radioimmunoprecipitation assay (RIPA) lysis buffer buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.25% Sodium deoxycholate and 1 mM EDTA) with protease inhibitor cocktail, 1 mM NaF and 1 mM Na3VO4. Equal amounts (~20 μg) of whole cell lysates were resolved on a 10% SDS-PAGE gel (Bio-Rad). After electrophoresis, samples were electrotransferred to a nitrocellulose membrane (Bio-Rad), probed with relevant primary antibodies at 4 °C overnight, incubated with HRP (horseradish peroxidase) conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA), and detected with an enhanced chemiluminescence substrate (Amersham, Piscataway, NJ). The following primary antibodies were used: p-Src (Y416), total Src, p-Lyn (Y507), total Lyn, p-Fyn (Y530) and Fyn (Cell Signaling, Danvers, MA); HIF-1α (Genetex, Irvine, CA); β-actin (Sigma). To quantify the band intensity,
Overall survival analysis in patients
To analyze the association of SFKs’ gene expression levels with overall survival in patients with prostate adenocarcinoma, TCGA_PRAD_exp_HiSeqV2_PANCAN-2015-02-24.tgz datasets (
Statistical analysis
Two-tailed Student’s t-test was employed for analyzing functional and Western blotting data in cells for pairwise comparisons, while for tissue Western blotting data, non-parametric Mann-Whitney U test was applied, using GraphPad Prism 6.0 software (San Diego, CA). In the univariate survival analyses, the Kaplan-Meier method and log-rank test were used to compare overall survival curves between high- and low-gene expression groups. A threshold of P<0.05 was defined as statistically significant.
Results
SFKs are activated in prostate cancer
Previous studies have indicated the important role of SFKs in prostate cancer; however, which SFK member is the most critical remains unclear. To elucidate the individual SFK protein that may be the key player, we analyzed the relationships between gene expression of three key SFKs, eg, c-Src, Lyn and Fyn, and overall survival in a cohort of prostate cancer patients using the TCGA database. Our data show that patients with highly expressed SRC, but not LYN and FYN, exhibited significantly shorter overall survival than patients with lower SRC expression (Figure 1A). To detect the activities of these proteins at the phosphorylation level, we tested 6-paired frozen tumors and non-malignant adjacent normal tissues from prostate cancer patients. All three phospho-proteins – p-Src (Y416), p-Lyn (Y507) and p-Fyn (Y530) – showed greater expression in tumors than adjacent normal tissues (Figure 1B, upper). Quantification analysis indicated that p-Src, but not p-Lyn and p-Fyn, was significantly increased in tumors compared to adjacent normal tissues (Figure 1B, below). These data suggest that although SRC appears to be the only SFK gene that could predict the overall survival in patients, all three SFKs are activated in malignant tissues.
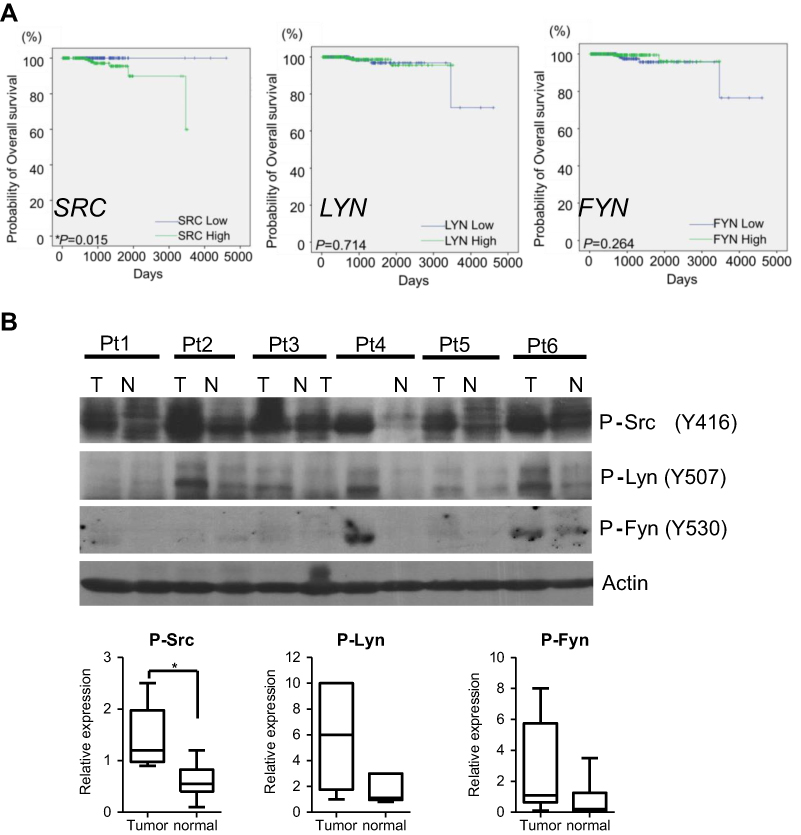 | Figure 1 SFKs are activated in prostate cancer. (A) Gene expression of SRC, FYN and LYN and overall survival curve in prostate cancer patients analyzed by TCGA data-mining. Kaplan-Meier method and log-rank test were used for survival curve plotting and comparison between high- and low-gene expression groups, respectively. *P<0.05 (n=550). (B) Lysates from prostate cancer patient tissues and adjacent normal tissues were processed for Western blotting. Actin is a loading control. Bands were quantified by Image J and plotted as “Relative Expression” by dividing each band’s density to that of the first sample (T of Pt1) in the blot. Data are presented as median with 95% CI. **P<0.01; ***P<0.001 (Mann-Whitney U test).Abbreviations: Pt, patient; T, tumor; N, adjacent normal tissue. |
Hypoxia induces Src activation
In PC-3ML cells, short term hypoxic exposure (2–6 h) clearly facilitated c-Src phosphorylation, while expression of p-Lyn and p-Fyn was reduced (Figure 2, left). After a very short exposure to hypoxia (30 min), p-Lyn and p-Fyn, but not p-Src, tended to show a mild increase, however the expression level of total Lyn and Fyn was not consistent. After 24 h oxygen depletion, p-Lyn and p-Fyn were significantly decreased, whereas p-Src remained unchanged (Figure 2, left). In addition, HIF-1α was clearly accumulated after 2–6 h of hypoxic exposures, showing a consistent profile with p-Src, but not p-Lyn and p-Fyn. C4-2B cells also showed time dependent increases in p-Src and HIF-1α, over the period of 2–24 h. In contrast, both p-Lyn and p-Fyn were significantly decreased after 24 h of hypoxic exposure (Figure 2, right). These data indicate, at least for the two prostate cancer lines investigated here, that hypoxia primarily induces Src activation, while Fyn and Lyn are not significantly affected.
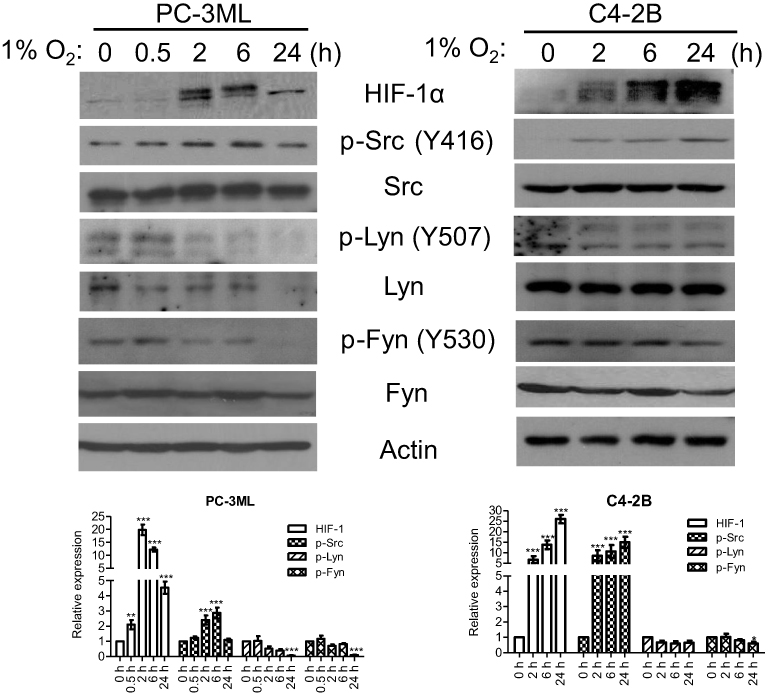 | Figure 2 Hypoxia induces Src activation. PC-3ML and C4-2B cells were exposed to 1% O2 for 0, 0.5, 2, 6 and 24 h. Whole cell lysates were processed for Western blotting. p-Src (Y416), p-Lyn (Y507) and p-Fyn (Y530), and their total proteins were visualized. Actin is a loading control. The data are one typical data from three independent experiments. Bands from 3 experiments were quantified by Image J and plotted as a bar graph. *P<0.05; **P<0.01; ***P<0.001 compared to “0 h”, which indicates normal control (20% O2) in each group. |
Hypoxia facilitates metastasis-associated functions in prostate cancer cells
Migration and invasion are two key steps in metastatic spread, while clonogenic survival has been indicated as a crucial event in metastatic colonization at the secondary site. In PC-3ML cells, the “wound-healing” assay showed that short-term exposure to hypoxia (2 or 6 h) significantly increased cell movement, with the greatest increase in cell migration occurred following a 6 h exposure, while prolonged hypoxia (24 h) did not enhance cell migration (Figure 3A). PC-3ML cells also showed the greatest increase in invasive capacity (Figure 3B) and clonogenic survival (Figure 3D) after 6 h hypoxic exposure, compared to 2 or 24 h exposure. In C4-2B cells however, significant enhancement of invasion (Figure 3C) and survival (Figure 3E) was observed following 6–24 h exposures to hypoxia, while acute exposure (2 h) was not sufficient to trigger functional changes (Figure 3C). In both cell lines, chronic oxygen depletion (48 h) decreased clonogenic survival (Figure 3D and E).
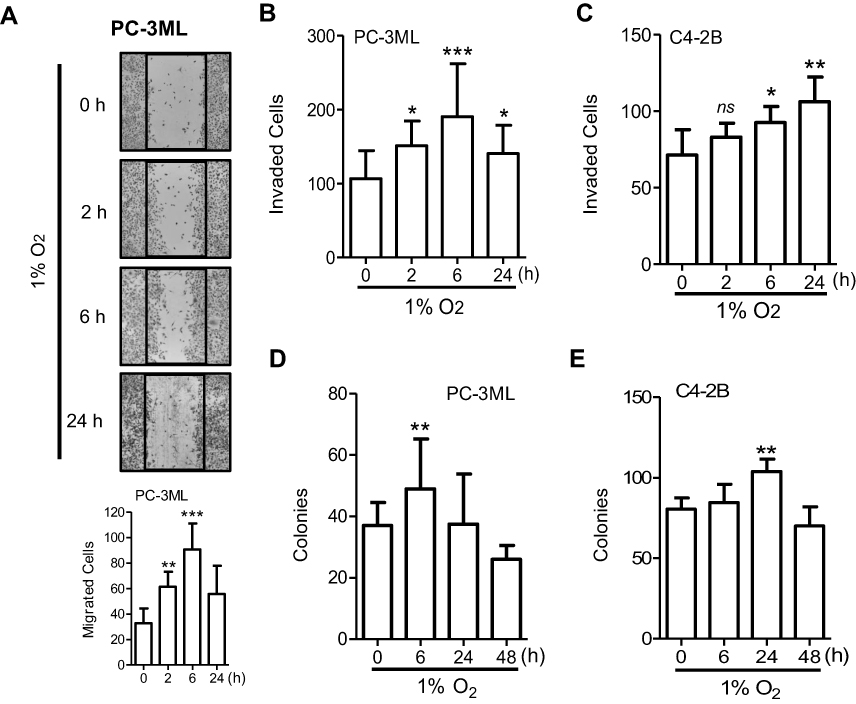 | Figure 3 Hypoxia facilitates metastasis-associated functions in prostate cancer cells. Cells were exposed to 1% O2 for 0, 2, 6 and 24 h prior to functional assessment. (A) PC-3ML cell monolayers were scratched immediately after hypoxic exposure, and photographed 24 h later. Cells migrated into the denuded area of the “wound” are shown. Original magnification, ×50. Data are one typical example from 3 independent experiments. (B and C), PC-3ML (B) and C4-2B (C) cells after hypoxic exposure were seeded into Matrigel-coated transwell inserts (5×103/insert) in serum-free medium. Complete medium was loaded on the bottom chamber as a chemo-attractant. The underside of the filter was stained 24 h after cell loading. Invaded cells were scored from 8 random fields (original magnification, ×100). Data are from 3 independent experiments. Bars, SD (n=6). (D and E) PC-3ML (D) and C4-2B (E) cells exposed to hypoxia were seeded into 6-well plates. After 14 days’ incubation, cell colonies (>50 cells) were counted. Columns, mean; bars, SD (n=3). *P<0.05; **P<0.01; ***P<0.001 compared to “0 h”, which indicates normal control (20% O2) in each group.Abbreviation: ns, not significant. |
SRC knockdown attenuates hypoxia-induced cell functions
We have shown that hypoxia is able to increase Src autophosphorylation at Y416 (Figure 2). Since this autophosphorylation site in c-Src protein can cross-react with the site in other non-Src SFKs, the exact SFK member that is phosphorylated under hypoxia is not clear. To shed light on the possible contributors, we applied siRNA to transiently knockdown c-Src, Fyn and Lyn. As shown in Figure 4A (left), PC-3ML cells with SRC knockdown for 48 h showed strong protein inhibition of both Src and p-Src. Interestingly, hypoxia failed to induce p-Src in SRC knockdown cells, while in control knockdown cells; p-Src activation was significantly induced with a 6 h hypoxic exposure (Figure 4A, right). Consistent with the results shown in Figure 3, short-term hypoxic exposure significantly enhanced cell invasion (Figure 4B) and migration (Figure 4C) in control knockdown cells. However, in SRC knockdown cells, such enhancement was not observed (Figure 4B and C, right).
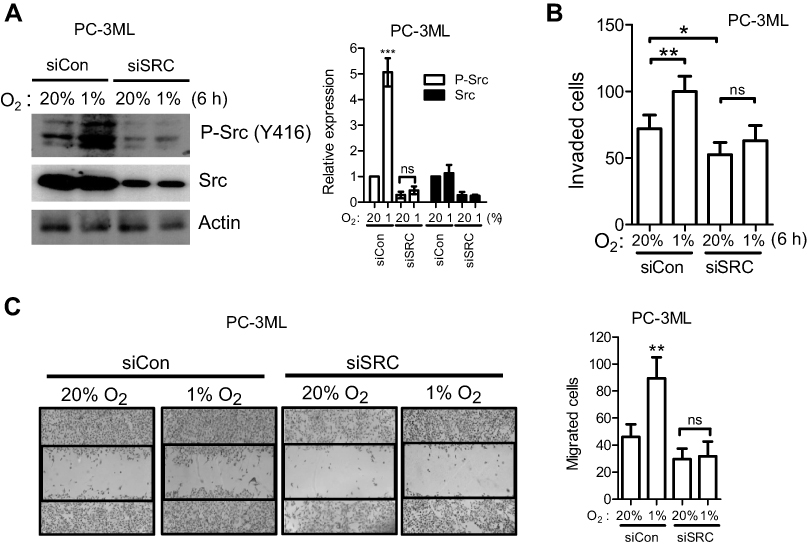 | Figure 4 SRC knockdown attenuates hypoxia-induced cell functions. PC-3ML cells were transiently transfected with siSRC or control siRNA for 24 h, and exposed to 1% O2 for 6 h. (A) Knockdown effects were examined 48 h after hypoxia by Western blotting. (B) hypoxic cells were seeded into Matrigel-coated transwell inserts and cell invasion was detected 24 h after seeding. (C) hypoxic cell monolayers were scratched and the “wound” was photographed 24 h after the scratch. Original magnification, ×50. Data are from 3 independent experiments. Bars, SD (n=3). *P<0.05; **P<0.01.Abbreviation: ns, not significant. |
LYN or FYN knockdown does not attenuate hypoxia-induced functions
In C4-2B cells, individual silencing of the SRC and LYN gene showed clear inhibition of total protein (Figure 5A). Similar to the results observed in PC-3ML cells (Figure 4A), SRC knockdown significantly attenuated hypoxia-inducedSrc phosphorylation in C4-2B cells, while p-Lyn was not elevated under hypoxia (Figure 5A). In line with the molecular data, hypoxia-induced clonogenic cell survival was blocked by knockdown of SRC, but not LYN (Figure 5B). Similarly, in PC-3ML cells, neither Fyn nor Lyn was phosphorylated under hypoxia, with or without gene manipulation (Figure 5C). Also, knockdown of either gene did not reduce hypoxia-induced clonogenic cell survival (Figure 5D). Taken together these data suggest that c-Src may be the most important SFK protein modulated by hypoxia resulting functional activation in prostate cancer cells.
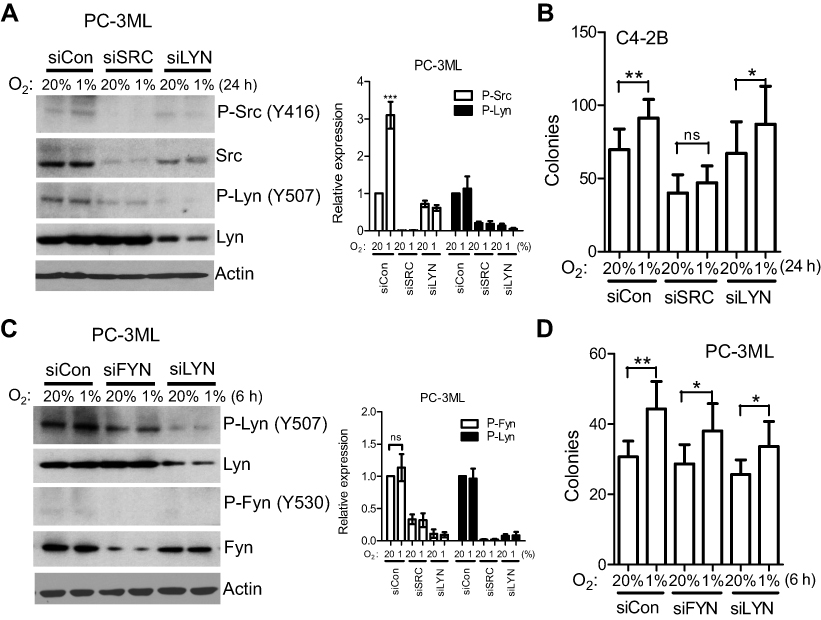 | Figure 5 LYN or FYN knockdown does not attenuate hypoxia-induced functions. (A and B), C4-2B cells were transiently transfected with SRC, LYN or control siRNA for 24 h, and exposed to 1% O2 for 24 h. Protein expression (A) and colony formation (B) were determined. Columns, mean; bars, SD (n=3). *P<0.05; **P<0.01. C & D, PC-3ML cells were transiently transfected with FYN, LYN or control siRNA for 24 h, and exposed to 1% O2 for 6 h. Knockdown effects were examined 48 h after hypoxia by Western blotting (C), and colony formation assay was detected after hypoxic exposure (D). Columns, mean; bars, SD (n=9). *P<0.05; **P<0.01. Data are from 3 independent experiments.Abbreviation: ns, not significant. |
Saracatinib inhibits hypoxia-induced cell phenotypes
To determine whether small molecule agents targeting SFKs can inhibit the enhanced effects on hypoxia-mediated functions, cells were pre-treated with Src inhibitors followed by hypoxic incubation for 6 h. Following both 20% and 1% O2 exposure, saracatinib inhibited cell invasion in a dose-dependent manner, but the inhibition was most striking under hypoxic conditions (Figure 6A). For example, at a drug concentration of 333 nM, the inhibition of invasion was 26±7.2% (P<0.05 vs DMSO) and 50±5.7% (P<0.001 vs DMSO) under normoxic and hypoxic conditions, respectively (Figure 6A). Similar effects were observed when assessing PC-3ML cell migration that saracatinib significantly reduced cell migration in a greater extent under hypoxic than normal conditions (Figure 6B). Similarly, C4-2B cells treated with another Src inhibitor dasatinib showed comparable decrease in clonogenic survival under both 20% and 1% O2 (Figure 6C). These data indicate that hypoxic tumor cells may be more sensitive to Src inhibitors than aerobic tumor cells.
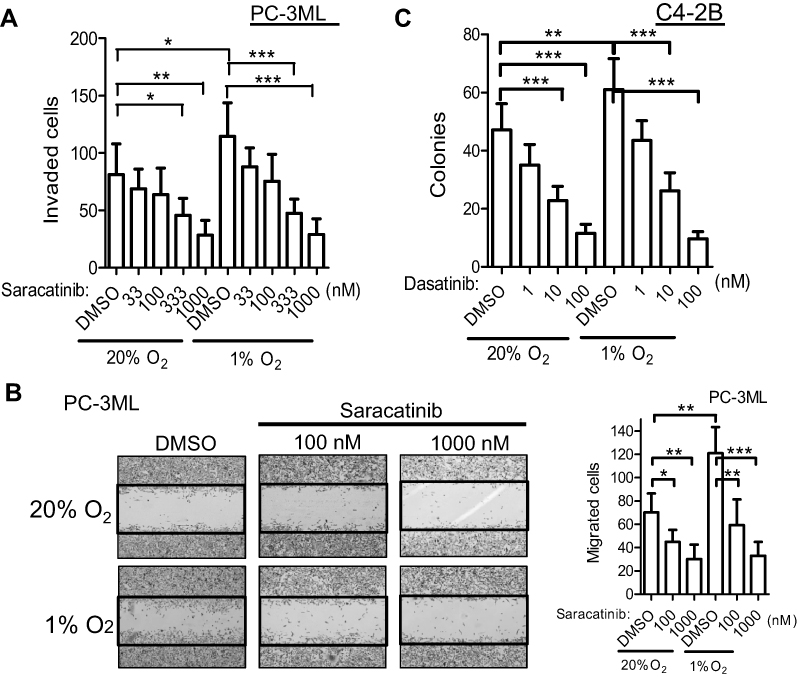 | Figure 6 Treatment effects of Src inhibitors on hypoxia-induced cell functions. (A and B), PC-3ML cells were pre-treated with DMSO or saracatinib with indicated concentrations for 1 h and exposed to 1% O2 for 6 h. Cell invasion (A) and migration (B) were detected as previously described. C, C4-2B cells were pre-treated with DMSO or dasatinib for 1 h before exposing to 1% O2 for 24 h. Clonogenic survival assay was performed. Data are from 3 independent experiments. Bars, SD (n=3). *P<0.05; **P<0.01; ***P<0.001. |
Saracatinib inhibits hypoxia-induced Src activation
To explore the molecular events occurring when tumor cells were treated with Src inhibitors under hypoxic conditions, lysates from PC-3ML cells, treated under identical hypoxia treatment protocols as the functional assays, were analyzed by Western blotting. Pre-treatment of saracatinib inhibited hypoxia-induced Src phosphorylation and HIF-1α accumulation (Figure 7). With a moderate drug concentration (333 nM), suppression of p-Src and HIF-1α were clearly observed only after 1% O2 exposure, with no decrease under normal conditions (Figure 7). In comparison, high concentration (1000 nM) of saracatinib resulted in significant reduction of both HIF-1 and p-Src under both hypoxic and normal conditions (Figure 7). These data suggest that Src inhibitors function equally under hypoxia.
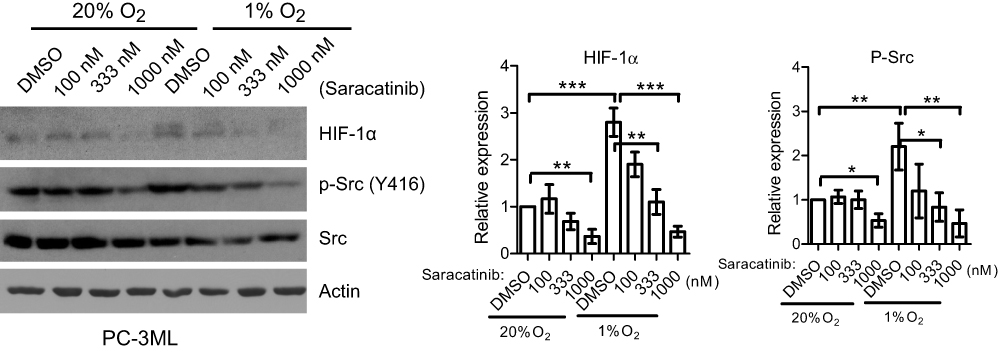 | Figure 7 Saracatinib inhibits hypoxia-induced cell phenotypes. PC-3ML cells were treated with saracatinib for 1 h and incubated under 1% O2 for 6 h. Whole cell lysates were probed for HIF-1α, p-Src (Y416) and total Src by Western blotting. Actin was used as a loading control. Bands from 3 independent experiments were quantified by Image J. Bars, SD (n=3). *P<0.05; **P<0.01; ***P<0.001. |
SRC knockdown attenuates saracatinib-mediated events under hypoxia
To further determine whether c-Src is required for drug-mediated effects under hypoxia, PC-3ML cells were knocked down by siSRC before drug treatment. Cells were then exposed to 1% O2 for 6 h and cell invasion and protein expression were tested as described previously. In control knockdown cells, saracatinib was able to significantly suppress cell invasion (P<0.01), while in SRC knockdown cells, significant inhibition was not observed (Figure 8A). Consistently, under hypoxic conditions, saracatinib failed to suppress Src phosphorylation in SRC-knockdown cells, while in control knockdown cells, p-Src inhibition by drug treatment clearly was observed (Figure 8B), indicating that the drug-mediated effects are due to c-Src inhibition. Interestingly, HIF-1α showed similar changes in response to SRC knockdown (Figure 8B), suggesting HIF-1α signaling functions as a downstream component of the Src pathway.
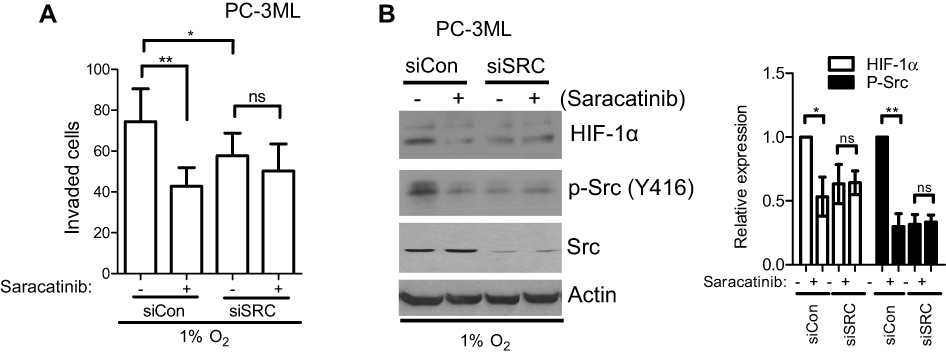 | Figure 8 SRC knockdown attenuates saracatinib-mediated events under hypoxia. PC-3ML cells were transfected with Control or SRC siRNA. Twenty-four hours after transfection, cells were treated with saracatinib (333 nM) for 1 h and incubated under 1% O2 for 6 h. (A) Cell invasion were detected 24 h after cell seeding. Bars, SD (n=3). *P<0.05; **P<0.01. (B) Whole cell lysates were probed for HIF-1α, p-Src (Y416) and total Src by Western blotting. Actin was used as a loading control. Bands from 3 independent experiments were quantified by Image J. Columns, mean; bars, SD (n=9). *P<0.05; **P<0.01.Abbreviation: ns, not significant. |
Discussion
Hypoxia is an important microenvironmental factor in tumors that can enhance many cellular functions associated with tumor progression and dissemination including invasion, migration, and survival. In the current study, we found that c-Src, the major member of SFKs, plays an essential role in disease progression in prostate cancer patients. All three key SFKs, c-Src, Fyn and Lyn, were observed to be activated in prostate tumors compared to adjacent normal tissues. However, unlike p-Src, we found that hypoxia was unable to induce p-Lyn and p-Fyn in prostate cancer cells. Knockdown of SRC, but not LYN and FYN, significantly attenuated hypoxia-mediated functional changes at the cellular level and protein expression at the molecular level. Furthermore, small molecular Src inhibitors counteracted the enhanced metastatic-associated cell functions induced by low oxygen tensions, and such drug effects were mediated by c-Src. Taken together, these data suggest that c-Src is a key player in hypoxia-mediated prostate cancer progression.
Hypoxia in clinically localized primary prostate tumors is negative prognostic factor associated with disease progression and poor outcomes.30 Previous studies in our lab have demonstrated that hypoxia and HIF-1a are strongly associated with invasiveness and disease progression in prostate cancer.31–33 Interestingly, although we have shown the critical role of several oncogenic TKs including Src in prostate cancer cell migration and invasion,23,34 whether Src or other non-Src SFKs could be regulated under oxygen-depleted environments remained largely unknown. The current study reports that c-Src, the major SFK member, is significantly upregulated by hypoxia that triggers elevated functions, indicating the potential role of c-Src in prostate cancer metastasis, but not cell proliferation, particularly under adverse tumor microenvironments. Lyn and Fyn, two non-Src SFKs that have been implicated in prostate cancer metastasis and castration resistance,15,18 are not activated under hypoxia. A prior study did show elevated Lyn’s phosphorylation after a 6 h hypoxic exposure in PC-3 cells.35 However, that study examined phosphorylation at tyrosine 416, which is believed to be the pan-tyrosine site for all SFKs. In contrast, we detected the specific autophosphorylation site of Lyn at Y507.
One limitation of the current study is the lack of sophisticated genetic manipulations to elucidate the molecular underpinnings of Src signaling. Although knockdown methods were utilized in high Src-expressing cells in the current study, overexpressing Src in low Src-expressing cells may help confirm its molecular functions. In addition, to obtain direct evidence for whether Src kinase activity is required for hypoxia-mediated functions, dominant negative c-Src (K295R) can be used in high p-Src-expressing cells; alternatively, constitutively active c-Src (Y529F) in low p-Src-expressing cells. We are currently pursuing these experiments in order to validate the functional role of c-Src under hypoxic conditions.
Src signaling has been demonstrated to play a pleiotropic role in mediating various malignant functions. Several previous studies have suggested a tight association between Src and hypoxia and its associated molecular markers, such as HIF-1α. For example, Src activity has been shown to be associated with hypoxic marker EF5 positive regions in pre-clinical cancer models, suggesting that the Src signaling pathway is responsive to tumor hypoxia in vivo.26 Hanna et al reported that HIF-1α independently activates Src to promote metastases in melanoma, suggesting that in some preclinical models Src may function as a downstream effector of HIF-1, even in the absence of hypoxia.27 Interestingly, other studies have shown that Src can regulate HIF-1α/VEGF pathway, leading to enhanced invasion and metastasis, suggesting it as an upstream regulator of HIF-1.29,36 Our data suggest that although HIF-1α can be accumulated with only a few hours hypoxic exposure, the Src inhibitor saracatinib can still inhibit such HIF-1α accumulation (Figure 7), while knockdown SRC leads to attenuated HIF-1α expression (Figure 8B), suggesting that HIF-1α may reflect functional inhibition of Src. Future studies will focus on identifying the detailed mechanism of HIF-1/Src pathway in our current models.
In summary, our current findings demonstrate that hypoxia is able to enhance metastatic phenotypes by activating Src in prostate cancer cells, and that c-Src may be the most important SFK member for hypoxia-mediated cancer cell behaviors. Importantly, SFK inhibitors were able to suppress the hypoxia-induced cell functions, suggesting that such agents may have therapeutic potential to control tumor metastasis that is driven by hypoxia in prostate cancer.
Acknowledgments
The authors gratefully acknowledge Dr. Shuang Huang for TCGA data analysis. This study was financially supported by a grant from the National Institute of Health (1R01CA197477-01A1).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Niu Y, Chang TM, Yeh S, Ma WL, Wang YZ, Chang C. Differential androgen receptor signals in different cells explain why androgen-deprivation therapy of prostate cancer fails. Oncogene. 2010;29:3593–3604. doi:10.1038/onc.2010.121
2. Yap TA, Zivi A, Omlin A, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8:597–610. doi:10.1038/nrclinonc.2011.117
3. Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8:180–192. doi:10.1038/nrc2344
4. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi:10.1038/nrc1187
5. Pipinikas CP, Carter ND, Corbishley CM, Fenske CD. HIF-1alpha mRNA gene expression levels in improved diagnosis of early stages of prostate cancer. Biomarkers. 2008;13:680–691. doi:10.1080/13547500802591992
6. Kimbro KS, Simons JW. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr Relat Cancer. 2006;13:739–749. doi:10.1677/erc.1.00728
7. Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–480. doi:10.1038/nrc1366
8. Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358.
9. Fizazi K. The role of Src in prostate cancer. Ann Oncol. 2007;18:1765–1773. doi:10.1093/annonc/mdm086
10. Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res. 2009;15:3540–3549. doi:10.1158/1078-0432.CCR-08-1857
11. Sturge J, Caley MP, Waxman J. Bone metastasis in prostate cancer: emerging therapeutic strategies. Nat Rev Clin Oncol. 2011;8:357–368. doi:10.1038/nrclinonc.2011.67
12. Chattopadhyay I, Wang J, Qin M, et al. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget. 2017;8:10324–10347. doi:10.18632/oncotarget.14401
13. Posadas EM, Al-Ahmadie H, Robinson VL, et al. FYN is overexpressed in human prostate cancer. BJU Int. 2009;103:171–177. doi:10.1111/j.1464-410X.2008.08009.x
14. Wang X, Wen J, Li R, Qiu G, Zhou L, Wen X. Gene expression profiling analysis of castration-resistant prostate cancer. Med Sci Monit. 2015;21:205–212. doi:10.12659/MSM.891193
15. Gururajan M, Cavassani KA, Sievert M, et al. SRC family kinase FYN promotes the neuroendocrine phenotype and visceral metastasis in advanced prostate cancer. Oncotarget. 2015;6:44072–44083. doi:10.18632/oncotarget.6398
16. Chatterji T, Varkaris AS, Parikh NU, et al. Yes-mediated phosphorylation of focal adhesion kinase at tyrosine 861 increases metastatic potential of prostate cancer cells. Oncotarget. 2015;6:10175–10194. doi:10.18632/oncotarget.3391
17. Goldenberg-Furmanov M, Stein I, Pikarsky E, et al. Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004;64:1058–1066.
18. Zardan A, Nip KM, Thaper D, et al. Lyn tyrosine kinase regulates androgen receptor expression and activity in castrate-resistant prostate cancer. Oncogenesis. 2014;3:e115. doi:10.1038/oncsis.2014.30
19. Cai H, Babic I, Wei X, Huang J, Witte ON. Invasive prostate carcinoma driven by c-Src and androgen receptor synergy. Cancer Res. 2011;71:862–872. doi:10.1158/0008-5472.CAN-10-1605
20. Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008;27:6365–6375. doi:10.1038/onc.2008.250
21. Nam S, Kim D, Cheng JQ, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65:9185–9189. doi:10.1158/0008-5472.CAN-05-1731
22. Dong M, Rice L, Lepler S, Pampo C, Siemann DW. Impact of the Src inhibitor saracatinib on the metastatic phenotype of a fibrosarcoma (KHT) tumor model. Anticancer Res. 2010;30:4405–4414.
23. Rice L, Lepler S, Pampo C, Siemann DW. Impact of the SRC inhibitor dasatinib on the metastatic phenotype of human prostate cancer cells. Clin Exp Metastasis. 2012;29:133–142. doi:10.1007/s10585-011-9436-2
24. Saffran V, Siemann DW Src inhibitor dasatinib reduces the metastatic phenotype of breast cancer cells.
25. Siemann DW, Dong M. Targeting metastatic tumor cell functions by Src signaling interference using the small molecule Src inhibitor, saracatinib (AZD0530). Mol Cancer Ther. 2009;8(12 Suppl):B107. doi:10.1158/1535-7163.TARG-09-B107
26. Pham NA, Magalhaes JM, Do T, et al. Activation of Src and Src-associated signaling pathways in relation to hypoxia in human cancer xenograft models. Int J Cancer. 2009;124:280–286. doi:10.1002/ijc.23912
27. Hanna SC, Krishnan B, Bailey ST, et al. HIF1alpha and HIF2alpha independently activate SRC to promote melanoma metastases. J Clin Invest. 2013;123:2078–2093. doi:10.1172/JCI66715
28. Park JJ, Jin YB, Lee YJ, et al. KAI1 suppresses HIF-1alpha and VEGF expression by blocking CDCP1-enhanced Src activation in prostate cancer. BMC Cancer. 2012;12:81. doi:10.1186/1471-2407-12-81
29. Gray MJ, Zhang J, Ellis LM, et al. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005;24:3110–3120. doi:10.1038/sj.onc.1208513
30. Marignol L, Coffey M, Lawler M, Hollywood D. Hypoxia in prostate cancer: a powerful shield against tumour destruction? Cancer Treat Rev. 2008;34:313–327. doi:10.1016/j.ctrv.2008.01.006
31. Bae KM, Dai Y, Vieweg J, Siemann DW. Hypoxia regulates SOX2 expression to promote prostate cancer cell invasion and sphere formation. Am J Cancer Res. 2016;6:1078–1088.
32. Dai Y, Shi W, Molnar N, Siemann DW. Impact of tumor hypoxia, Src, and Met signaling in the dissemination of tumor cells (Chapter 7). In: Fatatis A, editor. Signaling Pathways and Molecular Mediators in Metastasis.
33. Dai Y, Bae K, Siemann DW. Impact of hypoxia on the metastatic potential of human prostate cancer cells. Int J Radiat Oncol Biol Phys. 2011;81:521–528. doi:10.1016/j.ijrobp.2011.04.027
34. Siemann DW, Dong M, Pampo C, Shi W. Src-signaling interference impairs the dissemination of blood-borne tumor cells. Cell Tissue Res. 2012;349:541–550. doi:10.1007/s00441-012-1415-7
35. Shiozawa N, Sugahara R, Namiki K, et al. Inhibitory effect of a redox-silent analogue of tocotrienol on hypoxia adaptation in prostate cancer cells. Anti-Cancer Drugs. 2017;28:289–297. doi:10.1097/CAD.0000000000000460
36. Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–5335.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.