Back to Journals » Journal of Hepatocellular Carcinoma » Volume 2
c-MET receptor tyrosine kinase as a molecular target in advanced hepatocellular carcinoma
Authors Granito A ![]() , Guidetti E, Gramantieri L
, Guidetti E, Gramantieri L
Received 5 November 2014
Accepted for publication 6 January 2015
Published 24 April 2015 Volume 2015:2 Pages 29—38
DOI https://doi.org/10.2147/JHC.S77038
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Ahmed Kaseb
Alessandro Granito,1 Elena Guidetti,1 Laura Gramantieri2,3
1Dipartimento di Scienze Mediche e Chirurgiche Università di Bologna, Bologna, Italy; 2Dipartimento dell'Apparato Digerente, Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy; 3Centro di Ricerca Biomedica Applicata (CRBA), Azienda Ospedaliero-Universitaria Policlinico S Orsola-Malpighi e Università di Bologna, Bologna, Italy
Abstract: c-MET is the membrane receptor for hepatocyte growth factor (HGF), also known as scatter factor or tumor cytotoxic factor, a mitogenic growth factor for hepatocytes. HGF is mainly produced by cells of mesenchymal origin and it mainly acts on neighboring epidermal and endothelial cells, regulating epithelial growth and morphogenesis. HGF/MET signaling has been identified among the drivers of tumorigenesis in human cancers. As such, c-MET is a recognized druggable target, and against it, targeted agents are currently under clinical investigation. c-MET overexpression is a common event in a wide range of human malignancies, including gastric, lung, breast, ovary, colon, kidney, thyroid, and liver carcinomas. Despite c-MET overexpression being reported by a large majority of studies, no evidence for a c-MET oncogenic addiction exists in hepatocellular carcinoma (HCC). In particular, c-MET amplification is a rare event, accounting for 4%–5% of cases while no mutation has been identified in c-MET oncogene in HCC. Thus, the selection of patient subgroups more likely to benefit from c-MET inhibition is challenging. Notwithstanding, c-MET overexpression was reported to be associated with increased metastatic potential and poor prognosis in patients with HCC, providing a rationale for its therapeutic inhibition. Here we summarize the role of activated HGF/MET signaling in HCC, its prognostic relevance, and the implications for therapeutic approaches in HCC.
Keywords: hepatocellular carcinoma, c-MET, clinical trials
Introduction to c-MET and brief overview of physiological functions
c-MET is the tyrosine kinase receptor for hepatocyte growth factor (HGF),1,2 also known as Scatter Factor3 or Tumor Cytotoxic Factor.4 It is a single-pass heterodimer made of an extracellular alpha subunit containing three functional domains (the semaphorin, plexin–semaphorin–integrin, and immunoglobulin–plexin–transcription domains) linked to a transmembrane beta subunit by a disulphide bond. The transmembrane and the intracellular subunits are made of three portions controlling the kinase activity. c-MET pathway activation may occur either upon HGF binding (canonical pathway) or following interaction with other signaling cascades, such as that triggered by epidermal growth factor receptor (EGFR) or by binding with other circulating factors such as des-gamma carboxyprothrombin.5 In the canonical pathway, HGF binding leads to c-MET receptor homodimerization and autophosphorylation of tyrosine residues of the carboxy terminal domain of c-MET. These events lead to the activation of mitogen-activated protein kinase, phosphoinositide 3-kinase (PI3K)-dependent, rat sarcoma (RAS)-dependent, extracellular-signal-regulated kinase (ERK)-dependent, and RAC1-cell division control protein 42 homolog cascades,6 as summarized in Figure 1, which promote cell proliferation, survival, and cell motility. Another important function of c-MET is the prevention of apoptosis, which occurs through sequestering of the death receptor FAS, thus preventing its binding to FAS ligand.7 The physiological activation of the c-MET–driven program occurs in embryogenesis, wound healing, and tissue repair, and it is self limiting. c-MET’s activation promotes the “invasive growth” program, which is characterized by enhanced cell motility, invasion, and reduced apoptosis.
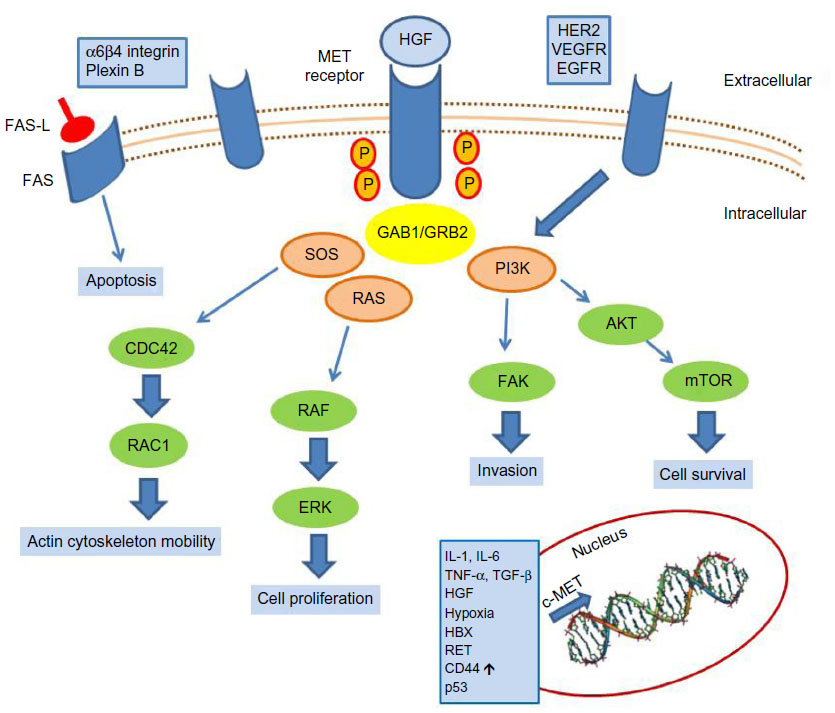 | Figure 1 c-MET activation signaling pathways. |
During embryogenesis, the HGF/MET axis sustains hepatocyte proliferation and liver and placenta development. While homozygous null mice for either HGF or c-MET die in utero at day 13 and 16, respectively, due to impaired organogenesis,8,9 when HGF or MET are knocked down at later phases during the development, the livers of these mice are reduced in size as a result of decreased hepatocyte proliferation and increased susceptibility to apoptosis. In the adult animals, under physiological conditions, loss of c-MET is not critical for hepatocyte function.10 Conversely, the role of c-MET appears to be critical when a response to injuries is required. In this regard, several experimental models have confirmed the pivotal role of MET in liver regeneration and restoration of the liver mass after partial hepatectomy.10 In the setting of fulminant hepatitis in mice treated by the agonistic antibody of FAS receptor, HGF was able to prevent the onset of fulminant hepatitis by suppressing hepatocytes apoptosis.11 When liver injury is induced as in FAS-induced apoptosis, the adaptive response of the liver is strongly reduced in the absence of c-MET. Mice lacking c-MET gene in hepatocytes are hypersensitive to FAS-induced apoptosis, dying as a result of a massive liver apoptosis.12
Another pathological condition in which c-MET exerts its cytoprotective role is cholestasis13 induced by bile duct ligation in mice. Indeed, during cholestasis the HGF/c-MET signaling provides cytoprotective effects in hepatocytes.13 In line with these findings, the role of c-MET in the maintenance of the structural integrity and adaptive plasticity of the liver under adverse conditions was reported by Marquardt et al,14 who explored the effects of c-MET inhibition (in c-MET conditional knockout mice) in the presence of carbon tetrachloride-induced liver damage. Loss of hepatocyte c-MET signaling altered the hepatic microenvironment and was associated with more pronounced fibrogenesis and liver damage, decreased hepatocyte proliferation, stellate cell activation, and rapid dystrophic calcification of necrotic areas. In the same setting, a transcriptomic analysis revealed an impact of c-MET on signaling pathways leading to fibrosis, chemotactic and inflammatory signaling, reorganization of the cytoskeletal network, intercellular communications and adhesion, proliferation, damage, and stress response.
Very recently, Kroy et al15 showed that c-MET deletion in the methionine–choline-deficient mouse model of non-alcoholic steato-hepatitis (NASH) triggers NASH progression, due to fatty acid accumulation, early progression of fibrosis, and increased apoptosis. Hepatocyte-specific deletion of c-MET (occurring in the postnatal period in a conditional knockout mice) leads to the development of severe NASH in mice.15 One of the molecular mechanisms linking c-MET to NASH is the ability of c-MET to sequester the death receptor FAS, preventing its binding to FAS ligand. In NASH, FAS ligand is produced in excess and the protective effect of MET is not effective, resulting in increased apoptotic death of hepatocytes.16 In liver cirrhosis, Kim et al17 showed how HGF/MET activation is able to suppress hepatocyte apoptosis and, at the same time, to trigger apoptosis of alpha-smooth muscle positive and portal myofibroblasts, outlining the contribution of this signaling to the resolution of cirrhotic changes in animal models of cirrhosis. This study showed that, while c-MET is undetectable in quiescent hepatic stellate cells, its expression becomes relevant in activated hepatic stellate cells and in liver myofibroblasts expressing alpha-smooth muscle actin. In vitro, HGF inhibited the activation of ERK1/2 pathway, induced c-Jun N-terminal kinase (JNK)1 phosphorylation, and promoted apoptosis in cultures of rat portal myofibroblasts. Similarly, in vivo, during diethylnitrosamine-induced rat liver injury, HGF inhibited proliferation and induced apoptosis of alpha-smooth muscle actin-positive portal myofibroblasts, reducing liver fibrosis.
All of these findings are in line with a critical role of the HGF/c-MET axis in the regulation of liver regeneration and survival, in the adaptive response of the liver, and in tissue remodeling.
Brief overview of physiological functions and patient outcomes
The role of the HGF/MET axis in liver development has been underscored by c-MET and HGF knockout mice, which display an impaired liver development with reduced size. Similarly, experimental models of liver regeneration in response to acute and chronic damage outline the role of c-MET as a strong mitogenic and antiapoptotic stimulus.18,19 In line with these findings, deregulated c-MET signaling was observed in hepatocelluar carcinoma (HCC) tissues and its deregulated expression was associated with clinical and pathological characteristics in HCC patients.20 c-MET and HGF expression in tumor tissue has been evaluated in many studies, with contrasting findings probably due to the heterogeneous and small populations assayed, the different etiologies, the intrinsic heterogeneous nature of HCC (whose molecular classification is still poorly defined) and the use of different techniques, such as Northern blot, Western blot, reverse-transcription polymerase chain reaction (RT-PCR), and immunohistochemistry (IHC) – this last being the most commonly used method for examination of c-MET expression in human tissues.
c-MET overexpression was observed in human HCC samples by means of Northern blot analysis and an IHC study. Northern blot analysis revealed c-MET mRNA expression in the tumors of 6/19 patients (31.6%); in the IHC study, high c-MET expression was detected in 16/23 patients (69.9%). Both methods revealed c-MET overexpression in HCC compared with the surrounding normal liver.20 Tavian et al21 performed an IHC study and RT-PCR in 24 patients with HCC and showed c-MET overexpression in most of the cases, but low levels of HGF. c-MET overexpression was observed by Western blot analysis in 62 patients with HCC and was associated with an increased incidence of intrahepatic metastases and worse survival. Patients with high expression of c-MET in HCC tissue had a significantly shorter survival than patients with low c-MET expression (33% versus 80.3%, respectively).22 These findings were further confirmed by Kaposi-Novak et al,23 who identified a c-MET-driven gene-expression signature in all HCC metastases but in only a subgroup of primary HCCs.
Most reports suggest that c-MET overexpression is significantly associated with clinicopathological features of HCC, such as tumor grade,20,24 vascular invasion or thrombosis, tumor recurrence,25 metastases,25,26 and worse prognosis with impaired 5-year survival.22,27 However, other studies reported contradictory findings, in particular with respect to tumor stage.28,29 A recent large retrospective study of 194 patients with HCC treated by hepatectomy or microwave ablation revealed that c-MET overexpression was associated with unfavorable clinical outcomes.27 Recently, Lee et al30 assayed c-MET expression and amplification in 287 patients with HCC that was associated with hepatitis B virus infection in 75% of cases. They reported a c-MET overexpression in about 30% of patients with HCC that was not associated with any clinicopathological variable (histopathological grade, size, microvascular and macrovascular invasion, stage, recurrence-free survival, overall survival).
A higher consensus can be found among studies testing genetic alterations of c-MET genomic region, including c-MET amplification and activating point mutations. c-MET amplification and mutation seem to be rare events in most of the studies performed in HCC. A low frequency of c-MET amplification was reported in the HCC series examined by Takeo et al,31 who identified only one case out of 20 HCCs with c-MET amplification, as well as in the 59 patients with HCC examined by Kondo et al26 and in the large series of 286 patients with HCC assayed by Wang et al, where only 4%–5% of cases displayed c-MET amplification.32 Conversely, in this same study, no patient with HCC displayed an amplification of HGF. Concerning activating point mutations, Guichard et al33 did not identify any c-MET point mutations in 24 patients with HCC analyzed by whole exome sequencing.
On the other hand, HGF expression was not increased in tumor tissues in most of the studies performed on human HCCs.21,22 This last evidence suggests an HGF-independent c-MET activation. Remarkably, abnormal c-MET pathway activation can occur from the interaction with adhesive receptors, tyrosine kinases receptors, such as EGFR and vascular endothelial growth factor receptor (VEGFR), proapoptotic FAS, and, finally, by binding with des-gamma-carboxy prothrombin secreted from HCC cells.5
Because no standardized quantification approach can be recommended to assay c-MET expression in HCC, and no scoring system can be suggested, c-MET quantification in HCC largely depends on the analytical approach adopted by each individual study, assessing either mRNA or protein expression. In addition, because HCC is an extremely heterogeneous cancer, the analysis of small groups with different etiologies and different clinical and pathological characteristics may contribute at least in part to the contradictory results reported in the literature. Meanwhile, the discovery of microRNAs as regulators of c-MET expression makes the quantification of mRNA questionable as a biomarker to assess c-MET expression in HCC. In this perspective, the use of c-MET–dependent downstream factors or pathways as biomarkers reflecting c-MET pathway activation might help in the choice of patients more likely to benefit from c-MET-inhibitors. Moreover, most of the data reported in the literature are derived primarily from surgical series of patients who underwent hepatectomy for early HCC32 and who were usually previously untreated.
Remarkably, few data on c-MET expression are available in advanced HCC and, especially, in previously treated patients, as several therapeutic approaches (such as transarterial chemoembolization or antiangiogenic drugs) induce hypoxia and might be responsible for increased c-MET expression in tumor tissue. Hypoxia, occurring during tumor progression or as a result of specific treatments, is a well-known factor able to activate the transcription of the c-MET proto-oncogene, as proven both in vitro and in vivo.34 Although low partial oxygen tension represents a limiting factor for tumor growth, it nonetheless acts as a positive stimulus by inducing neoangiogenesis,35 by selecting cells that are more resistant to apoptosis,36 and by triggering invasive growth through the increased transcription of the c-MET proto-oncogene and the HGF signaling stimulation.
Role of c-MET in cancer
c-MET overexpression is a common event in a wide range of human malignancies, including gastric, lung, breast, ovary, colon, kidney, thyroid and liver carcinomas.37 The first evidence underscoring the driver effect of c-MET in tumorigenesis was the discovery of the germline activating mutation in patients with hereditary papillary renal cancer.38 Similarly, activating germline point mutations have been identified in patients affected by childhood HCC,39 gastric carcinoma,40 and squamous cell cancers.41 Spontaneous somatic mutations remain a rare event, accounting for no more than 3%–4% of cases.42 Another relevant finding confirming the role of c-MET in cancer was represented by the identification of c-MET as the protein product of a chromosomal rearrangement in an osteosarcoma cell line treated with a chemical carcinogen.43 This rearrangement results in a constitutively active fused oncogene, the translocated promoter region (TPR)-MET. In a transgenic mouse model, the enforced expression of TPR-MET leads to the development of mammary tumors.44 In humans, the TPR-MET rearrangement has been detected in some cases of gastric cancer.45
Different molecular alterations were found to determine c-MET activation in human tumors: point mutations, gene amplifications, enhanced transcription, autocrine activation. In HCC, the aberrant activation of c-MET signaling results mostly from ligands binding or from its overexpression due to enhanced transcription, rather than from gene mutations.46,47 In turn, c-MET enhanced transcription can be triggered by various factors, including: 1) cytokines such as interleukin (IL)-1, IL-6, tumor necrosis factor-α, and transforming growth factor-β;48 2) stimulation by HGF;49 3) hypoxia and in particular by HIF1-α;34 4) cross-talking with oncogenic pathways such as those of RAS,50 ETS,51 and RET;52 5) adhesive receptors such as CD4453 and α6β4 integrins;54 6) cross-talk with the downstream pathways regulated by receptor tyrosine kinases such as EGFR, human epidermal growth factor receptor 2, and VEGFR;55,56 7) cross-talk with plasma membrane proteins such as Caveolin1, involved in the modulation of signal transduction;57 or, 8) as a result of tumor-suppressor gene inactivation, such as TP53.58
Besides these events responsible for c-MET overexpression, the contribution of microRNAs as modulators of c-MET expression was recently outlined. In particular, miR-1, miR-34b and c,59 and miR-199a60 were demonstrated to directly bind c-MET transcript and to modulate its expression as well as its biological effects. miR-199a downregulation is a common and quantitatively relevant event in HCC61 resulting from abnormal histone methylation. In HCC, c-MET overexpression resulting from miR-199a downregulation is responsible for increased proliferation and increased invasion capability.60 Korhan et al62 recently demonstrated that c-MET is a direct target of miR-181a-5p, whose downregulation in HCC leads to enhanced motility, invasion, and branching morphogenesis. Another microRNA-dependent modulation of the HGF/c-MET pathway is operated by miR-26a in HCC. Indeed miR-26a directly targets HGF, thus suppressing angiogenesis.63 Recently, Takeda et al64 demonstrated that mixed-lineage leukemia (MLL), the human homologue of the trithorax in Drosophila, activates matrix metalloproteinase (MMP)-1 and MMP-3 transcription via H3K4 methylation. MLL-ETS2 complex, which is responsible for histone H3K4 methylation, is stabilized by HGF/MET signaling, conferring invasive and metastatic potential to HCC cells. Indeed, deregulated extracellular proteolysis is an essential element contributing to cancer-cell metastatic spread and it mainly relies upon MMP activity. Ozaki et al65 described an upregulation of MMP-1, MMP-3, MMP-7 as well as c-MET both in primary HCCs and in HCC-derived cell lines. They also determined the role of HGF as an inducer of MMP-1, MMP-3, MMP-7, and c-MET via ETS-1 binding to MMP promoters in HCC.65 Conversely, no increase of MMP-2, MMP-9 or HGF mRNA expression could be found.
To date it is commonly accepted that the HGF/c-MET signaling pathway regulates multiple cellular processes, notably leading to increased cell growth, protection from apoptosis, scattering and migration, invasion, and angiogenesis, through interaction with a plethora of downstream stress and survival association molecules.66 These complex events, defined as “invasive growth” program,67 are involved in a wide variety of physiological and pathological contexts, such as embryonic development during gastrulation and nervous system expansion, adult tissue regeneration after injuries and organ failure,68,69 and, finally, in pathological conditions, in the mechanisms of growth and invasion that occur during tumor development and progression.70 While in physiological events c-MET activation is a transient occurrence, during tumor onset and progression c-MET signaling can be constitutively active. Experimental evidence linking the constitutive activation of c-MET to the malignant phenotype and in particular to liver cancer have been reported both in vitro and in vivo. Indeed, c-MET overexpression increases the tumorigenic potential of HCC cell lines injected into nude mice. Xenografts obtained with HCC-derived cell lines manipulated for enhanced or inhibited expression of c-MET clearly outline the relevance of MET overexpression as an event promoting the tumorigenic properties of these cells.29 Experimental conditions that mimic the spontaneous amplification of the c-MET proto-oncogene observed in human tumors, such as transgenic mice overexpressing c-MET, confirmed the development of hepatocellular carcinomas.71,72 In addition, c-MET cooperates with other known oncogenes involved in HCC development, such as c-Myc and beta-catenin, to generate more aggressive tumors in mice.73
The advantage conferred by the activation of c-MET pathway to neoplastic cells during tumor progression has been linked mainly to their increased capability to disaggregate from surrounding tumor cells, destroy the basement membranes, and enhance cell motility and metastatic potential. The involvement of the HGF/MET axis in facilitating tumor metastasis is sustained by experimental evidence obtained in vitro, in animal models, and indirectly confirmed by studies on human HCC specimens. The induction of proteases such as urokinase-type plasminogen activator and MMPs is responsible for the breakdown of the extracellular matrix which, in turn, facilitates the invasion capability of cancer cells. Several studies performed on human HCC samples seem to confirm the possible role of c-MET in tumor progression and metastasis, since MET overexpression is associated with poor-to-moderate differentiation,20,25 presence of intrahepatic metastasis,22,25 and shorter survival.21 In line with these findings, c-MET inhibition in experimental settings was shown to reduce HCC growth and invasion capability.74,75
However, the studies analyzing the role of HGF/MET signaling in hepatocarcinogenesis have also provided contrasting evidence. Even though it is widely accepted that c-MET is required for normal liver regeneration,10,12 Takami et al76 reported that liver-specific c-MET-/- mice displayed a greater number of HCCs that were also larger in size and earlier in the development following a treatment with N-nitrosodiethylamine.76 Interestingly, the growth advantage conferred by the abrogation of c-MET signaling was observed in the early stages of hepatocarcinogenesis. In this model of HCC induced by N-nitrosodiethylamine, c-MET knockout was associated with increased lipid peroxidation, decreased ratio of reduced glutathione to oxidized glutathione, and upregulation of superoxide dismutase 1 and heat shock protein 70, all consistent with a compensatory response to increased oxidative stress and with a role of MET in the maintenance of a normal redox homeostasis. In addition, the transcriptomic analysis performed by microarray confirmed an upregulation of genes associated with cell proliferation and stress responses in c-MET mutant livers.
Thus, HGF/MET signaling may elicit opposing proliferative responses in normal and transformed hepatocytes.77 The loss of functional c-MET in hepatocytes was thus responsible for a state of chronic oxidative stress with increased reactive oxygen species production triggering hepatocarcinogenesis. Indeed, in this experimental setting, dietary supplementation with the thiol-containing antioxidant N-acetyl-L-cysteine rescued the pro-oxidative effect of c-MET deficiency, reducing hepatocarcinogenesis. Similar findings were reported by Marx-Stoelting et al78 in a N-nitrosodiethylamine and phenobarbital-induced model of HCC obtained in a different strain of mice that lacked a functional c-MET. As reported in the previous studies, conditional c-MET knockout mice developed an increased number of preneoplastic and neoplastic liver lesions when compared to controls, again outlining how impaired c-MET signaling participates in HCC induction. These findings supporting tumor-promoting effects of c-MET deficiency should be taken into consideration for their possible implications in the planning of c-MET targeted treatment in specific subgroups of patients.
Even more controversial data have been reported in studies exploring the functions of HGF in in vitro and in vivo models of HCC. Indeed, HGF, originally identified as a factor sustaining mitogenic, morphogenic, antiapoptotic, and motogenic properties of hepatocytes, is also able to inhibit proliferation and cell growth in hepatic stellate cells. It is likely that the final effects of HGF/MET axis activation mainly rely upon the cell type, the simultaneous functional status of other signaling pathways cross-talking with it, such as ERK phosphorylation,79 or activation of JNK1.80
Critical analysis of the potential for targeting c-MET in hepatocellular carcinoma
Based on the therapeutic rationale to target c-MET, various c-MET inhibitors are currently being developed as potential treatments for a variety of cancers.81 Clinical trials targeting c-MET in hepatocellular carcinoma are summarized in Table 1.
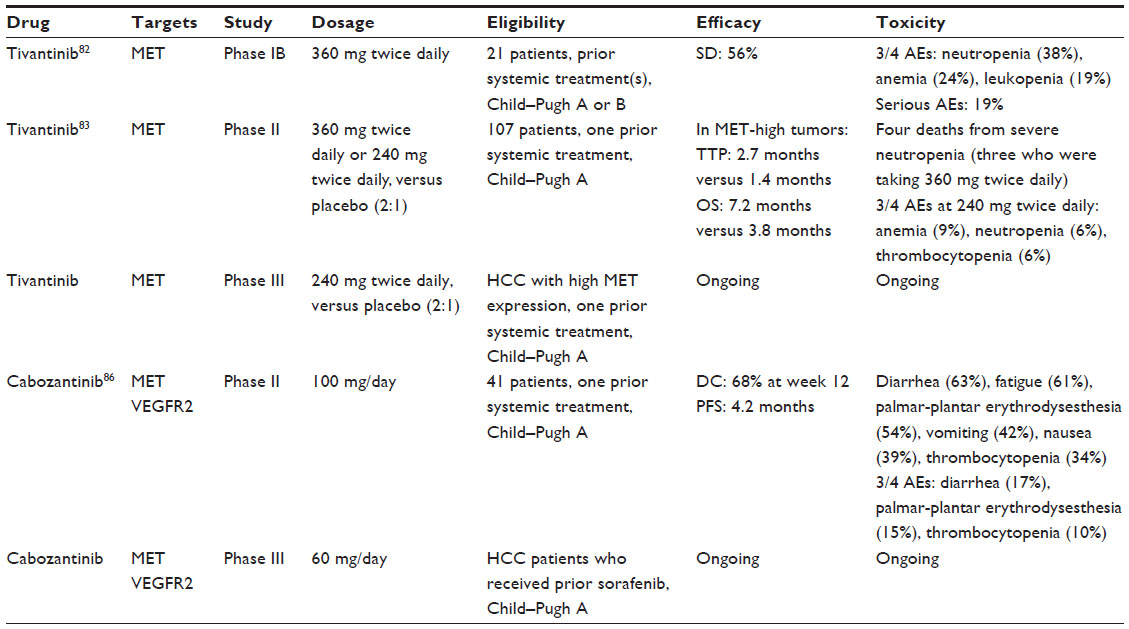 | Table 1 Clinical trials targeting c-MET in hepatocellular carcinoma |
Tivantinib, an oral selective c-MET receptor tyrosine kinase inhibitor, has shown promising results in Phase I and II studies as monotherapy for the treatment of advanced HCC.82,83
In a multicenter, single-arm, Phase IB study, 21 cirrhotic patients (Child–Pugh A or B) with advanced HCC for whom prior systemic therapy had failed were treated with tivantinib at a dose of 360 mg twice daily.82 Treatment was associated with disease stabilization in 56% of 16 evaluable patients. Twenty patients (95%) had at least one drug-related adverse event (AE); the most common of any grade were neutropenia (52%), anemia (48%), asthenia (48%), leukopenia (38%), anorexia (38%), and diarrhea (29%). The most-frequent grade 3/4 AEs were neutropenia (38%), anemia (24%), and leukopenia (19%). Serious AEs occurred in four (19%) patients and included anemia, neutropenia, and one fatal septic shock secondary to neutropenia.
More recently, tivantinib was studied in a randomized Phase II trial in patients with advanced HCC and Child–Pugh A cirrhosis who had radiological progression or intolerance to first line systemic therapy.83 Patients were randomized (2:1) to receive tivantinib (360 mg twice daily) or placebo until disease progression. The primary endpoint was time to progression (TTP) in the intention-to-treat population. c-MET expression was assessed in archival or recent tumor samples by IHC, and samples that scored ≥2 in at least 50% of tumor cells were considered as having high c-MET expression. Importantly, the tivantinib dose was amended to 240 mg twice daily because of high incidence of treatment-related grade ≥3 neutropenia (21%) observed with the starting dose of 360 mg twice daily. Four deaths occurred in patients receiving tivantinib, three in the 360 mg twice daily group and one in the 240 mg twice daily group, all of which were related to severe neutropenia.
From an antitumor standpoint, in molecularly unselected patients (71 tivantinib versus 36 placebo), median TTP was longer for those treated with tivantinib (1.6 months [95% confidence interval {CI} =1.4–2.8]) than placebo (1.4 months [95% CI =1.4–1.5]; hazard ratio [HR] =0.64; P=0.04) with no significantly different survival (median overall survival was 6.6 months for patients in the tivantinib group and 6.2 months for patients in the placebo group; HR =0.90; P=0.63).
Interestingly, in the post hoc analysis of a c-MET-high subgroup, the tivantinib group (n=22) had a median TTP of 2.7 months (95% CI =1.4–8.5 months) compared to 1.4 months (95% CI =1.4–1.6 months; HR =0.43; P=0.03) in the placebo group (n=15), with a significantly longer median overall survival (7.2 months [95% CI =3.9–14.6 months] versus 3.8 months [95% CI =2.1–6.8 months], respectively; HR =0.38; P=0.01). Furthermore, the comparison of outcomes by c-MET status in the placebo group showed that the c-MET-high subgroup had a significantly shorter median overall survival than did c-MET-low patients (3.8 months [95% CI =2.1–6.8 months] versus 9.0 months [95% CI =3.7–24.0 months], respectively; HR =2.94; P=0.02), thus suggesting that c-MET overexpression is an independent negative prognostic factor for overall survival in a population of previously treated HCC patients.
Given these promising results, a Phase III, randomized, placebo-controlled trial to evaluate the efficacy of tivantinib in patients with c-MET-high HCC for whom sorafenib therapy had failed was initiated. The confirmation that HCC patients with high c-MET expression treated with tivantinib have a better outcome than those with low MET expression might offer the background for the first attempt at personalized therapy in HCC patients selected on a molecular basis.
Other c-MET inhibitors are currently under investigation in HCC.84 Cabozantinib is an oral small-molecule tyrosine kinase inhibitor that blocks phosphorylation of c-MET and VEGFR2.85 In a randomized discontinuation Phase II study, 41 HCC patients (Child–Pugh A) who experienced tumor progression after one prior systemic therapy received cabozantinib at a dose of 100 mg daily over a 12 week lead-in stage.86 Treatment continuation past week 12 was based on response: patients with partial response (PR) continued open-label cabozantinib, patients with stable disease (SD) were randomized to cabozantinib versus placebo, and those with progressive disease discontinued the treatment. The primary endpoint in the randomized phase was progression-free survival.
Twenty-nine (71%) patients completed the lead-in stage. The overall disease control rate (DCR = PR + SD) at week 12 was 68%. Median progression-free survival was 4.2 months (95% CI =3.0–5.6 months). Most common grade 3/4 AEs were diarrhea (17%), palmar-plantar erythrodysesthesia (15%), and thrombocytopenia (10%). Based on the encouraging antitumor activity of cabozantinib, a Phase III evaluation in HCC patients where sorafenib failed or could not be tolerated is currently underway.
Conclusion
Understanding the signaling pathways driving the malignant phenotype opens the possibility of their therapeutic inhibition. The molecular events contributing to the c-MET–induced “invasive growth” program are being identified in different experimental settings. Conversely, the full understanding of their role and their therapeutic targeting in HCC patients is still in progress. This has to be ascribed to several factors, among which is the high molecular heterogeneity of HCC, which is even more complex when advanced and previously treated tumors are considered. Anticancer treatments are in fact well-known triggers of genetic, epigenetic, and transcriptomic changes. In addition, a consensus on tissue biomarkers identifying tumors in which the c-MET–dependent “invasive growth” program is a driver event are still poorly defined. Indeed, activating point mutations, gene amplification, c-MET–enhanced mRNA and protein expression, as well as c-MET–dependent signatures, have been assayed in different studies, but a correlation between a specific biomarker(s) and the likelihood of response to c-MET inhibition is still under evaluation. The identification of shared assays reflecting the activation of c-MET signaling will give useful information, especially if the final effectors of the signaling pathways will be tested.
Indeed, the complexity of cross-talk with other intracellular cascades makes the molecular scenario quite complex. This is more and more evident if we consider that HCC candidates for c-MET inhibition are those previously treated by both locoregional and systemic approaches. In these HCCs, a molecular classification is not available. Preclinical findings, however, clearly outline the role of hypoxia-inducing treatments in the selection of more aggressive clones with high migration and invasion capability. In these settings it will be crucial to investigate whether the combinations between c-MET inhibitors or HGF antagonists might prevent the escape mechanisms described under hypoxic conditions.87 In addition, it should be kept in mind that in several cases a lack of correlation was reported between HGF levels and c-MET expression and activation. This finding outlines the role of alternative ways of triggering of c-MET signaling, other than HGF, and might play a role in the resistance to c-MET receptor targeted treatments.
Disclosure
The authors have no competing interest to declare. The authors report no other conflicts of interest in this work.
References
Nakamura T, Nawa K, Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122(3):1450–1459. | |
Russell WE, McGowan JA, Bucher NL. Partial characterization of a hepatocyte growth factor from rat platelets. J Cell Physiol. 1984;119(2):183–192. | |
Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327(6119):239–242. | |
Shima N, Nagao M, Ogaki F, Tsuda E, Murakami A, Higashio K. Tumor cytotoxic factor/hepatocyte growth factor from human fibroblasts: cloning of its cDNA, purification and characterization of recombinant protein. Biochem Biophys Res Commun. October 31, 1991; 180(2):1151–1158. | |
Suzuki M, Shiraha H, Fujikawa T, et Al. Des-gamma-carboxy prothrombin is a potential autologous growth factor for hepatocellular carcinoma. J Biol Chem. February 25, 2005;280(8):6409–6415. | |
Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. January 24, 2012;12(2):89–103. | |
Wang X, DeFrances MC, Dai Y,et Al. A mechanism of cell survival: sequestration of Fas by the HGF receptor Met. Mol Cell. February 2002; 9(2):411–421. | |
Schmidt C, Bladt F, Goedecke S, et Al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. February 23, 1995;373(6516):699–702. | |
Uehara Y, Minowa O, Mori C, et Al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. February 23, 1995;373(6516):702–705. | |
Borowiak M, Garratt AN, Wüstefeld T, Strehle M, Trautwein C, Birchmeier C. Met provides essential signals for liver regeneration. Proc Natl Acad Sci U S A. July 20, 2004;101(29):10608–10613. | |
Kosai K, Matsumoto K, Nagata S, Tsujimoto Y, Nakamura T. Abrogation of Fas-induced fulminant hepatic failure in mice by hepatocyte growth factor. Biochem Biophys Res Commun. March 27, 1998;244(3):683–690. | |
Huh CG, Factor VM, Sánchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. March 30, 2004;101(13):4477–4482. | |
Giebeler A, Boekschoten MV, Klein C, et Al.c-Met confers protection against chronic liver tissue damage and fibrosis progression after bile duct ligation in mice. Gastroenterology. July 2009;137(1):297–308, 308.e1–4. | |
Marquardt JU, Seo D, Gómez-Quiroz LE, et Al.Loss of c-Met accelerates development of liver fibrosis in response to CCl(4) exposure through deregulation of multiple molecular pathways. Biochim Biophys Acta. June 2012;1822(6):942–951. | |
Kroy DC, Schumacher F, Ramadori P,et Al. Hepatocyte specific deletion of c-Met leads to the development of severe non-alcoholic steatohepatitis in mice. J Hepatol. October 2014;61(4):883–890. | |
Zou C, Ma J, Wang X, et Al.Lack of Fas antagonism by Met in human fatty liver disease. Nat Med. September 2007;13(9):1078–1085. | |
Kim WH, Matsumoto K, Bessho K, Nakamura T. Growth inhibition and apoptosis in liver myofibroblasts promoted by hepatocyte growth factor leads to resolution from liver cirrhosis. Am J Pathol. April 2005;166(4):1017–1028. | |
Zarnegar R, Michalopoulos G. Purification and biological characterization of human hepatopoietin A, a polypeptide growth factor for hepatocytes. Cancer Res. June 15, 1989;49(12):3314–3320. | |
Nakamura T, Nishizawa T, Hagiya M, et Al. Molecular cloning and expression of human hepatocyte growth factor. Nature. November 23, 1989;342(6248):440–443. | |
Suzuki K, Hayashi N, Yamada Y, et Al. Expression of the c-met protooncogene in human hepatocellular carcinoma. Hepatology. November 1994;20(5):1231–1236. | |
Tavian D, De Petro G, Benetti A, Portolani N, Giulini SM, Barlati S. u-PA and c-MET mRNA expression is co-ordinately enhanced while hepatocyte growth factor mRNA is down-regulated in human hepatocellular carcinoma. Int J Cancer. September 1, 2000;87(5):644–649. | |
Ueki T, Fujimoto J, Suzuki T, Yamamoto H, Okamoto E. Expression of hepatocyte growth factor and its receptor c-met proto-oncogene in hepatocellular carcinoma. Hepatology. April 1997;25(4):862–866. | |
Kaposi-Novak P, Lee JS, Gòmez-Quiroz l, Coulouarn C, Factor VM, Thorgeirsson SS. Met – regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest. June 2006; 116(6):1582–1595. | |
D’Errico A, Fiorentino M, Ponzetto A, et Al. Liver hepatocyte growth factor does not always correlate with hepatocellular proliferation in human liver lesions: its specific receptor c-met does. Hepatology. July 1996; 24(1):60–64. | |
Daveau M, Scotte M, François A, et Al. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog. March 2003;36(3):130–141. | |
Kondo S, Ojima H, Tsuda H, et Al. Clinical impact of c-Met expression and its gene amplification in hepatocellular carcinoma. Int J Clin Oncol. April 2013;18(2):207–213. | |
Wang ZL, Liang P, Dong BW, Yu XL, Yu de J. Prognostic factors and recurrence of small hepatocellular carcinoma after hepatic resection or microwave ablation: a retrospective study. J Gastrointest Surg. February 2008;12(2):327–337. | |
Shimomura T, Kondo J, Ochiai M, et Al. Activation of the zymogen of hepatocyte growth factor activator by thrombin. Journal of Biological Chemistry. 1993;268(30):22927–22932. | |
Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nature Reviews Molecular Cell Biology. 2003;4(12):915–925. | |
Lee SJ, Lee J, Sohn I, Mao M, Kai W, Park CK, Lim HY. A survey of c-MET expression and amplification in 287 patients with hepatocellular carcinoma. Anticancer Res. November 2013;33(11):5179–5186. | |
Takeo S, Arai H, Kusano N, et Al. Examination of oncogene amplification by genomic DNA microarray in hepatocellular carcinomas: comparison with comparative genomic hybridization analysis. Cancer Genet Cytogenet. October 15, 1996;130(2):127–132. | |
Wang K, Lim HY, Shi S,et Al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. August 2013; 58(2):706–717. | |
Guichard C, Amaddeo G, Imbeaud S, et Al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. May 6, 2012;44(6):694–698. | |
Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM.Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003 April; 3(4):347–61. | |
A.L. Harris. Hypoxia-a key regulatory factor in tumor growth. Nature Rev. Cancer (2002),pp 38–48. | |
J.L. Yu, J.W. Rak, B.L. Coomber et Al. Effect of p53 status on tumor response to antiangiogenic therapy. Science, 295 (2002), pp. 1526–1528. | |
Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. November 2011; 3(1 Suppl):S21–35. | |
Schmidt L, Duh FM, Chen F,et Al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. May 1997; 16(1):68–73. | |
Park WS, Dong SM, Kim SY, et Al. Somatic mutations in the kinase domain of the Met/hepatocyte growth factor receptor gene in childhood hepatocellular carcinomas. Cancer Res. January 15, 1999;59(2):307–310. | |
Lee JH, Han SU, Cho H,et Al. A novel germ line juxtamembrane Met mutation in human gastric cancer. Oncogene. October 12, 2000;19(43):4947–4953. | |
Aebersold DM, Landt O, Berthou S, et Al. Prevalence and clinical impact of Met Y1253D-activating point mutation in radiotherapy-treated squamous cell cancer of the oropharynx. Oncogene. November 20, 2003;22(52):8519–8523. | |
Wellcome Trust Sanger Institute. COSMIC-Catalogue of Somatic Mutations in Cancer [webpage on the Internet]. Hinxton, Cambridge, UK: Wellcome Trust Sanger Institute, Genome Research limited, 2010. Available from: http//www.sanger.ac.uk/genetics/CGP/cosmic/. Accessed January 5, 2015. | |
Cooper CS, Park M, Blair DG, et Al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. September 6–11, 1984;311(5981):29–33. | |
Liang TJ, Reid AE, Xavier R, Cardiff RD, Wang TC. Transgenic expression of tpr-met oncogene leads to development of mammary hyperplasia and tumors. J Clin Invest. June 15, 1996;97(12):2872–2877. | |
Soman NR, Correa P, Ruiz BA, Wogan GN. The TPR-MET oncogenic rearrangement is present and expressed in human gastric carcinoma and precursor lesions. Proc Natl Acad Sci U S A. June 1, 1991;88(11):4892–4896. | |
Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors.[Review]Clin Invest. April 2002;109(7):863–867. Review. | |
Corso S, Comoglio PM, Giordano S. Cancer therapy: can the challenge be MET? [Review]Trends Mol Med. June 2005;11(6):284–292. | |
Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer.[Review] Nat Rev Mol Cell Biol. December 2010;11(12):834–848. | |
Boccaccio C, Gaudino G, Gambarotta G, Galimi F, Comoglio PM. Hepatocyte growthn factor (HGF) receptor expression is inducible and is part of the delayed-early response to HGF. J Biol Chem. April 29, 1994;269(17):12846–12851. | |
Webb CP, Taylor GA, Jeffers M, et Al. Evidence for a role of Met-HGF/SF during Ras-mediated tumorigenesis/metastasis. Oncogene. October 22, 1998;17(16):2019–2025. | |
Gambarotta G, Boccaccio C, Giordano S, Ando M, Stella MC, Comoglio PM. Ets up-regulates MET transcription. Oncogene. November 7, 1996;13(9):1911–1917. | |
Ivan M, Bond JA, Prat M, Comoglio PM, Wynford-Thomas D. Activated ras and ret oncogenes induce over-expression of c-met (hepatocyte growth factor receptor) in human thyroid epithelial cells. Oncogene. May 22, 1997;14(20):2417–2423. | |
Orian-Rousseau V, Chen L, Sleeman JP, Herrlich P, Ponta H. CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. December 1, 2002;16(23):3074–3086. | |
Trusolino L, Bertotti A, Comoglio PM. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell. November 30, 2001;107(5):643–654. | |
Bergström JD, Westermark B, Heldin NE. Epidermal growth factor receptor signaling activates met in human anaplastic thyroid carcinoma cells. Exp Cell Res. August 25, 2000;259(1):293–299. | |
Lu KV, Chang JP, Parachoniak CA, et Al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. July 10, 2012;22(1):21–35. | |
Korhan P, Erdal E, Kandemis E, et Al. Reciprocal activating crosstalk between c-Met and caveolin 1 promotes invasive phenotype in hepatocellular carcinoma. PLoS One. August 22, 2014;9(8):e105278. | |
Rong S, Donehower LA, Hansen MF, et Al. Met proto-oncogene product is overexpressed in tumors of p53-deficient mice and tumors of Li-Fraumeni patients. Cancer Res. May 1, 1995;55(9):1963–1970. | |
Migliore C, Petrelli A, Ghiso E, et Al. MicroRNAs impair MET-mediated invasive growth. Cancer Res. December 15, 2008;68(24):10128–10136. | |
Fornari F, Milazzo M, Chieco P, et Al. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. June 15, 2010;70(12):5184–5193. | |
Hou J, Lin L, Zhou W, et Al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell. February 15, 2011;19(2):232–243. | |
Korhan P, Erdal E, Atabey N. MiR-181a-5p is downregulated in hepatocellular carcinoma and suppresses motility, invasion and branching-morphogenesis by directly targeting c-Met. Biochem Biophys Res Commun. August 8, 2014;450(4):1304–1312. | |
Yang X, Zhang XF, Lu X, et Al. MicroRNA-26a suppresses angiogenesis in human hepatocellular carcinoma by targeting hepatocyte growth factor-cMet pathway. Hepatology. May 2014;59(5):1874–1885. | |
Takeda S, Liu H, Sasagawa S,et Al. HGF-MET signals via the MLL-ETS2 complex in hepatocellular carcinoma. J Clin Invest. July 1, 2013;123(7):3154–3165. | |
Ozaki I, Mizuta T, Zhao G. Induction of multiple matrix metalloproteinase genes in human hepatocellular carcinoma by hepatocyte growth factor via a transcription factor Ets-1. Hepatol Res. December 2003;27(4):289–301. | |
Comoglio PM, Trusolino L. Invasive growth: from development to metastasis. [Review] J Clin Invest. April 2002;109(7):857–862. | |
Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. [Review] Nat Rev Cancer. April 2002; 2(4):289–300. | |
Michalopoulos GK, DeFrances MC. Liver regeneration.[Review] Science.April 4, 1997;276(5309):60–66. | |
Rabkin R, Fervenza F, Tsao T, et Al. Hepatocyte growth factor receptor in acute tubular necrosis. J Am Soc Nephrol. March 2001; 12(3):531–540. | |
Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. [Review] Nat Rev Cancer. August 2006; 6(8):637–645. | |
Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol. May 28, 2001;153(5):1023–1034. | |
Sakata H, Takayama H, Sharp R, Rubin JS, Merlino G, LaRochelle WJ. Hepatocyte growth factor/scatter factor overexpression induces growth, abnormal development, and tumor formation in transgenic mouse livers. Cell Growth Differ. November 1996; 7(11):1513–1523. | |
Amicone L, Terradillos O, Calvo L, et Al. Synergy between truncated c-Met (cyto-Met) and c-Myc in liver oncogenesis: importance of TGF-beta signalling in the control of liver homeostasis and transformation. Oncogene. February 21, 2002;21(9):1335–1345. | |
Zhang SZ, Pan FY, Xu JF, et Al. Knockdown of c-Met by adenovirus-delivered small interfering RNA inhibits hepatocellular carcinoma growth in vitro and in vivo. Mol Cancer Ther. October 2005; 4(10):1577–1584. | |
Xie B, Xing R, Chen P, et Al. Down-regulation of c-Met expression inhibits human HCC cells growth and invasion by RNA interference. J Surg Res. August 2010; 162(2):231–238. | |
Takami T, Kaposi-Novak P, Uchida K, et Al. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine induced hepatocarcinogenesis. Cancer Res. October 15, 2007;67(20):9844–9851. | |
Liu ML, Mars WM, Michalopoulos GK. Hepatocyte growth factor inhibits cell proliferation in vivo of rat hepatocellular carcinomas induced by diethylnitrosamine. Carcinogenesis. April, 1995; 16(4):841–843. | |
Marx-Stoelting P, Borowiak M, Knorpp T, Birchmeier C, Buchmann A, Schwarz M. Hepatocarcinogenesis in mice with a conditional knockout of the hepatocyte growth factor receptor c-Met. Int J Cancer. April 15, 2009;124(8):1767–7672. | |
Kim WH, Matsumoto K, Bessho K, Nakamura T. Growth inhibition and apoptosis in liver myofibroblasts promoted by hepatocyte growth factor leads to resolution from liver cirrhosis. Am J Pathol. April 2005; 166(4):1017–1028. | |
Conner EA, Teramoto T, Wirth PJ, Kiss A, Garfield S, Thorgeirsson SS. HGF-mediated apoptosis via p53/bax-independent pathway activating JNK1. Carcinogenesis. April 1999; 20(4):583–590. | |
Smyth EC, Sclafani F, Cunningham D. Emerging molecular targets in oncology: clinical potential of MET/hepatocyte growth-factor inhibitors.[Review]Onco Targets Ther. June 12, 2014;7:1001–1014. | |
Santoro A, Simonelli M, Rodriguez-Lope C, et Al. A Phase-1b study of tivantinib (ARQ 197) in adult patients with hepatocellular carcinoma and cirrhosis. Br J Cancer. January 15, 2013;108(1):21–24. | |
Santoro A, Rimassa L, Borbath I, at Al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. January 2013; 14(1):55–63. | |
Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c-MET pathway in hepatocellular carcinoma.[Review]Clin Cancer Res. May 1, 2013;19(9):2310–2318. | |
Xiang Q, Chen W, Ren M, Wang J, Zhang H, Deng DY, Zhang L, Shang C, Chen Y. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin Cancer Res. June 1, 2014;20(11):2959–2957. | |
Verslype C, Cohn AL, Kelley RK, et Al. Activity of cabozantinib (XL184) in hepatocellular carcinoma: Results from a phase II randomized discontinuation trial (RDT). American Society of clinical oncology 2012 Annual Meeting; June 1-5, 2012; Chicago, IL. J Clin Oncol, 2012;30(suppl);Abstract #4007. | |
Knudsen BS, Vande Woude G. Showering c-MET-dependent cancers with drugs. Curr Opin Genet Dev. February 2008; 18(1):87–96. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.