Back to Journals » Cancer Management and Research » Volume 13
By Increasing the Expression and Activation of STAT3, Sustained C5a Stimulation Increases the Proliferation, Migration, and Invasion of RCC Cells and Promotes the Growth of Transgrafted Tumors
Authors Zheng JM, Zhou HX, Yu HY, Xia YH ![]() , Yu QX
, Yu QX ![]() , Qu HS, Bao JQ
, Qu HS, Bao JQ
Received 24 June 2021
Accepted for publication 11 September 2021
Published 4 October 2021 Volume 2021:13 Pages 7607—7621
DOI https://doi.org/10.2147/CMAR.S326352
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Jing-Min Zheng,1,* Han-Xi Zhou,1,* Hong-Yuan Yu,1,* Yu-Hui Xia,2 Qing-Xin Yu,2 Hang-Shuai Qu,1 Jia-Qian Bao1
1Department of Urology, Taizhou Hospital, Wenzhou Medical University, Linhai, Zhejiang, People’s Republic of China; 2Department of Pathology, Taizhou Hospital, Wenzhou Medical University, Linhai, Zhejiang, People’s Republic of China
*These authors contributed equally to this work.
Correspondence: Jing-Min Zheng
Department of Urology, Taizhou Hospital, Wenzhou Medical University, 150 Ximen Road, Linhai, Zhejiang, 317000, People’s Republic of China
Tel +86-576-85133065
Fax +86-576-85199800
Email [email protected]
Background: Contradictive results about the direct role of C5a/C5aR1 axis in different cancer cells have been reported. The direct effect of C5a on human renal cell carcinoma (RCC) cells and the underlying mechanism are not clear. The aim of this study is to investigate the role of C5a/C5aR1 axis in RCC cells and its working mechanism.
Methods: RCC cells were infected with lentivirus Lenti-C5a, which was designed to over-express secretory C5a in the cells, or directly treated with recombinant C5a, the influence of these treatments in the cells and the underlying mechanism were explored.
Results: Transfection of RCC cells with Lenti-C5a markedly increased the production of C5a and significantly increased the proliferation, migration, and invasion of RCC cells, but direct addition of C5a to the cell culture medium had no such effects though it indeed induced a transient intracellular calcium rise. RCC cells were found to express carboxypeptidase D and M, which reportedly to inactivate C5a. Also, the RCC cells stably transfected with Lenti-C5a produced larger transgrafted tumors in nude mice compared with the non-transfected or control virus transfected cells. In addition, over-expression of C5a significantly increased the expression and phosphorylation of STAT3 as well as the phosphorylated JNK level. Furthermore, the effect of C5a over-expression on RCC cells’ proliferation, migration, and invasion could be blocked by Stattic, a STAT3-specific inhibitor.
Conclusion: Chronic over-activation of C5a/C5aR1 axis could directly increase RCC cells’ proliferation, migration, and invasion and thus contribute directly to the progression of the disease. Over-activation of STAT3 pathway is among the underlying mechanism.
Keywords: C5a, C5aR1, renal cell carcinoma, STAT3
Introduction
Renal cell carcinoma (RCC) is one of the 10 most commonly diagnosed malignancies. Worldwide, more than 140,000 people die of RCC annually.1,2 Clear cell renal cell carcinoma (ccRCC) and papillary renal cell carcinoma are the major subtypes of the disease, with the former accounting for approximately 70–80% and the latter approximately 10–15% of all RCC cases.3 Due to its resistance to commonly used chemotherapy agents and radiation,4 early surgical resection is the primary therapy for RCC. Currently, the 5-year overall survival rate for localized ccRCC is approximately 92.6% but declines markedly to only 66.7% in patients with regional spread and 11.5% in patients with metastatic disease. In addition, approximately 20–30% of ccRCC patients with localized disease even after nephrectomy.4,5 In recent years, a number of targeted therapy approaches to treat RCC have been introduced, such as immunotherapies and the use of receptor tyrosine kinase and mTOR inhibitors; however, none of these therapies provide durable responses, and the degree of prognosis improvement is fairly limited.4,6 Therefore, further investigations of the pathogenic mechanism of RCC are needed to facilitate the development of novel therapies to improve disease prognosis.
C5a, a 74–amino acid peptide, is a split-product of complement component C5 generated during the complement activation cascade.7 The C5a receptor, C5aR1, was originally identified in cells of myeloid origin. As a major immune response regulator, the C5a-C5aR1 axis has been extensively studied. However, C5aR1 is also expressed by various non-myeloid cells, including cells of normal and malignant tissues.8 Emerging evidence suggests that C5a plays an important role in the initiation and progression of various cancers. The C5a-C5aR1 axis might therefore be a useful therapeutic target in cancer treatment. However, details regarding the role of C5a in the underlying mechanism of cancer pathogenesis remain unclear. In particular, data pertaining to the direct role of C5a in different cancers are contradictory.
In kidney cancer, two research groups have reported an association between C5a/C5aR1 and RCC prognosis. Maeda et al reported that C5aR1 is expressed more frequently in metastatic RCC tumors, whereas Xi et al demonstrated that high levels of C5a and C5aR1 are associated with poor postoperative prognosis in ccRCC patients.9–11 However, direct evidence demonstrating the involvement of C5a-C5aR1 signaling in RCC remains scarce. To the best of our knowledge, no studies have reported the effect of C5a on human RCC cells. Therefore, in the present study, we examined the effect of over-expression of secretory C5a on the phenotype of two human RCC cell lines, 786-O and ACHN, in order to elucidate the direct role of C5a in the mechanism underlying RCC.
Materials and Methods
Cell Culture, Transfection, and Treatment
The 786-O and ACHN RCC cell lines were obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China). The cells were cultured in RPMI-1640 medium (Meilun Biology Technology, Dalian, China) supplemented with 10% fetal bovine serum. In order to obtain RCC cells stably over-expressing secretory C5a, the cells were transfected with a recombinant lentivirus, Lenti-C5a. To construct Lenti-C5a, a recombinant IL6-C5a gene carrying the IL-6 signal peptide sequence (117-203 of NM_000600.3) and C5a coding sequence (2079-2300 of NM_001317163) was inserted into the BamHI/AscI site of the lentivirus expression vector, pLenti6.3-MCS-IRES2-EGFP (Thermo Fisher Scientific, MA, USA), thus generating the recombinant C5a expression vector, pLenti6.3-IL6C5a-IRES2-EGFP. pLenti6.3-MCS-IRES2-EGFP was used to produce the control virus.
RCC cells were seeded at a density of about 1×105 cells/well in a 12-well cell culture plate and transfected. When the cells reached approximately 50% confluence, the medium was removed and exchanged with 0.5 mL of fresh medium containing 5×105 virus particles and 8 μg/mL polybrane. After 12 h, the medium was replaced with fresh medium. Stably transfected cells were screened out by addition of puromycin (5 μg/mL) beginning 48 h after transfection and incubation for 1 week. In C5a stimulation tests, recombinant C5a (CR52, Novoprotein, Shanghai, China) was directly added to the cell culture medium at a final concentration of 100 nM. Blockage of the C5aR1 and STAT3 pathways was carried out by direct addition of 1 μM PMX-53 (5336830001, Sigma-Aldrich, MA, USA), and 20 μM Stattic (T6308, Targetmol, MA, USA), respectively.
RNA Isolation and RT-PCR Analysis
According to the manufacturer’s protocol, total cellular RNA was isolated with TRIzol® reagent (9109, Thermo Fisher Scientific, MA, USA) and cDNA was generated using a PrimeScriptTM RT Master Mix kit (RR820A, Takara Biotechnology, Dalian, China). A fragment of 118 bp C5aR1 cDNA was amplified by using the following primers: C5aR1 forward, 5’-ATCTTTGCAGTCTTCCTG-3’ and C5aR1 reverse, 5’-CGGCTACCGCCAAGTTGAG-3’. The thermal cycle condition was: Pre-denatured at 94°C for 5 min, followed by 30 cycles of 94°C for 20 s and 60°C for 30s. The presence of the C5aR1 fragment was analyzed by agarose gel electrophoresis.
Western Blot Analysis
Total protein was extracted using RIPA lysis buffer (R0020, Solarbio Technology, Beijing, China) containing Phosphatase Inhibitor and Protease Inhibitor Cocktail (Targetmol, MA, USA) at 4°C. About 10 µg of total protein was separated by 10% SDS-PAGE and transferred to a PVDF membrane (Merck KgaA, Darmstadt, Germany). Following blocking with the PBS buffer containing 5% skimmed milk powder, the membranes were incubated with various primary antibodies respectively, overnight at 4°C. These antibodies include rabbit anti-human C5aR1 (Ls-B5559, dilution of 1:1000; Lifespan, Seattle, WA, USA), rabbit anti-human AKT (4691S, dilution of 1:1000; Cell Signaling Technology, MA, USA), rabbit anti-human p-AKT (4060S, dilution of 1:1000; Cell Signaling Technology, MA, USA), rabbit anti-human β-actin (YT0099, dilution of 1:5000; Immunoway, TX, USA), rabbit anti-human ERK1/2 (4695S, dilution of 1:1000; Cell Signaling Technology, MA, USA), rabbit anti-human p-ERK1/2 (4370S, dilution of 1:1000; Cell Signaling Technology, MA, USA), rabbit anti-human p38 (8690S, dilution of 1:1000; Cell Signaling Technology, MA, USA), rabbit anti-human p-p38 (4511S, dilution of 1:1000; Cell Signaling Technology, MA, USA), mouse anti-human STAT3 (9139T, dilution of 1:1000; Cell Signaling Technology, MA, USA), rabbit anti-human p-STAT3 (9145T, dilution of 1:1000; Cell Signaling Technology, MA, USA), rabbit anti-human p-SAPK/JNK (4668T, dilution of 1:1000; Cell Signaling Technology, MA, USA), mouse anti-human GAPDH (YM3029, dilution of 1:10000; Immunoway, TX, USA), rabbit anti-human CPD (NBP1-91447, dilution of 1:1000; Novus Biologicals, Centennial, CO, USA), rabbit anti-human CPM (NBP1-87403, dilution of 1:1000; Novus Biologicals, Centennial, CO, USA), rabbit anti-human CPN (Ls-c166993, dilution of 1:1000; Lifespan, Seattle, WA, USA) and rabbit anti-human CPZ (Ls-199274, dilution of 1:1000; Lifespan, Seattle, WA, USA). Then the membrane was washed with TBST three times and incubated with HRP-conjugated goat anti-rabbit (B0201, dilution of 1:10000; Immunoway, Texas, USA) or rabbit anti-mouse (B0101, dilution of 1:10000; Immunoway, Texas, USA) IgG antibody for 2 h at 37°C. The immunolabeled proteins were detected by chemiluminescence using the Chemiluminescent hRP substrate (WBULS0100, Merck KgaA, Darmstadt, Germany). Densitometric analysis was performed using ImageJ software, version 1.52a (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence Analysis
The cells growing on glass coverslips were washed with PBS, fixed in 4% paraformaldehyde at 4°C for 30 min and permeabilized in 0.5% Triton X-100 at room temperature for 10 min. After blocking with 3% BSA (Beyotime Institute of Biotechnology, Shanghai, China) for 30 min, the cells were incubated with rabbit anti-human C5aR1 antibodies (Ls-B5559, dilution of 1:100; Lifespan. Seattle, WA, USA) at room temperature for 2 h, washed with PBS three times, and incubated with Cy3-labeled goat anti-rabbit IgG antibody (33108ES60, dilution of 1:200; Yeasen Biotechnology, Shanghai, China) at room temperature for 30 min. Washed with PBS again, the cells were mounted, and fluorescent images were captured with a confocal microscope (LSM-800, Zeiss GmbH, Jena, Germany). For negative control, 10% FCS was used instead of the primary antibody.
Intracellular Ca2+ Assay
Intracellular Ca2+ was measured by using Fluo-3 AM (S1056, Beyotime Institute of Biotechnology, Shanghai, China). Briefly, the cells were incubated with 5 μM Fluo-3 AM for 45 min at 37°C in serum-free medium. Then, the cells were washed with serum-free medium three times and incubated for another 30 min. The relative intracellular Ca2+ level was examined by Fluorescence Microscope (Zeiss GmbH, Jena, Germany).
Cell Proliferation Assay
A Cell Counting Kit-8 kit (Beyotime Institute of Biotechnology, Shanghai, China) was used in the analysis of cell proliferation according to the manufacturer’s instruction. Briefly, 786-O or ACHN cells were seeded into the 96-well plates at a density of 3×103/well and 5×103/well, respectively. After 6, 24, 48 and 72 h, the number of the cells was detected. Here, the results of 6 h were used as a baseline.
Cell Invasion Assay
Cell invasion was assessed using BioCoat Matrigel invasion chambers (24-well plates, 8-μm pores; BD Biosciences, CA, USA). Cells were starved by incubating in serum-free medium for 24 h prior to the invasion assay. A total of 5×104 ACHN cells and 1.5×104 786-O cells with 200 μL of serum-free medium were added to the upper chamber, and 500 μL of medium containing 10% fetal bovine serum was added to the lower chamber. After 24 h, the cells on the upper-chamber side of the membrane were scraped off, and the cells on the underside of the membrane (which passed through the Matrigel and membrane) were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet, and counted under a microscope (Leica Dmi1, Leica, Wetzlar, Germany). For each chamber, the number of invaded cells was determined for at least 10 fields (100×), and the average number of invaded cells per field was used as the number of invaded cell in the chamber. Each experiment was repeated at least three times.
Cell Migration Assay
Cells were seeded in 6-well plates at a density of 5×106/well. Upon reaching near confluence, the cells were starved by incubation in serum-free medium for 24 h. Scratches were then made on the cell monolayer using a sterile pipette tip. The detached cells were removed by washing with PBS and then maintained in serum-free RPMI-1640 medium. Images of the scratches were captured at indicated time points using a camera (Leica S80/0.30) attached to the microscope (Leica DMi1). The area of the wound made by the scratches was measured at each time point using ImageJ software, version 1.52a (National Institutes of Health, Bethesda, MD, USA). The wound healing ratio was calculated as follows: wound healing ratio at a specific time point = (wound area at time 0 − wound area at specific time point)/wound area at time 0. In comparing the wound healing ratio between different groups, the wound healing ratio of the normal control group was defined as 1.0.
Allogeneic Transplantation of Tumors
Five-week-old male nude mice (Slac Laboratory Animal, Shanghai, China) were divided randomly into three groups: untransfected group, control virus group and Lenti-C5a group. A total of 1×106 different cells (untransfected 786-O or ACHN cells for untransfected group; control virus transfected 786-O or ACHN cells for control virus group; Lenti-C5a transfected 786-O or ACHN cells for Lenti-C5a group) were inoculated subcutaneously into the flank of each mouse. The growth of the tumor was monitored regularly. Eight weeks later, the mice were euthanized and the tumors’ size and weight were measured. Six mice were used in each group. All the animal experimental procedures were approved by the Experimental Animal Ethical Committee of Taizhou Hospital and were performed according to the guidelines of the Experimental Animal Care Committee of Taizhou Hospital (tzy-2019047). All efforts were made to minimize the number of animals used and their suffering.
C5a Assay
According to the manufacturer’s instruction, the level of C5a in the cell culture supernatant was measured using a human C5a ELISA kit (E-EL-H0190c, Elabscience, Houston, TX, USA) in an ELISA read (Mltiskan FC, Thermo Scientific, USA).
Statistical Analyses
Data are presented as the mean ± SEM. SPSS software (version no. 19.0; IBM, CHI, USA) was used for data analysis. Multiple comparisons were performed using one-way ANOVA and Tukey’s post hoc test was used in the comparison between two groups. All statistical tests were two tailed, p < 0.05 was considered statistically significant in all the cases.
Results
Expression of C5aR1 and C5a in 786-O and ACHN Cells
As shown in Figure 1, C5aR1 was expressed in both 786-O and ACHN cells. However, C5a was detected at only a very low level in the culture supernatant of 786-O and ACHN cells (0.16±0.19 ng/mL and 0.18±0.16 ng/mL, respectively).
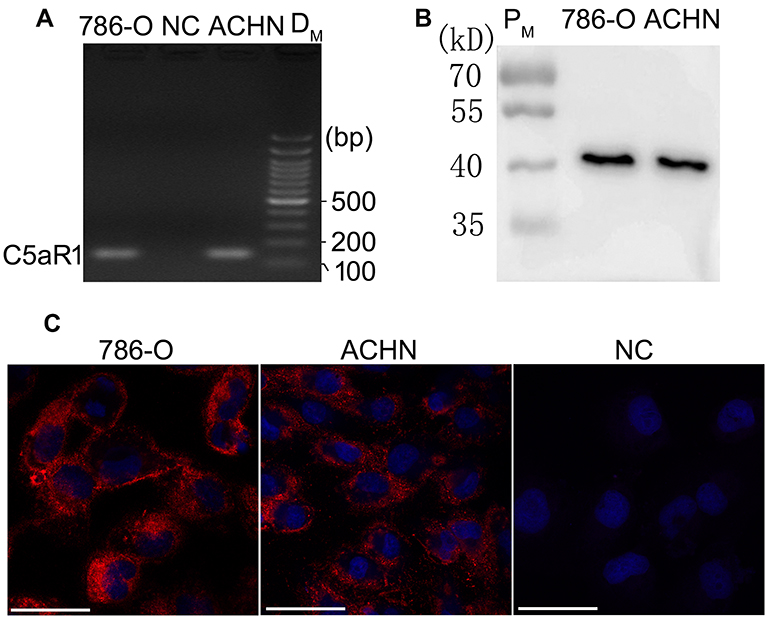 |
Figure 1 C5aR1 was expressed in both 786-O and ACHN cells. The expression of C5aR1 in 786-O and ACHN cells was examined by RT-PCR (A), Western blotting (B) and immunofluorescence analysis (C). Scale bar, 100µm. Abbreviations: 786-O, 786-O cells; ACHN, ACHN cells; NC in (A), Negative control in RT-PCR analysis; NC in (C), Negative control in immunofluorescence analysis (786-O cells were incubated with 10% fetal calf serum instead of anti-C5aR1 antibody); DM, DNA mark; PM, protein mark; RT-PCR, reverse transcription-polymerase chain reaction. |
Addition of C5a to the Culture Medium Had No Obvious Effect on the Proliferation, Migration, or Invasion of RCC Cells
To determine the direct effect of C5a on RCC cells, recombinant C5a was added directly to the culture medium of 786-O and ACHN cells. As shown in Figure 2, addition of C5a had no significant effect on the proliferation, migration, or invasion of these cells. However, addition of C5a induced a rapid increase in the cellular Ca2+ level, thus excluding the possible ineffectiveness of recombinant C5a itself (Figure 2K).
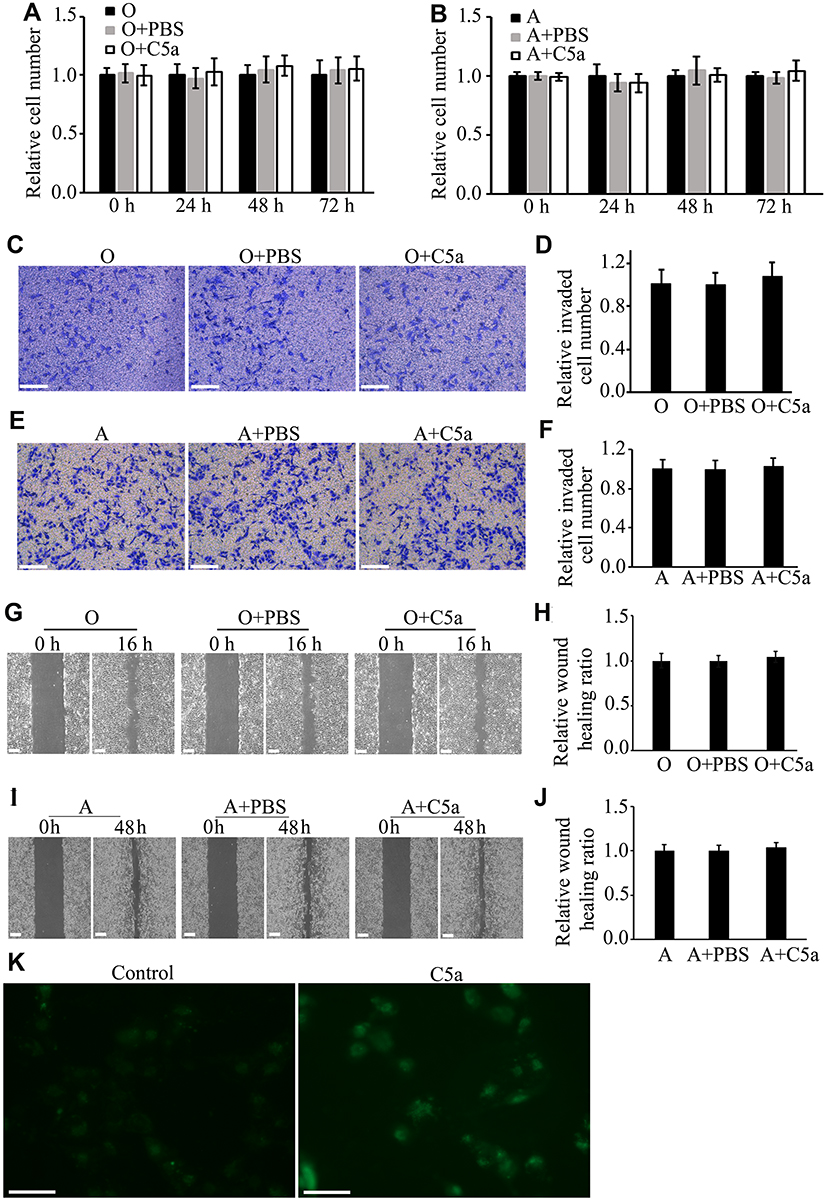 |
Figure 2 Addition of recombinant C5a to the cell culture medium had no significant influence on the proliferation, migration, or invasion of RCC cells. RCC cells (786-O and ACHN) were treated with 100 nM of recombinant C5a or not and the effects of C5a on cell proliferation, migration, and invasion were determined by CCK8, scratch healing and Matrigel invasion chamber methods. The influence of intracellular Ca2+ level was determined 5 minutes after addition of 100 nM recombinant C5a. All the experiments were repeated at least three times. (A and B) Results of proliferation analysis. (C–F) Results of invasion analysis. (G–J) Results of migration analysis. (K) Representative pictures under Fluorescence Microscope showing that addition of C5a obviously increased intracellular Ca2+ level of 786-O cells as represented by the increased fluorescence. Scale bar: 100 µm. Abbreviations: A, ACHN cells; O, untreated 786-O cells; O+PBS, 786-O cells treated with the same volume of phosphate buffer saline (PBS) as C5a that used in C5a treated group; O+C5a, 786-O cells treated with 100 nM of recombinant C5a; A+PBS, ACHN cells treated with the same volume of PBS as C5a that used in C5a treated group; A+C5a, ACHN cells treated with 100 nM of recombinant C5a; CCK8, Cell Counting Kit-8 kit; RCC, renal cell carcinoma; PBS, phosphate buffered saline. |
RCC Cells Express Carboxypeptidases
C5a is readily inactivated in vivo by carboxypeptidases via cleavage of the C-terminal arginine residue.12,13 To determine whether carboxypeptidases also inactivate recombinant C5a added to the culture medium, we assayed 786-O and ACHN cells for the expression of carboxypeptidase D (CPD), carboxypeptidase M (CPM), carboxypeptidase N (CPN), and carboxypeptidase Z (CPZ), each of which reportedly inactivates C5a. Bands corresponding to CPN1 and CPZ were not detected in Western blot analysis. However, expression of CPD and CPM was detected (Figure 3).
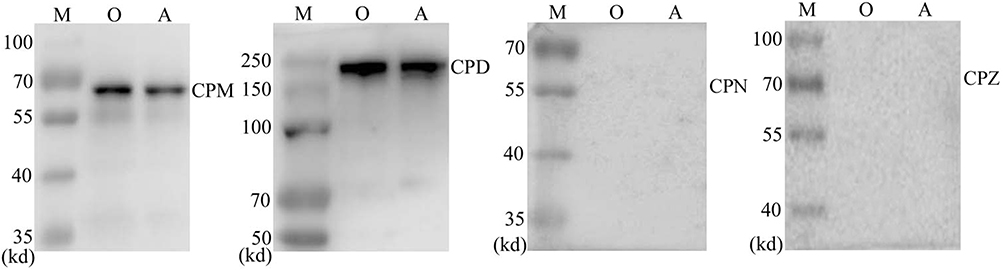 |
Figure 3 RCC cells were found to express carboxypeptidase M and carboxypeptidase D, but not carboxypeptidase N or carboxypeptydase Z. Abbreviations: M, protein mark; O, 786-O cells; A, ACHN cells; RCC, renal cell carcinoma; CPD, carboxypeptidase D; CPM, carboxypeptidase M; CPN, carboxypeptidase N; CPZ, carboxypeptidase Z. |
Secretory Over-Expression of C5a Increased the Proliferation, Migration, and Invasion of RCC Cells
Over-expression of secretory C5a was demonstrated by measuring C5a and its inactivated product C5a-des Arg in cell culture supernatants (Figure 4A and B). As shown in Figure 4C–L, significantly increased proliferation (Figure 4C and D), migration (Figure 4E–H) and invasion (Figure 4I–L) were observed in secretory C5a–over-expressing 786-O (786-O-C5a) and ACHN (ACHN-C5a) cells. This effect could be blocked by addition of 1 μM PMX-53, a C5aR1-specific inhibitor.
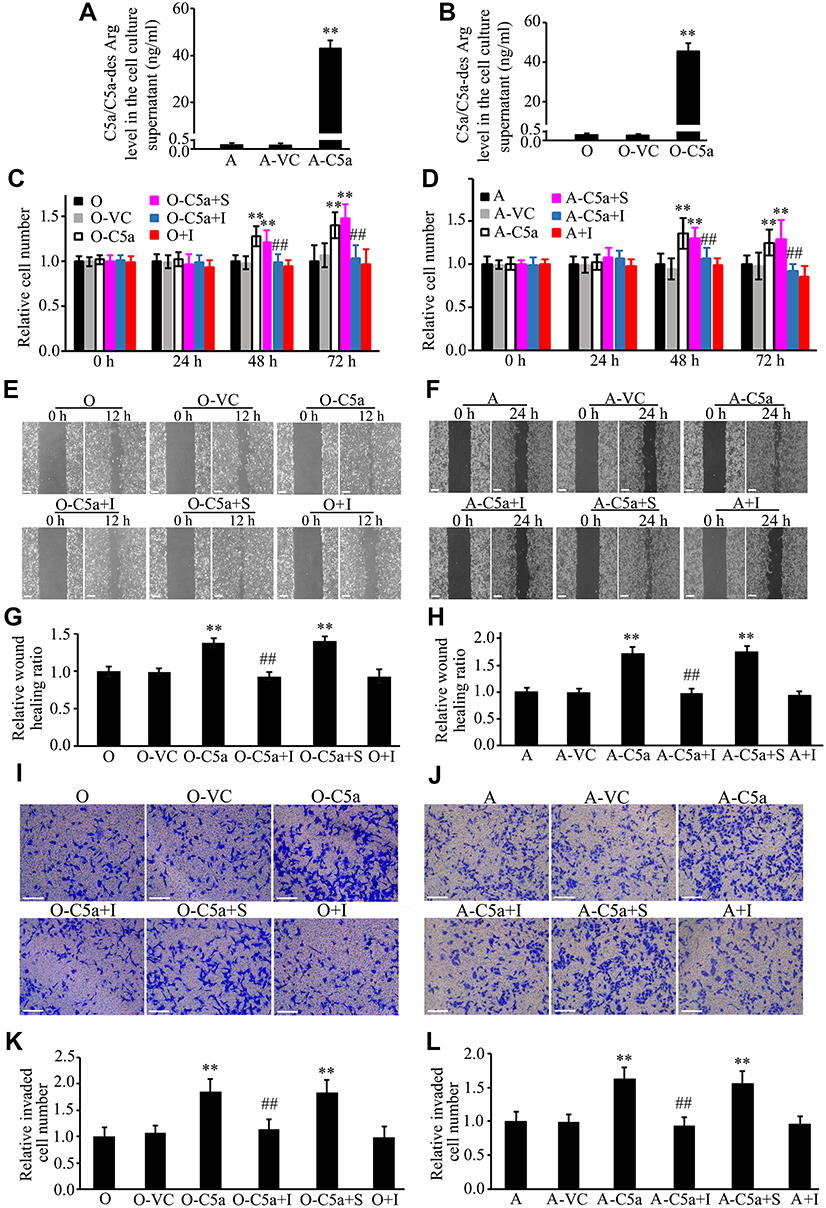 |
Figure 4 Secretory over-expression of C5a significantly increased the proliferation, migration, and invasion of RCC cells. Secretory over-expression of C5a in Lenti-C5a transfected cells was confirmed through measuring the C5a/C5a-des Arg level in the cell culture supernatant (A and B). The influence of secretory C5a over-expression on the cell proliferation (C and D), migration (E–H), and invasion (I–L) were determined by CCK8, scratch healing and Matrigel invasion chamber methods. All the experiments were repeated at least three times. **P<0.01 vs O group in (B, C, G and K), or vs A group in (A, D, H and L). ##P<0.01 vs O-C5a group in (C, G and K), or vs A-C5a group in (D, H and L). Scale bar: 100 µm. Abbreviations: O, 786-O cells; O-VC, 786-O cells transfected with control virus; O-C5a, 786-O cells transfected with the secretory C5a expression lentivirus Lenti-C5a; O-C5a+S, O-C5a cells treated with the same volume of DMSO as PMX-53 used in the O-C5a+I group; O-C5a+I, O-C5a cells treated with 1 μM of PMX-53; O+I, 786-O cells treated with 1 μM of PMX-53; A, ACHN cells; A-VC, ACHN cells transfected with control virus; A-C5a, ACHN cells transfected with the secretory C5a expression lentivirus Lenti-C5a; A-C5a+S, A-C5a cells treated with the same volume of DMSO as PMX-53 used in the A-C5a+I group; A-C5a+I, A-C5a cells treated with 1 μM of PMX-53; A+I, ACHN cells treated with 1 μM of PMX-53; CCK8, Cell Counting Kit-8 kit; RCC, renal cell carcinoma; DMSO, dimethyl sulfoxide. |
Secretory Over-Expression of C5a Significantly Increased the Growth of Allogeneic Transgrafted Tumors
As shown in Figure 5, compared with un-transfected and control virus-transfected RCC cells, cells over-expressing secretory C5a produced significantly larger tumors in nude mice.
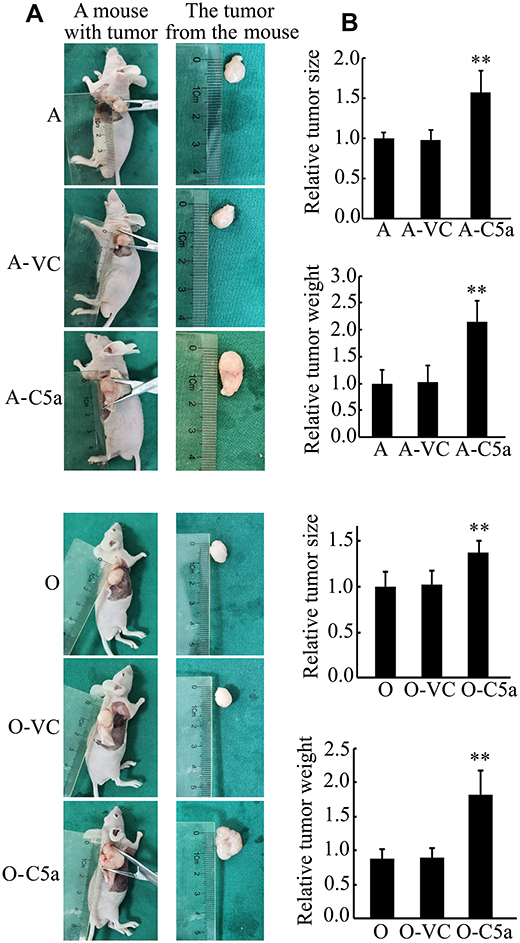 |
Figure 5 Secretory over-expression of C5a significantly increased the growth of transgrafted tumors in nude mice. (A) Representative pictures of the mice with tumor and the tumor isolated. (B) Results of statistical analysis of the tumor size and weight. **P<0.01 vs A or O group. Abbreviations: A, ACHN cell group (mice were inoculated with untransfected ACHN cells, n = 6); A-VC, control virus transfected ACHN cell group (mice were inoculated with control virus transfected ACHN cells, n = 6); A-C5a, C5a over-expression ACHN cell group (mice were inoculated with Lenti-C5a transfected ACHN cells, n = 6); O, 786-O cell group (mice were inoculated with untransfected 786-O cells, n = 6); O-VC, control virus transfected 786-O cell group (mice were inoculated with control virus transfected 786-O cells, n = 6); O-C5a, C5a over-expression 786-O cell group (mice were inoculated with Lenti-C5a transfected 786-O cells, n = 6). |
Secretory Over-Expression of C5a Significantly Increased STAT3 Pathway Expression and Activation
To investigate the mechanisms underlying the effects of C5a over-expression, we first examined the effect of C5a over-expression on the AKT, ERK, p38, and JNK/STAT3 pathways. Given the similar effects of secretory C5a over-expression in 786-O and ACHN cells with regard to cellular proliferation, migration, and invasion and the growth of allogeneic transgrafted tumors, we examined only 786-O cells in the subsequent study. As shown in Figure 6, compared with un-transfected and control virus-transfected 786-O cells, levels of STAT3, phosphorylated STAT3, and phosphorylated JNK increased significantly. However, no significant changes in the expression or phosphorylation levels of AKT, ERK, and p38 were observed.
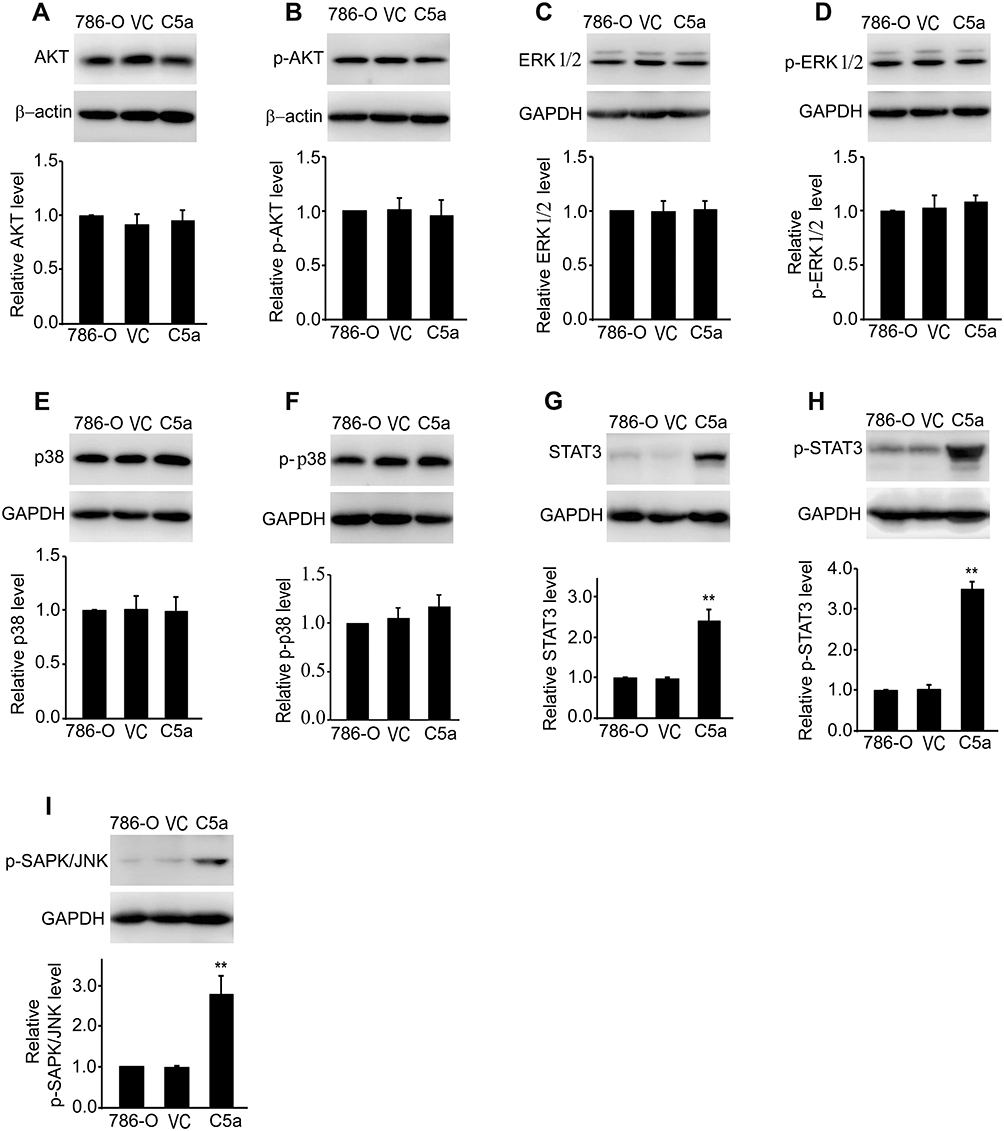 |
Figure 6 Secretory over-expression of C5a significantly increased the expression and activation of JNK/STAT3 pathway. The level of AKT (A), phosphorylated AKT (p-AKT, (B), ERK1/2 (C), phosphorylated ERK1/2 (p-ERK1/2, (D), p38 (E), phosphorylated p38 (p-p38, (F), STAT3 (G), phosphorylated STAT3 (p-STAT3, (H) and phosphorylated JNK (p-SAPK/JNK, (I) were measured by Western blot method. All the experiments were repeated three times. **P<0.01 vs 786-O group in (G, H and I). Abbreviations: 786-O, untransfected 786-O cells; VC, control virus transfected 786-O cells; C5a, C5a over-expression 786-O cells. |
Inhibition of STAT3 Blocked the Effect of Secretory C5a Over-Expression on the Proliferation, Migration, and Invasion of 786-O Cells
To confirm the involvement of the STAT3 pathway in the observed changes in the proliferation, migration, and invasion of RCC cells, a STAT3 pathway blocking test was conducted using the STAT3-specific inhibitor Stattic. As shown in Figure 7, Stattic treatment significantly reduced the proliferation, migration, and invasion of 786-O cells over-expressing C5a. Stattic treatment also significantly reduced the proliferation of un-transfected 786-O cells. Stattic treatment also reduced the migration and invasion of un-transfected 786-O cells, albeit to a much lower and non-significant degree.
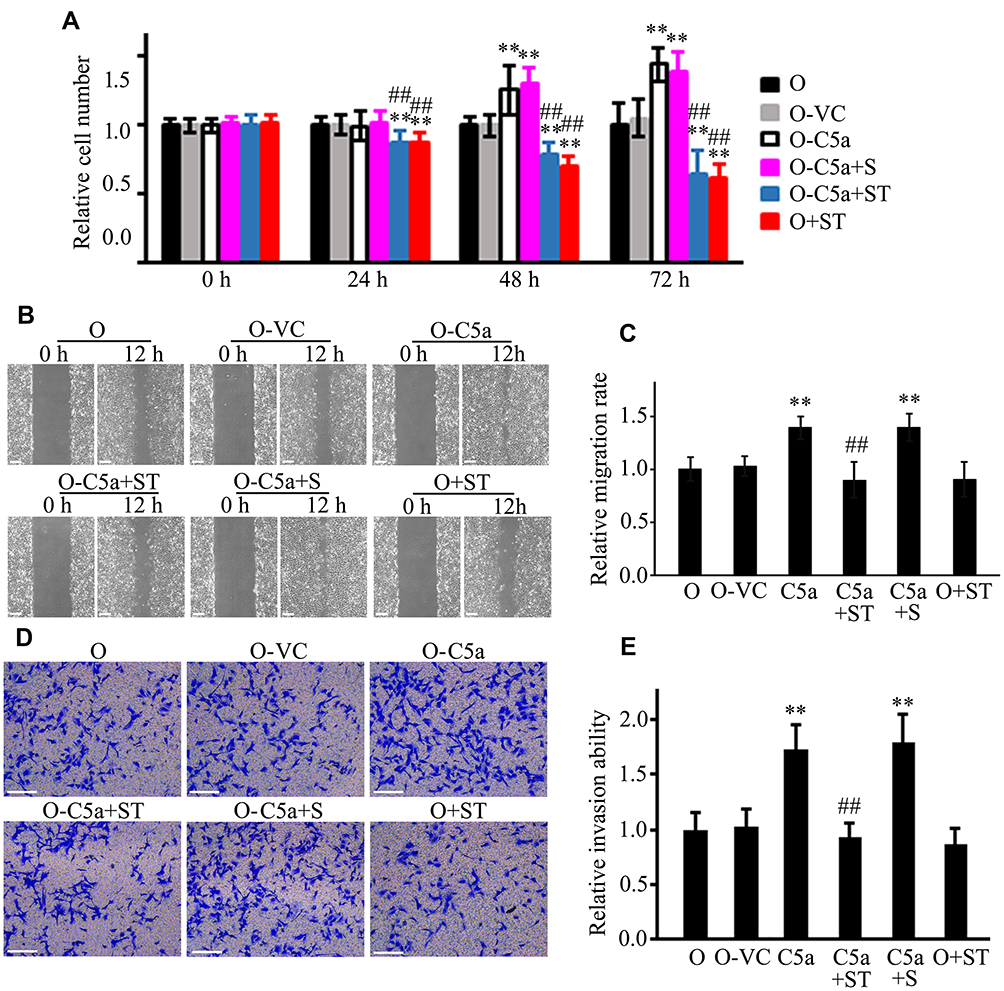 |
Figure 7 Inhibition of STAT3 blocked the effect of secretory C5a over-expression on the proliferation, migration, and invasion of RCC cells. The STAT3 specific inhibitor Stattic (20 μM) was used in the assay. The influence of Stattic on cellular proliferation (A), migration (B and C), and invasion (D and E) of untransfected 786-O and secretory C5a over-expression 786-O cells were analyzed by using CCK8, scratch healing and Matrigel invasion chamber methods. All the experiments were repeated at least three times. **P<0.01 vs O group. ##P<0.01 vs O-C5a group. Scale bar: 100 µm. Abbreviations: O, 786-O cells; O-VC, 786-O cells transfected with control virus; O-C5a, 786-O cells over-expressing secretory C5a; O-C5a+S, O-C5a cells treated with the same volume of DMSO as Stattic used in the O-C5a+ST group; O-C5a+ST, O-C5a cells treated with 20 μM of Stattic; O+ST, 786-O cells treated with 20 μM of Stattic; CCK8, Cell Counting Kit-8 kit; RCC, renal cell carcinoma; DMSO, dimethyl sulfoxide. |
Discussion
The present study is the first to report the effect of secretory over-expression of C5a on the phenotype of RCC cells. Over-expression of C5a significantly increased the proliferation, migration, and invasion of RCC cells, and this effect was blocked by treatment with the C5aR1-specific inhibitor PMX-53. Secretory over-expression of C5a also induced a significant increase in the growth of transgrafted RCC tumors. In addition, secretory over-expression of C5a significantly increased the expression and phosphorylation levels of STAT3 and the level of phosphorylated JNK, whereas inhibition of the JNK/STAT3 pathway using the STAT3-specific inhibitor Stattic reversed the effect induced by C5a over-expression. These results suggest that C5a contributes to the progression of RCC by directly regulating specific functions of cancer cells. Two receptors for C5a have been identified, C5aR1 and C5aR2 (also designated C5L2).14 Given that the effect of C5a over-expression on the phenotype of RCC cells could be blocked by treatment with a C5aR1-specific inhibitor, we believed that constitutively expressed C5a exerts its effects on RCC cells via activation of C5aR1. That is, over-expression of secretory C5a activates C5aR1, leading to subsequent activation of the JNK/STAT3 pathway and promotion of the proliferation, migration, and invasion of RCC cells.
Interestingly, direct addition of C5a to the cell culture medium had no significant effect on the proliferation, migration, and invasion of RCC cells in the present study. Conditions such as improper preservation may cause inactivation of recombinant C5a. To exclude this possibility, an experiment was performed to determine whether addition of the recombinant C5a could stimulate increase of cellular Ca2+ level, a phenomenon being well-proved in a variety of cells. Indeed, addition of the C5a stimulated a rapid increase in the cellular Ca2+ level, excluding the possibility of the ineffectiveness of the C5a itself. Given that C5a is readily inactivated in vivo by carboxypeptidases via cleavage of the C-terminal arginine residue, thus converting C5a into its inactive form, C5a-des Arg, we then examined the expression of various carboxypeptidases in RCC cells. Indeed, both of the RCC cell lines used in the study, 786-O and ACHN, expressed CPM and CPD. As an extracellular peptidase bound to the outer membrane via a glycosyl-phosphatidylinositol anchor, CPM reportedly cleaves C-terminal arginine residues from peptides and proteins and is believed to function as CPN, the major circulating carboxypeptidase that mediates the rapid inactivation of anaphylatoxins such as C3a, C4a, and C5a in the extracellular space.15 CPD is localized in the plasma membrane16 and reportedly cleaves C-terminal arginine residues from extracellular substrates for uptake of arginine into cells for nitric oxide production.17,18 The present finding that 786-O and ACHN RCC cells express CPD and CPM thus supports the hypothesis that exogenous C5a is rapidly inactivated by carboxypeptidases expressed by RCC cells. Therefore, direct addition of C5a leads to only transient activation of C5aR1 on the cell membrane. This situation differs markedly from secretory over-expression of C5a by RCC cells, in which C5a is sustainably produced and may induce sustained activation of C5aR1 on the cell membrane. This mechanism could explain the different effects we observed following direct addition of C5a to the cell culture medium and over-expression of secretory C5a by the cells.
As it is believed to be a major component of the complement system that influences both tumor initiation and progression, C5a and its receptor, C5aR1, have attracted considerable attention from tumor investigators in recent years. A high level of C5aR1 in RCC tumors is reportedly associated with high rates of tumor cell metastasis, advanced-stage disease, and poor clinical prognosis.11 Both indirect and direct mechanisms have been proposed to explain the role of C5a/C5aR1 signaling in the pathogenesis of malignancy. The indirect mechanism postulates that C5a exerts its effects via regulation of the recruitment and function of various immune cells in the tumor. This mechanism has been extensively investigated, resulting in nearly unanimous consensus. In contrast, relatively few studies have examined the direct role of C5a in cancer cells, and these studies have produced contradictory results. For example, Zhao et al, Zhang et al, Lu et al, and Cho et al reported that C5a induces the proliferation of A549 and PC9 lung cancer cells, C666-1 human nasopharyngeal carcinoma cells, MCF-7 and MDA-MB-231 breast cancer cells, SKOV3ip1 human ovarian cancer cells, Hec265 endometrial cancer cells, and H226 lung squamous cancer cells, respectively.19–22 However, Ajona et al, Nabizadeh et al, and Zha et al reported that C5a had no effect on the proliferation of A549 and H460M5 lung cancer cells, B16.F0 mouse melanoma cells, CT26 mouse colon cancer cells, or LLC mouse lung adenocarcinoma cells, respectively.23–25 Maeda et al reported that although C5aR1 signaling plays a crucial role in the invasion of RCC cells, it does not affect the proliferation of these cells.9 The differing results reported by these various studies might reflect the complex role C5a signaling plays in cancer cells. Differences in cell types used, cellular context, and experimental conditions could help explain the apparent functional complexity of C5a observed in various studies. Given that the level of C5aR1 expression varies greatly in cancer cells,19–23 this might be quite important in explaining differences in results, as under most experimental conditions, this could represent the level of C5a/C5aR1 axis signaling in cancer cells. As shown in the present study, 786-O and ACHN cells also express carboxypeptidases. At present, we do not know whether other cancer cells commonly express carboxypeptidases. If they do, the level of carboxypeptidase expression should also be taken into account in explaining the effect of C5a on the phenotype of cancer cells.
Of note, in the microenvironment of in vivo tumors, C5a is continually produced through various pathways. C5aR1 should therefore be chronically activated in tumor cells. At least in this regard, the present study based on over-expression of secretory C5a in cancer cells might have more realistically mimicked the actual in vivo tumor microenvironment and better reflected the true effects of chronic activation of the C5a/C5aR1 axis in RCC.
Pathways such as ERK, AKT, p38, and STAT3 are reportedly involved in a variety of cellular processes induced by C5a/C5aR1 activation. For example, by increasing the phosphorylation of ERK1/2 and p38 MAPK, C5a-induced C5aR1 activation was shown to promote osteoclast formation and inflammatory responses.26 In addition, C5a induces neutrophil apoptosis by inhibiting the AKT pathway.27 In cancer cells, C5a reportedly suppresses p21 expression via activation of the PI3K/AKT pathway and thus functions in the pathogenesis of gastric cancer.28,29 C5a also reportedly increases ZEB1 expression and enhances the invasion of human glioblastoma cells via activation of p38 MAPK.30 Using C5aR1-transfected mouse RCC cells (Renca cells), Maeda et al reported that C5a elicited cytoskeletal rearrangements and morphological changes in the cells and ultimately increased their invasiveness through activation of the ERK and AKT pathways.9 Unexpectedly, in the present study, no significant changes were observed in the ERK, AKT, or p38 pathways. Thus, our results do not support the involvement of these pathways in the pathogenic role of chronic C5a/C5aR1 activation in RCC.
In the present study, over-expression of secretory C5a increased the expression and activation of the STAT3 pathway. STAT3 is an important signaling molecule and reportedly involved in a variety of biological processes, including immune regulation, angiogenesis, cellular proliferation, differentiation, survival, and apoptosis.31–33 Constitutive STAT3 activation has been reported in a variety of human cancers, including RCC.34–40 A broad spectrum of factors reportedly activate STAT3, including various growth factors and cytokines.41,42 However, to our knowledge, no study has reported the role of C5a/C5aR1 signaling in activation of the STAT3 pathway in renal cancer cells. Given the similarity between over-expression of secretory C5a in RCC cells and the sustained C5a production resulting from chronic complement activation in renal tumor tissues, the present finding that sustained C5a stimulation induces STAT3 expression and activation might provide a novel explanation for the cause of constitutive STAT3 activation in renal cancer.
The present study does have some limitations. First, as our results were based on only two RCC cell lines, further in vivo investigations are needed to confirm our findings. Second, the level of C5a expression could not be accurately controlled like addition of recombinant C5a in the present study, the exact effect of certain C5a level in RCC cells remains to be evaluated.
In conclusion, we used recombinant C5a and over-expression of secretory C5a to examine the direct effect of C5a/C5aR1 activation in RCC cells and explored the underlying mechanism. Over-expression of secretory C5a promoted the proliferation, migration, and invasion of RCC cells, whereas direct addition of C5a did not. Because RCC cells expressed carboxypeptidases that are known to inactivate C5a, we believe that direct addition of C5a only transiently activates C5aR1, whereas over-expression of C5a causes sustained stimulation of the cells, which is the reason for the different results we observed using the two different methods. Given that C5a is produced continually in actual tumor tissue due to chronic complement activation, over-expression of secretory C5a in RCC cells might better mimic the situation in RCC tumors. Thus, we deduced that sustained activation of C5aR1 by C5a produced as a result of chronic complement activation might play an important role in promoting the progression of RCC; patients with RCC might therefore benefit from blockade of the C5a/C5aR1 axis. In addition, in the present study, over-expression of C5a exerted effects on RCC cells by increasing STAT3 expression and activation. Given the key role of the STAT3 pathway in the regulation of cancer cell phenotype and the observation that constitutive STAT3 activation occurs in RCC tumor tissues, this finding not only provides a reasonable explanation for the observed effects of C5a over-expression, it also suggests a novel mechanism for the constitutive STAT3 activation in RCC tumors. That is, our findings support the hypothesis that chronic C5a/C5aR1 over-activation contributes directly to the progression of RCC and that blockade of this axis could be an effective therapeutic strategy for treating the disease.
Abbreviations
BSA, bovine serum albumin; ccRCC, clear cell renal cell carcinoma; CPD, carboxypeptidase D; CPM, carboxypeptidase M; CPN, carboxypeptidase N; CPZ, carboxypeptidase Z; FCS, fetal calf serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HRP, horseradish peroxidase; PVDF, polyvinylidene fluoride; p-AKT, phosphorylated AKT; PBS, phosphate buffer saline; p-ERK1/2, phosphorylated ERK1/2; p-p38, phosphorylated p38; p-SAPK/JNK, phosphorylated p-SAPK/JNK; p-STAT3, phosphorylated STAT3; RCC, renal cell carcinoma; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Data Sharing Statement
The data generated for this study are available on request to the corresponding author.
Ethics Approval and Consent to Participate
All the animal experimental procedures were approved by the Experimental Animal Ethical Committee of Taizhou Hospital and were performed according to the guidelines of the Experimental Animal Care Committee of Taizhou Hospital (tzy-2019047). The experiments were carried out following the Declaration of Helsinki.
Consent for Publication
The consents for publication from all authors were obtained.
Author Contributions
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This work was supported by a Doctor funding from Enze Medical Center (Grant No. 2017BSKYQDJJ01).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Srivastava A, Rivera-Núñez Z, Kim S, et al. Impact of pathologic lymph node-positive renal cell carcinoma on survival in patients without metastasis: evidence in support of expanding the definition of stage IV kidney cancer. Cancer. 2020;126(13):2991–3001. doi:10.1002/cncr.32912
2. Roumenina LT, Daugan MV, Noé R, et al. Tumor cells hijack macrophage-produced complement C1q to promote tumor growth. Cancer Immunol Res. 2019;7(7):1091–1105. doi:10.1158/2326-6066.Cir-18-0891
3. Roncati L, Maiorana A. Biological characterization of metastatic renal cell carcinoma. Urologia. 2010;77(Suppl 16):37–41. doi:10.1177/0391560310077016S09
4. Netti GS, Lucarelli G, Spadaccino F, et al. PTX3 modulates the immunoflogosis in tumor microenvironment and is a prognostic factor for patients with clear cell renal cell carcinoma. Aging. 2020;12(8):7585–7602. doi:10.18632/aging.103169
5. Battaglia M, Lucarelli G. The role of renal surgery in the era of targeted therapy: the urologist’s perspective. Urologia. 2015;82(3):137–138. doi:10.5301/uro.5000105
6. Di Lorenzo G, De Placido S, Pagliuca M, et al. The evolving role of monoclonal antibodies in the treatment of patients with advanced renal cell carcinoma: a systematic review. Expert Opin Biol Ther. 2016;16(11):1387–1401. doi:10.1080/14712598.2016.1216964
7. Liu Y, Xu SQ, Long WJ, Zhang XY, Lu HL. C5aR antagonist inhibits occurrence and progression of complement C5a induced inflammatory response of microglial cells through activating p38MAPK and ERK1/2 signaling pathway. Eur Rev Med Pharmacol Sci. 2018;22(22):7994–8003. doi:10.26355/eurrev_201811_16428
8. Ajona D, Ortiz-Espinosa S, Pio R. Complement anaphylatoxins C3a and C5a: emerging roles in cancer progression and treatment. Semin Cell Dev Biol. 2019;85:153–163. doi:10.1016/j.semcdb.2017.11.023
9. Maeda Y, Kawano Y, Wada Y, et al. C5aR is frequently expressed in metastatic renal cell carcinoma and plays a crucial role in cell invasion via the ERK and PI3 kinase pathways. Oncol Rep. 2015;33(4):1844–1850. doi:10.3892/or.2015.3800
10. Xi W, Liu L, Wang J, et al. High level of anaphylatoxin C5a predicts poor clinical outcome in patients with clear cell renal cell carcinoma. Sci Rep. 2016;6:29177. doi:10.1038/srep29177
11. Xi W, Liu L, Wang J, et al. Enrichment of C5a-C5aR axis predicts poor postoperative prognosis of patients with clear cell renal cell carcinoma. Oncotarget. 2016;7(49):80925–80934. doi:10.18632/oncotarget.13108
12. Bokisch VA, Müller-Eberhard HJ. Anaphylatoxin inactivator of human plasma: its isolation and characterization as a carboxypeptidase. J Clin Invest. 1970;49(12):2427–2436. doi:10.1172/jci106462
13. Matthews KW, Mueller-Ortiz SL, Wetsel RA. Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol. 2004;40(11):785–793. doi:10.1016/j.molimm.2003.10.002
14. Li XX, Lee JD, Kemper C, Woodruff TM. The complement receptor C5aR2: a powerful modulator of innate and adaptive immunity. J Immunol. 2019;202(12):3339–3348. doi:10.4049/jimmunol.1900371
15. Garcia-Pardo J, Tanco S, Garcia-Guerrero MC, et al. Substrate specificity and structural modeling of human carboxypeptidase Z: a unique protease with a frizzled-like domain. Int J Mol Sci. 2020;21(22):8687. doi:10.3390/ijms21228687
16. MacDonald TM, Thomas LN, Gupta A, Barnes PJ, Too CK. Prolactin and androgen R1881 induce pro-survival carboxypeptidase-D and EDD E3 ligase in triple-negative and HER2+ breast cancer. Am J Cancer Res. 2020;10(5):1321–1343.
17. Skidgel RA, Erdös EG. Cellular carboxypeptidases. Immunol Rev. 1998;161:129–141. doi:10.1111/j.1600-065x.1998.tb01577.x
18. Song L, Fricker LD. Purification and characterization of carboxypeptidase D, a novel carboxypeptidase E-like enzyme, from bovine pituitary. J Biol Chem. 1995;270(42):25007–25013. doi:10.1074/jbc.270.42.25007
19. Zhao C, Li Y, Qiu W, et al. C5a induces A549 cell proliferation of non-small cell lung cancer via GDF15 gene activation mediated by GCN5-dependent KLF5 acetylation. Oncogene. 2018;37(35):4821–4837. doi:10.1038/s41388-018-0298-9
20. Zhang Y, Cao Y, Zhang L, et al. Apigenin inhibits C5a-induced proliferation of human nasopharyngeal carcinoma cells through down-regulation of C5aR. Biosci Rep. 2018;38(3). doi:10.1042/bsr20180456
21. Lu Y, Hu XB. C5a stimulates the proliferation of breast cancer cells via Akt-dependent RGC-32 gene activation. Oncol Rep. 2014;32(6):2817–2823. doi:10.3892/or.2014.3489
22. Cho MS, Vasquez HG, Rupaimoole R, et al. Autocrine effects of tumor-derived complement. Cell Rep. 2014;6(6):1085–1095. doi:10.1016/j.celrep.2014.02.014
23. Ajona D, Zandueta C, Corrales L, et al. Blockade of the complement C5a/C5aR1 axis impairs lung cancer bone metastasis by CXCL16-mediated effects. Am J Respir Crit Care Med. 2018;197(9):1164–1176. doi:10.1164/rccm.201703-0660OC
24. Nabizadeh JA, Manthey HD, Panagides N, et al. C5a receptors C5aR1 and C5aR2 mediate opposing pathologies in a mouse model of melanoma. FASEB J. 2019;33(10):11060–11071. doi:10.1096/fj.201800980RR
25. Zha H, Wang X, Zhu Y, et al. Intracellular activation of complement C3 leads to PD-L1 antibody treatment resistance by modulating tumor-associated macrophages. Cancer Immunol Res. 2019;7(2):193–207. doi:10.1158/2326-6066.Cir-18-0272
26. Bergdolt S, Kovtun A, Hägele Y, et al. Osteoblast-specific overexpression of complement receptor C5aR1 impairs fracture healing. PLoS One. 2017;12(6):e0179512. doi:10.1371/journal.pone.0179512
27. Perianayagam MC, Balakrishnan VS, King AJ, Pereira BJ, Jaber BL. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3-kinase-signaling pathway. Kidney Int. 2002;61(2):456–463. doi:10.1046/j.1523-1755.2002.00139.x
28. Chen J, Li GQ, Zhang L, et al. Complement C5a/C5aR pathway potentiates the pathogenesis of gastric cancer by down-regulating p21 expression. Cancer Lett. 2018;412:30–36. doi:10.1016/j.canlet.2017.10.003
29. Chen J, Sun ZH, Chen LY, et al. C5aR deficiency attenuates the breast cancer development via the p38/p21 axis. Aging. 2020;12(14):14285–14299. doi:10.18632/aging.103468
30. Lim EJ, Kim S, Oh Y, et al. Crosstalk between GBM cells and mesenchymal stemlike cells promotes the invasiveness of GBM through the C5a/p38/ZEB1 axis. Neuro-Oncology. 2020;22(10):1452–1462. doi:10.1093/neuonc/noaa064
31. Huang Q, Zhong Y, Dong H, et al. Revisiting signal transducer and activator of transcription 3 (STAT3) as an anticancer target and its inhibitor discovery: where are we and where should we go? Eur J Med Chem. 2020;187:111922. doi:10.1016/j.ejmech.2019.111922
32. Hillmer EJ, Zhang H, Li HS, Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016;31:1–15. doi:10.1016/j.cytogfr.2016.05.001
33. Kujawski M, Kortylewski M, Lee H, et al. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest. 2008;118(10):3367–3377. doi:10.1172/jci35213
34. Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer. 2019;19(2):82–96. doi:10.1038/s41568-018-0090-8
35. Beebe JD, Liu JY, Zhang JT. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol Ther. 2018;191:74–91. doi:10.1016/j.pharmthera.2018.06.006
36. Laudisi F, Cherubini F, Monteleone G, Stolfi C. STAT3 interactors as potential therapeutic targets for cancer treatment. Int J Mol Sci. 2018;19(6). doi:10.3390/ijms19061787
37. O’Sullivan KE, Michielsen AJ, O’Regan E, et al. pSTAT3 levels have divergent expression patterns and associations with survival in squamous cell carcinoma and adenocarcinoma of the oesophagus. Int J Mol Sci. 2018;19(6):1720. doi:10.3390/ijms19061720
38. Chong PSY, Chng WJ, de Mel S. STAT3: a promising therapeutic target in multiple myeloma. Cancers. 2019;11(5):731. doi:10.3390/cancers11050731
39. Qin JJ, Yan L, Zhang J, Zhang WD. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. 2019;38(1):195. doi:10.1186/s13046-019-1206-z
40. Horiguchi A, Oya M, Shimada T, et al. Activation of signal transducer and activator of transcription 3 in renal cell carcinoma: a study of incidence and its association with pathological features and clinical outcome. J Urol. 2002;168(2):762–765. doi:10.1016/S0022-5347(05)64741-6
41. Siveen KS, Sikka S, Surana R, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136–154. doi:10.1016/j.bbcan.2013.12.005
42. Khan MW, Saadalla A, Ewida AH, et al. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol Immunother. 2018;67(1):13–23. doi:10.1007/s00262-017-2057-0
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.