Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Buprenorphine/samidorphan combination for the adjunctive treatment of major depressive disorder: results of a phase III clinical trial (FORWARD-3)
Authors Zajecka JM ![]() , Stanford AD, Memisoglu A, Martin WF
, Stanford AD, Memisoglu A, Martin WF ![]() , Pathak S
, Pathak S
Received 22 December 2018
Accepted for publication 11 February 2019
Published 4 April 2019 Volume 2019:15 Pages 795—808
DOI https://doi.org/10.2147/NDT.S199245
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
John M Zajecka,1,2 Arielle D Stanford,3 Asli Memisoglu,4 William F Martin,5 Sanjeev Pathak3
1Department of Psychiatry, Rush University Medical Center, Chicago, IL, USA; 2Psychiatric Medicine Associates, LLC, Skokie, IL, USA; 3Department of Clinical Research, Alkermes, Inc., Waltham, MA, USA; 4Department of Biostatistics, Alkermes, Inc., Waltham, MA, USA; 5Clinical Operations, Alkermes, Inc., Waltham, MA, USA
Background: The endogenous opioid system is a fundamental regulator of mood in humans. Previously reported clinical trials have demonstrated the efficacy of the investigational agent buprenorphine/samidorphan (BUP/SAM) combination, an opioid-system modulator, for the adjunctive treatment of major depressive disorder. We present here a third phase III study of different design.
Methods: Adult patients with major depressive disorder and inadequate response to antidepressant therapy were enrolled in this double-blind, placebo-controlled, placebo run-in study to evaluate the efficacy, safety, and tolerability of adjunctive BUP/SAM 2 mg/2 mg. Patients with baseline Hamilton Depression Rating Scale score ≥20 received double-blind placebo in addition to background antidepressant therapy for 4 weeks. Nonresponders were randomized to receive adjunctive BUP/SAM 2 mg/2 mg or placebo for 6 weeks. The primary end point was change in Montgomery–Åsberg Depression Rating Scale (MADRS)-10 total score from randomization at baseline to the end of the 6-week treatment period.
Results: Least-squares mean change in MADRS-10 score at end of treatment was -4.8 (SE 0.67) in the BUP/SAM 2 mg/2 mg group and -4.6 (SE 0.66) in the placebo group (mean difference -0.3 [SE 0.95], P=0.782). There were no differences in MADRS-based response or remission rates. Overall, 42.9% of the BUP/SAM 2 mg/2 mg group and 34.5% of the placebo group experienced at least one treatment-emergent adverse event during the 6-week treatment period, most of which were mild or moderate in severity. There were no clinically important changes in laboratory parameters, weight, or vital signs and no evidence of abuse potential during treatment or opiate-withdrawal symptoms post treatment.
Conclusion: Efficacy results in FORWARD-3 measured by change in MADRS-10 score did not meet the primary end point, but postbaseline improvement in MADRS-10 in the BUP/SAM 2 mg/2 mg group was consistent with that seen in previously reported trials. BUP/SAM 2 mg/2 mg was well tolerated.
Keywords: buprenorphine, samidorphan, randomized clinical trial, adjunctive therapy, study design, placebo response, opioid system modulator
Introduction
Major depressive disorder (MDD) is a leading cause of disability worldwide and represents a substantial persistent burden to affected individuals.1,2 Currently approved antidepressant therapies (ADTs) target the monoaminergic pathways, consistent with the monoamine hypothesis of depression. These agents provide important therapeutic benefit to some patients, but for a substantial proportion of others they offer inadequate symptom relief.3–6 In addition to the symptomatic burden, patients with partial or minimal response to treatment have higher rates of unemployment and productivity loss, as well as greater health care utilization.2 The mainstay of adjunctive pharmacologic treatment for MDD is atypical antipsychotics, which are also monoaminergic and associated with serious and sometimes treatment-limiting adverse effects, including significant metabolic abnormalities and tardive dyskinesia.7 Nonpharmacological, neuromodulatory therapies, such as electroconvulsive therapy, repetitive transcranial magnetic stimulation, and vagal nerve stimulation, are additional treatment options that can be beneficial;8,9 however, their use is constrained by the need for specialized equipment and significant time commitment from patients.10 Pharmacotherapies with alternative mechanisms of action and different safety and tolerability profiles are needed to address the complex, debilitating symptoms characteristic of MDD.3–5
Opioid modulation is a promising target in the treatment of MDD, based on several lines of evidence.11–13 The endogenous opioid system is known as a fundamental regulator of mood,14–16 and dysregulation of circuits in this system is implicated in the development of symptoms associated with MDD.17–19 Historically, prior to the advent of monoaminergic antidepressants, opioids were used to treat MDD, a practice sometimes referred to as the “opium cure”. 20 More recently, buprenorphine (BUP) has been shown in clinical trials to possess antidepressant activity, particularly in treatment-refractory patients.20–24 However, the clinical utility of opioids has been limited by the risk of abuse and dependence.22
The buprenorphine/samidorphan (BUP/SAM; ALKS 5461) combination is an opioid-system modulator under investigation as an adjunctive treatment of MDD. BUP is a μ-opioid-receptor partial agonist and κ-opioid-receptor antagonist with antidepressant activity.21,23,25–27 SAM is a μ-opioid-receptor antagonist coformulated with BUP in a tablet for sublingual administration. The purpose of SAM in the combination is to address the potential for abuse of and dependence on BUP28 while preserving its antidepressant effects.29,30
High placebo-response rates are a major factor affecting the ability to detect efficacy in psychiatric studies, and present a challenge in the development of new ADTs.31 Clinical trials in MDD have shown a direct correlation over time between rising placebo-response rates and a decline in trial successes, even in investigations of agents with established efficacy.32–35 Various study-design approaches have attempted to minimize the impact of placebo response on MDD-trial outcomes.31,36 One such strategy is the single-blind, placebo run-in design, where patients are unaware of the placebo run-in phase and those not responding to placebo are randomized to treatment. The single-blind approach has had limited success in the acute-phase efficacy setting, attributable to factors relating both to selection of patients for study entry and to study design.37,38
The double-blind, placebo run-in design was developed to address the limitations of the single-blind approach.37 By this design, both patients and study personnel are blinded to the existence of the placebo run-in period and the point of initiation of active treatment. This double-blind, placebo run-in design is expected to identify a greater proportion of placebo responders compared with the single-blind approach, and this exclusion of placebo responders from randomization should theoretically enhance the ability to detect study drug–placebo treatment differences in the subsequent treatment period.37
The main objective of the current study was to evaluate the efficacy, safety, and tolerability of BUP/SAM for the adjunctive treatment of MDD in adults who have an inadequate response to ADT. The study adopted a double-blind, placebo run-in design to investigate BUP/SAM in patients with MDD. Herein we present the details of this methodology, describe the results of the study, and compare them with the other phase III studies of BUP/SAM, in which active treatment with BUP/SAM demonstrated a significant difference in efficacy compared with placebo in one study and was supported by post hoc analysis of efficacy results in the other.39
Methods
Study population and design
FORWARD-3 (ClinicalTrials.gov NCT02158546) was a phase III, multicenter, randomized, placebo-controlled study conducted at 58 study sites: 49 in the US and nine in Bulgaria. The study enrolled male and female patients aged 18–70 years with a diagnosis of MDD based on the Diagnostic and Statistical Manual for Mental Disorders (DSM-IV-TR) criteria and a current major depressive episode of 8 weeks to 24 months. Additionally, all patients had a 17-item Hamilton Rating Scale for Depression (HAM-D) total score ≥18 and a Clinical Global Impression – severity (CGI-S) score ≥4.
Patients were eligible to enter the double-blind period if they demonstrated an inadequate response to one or two courses of ADT during the current episode. This criterion could be met either historically or prospectively. Patients who did not have sufficient historical evidence of one or two inadequate responses to ADT and whose HAM-D total score was ≥22 were allowed to enter the prospective lead-in period, during which ADT was administered open-label for 8 weeks. Patients who demonstrated <50% reduction from screening in HAM-D total score and had a HAM-D total score ≥15 at all visits during this prospective lead-in period were eligible to continue to the double-blind study. At randomization, all patients would have then received ADT treatment for ≥8 weeks at an adequate dose that was stable over the last 4 weeks.
Key exclusion criteria included any primary axis I disorder besides MDD, the use of adjunctive treatments during the current episode (except as noted in the Supplementary material), imminent suicide risk, and evidence of an alcohol- or substance-use disorder within the past year. Suicide risk was deemed imminent based on one or more of a recent history of suicide attempt (past 2 years), acknowledgment of current suicidal ideation with intent, with or without a plan (based on the Columbia Suicide Severity Rating Scale [CSSRS]), and investigator clinical assessment. Patients were also excluded if they had a current axis II diagnosis of borderline, antisocial, paranoid, schizoid, schizotypal, or histrionic personality disorder. Detailed patient-inclusion and -exclusion criteria are included in the Supplementary material.
At the start of the double-blind treatment period, patients were stratified according to their baseline HAM-D total score. Patients with a baseline HAM-D total score ≥20 (group 1) received double-blind placebo for 4 weeks, and thereafter placebo nonresponders were randomized to BUP/SAM 2 mg/2 mg or placebo as a sublingual tablet once daily, in addition to continuing their current ADT, for a further 6 weeks. Data from these patients were used to evaluate efficacy. Placebo responders remained on placebo for the duration of the double-blind treatment period.
Patients with a baseline HAM-D total score of 18–19 (group 2) were randomized to BUP/SAM 2 mg/2 mg or placebo, in addition to continuing their current ADT, for the duration of the 10-week double-blind treatment period. Group 2 patients were included in the study to provide additional blinding of the existence of the placebo run-in period for group 1 patients and to minimize baseline-score inflation by allowing patients with lower HAM-D scores into the study. After the double-blind treatment period, patients in both groups entered a long-term safety study (FORWARD-2, ClinicalTrials.gov NCT02141399) or completed a 2-week safety follow-up period. See Figure S1 for a schema of the FORWARD-3 study design. All investigative staff were blinded to the existence of two groups, the existence of the placebo run-in, the timing of randomization, and the precise criteria by which nonresponse to placebo was assessed.
The study protocol and informed-consent document were reviewed by an independent ethics committee or institutional review board (IRB) at each site: in the US, these were the Copernicus Group IRB Office of Regulatory Affairs, University of Pennsylvania IRB, University of Cincinnati IRB, or University of Texas Southwestern IRB, and in Bulgaria the Ethics Committee for Multicenter Trials. The study was conducted following the principles of good clinical practice derived from the Declaration of Helsinki (1964) and in accordance with local regulations and International Council for Harmonisation guidelines. All patients provided written informed consent prior to study entry.
Efficacy and safety evaluations
The primary efficacy end point was change in Montgomery–Åsberg Depression Rating Scale (MADRS) 10 score from randomization at baseline to the end of the treatment period among placebo nonresponders in group 1 who were randomized to double-blind treatment. Secondary efficacy end points were proportions of patients demonstrating a treatment response (ie, ≥50% reduction in MADRS-10 score from randomization baseline to the end of the efficacy period) and achieving remission (ie, MADRS-10 score ≤10 at the end of the treatment period).
Safety and tolerability evaluations included treatment-emergent adverse events (AEs; coded using Medical Dictionary for Regulatory Activities version 16.1 and recorded at each study visit), laboratory assessments of chemistry and hematology parameters, vital signs, electrocardiography parameters, level of opiate withdrawal assessed by the Clinical Opiate Withdrawal Scale, and suicidal behavior and suicidal ideation assessed by the CSSRS. An AE was considered treatment-emergent if it had started after receiving the study drug or worsened (if present at baseline) after baseline. The relationship of AEs to study drug was assessed by the investigators: drug-related AEs were defined as definitely, probably, or possibly related.
AEs of special interest to evaluate abuse potential included preferred terms suggestive of abuse behavior and euphoria and nonspecific terms possibly related to abuse potential (eg, dizziness, somnolence). AEs of special interest to evaluate for opioid withdrawal were assessed during the 2-week safety follow-up period. A complete list of preferred terms is included in the Supplementary material. An analysis of AEs potentially associated with suicidal ideation and/or behavior was also performed. Similar safety and efficacy evaluations were performed separately for group 2, which had no placebo run-in and thus a longer randomized treatment period of 10 weeks.
Statistics
The study sample size was designed to ensure that at least 260 patients were randomized at the end of the 4-week placebo run-in period and included in the analysis population. This sample size was estimated to provide >80% and >90% power to detect a treatment difference of 3.5 and 4.0 points, respectively, between BUP/SAM 2 mg/2 mg and placebo for the primary end point. Analysis employed mixed model repeated measures (MMRM) to estimate change from baseline in MADRS-10 score in BUP/SAM 2 mg/2 mg compared to placebo (least-squares [LS] mean difference and standard error). Hypothesis tests were two-sided with α=0.05.
Efficacy was assessed in randomized group 1 patients (ie, placebo nonresponders) who had received at least one dose of the study drug and at least one postrandomization assessment of MADRS-10 score (analysis population). Any duplicate patients who had participated in a prior clinical study of BUP/SAM were identified and excluded from efficacy analysis during the study (n=2, BUP/SAM 2 mg/2 mg; n=3, placebo), but included in the safety analysis. Data from one of the 58 sites were excluded from analysis of efficacy and safety when prior to study completion aggregate review of blinded data from the patients enrolled from this site revealed evidence of data-integrity issues.
Efficacy was evaluated at weekly visits from randomized baseline to the end of the double-blind treatment period, ie, 6 weeks for the analysis population. For continuous end points with repeated postrandomization measurements, an MMRM model was used to analyze change from baseline. The model included variables for treatment group, visit, and a treatment group-by-visit interaction term as categorical fixed effects, and baseline value and baseline-by-visit interaction were included as covariates. Unstructured variance–covariance was specified. Statistical hypothesis tests were two sided with α=0.05% and 95% CI.
For analysis of response and remission, the proportion of patients meeting the criteria of response or remission at the end of the 6-week efficacy period was estimated using last-observation-carried-forward imputation. BUP/SAM 2 mg/2 mg group vs placebo-group differences in rates of response and remission and associated 95% CIs and P-values were reported based on normal approximation to binominal distributions.
Safety was assessed in randomized group 1 patients who had received at least one dose of the study drug during the double-blind treatment period. AEs occurring during the placebo lead-in period are not presented here. All safety assessments were summarized using descriptive statistics. Data for group 2 were analyzed separately and were not part of the primary efficacy or safety evaluation, as prespecified in the protocol.
Results
Patients
A total of 399 and 30 patients were enrolled in group 1 and group 2, respectively (Figure 1). A total of 297 (74.4%) group 1 patients were placebo nonresponders and randomized into the double-blind treatment period. Of the remaining 102 patients, 77 (19.3%) were placebo responders and 25 discontinued (6.3%). Two randomized patients did not receive the study drug, leaving 295 patients in the safety-analysis population.
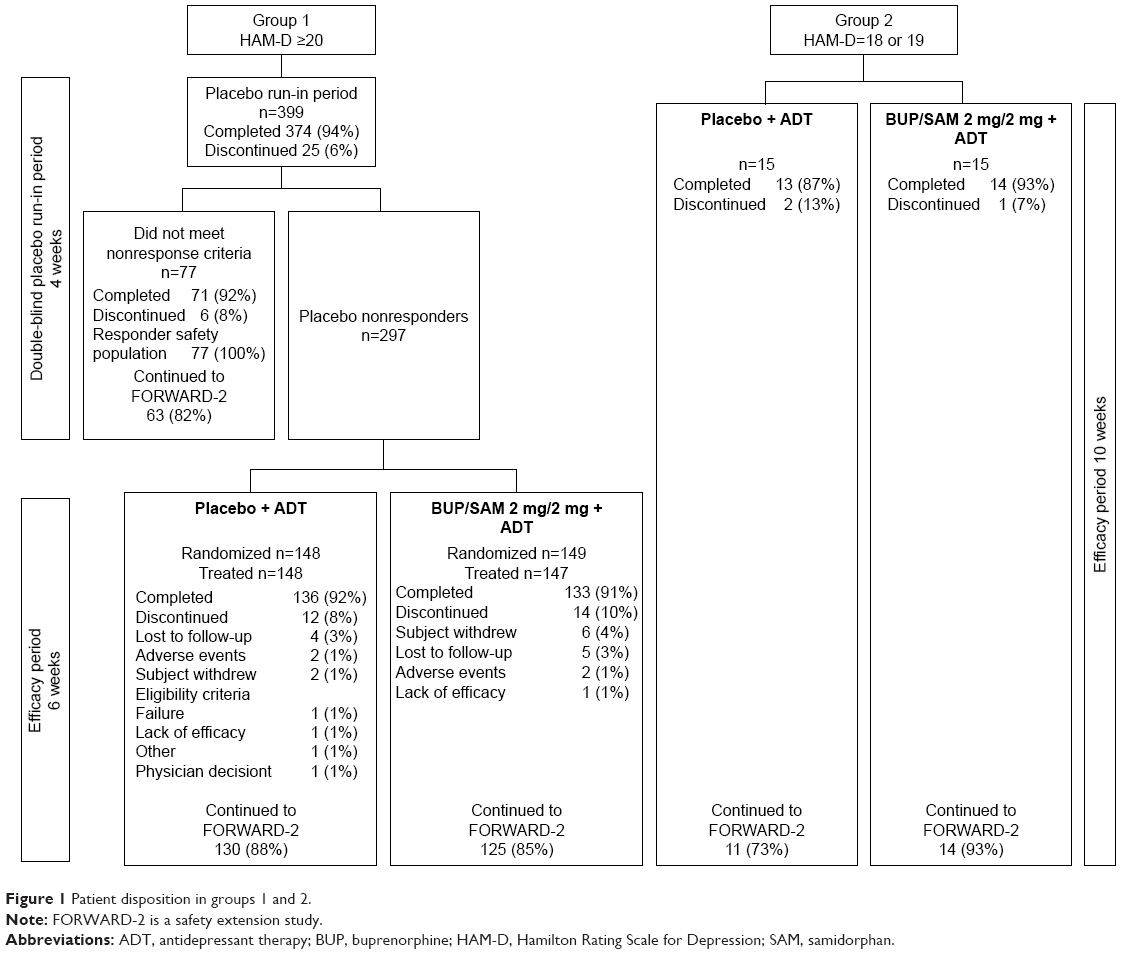 | Figure 1 Patient disposition in groups 1 and 2. |
The study was completed by 133 (90.5%) and 136 (91.9%) patients in BUP/SAM 2 mg/2 mg and placebo groups, respectively. The most common reasons for discontinuation were loss to follow-up (3.4%, BUP/SAM 2 mg/2 mg; 2.7%, placebo) and withdrawal by patient (4.1%, BUP/SAM 2 mg/2 mg; 1.4%, placebo; Figure 1). Demographics and baseline disease characteristics were similar between the BUP/SAM 2 mg/2 mg and placebo groups (n=147 and 148, respectively; Table 1). The majority of patients were female (61.7%) and white (74.9%). Mean (SE) duration of the current major depressive episode was 8.6 (5.73) months, and selective serotonin-reuptake inhibitors were the most frequently reported ADT (67.8%).
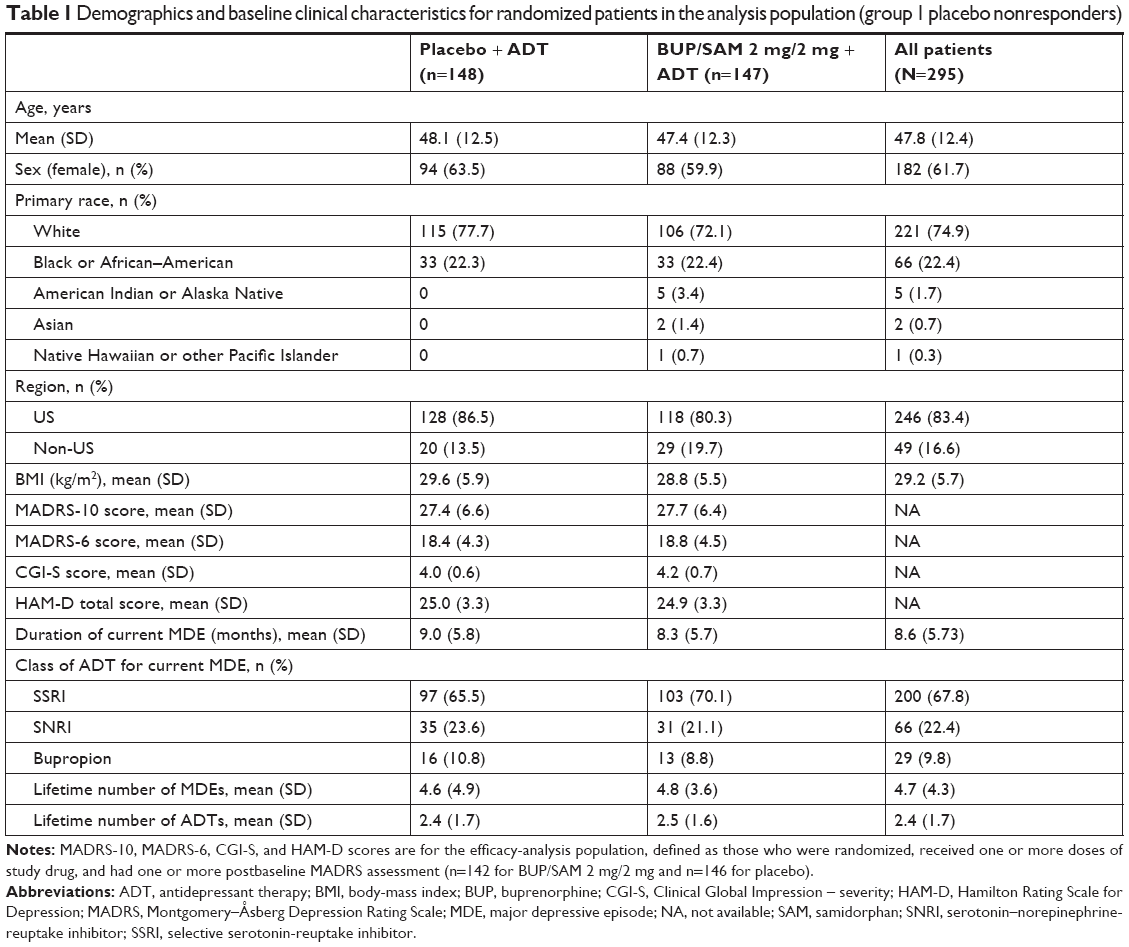 | Table 1 Demographics and baseline clinical characteristics for randomized patients in the analysis population (group 1 placebo nonresponders) |
Efficacy
LS mean change in MADRS-10 score at the end of the 6-week treatment period (the primary study end point) in the efficacy-analysis population was −4.8 (SE 0.67) in BUP/SAM 2 mg/2 mg and −4.6 (SE 0.66) in the placebo group. LS mean difference in MADRS-10 score between BUP/SAM 2 mg/2 mg and placebo was −0.3 (SE 0.95, 95% CI −2.1 to 1.6; P=0.782). There were no group differences in MADRS-10 score at any visit (Figure 2).
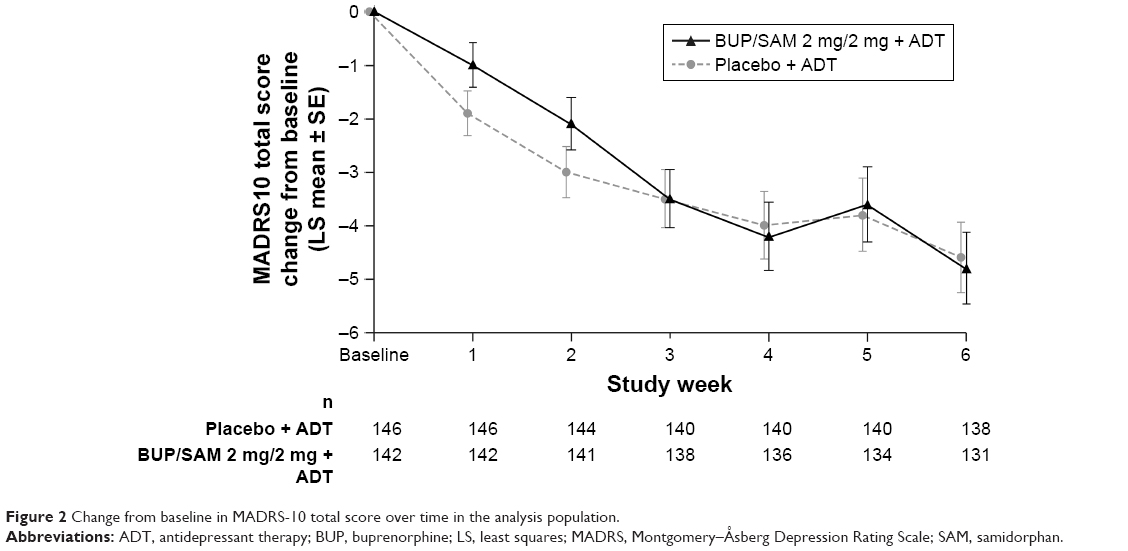 | Figure 2 Change from baseline in MADRS-10 total score over time in the analysis population. |
The response rate at the end of the 6-week treatment period was 16.9% for BUP/SAM 2 mg/2 mg and 14.4% for placebo (difference 2.5 [SE 4.3]; 95% CI −5.9 to 10.9; P=0.556), while remission rates were 14.1% for BUP/SAM 2 mg/2 mg and 12.3% for placebo (difference 1.8 [SE 4.0], 95% CI −6.1 to 9.6; P=0.660).
Safety
Of the 295 patients in the safety-analysis population, 42.9% (n=63) of the BUP/SAM 2 mg/2 mg group and 34.5% (n=51) of the placebo group experienced at least one treatment-emergent AE during the 6-week treatment period (Table 2). Most AEs in the treatment groups were mild or moderate (25.9% [n=38] and 15.6% [n=23], respectively, in the BUP/SAM 2 mg/2 mg group and 20.9% [n=31] and 12.2% [n=18], respectively, in the placebo group). The most frequently reported AEs in the BUP/SAM 2 mg/2 mg group were nausea (8.8%, placebo 0.7%), headache (4.1%, placebo 3.4%), increased blood creatine phosphokinase (2.7%, placebo 0), fatigue (2.7%, placebo 2.0%), upper respiratory tract infection (2.7%, placebo 2.0%), and vomiting (2.7%, placebo 1.4%). All others occurred in ≤2.0% of patients. Two patients (1.4%) in the BUP/SAM 2 mg/2 mg group (n=1 anxiety and depression, n=1 emotional disorder), and two patients (1.4%) in the placebo group (n=1 atrial fibrillation, n=1 renal carcinoma) discontinued the study due to AEs. The AE of renal carcinoma was initially reported during the placebo run-in period.
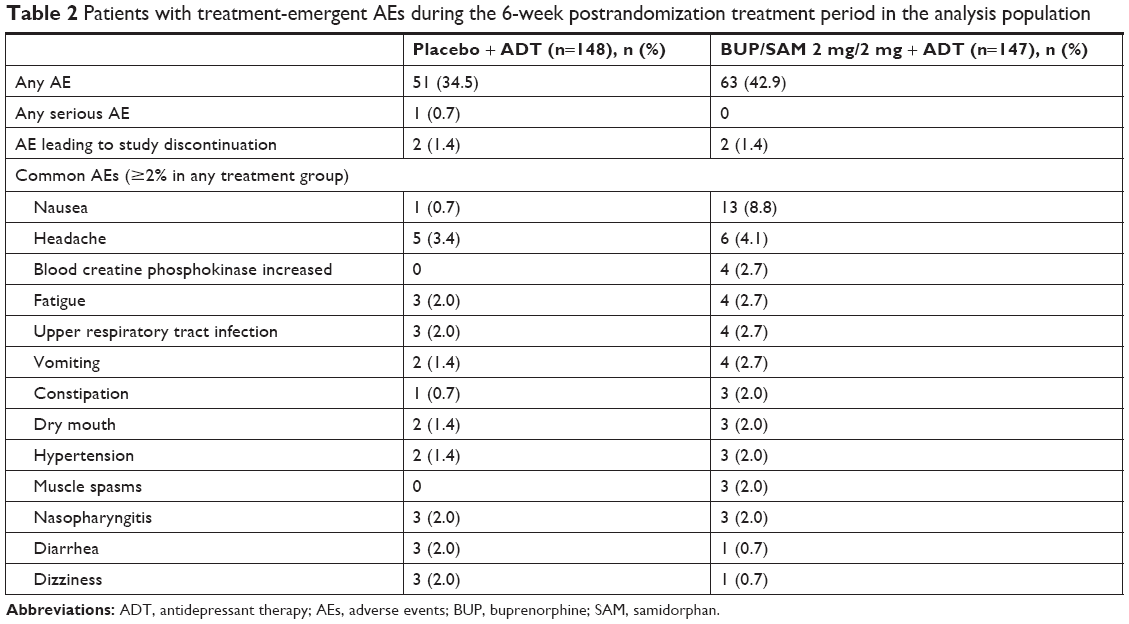 | Table 2 Patients with treatment-emergent AEs during the 6-week postrandomization treatment period in the analysis population |
No serious AEs occurred in the BUP/SAM 2 mg/2 mg group. AEs considered by the investigator to be related to the study drug were reported in 19.0% (n=28) and 12.2% (n=18) of the BUP/SAM 2 mg/2 mg and placebo groups, respectively, during the treatment period. Nausea was the most frequently reported drug-related AE in the BUP/SAM 2 mg/2 mg group (6.1%, placebo 0), followed by constipation (2.0%; placebo 0.7%).
There were no clinically important changes in laboratory parameters, weight, vital signs, or electrocardiography parameters (including QT prolongation). There were four cases of elevated blood creatine phosphokinase in the BUP/SAM 2 mg/2 mg group (Table 2), and three of these resolved. All four cases were asymptomatic, and were attributed to exercise or environmental factors and assessed as not drug-related.
No evidence of abuse behavior, euphoria, or dependence was noted from analysis of the AEs of special interest. Aspecific AEs of abuse potential occurred in two patients in the BUP/SAM 2 mg/2 mg group (n=1 dizziness, n=1 somnolence) and three patients in the placebo group (dizziness). There was no evidence of opioid withdrawal assessed by AEs of special interest during the 2-week postdiscontinuation follow-up period. Clinical Opiate Withdrawal Scale assessment of opiate-withdrawal signs and symptoms, which was performed during the postdiscontinuation follow-up period in only those patients who did not enter the long-term safety study after discontinuing the study drug (n=13 BUP/SAM 2 mg/2 mg, n=25 placebo), showed no evidence of withdrawal in either treatment group.
The incidence of suicidal ideation assessed by the CSSRS was 10.1% in the placebo group vs 6.1% in the BUP/SAM 2 mg/2 mg group at baseline and 12.8% and 10.2%, respectively, at any postbaseline visit. Suicidal behavior was not exhibited at any time in either treatment group. One patient receiving BUP/SAM 2 mg/2 mg had a treatment-emergent AE of “suicidal ideation without intent”, which resolved the same day and did not lead to study discontinuation.
Discussion
The main finding of this study was that patients treated with adjunctive BUP/SAM 2 mg/2 mg showed improvements in depressive symptom score vs baseline on the MADRS-10 scale comparable to those observed in other randomized controlled trials of adjunctive BUP/SAM for the treatment of MDD in similar patient populations,29,39 but the treatment effect was not statistically significantly different compared with placebo. Therefore, the study did not meet its primary end point.
BUP/SAM 2 mg/2 mg was generally well tolerated as an adjunctive treatment for MDD in FORWARD-3, and there was a very high study-retention rate. Most treatment-emergent AEs were of mild or moderate intensity. Of particular importance, there was no evidence of abuse potential or opioid withdrawal based on the safety measures used. These results are consistent with those previously reported from the other BUP/SAM 2 mg/2 mg phase III studies.39 FORWARD-3 thereby adds to the growing body of available evidence that supports the safety of BUP/SAM 2 mg/2 mg in the treatment of MDD.
Reasons for the failure to observe an antidepressant signal vs placebo cannot be known for certain. It is well established that approximately 50% of clinical trials with approved antidepressants fail to separate from placebo.35 One potential factor is the degree of placebo effect observed in this study. Comparison of results from FORWARD-3 to those with similar MDD populations in two other BUP/SAM studies (FORWARD-4 and FORWARD-5)39 shows that the reduction in MADRS-10 scores in the BUP/SAM arms was consistent across all three studies. In contrast, the reduction of MADRS-10 scores in the placebo arms was variable, with FORWARD-3 demonstrating the highest placebo response (Figure 3).
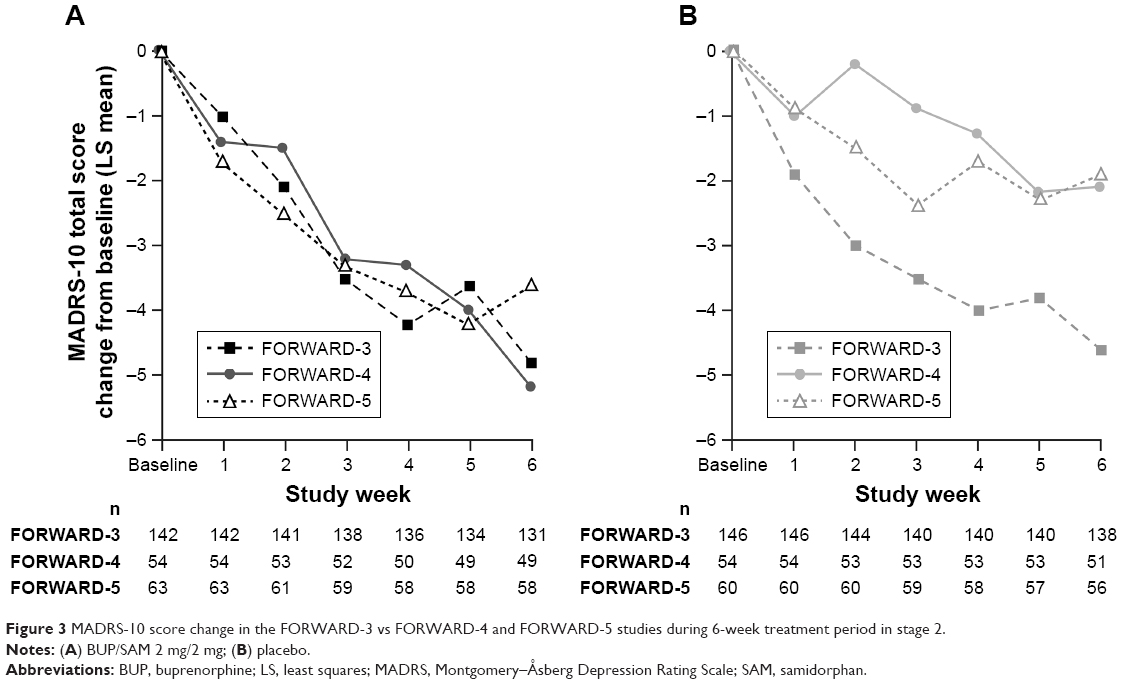 | Figure 3 MADRS-10 score change in the FORWARD-3 vs FORWARD-4 and FORWARD-5 studies during 6-week treatment period in stage 2. |
Reasons for the larger placebo response in FORWARD-3 cannot be definitively ascertained either. One potential contributor may have been the lower proportion of randomized patients who were identified as placebo responders during the placebo run-in period of FORWARD-3 (19.3%) compared with the proportion identified during stage 1 of FORWARD-4 (31.3%) and FORWARD-5 (24.6%), both of which used a different study design than FORWARD-3. FORWARD-4 and FORWARD-539 utilized a sequential parallel-comparison design (SPCD). The strategy of SPCD is to enhance signal detection in the presence of placebo response and obtain greater statistical power with fewer patients compared with the placebo run-in approach used in FORWARD-3.31 SPCD uses a two-stage approach. In the first stage, patients are randomized to active treatment and placebo in a standard parallel comparison. In the second stage, only placebo nonresponders are randomized to active treatment or placebo using a parallel comparison design, similar to the analysis population of a placebo run-in study like FORWARD-3 (Supplementary material). In SPCD, statistical analysis is performed on data from individual stages, and stage-specific estimates are combined using prespecified weights to estimate the overall drug–placebo difference.
A possible advantage of SPCD over double-blind placebo run-in for signal detection is suggested by the outcomes of the FORWARD-4 and FORWARD-5 trials, where the placebo response was better controlled and the antidepressant efficacy of BUP/SAM 2 mg/2 mg consistently greater than that of placebo.39 While clinical trials for other adjunctive treatments have used SPCD,40–42 we are aware of none that compares outcomes using different trial designs, highlighting the uniqueness of our observations.
This study had certain limitations, including those common with most short-term placebo-controlled trials. For instance, the short duration of evaluation does not inform the efficacy or safety of BUP/SAM with longer duration of treatment. Additional limitations apply when making comparison across BUP/SAM studies. The BUP/SAM studies compared in the discussion are few in number, and thus the potential advantages of SPCD must be viewed in the context of this limitation. Since clinical trials in MDD often fail irrespectively of study design, it is conceivable that the placebo run-in design may have delivered positive-efficacy results if more studies with this design had been conducted.
Conclusion
Efficacy results in FORWARD-3, measured by change in MADRS-10 total score, did not meet the primary end point, but postbaseline improvement in MADRS-10 in the BUP/SAM 2 mg/2 mg group was consistent with that seen in previously reported trials.29,39 The relatively larger placebo response may have obscured the ability to detect efficacy vs placebo in this study. The higher proportion of placebo responders detected in stage 1 of FORWARD-4 and FORWARD-5 suggests that SPCD may be more effective for improving signal detection. An additional advantage of SPCD is that it is a more efficient trial design because it utilizes data from all enrolled patients. BUP/SAM 2 mg/2 mg demonstrated a favorable safety and tolerability profile, with no evidence of abuse potential or opioid withdrawal based on the outcomes used in this study. Clinical development of the BUP/SAM combination, a promising novel adjunctive treatment for patients with MDD, is warranted and ongoing.
Abbreviations
ADT, antidepressant therapy; AE, adverse event; BUP, buprenorphine; CGI-S, Clinical Global Impression – severity; CSSRS, Columbia Suicide Severity Rating Scale; DSM-IV-TR, Diagnostic and Statistical Manual for Mental Disorders, fourth edition, text revision; HAM-D, Hamilton Rating Scale for Depression; LS, least squares; MADRS, Montgomery–Åsberg Depression Rating Scale; MDD, major depressive disorder; MDE, major depressive episode; MMRM, mixed-model repeated-measures; SAM, samidorphan; SPCD, sequential parallel-comparison design; SSRI, selective serotonin-reuptake inhibitor.
Availability of data and material
The data collected in this study are proprietary to Alkermes, Inc. Alkermes, Inc. is committed to public sharing of data in accordance with applicable regulations and laws.
Acknowledgments
The authors thank the patients who participated in the study, as well as the study investigators and research personnel. The study was sponsored by Alkermes, Inc. Medical writing assistance was provided by Bill Wolvey, BSc, and Bill Kadish, MD, CMPP, of PAREXEL International, and funded by Alkermes, Inc.
Author contributions
ADS, AM, JMZ, SP, and WFM each contributed substantially to data acquisition, data analysis, and data interpretation, critically revised the manuscript for important intellectual content at each stage of development, approved the version to be published, and agreed to be accountable for all aspects of the work. Substantial contributions to study conception and design were provided by ADS, AM, SP, and WFM.
Disclosure
JMZ has received grants/research support from Actavis, Alkermes, Inc., Allergan, Axsome, elminda, Forest, the Cheryl T Herman Foundation, Hoffman-La Roche, Intra-Cellular Therapies, Janssen, Johnson & Johnson, Lundbeck, Otsuka, National Institutes of Health, Taisho, and Takeda, and has served as a consultant/advisory board participant for Alkermes, Inc., elminda, Lundbeck, and Takeda. ADS, AM, WFM, and SP are employees of Alkermes, Inc. The authors report no other conflicts of interest in this work.
References
Ferrari AJ, Charlson FJ, Norman RE, et al. Burden of depressive disorders by country, sex, age, and year: findings from the Global Burden of Disease Study 2010. PLoS Med. 2013;10(11):e1001547. | ||
Knoth RL, Bolge SC, Kim E, Tran QV. Effect of inadequate response to treatment in patients with depression. Am J Manag Care. 2010;16(8):e188–e196. | ||
Machado-Vieira R, Henter ID, Zarate CA Jr. New targets for rapid antidepressant action. Prog Neurobiol. 2017;152:21–37. | ||
Otte C, Gold SM, Penninx BW, et al. Major depressive disorder. Nat Rev Dis Primers. 2016;2:16065. | ||
Bauer M, Bschor T, Pfennig A, et al. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of unipolar depressive disorders in primary care. World J Biol Psychiatry. 2007;8(2):67–104. | ||
Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–1917. | ||
Blier P. Atypical antipsychotics for mood and anxiety disorders: safe and effective adjuncts? J Psychiatry Neurosci. 2005;30(4):232–233. | ||
Serafini G, Pompili M, Belvederi Murri M, et al. The effects of repetitive transcranial magnetic stimulation on cognitive performance in treatment-resistant depression. A systematic review. Neuropsychobiology. 2015;71(3):125–139. | ||
Cusin C, Dougherty DD. Somatic therapies for treatment-resistant depression: ECT, TMS, VNS, DBS. Biol Mood Anxiety Disord. 2012;2(1):14. | ||
Berlim MT, van den Eynde F, Daskalakis ZJ. Efficacy and acceptability of high frequency repetitive transcranial magnetic stimulation (rTMS) versus electroconvulsive therapy (ECT) for major depression: a systematic review and meta-analysis of randomized trials. Depress Anxiety. 2013;30(7):614–623. | ||
Berrocoso E, Sánchez-Blázquez P, Garzón J, Mico JA. Opiates as antidepressants. Curr Pharm Des. 2009;15(14):1612–1622. | ||
Lutz P-E, Kieffer BL. Opioid receptors: distinct roles in mood disorders. Trends Neurosci. 2013;36(3):195–206. | ||
Williams NR, Heifets BD, Blasey C, et al. Attenuation of antidepressant effects of ketamine by opioid receptor antagonism. Am J Psychiatry. Epub 2018 Aug 29. | ||
Bérubé P, Laforest S, Bhatnagar S, Drolet G. Enkephalin and dynorphin mRNA expression are associated with resilience or vulnerability to chronic social defeat stress. Physiol Behav. 2013;122:237–245. | ||
Hsu DT, Sanford BJ, Meyers KK, et al. It still hurts: altered endogenous opioid activity in the brain during social rejection and acceptance in major depressive disorder. Mol Psychiatry. 2015;20(2):193–200. | ||
Nummenmaa L, Karjalainen T. Opioidergic regulation of pain and Pleasure in human social relationships. Neuropsychopharmacology. 2018;43(1):217–218. | ||
Kennedy SE, Koeppe RA, Young EA, Zubieta JK. Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Arch Gen Psychiatry. 2006;63(11):1199–1208. | ||
Knoll AT, Carlezon WA. Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. | ||
Ribeiro SC, Kennedy SE, Smith YR, Stohler CS, Zubieta JK. Interface of physical and emotional stress regulation through the endogenous opioid system and mu-opioid receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(8):1264–1280. | ||
Tenore PL. Psychotherapeutic benefits of opioid agonist therapy. J Addict Dis. 2008;27(3):49–65. | ||
Bodkin JA, Zornberg GL, Lukas SE, Cole JO. Buprenorphine treatment of refractory depression. J Clin Psychopharmacol. 1995;15(1):49–57. | ||
Ehrich E, Turncliff R, Du Y, et al. Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology. 2015;40(6):1448–1455. | ||
Karp JF, Butters MA, Begley AE, et al. Safety, tolerability, and clinical effect of low-dose buprenorphine for treatment-resistant depression in midlife and older adults. J Clin Psychiatry. 2014;75(8):e785–e793. | ||
Serafini G, Adavastro G, Canepa G, et al. The efficacy of buprenorphine in major depression, treatment-resistant depression and suicidal behavior: a systematic review. Int J Mol Sci. 2018;19(8):2410. | ||
Emrich HM, Vogt P, Herz A, Kissling W. Antidepressant effects of buprenorphine. Lancet. 1982;320(8300):709. | ||
Nyhuis PW, Gastpar M, Scherbaum N. Opiate treatment in depression refractory to antidepressants and electroconvulsive therapy. J Clin Psychopharmacol. 2008;28(5):593–595. | ||
Yovell Y, Bar G, Mashiah M, et al. Ultra-low-dose buprenorphine as a time-limited treatment for severe suicidal ideation: a randomized controlled trial. Am J Psychiatry. 2016;173(5):491–498. | ||
Bidlack JM, Knapp BI, Deaver DR, Plotnikava M, Arnelle D, Quinn AM. ALKS 5461: affinity, potency, and functional activity of buprenorphine and samidorphan alone and in combination. Presented at: 73rd Annual Meeting of the Society of Biological Psychiatry; May 10–12, 2018; New York, NY. | ||
Fava M, Memisoglu A, Thase ME, et al. Opioid modulation with buprenorphine/samidorphan as adjunctive treatment for inadequate response to antidepressants: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2016;173(5):499–508. | ||
Fava M, Thase M, Trivedi M, et al. ALKS 5461 (a buprenorphine-samidorphan combination) for adjunctive treatment of major depressive disorder: results from analyses of US patients from three independent placebo-controlled trials. Neuropsychopharmacology. 2017;42:2. | ||
Fava M, Evins AE, Dorer DJ, Schoenfeld DA. The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychother Psychosom. 2003;72(3):115–127. | ||
Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391(10128):1357–1366. | ||
Furukawa TA, Cipriani A, Atkinson LZ, et al. Placebo response rates in antidepressant trials: a systematic review of published and unpublished double-blind randomised controlled studies. Lancet Psychiatry. 2016;3(11):1059–1066. | ||
Undurraga J, Baldessarini RJ. Randomized, placebo-controlled trials of antidepressants for acute major depression: thirty-year meta-analytic review. Neuropsychopharmacology. 2012;37(4):851–864. | ||
Khin NA, Chen Y-F, Yang Y, Yang P, Laughren TP. Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry. 2011;72(4):464–472. | ||
Khan A, Schwartz K. Study designs and outcomes in antidepressant clinical trials. Essent Psychopharmacol. 2005;6(4):221–226. | ||
Faries DE, Heiligenstein JH, Tollefson GD, Potter WZ. The double-blind variable placebo lead-in period: results from two antidepressant clinical trials. J Clin Psychopharmacol. 2001;21(6):561–568. | ||
Trivedi MH, Rush H. Does a placebo run-in or a placebo treatment cell affect the efficacy of antidepressant medications? Neuropsychopharmacology. 1994;11(1):33–43. | ||
Fava M, Thase ME, Trivedi MH, et al. Opioid system modulation with buprenorphine/samidorphan combination for major depressive disorder: two randomized controlled studies. Mol Psychiatry. Epub 2018 Oct 29. | ||
Fava M, Freeman MP, Flynn M, et al. Double-blind, proof-of-concept (POC) trial of low-field magnetic stimulation (LFMS) augmentation of antidepressant therapy in treatment-resistant depression (TRD). Brain Stimul. 2018;11(1):75–84. | ||
Fava M, Mischoulon D, Iosifescu D, et al. A double-blind, placebo-controlled study of aripiprazole adjunctive to antidepressant therapy among depressed outpatients with inadequate response to prior antidepressant therapy (ADAPT-A study). Psychother Psychosom. 2012;81(2):87–97. | ||
Papakostas GI, Shelton RC, Zajecka JM, et al. L-methylfolate as adjunctive therapy for SSRI-resistant major depression: results of two randomized, double-blind, parallel-sequential trials. Am J Psychiatry. 2012;169(12):1267–1274. |
Supplementary materials
Inclusion and exclusion criteria
During enrollment and screening, patients meeting the following criteria were considered for admission to the study:
- willing and able to provide informed consent
- aged 18–70 years
- had a body-mass index of 18–40 kg/m2
- agreed to use an acceptable method of contraception for the duration of the study and at least 30 days after the last dose of study drug, unless surgically sterile or postmenopausal
- had a Diagnostic and Statistical Manual for Mental Disorders, fourth edition, text revision (DSM-IV-TR) major depressive disorder (MDD) primary diagnosis assessed and confirmed by the Mini-International Neuropsychiatric Interview (MINI; administered by qualified site staff); the primary diagnosis was defined as the primary source of current distress and functional impairment in the opinion of the investigator
- had a current major depressive episode (MDE) lasting 8 weeks to 24 months
- willing and able to follow the study procedures outlined in the protocol, including adherence with both the approved antidepressant therapy (ADT) and the study drug regimen.
In addition, for those patients entering the prospective lead-in period, the following criteria were considered for admission to the study:
- not currently taking any ADT
- had no more than one inadequate response in the current MDE, as of screening
- had a 17-item Hamilton Rating Scale for Depression (HAM-D) total score ≥22 at screening.
For those patients bypassing the prospective lead-in period (historic inadequate responders), the following criteria were considered for admission to the study:
- had been treated with an adequate dose of a selective serotonin-reuptake inhibitor (SSRI), selective serotonin–norepinephrine-reuptake inhibitor (SNRI), or bupropion during the current MDE for ≥8 weeks, with the same adequate dose over the last 4 weeks that was expected to remain stable throughout the study
- had had no more than two inadequate responses to ADT (inclusive of current inadequate response) in the current MDE, as of screening; current inadequate response must have been to an SSRI, SNRI, or bupropion; prior inadequate response within the current MDE, if applicable, may have been to any commercially available ADT
- had had an inadequate response (<50% reduction in depressive symptom severity during a course of treatment ≥8 weeks’ duration with an adequate dose of an SSRI, SNRI, or bupropion assessed by the Antidepressant Treatment Response Questionnaire)
- had a HAM-D total score ≥18 at screening.
Patients were excluded from the study if they met any of the following criteria:
- had had any finding that in the view of the investigator or medical monitor would compromise the safety of the patient or affect his/her ability to adhere to the protocol visit schedule or fulfill visit requirements
- had any current primary axis I diagnosis other than MDD, where primary diagnosis was defined as the primary source of current distress and functional impairment, in the opinion of the investigator
- had any of the following psychiatric conditions as per DSM-IV-TR criteria, as assessed by the MINI: lifetime history of an axis I diagnosis of delirium, dementia, schizophrenia, other psychotic disorder (including psychotic depression), or bipolar I or II disorder; history within the past 12 months of an axis I diagnosis of eating disorder, obsessive–compulsive disorder, panic disorder, posttraumatic stress disorder, or acute stress disorder; clinically significant current axis II diagnosis of borderline, antisocial, paranoid, schizoid, schizotypal, or histrionic personality disorder; or current diagnosis/clinical evidence of any cognitive disorder at screening
- had experienced hallucinations, delusions, or any psychotic symptoms in the current MDE
- had been hospitalized with a condition related to MDD within 3 months before screening
- had initiated psychotherapy within 6 weeks of screening or had an anticipated need for initiating psychotherapy during the study; a stable course of psychotherapy initiated >6 weeks prior to screening was permitted to continue throughout the study
- had used any of: an antipsychotic (eg, aripiprazole, quetiapine, and olanzapine) at any dose or duration for any indication within 1 year of screening or within the current MDE (whichever is longer); a nonantipsychotic adjunctive treatment (eg, lithium, bupropion, and psychostimulants) for the purpose of augmenting the effects of an ADT at any dose for a duration ≥6 weeks during the current MDE; any adjunctive treatment for the purpose of augmenting the effects of an ADT at any dose or duration within 30 days of screening; bupropion for smoking cessation at any dose for a duration ≥6 weeks during the current MDE or for any duration within 30 days of screening; an anticonvulsant (eg, topiramate, gabapentin, lamotrigine, oxcarbazepine) at any dose or duration for any indication within 1 year of screening; opioid agonists (eg, codeine, oxycodone, tramadol, morphine) or opioid antagonists (eg, naloxone, naltrexone) within 14 days prior to screening, had anticipated a need for opioid use at any point during the study (eg, planned surgery), or had used an extended-release formulation of an opioid antagonist within 2 months prior to screening
- had used a course of pharmacotherapy (including prescription or over-the-counter medications) or natural supplements for insomnia if initiated within 30 days of screening; prescription, over-the-counter medications, and natural supplements were permitted to treat insomnia if they had been used stably for ≥30 days prior to screening, not more than three times per week, and were expected to be used stably at no more than three times per week throughout the study; a hypnotic agent for any psychiatric indication other than insomnia within 30 days of screening; any pharmacotherapy (including prescription or over-the-counter medications) or natural supplements for anxiety within 30 days of screening
- had initiated or had had a dose adjustment to hormone-replacement therapy (including testosterone) or oral contraceptive within 30 days of screening
- had used inducers or moderate–strong inhibitors of CYP3A4 (prescription medications, over-the-counter medications, or dietary supplements) within 30 days prior to screening
- had received electroconvulsive therapy treatment within the last 5 years or received more than one course of electroconvulsive treatment during the patient’s lifetime
- posed a current suicide risk evidenced by any of: opinion of the investigator that the patient may have been at risk for suicide; responded yes to the baseline Columbia Suicide Severity Rating Scale question 4 (active suicidal ideation with some intent to act, without specific plan) or question 5 (active suicidal ideation with specific plan and intent) if the most recent episode had occurred within the past 12 months; had attempted suicide within the past 2 years
- had a QT interval >450 ms for males and >470 ms for females assessed in a relaxed state (corrected by the Fridericia formula) observed at visit 1 or 2
- had an aspartate-aminotransferase or alanine-transaminase measurement of more than double the upper limit of normal at visit 1
- had current evidence of or history of any of compromised respiratory function (eg, chronic obstructive pulmonary disease, respiratory depression, and signs or symptoms of hypoxia at screening), thyroid pathology (unless stabilized and euthyroid for >3 months at the time of screening), seizure disorder (excluding febrile seizure), hepatitis B, hepatitis C, HIV infection, myasthenia gravis, or any contraindicated medical condition as per the approved labeling for buprenorphine (BUP)
- had current evidence of or history (in the past 12 months) of alcohol or substance abuse or dependence (excluding nicotine) as per DSM-IV-TR criteria assessed by the MINI
- had a positive breath-alcohol test at screening
- had a positive test for drugs of abuse at screening or visit 2 (exception: a positive screen for benzodiazepine may not have been exclusionary if such medication was medically indicated for insomnia)
- pregnant, planning to become pregnant, or breastfeeding during the study
- had a history of either: intolerance, allergy, or hypersensitivity to opioid antagonists (eg, BUP, oxycodone) or opioid antagonists (eg, naltrexone, naloxone); or nausea and/or vomiting when taking an opioid agonist or opioid antagonist that interfered with the ability to continue on study drug
- had significant blood loss (>500 mL) or given blood (including platelets or plasma) within 60 days of screening or between screening and randomization or had anticipated blood donation at any time during the study
- had participated in any of clinical studies of more than two distinctive investigational products with a central nervous system indication in the past 4 years, any clinical study of an investigational product given as an adjunctive treatment for MDD at any time during the current MDE, and/or any clinical study of an investigational product and/or received an investigational drug or device within 30 days of screening
- had participated in a prior clinical study of BUP/samidorphan
- was an employee of the investigator or study center or immediate family of such employees or investigator
- was an employee or immediate family of an employee (permanent, temporary contract worker, or designee responsible for the conduct of the study) of Alkermes, Inc. or INC Research.
For inclusion in the double-blind treatment phase, all patients were required at visit 2 to:
- have a site-administered HAM-D score ≥18
- be deemed eligible by independent qualification, evidenced by an independent rater-administered SAFER (stands for the interview’s attention to the criteria of state vs trait; accessibility, face validity, ecological validity, and “rule of three Ps” [pervasive, persistent, and pathological]), Antidepressant Treatment Response Questionnaire, and HAM-D total score ≥18
- have a Clinical Global Impression – severity (CGI-S) score ≥4.
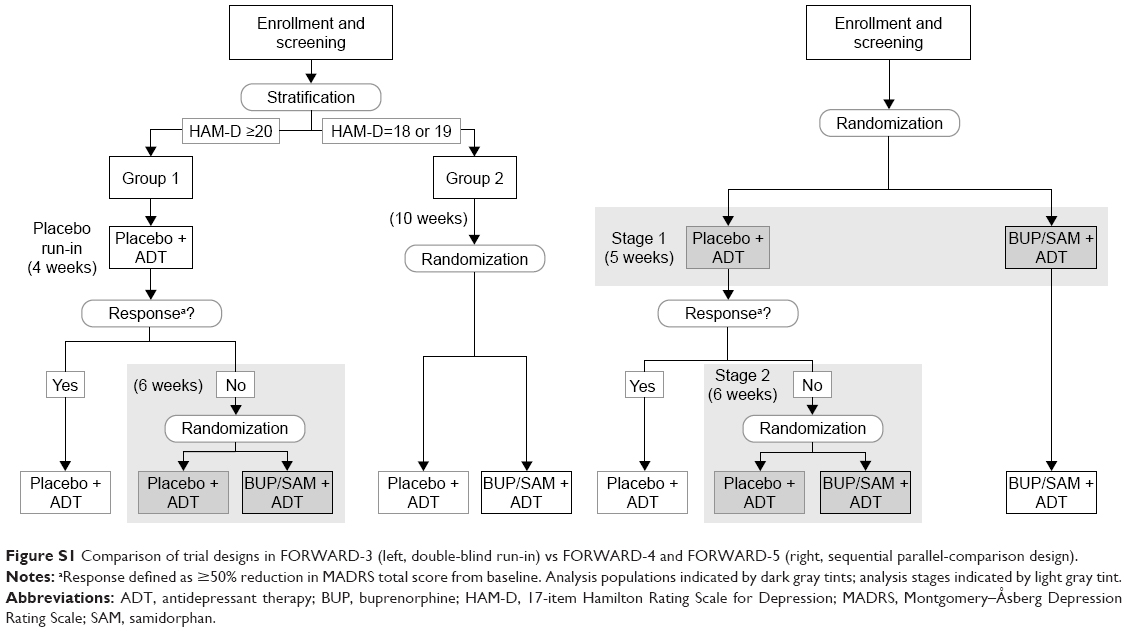 | Figure S1 Comparison of trial designs in FORWARD-3 (left, double-blind run-in) vs FORWARD-4 and FORWARD-5 (right, sequential parallel-comparison design). |
Treatment-emergent adverse events of special interest
Preferred terms for abuse potential
- euphoria-related: euphoric mood; feeling abnormal; feeling drunk; feeling of relaxation; hallucination; hallucination, auditory; hallucination, gustatory; hallucination, mixed; hallucination, olfactory; hallucination, synesthetic; hallucination, tactile; hallucination, visual; inappropriate affect; thinking abnormal.
- nonspecific: acute psychosis, aggression, cognitive disorder, confusional state, delirium, delusional disorder, depersonalization/derealization disorder, disorientation, dissociation, disturbance in attention, disturbance in social behavior, dizziness, dopamine-dysregulation syndrome, emotional disorder, flight of ideas, medication-overuse headache, mental impairment, mood altered, mood swings, narcotic bowel syndrome, paranoia, psychotic behavior, sedation, somnolence, stupor.
- abuse behavior: accidental overdose, drug abuser, drug diversion, drug level above therapeutic, drug screen, drug screen–positive, intentional overdose, intentional product misuse, intentional product-use issue, maternal use of illicit drugs, needle-track marks, neonatal complications of substance abuse, overdose, prescription drug used without a prescription, prescription form tampering, product tampering, substance abuse, substance abuser, substance use, substance-induced mood disorder, substance-induced psychotic disorder, toxicity to various agents.
Preferred terms for suicidal ideation and behavior
- completed suicide, intentional overdose, intentional self-injury, multiple-drug overdose, overdose, poisoning deliberate, self-injurious behavior, self-injurious ideation, self-mutilation, suicidal behavior, suicidal ideation, suicide attempt, suicide threat.
Preferred terms for withdrawal
- abdominal pain, agitation, anhedonia, anxiety, arthralgia, chills, depressed mood, depression, diarrhea, drug detoxification, drug rehabilitation, drug-withdrawal convulsions, drug-withdrawal headache, drug-withdrawal maintenance therapy, drug-withdrawal syndrome, dysphoria, dyssomnia, feeling of despair, headache, hyperhidrosis, insomnia, irritability, morose, mydriasis, nausea, negative thoughts, nervousness, obsessive thoughts, pain, persistent depressive disorder, piloerection, poor-quality sleep, rebound effect, reversal of opiate activity, rhinorrhea, steroid-withdrawal syndrome, syncope, tachycardia, terminal insomnia, tremor, vomiting, withdrawal arrhythmia, withdrawal syndrome, yawning.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.