Back to Journals » International Journal of Nanomedicine » Volume 13
Bufalin-loaded vitamin E succinate-grafted-chitosan oligosaccharide/RGD conjugated TPGS mixed micelles demonstrated improved antitumor activity against drug-resistant colon cancer
Authors Yuan Z, Yuan Y, Han L, Qiu Y, Huang X ![]() , Gao F
, Gao F ![]() , Fan G, Zhang Y, Tang X, He X, Xu K, Yin P
, Fan G, Zhang Y, Tang X, He X, Xu K, Yin P ![]()
Received 10 April 2018
Accepted for publication 9 August 2018
Published 15 November 2018 Volume 2018:13 Pages 7533—7548
DOI https://doi.org/10.2147/IJN.S170692
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Zeting Yuan,1,* Yuxia Yuan,1,* Lin Han,2 Yanyan Qiu,1 Xiaqin Huang,3 Feng Gao,3 Guohua Fan,1 Yixi Zhang,1 Xueyao Tang,1 Xue He,1 Ke Xu,1 Peihao Yin1,4,5
1Interventional Cancer Institute of Chinese Integrative Medicine, Putuo Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 200062, People’s Republic of China; 2Experimental Research Center, Putuo Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 200062, People’s Republic of China; 3Department of Pharmaceutics, School of Pharmacy, East China University of Science and Technology, Shanghai 200237, People’s Republic of China; 4Department of General Surgery, Putuo Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 200062, People’s Republic of China; 5Shanghai Putuo Central School of Clinical Medicine, Anhui Medical University, Anhui 230022, People’s Republic of China
*These authors contributed equally to this work
Background: Multidrug resistance (MDR) is the major reason for the failure of chemotherapy in colon cancer. Bufalin (BU) is one of the most effective antitumor active constituents in Chansu. Our previous study found that BU can effectively reverse P-glycoprotein (P-gp)-mediated MDR in colon cancer. However, the clinical application of BU is limited due to its low solubility in water and high toxicity. In the present study, a multifunctional delivery system based on vitamin-E-succinate grafted chitosan oligosaccharide (VES-CSO) and cyclic (arginine-glycine-aspartic acid peptide) (RGD)-modified d-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) was prepared by emulsion solvent evaporation method for targeted delivery of BU to improve the efficacy of drug-resistant colon cancer therapy.
Methods: The cytotoxicity of BU-loaded micelles against drug-resistant colon cancer LoVo/ADR and HCT116/LOHP cells was measured by CCK-8 assay. The cellular uptake, Rho123 accumulation, and cell apoptosis were determined by flow cytometry. The expression of apoptosis-related protein and P-gp was measured by Western blot assay. The antitumor activity of BU-loaded micelles was evaluated in LoVo/ADR-bearing nude mice.
Results: BU-loaded VES-CSO/TPGS-RGD mixed micelles (BU@VeC/T-RGD MM) were 140.3 nm in diameter with zeta potential of 8.66 mV. The BU@VeC/T-RGD MM exhibited good stability, sustained-release pattern, higher intracellular uptake, and greater cytotoxicity in LoVo/ADR cells. Furthermore, the mechanisms of the BU@VeC/T-RGD MM to overcome MDR might be due to enhanced apoptosis rate and P-gp efflux inhibition. Subsequently, in vivo studies confirmed an enhanced therapeutic efficiency and reduced side effects associated with BU@VeC/T-RGD MM compared with free BU, owing to the enhanced permeation and retention effect, improved pharmacokinetic behavior, and tumor targeting, which lead to MDR-inhibiting effect in LoVo/ADR-bearing nude mice.
Conclusion: Our results demonstrated that VeC/T-RGD MM could be developed as a potential delivery system for BU to improve its antitumor activity against drug-resistant colon cancer.
Keywords: bufalin, colon cancer, multidrug resistance, mixed micelle, tumor targeting, P-gp
Introduction
Colon cancer is a common malignant tumor, which is the third most common cause of tumor-related death, and therefore, a serious threat to human health.1 Surgery is the first choice for the treatment of colon cancer. However, 20%–30% of the patients suffer from terminal cancer and cannot be cured by surgery alone. Chemotherapy is the primary treatment for colon cancer patients who cannot be operated on. MDR is still a serious problem that restricts the clinical therapeutic efficiency of chemotherapy, thus affecting survival and prognosis.2,3 Therefore, overcoming drug resistance is critical for improving the antitumor effect and reducing the side effects of chemotherapy during treatment of colon cancer.
One of the major mechanisms conferring MDR in tumor cells is the overexpression of ABC, which results in an increase in drug efflux.4 P-gp is the most important member of ABC superfamily of transmembrane proteins, and is encoded by the MDR1 gene. P-gp, which is ATP dependent, can use the energy provided by hydrolysis of ATP to pump drugs out of the tumor cells, thus reducing the intracellular drug concentration.5–7 Modulation of cellular apoptosis pathway is another important mechanism responsible for MDR. Some anti-apoptotic pathways and anti-apoptotic signaling molecules in tumor cells are activated by chemotherapeutic drugs, allowing them to resist apoptosis and induce development of drug resistance.4,8 Therefore, inhibiting the P-gp-mediated drug efflux and/or inducing tumor cell apoptosis have recently become a major hotspot in drug resistance research.
The commonly used chemotherapeutic drugs for colon cancer, such as 5-FU, capecitabine, and oxaliplatin, are nontargeting, with low sensitivity and high side effects, and thus easily lead to MDR, which limits their wide clinical application. Developing new drugs and preparations with high antitumor activity and addressing MDR have an extremely important value for cancer therapy. BU is one of the most potent antitumor ligands extracted from Chansu. Its molecular formula is C24H34O4 with a relative molecular weight of 386 g/mol. It has strong hydrophobicity and extensive pharmacological activity. Recently studies have found that BU can produce significant antitumor activity in different cancers.9–12 In our previous study, we found that BU could inhibit P-gp-mediated MDR in colon cancer. Its mechanisms involved downregulating P-gp protein expression and competitive inhibition of the activity of P-gp ATP enzyme, thus, overcoming the P-gp-mediated drug efflux and inhibiting MDR in colon cancer.13 However, low solubility in water, toxicity, short elimination half-life, and fast oral metabolization rate limit its clinical application.
Nano-delivery systems have been shown to increase drug solubilization, stability, bioavailability, target delivery, and therapeutic efficacy, while reducing the side effects.14–18 Nanoparticulate drug delivery systems are making a remarkable contribution toward the improvement of active components of traditional Chinese medicine delivery in cancer therapy.19 It has been reported that nanocarriers could effectively load BU, prolong its retention time in vivo, improve its antitumor effect, and reduce its toxicity.20–26 However, there is no study on BU-loaded nano-delivery systems to overcome MDR. Polymer micelles are ideal carriers for both active and passive targeted drug delivery.27 VES is a water-soluble derivative of natural vitamin E, which can effectively encapsulate insoluble drugs, increase entrapment efficiency, and induce apoptosis of tumor cells, without any toxicity toward normal cells.28 TPGS is synthesized by esterification of carboxyl group of VES with PEG. TPGS-based nanocarriers show enhanced circulation behavior and help drugs target mitochondria.29,30 In addition, TPGS has been shown to be the most effective inhibitor among P-gp inhibitory surfactants.31 Moreover, it can induce cell apoptosis and develop a synergistic effect with chemotherapy drugs. RGD32 is an endogenous short peptide containing arginine, glycine, and aspartic acid (Arg–Gly–Asp). It can specifically bind to integrin receptor αvβ3, which is highly expressed on endothelial cells of vascularization, which is responsible for tumor growth. CSO33 has good biocompatibility, biodegradability, and nontoxicity, which can further increase the solubility and stability of drugs. VES-CSO and TPGS-RGD can self-assemble in water to form mixed micelle, with VES as the core and PEG as the shell. The hydrophilic targeted moiety, RGD, conjugates onto the surface of these nano-micelles.34
In this study, we explored whether VES-CSO/TPGS-RGD mixed micelles capable of delivering BU increased the antitumor therapeutic effects in drug-resistant colon cancer.
Materials and methods
Materials
BU was obtained from Chengdu Herbpurify Co., Ltd. (Sichuan, People’s Republic of China). Oxaliplatin (L-OHP) was sourced from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). DOX and C6 were purchased from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). CSO (MW 5 kDa, 90.0% deacetylation degree) was purchased from Aoxing Co. Ltd. (Zhejiang, People’s Republic of China). VES was obtained from TCI Development Co. Ltd. (Shanghai, People’s Republic of China). TPGS, c(RGDfK) was from GL Biochem Ltd. (Shanghai, People’s Republic of China). All the other solvents used were of analytical grade. RPMI 1640 and PBS were from Hyclone (Thermo Fisher Scientific, Waltham, MA, USA). Annexin V-FITC Apoptosis Detection Kit was purchased from BD Biosciences (Beijing, People’s Republic of China). CCK-8 was purchased from Dojindo (Kumamoto, Japan). FBS was obtained from Gibco BRL (Carlsbad, CA, USA). The primary antibodies for cleaved caspase-3, cleaved caspase-9, Bax, Bcl-2, and β-actin were sourced from Cell Signaling Technology (Boston, MA, USA). The secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Preparation and characterization of mixed micelles
Empty micelles and BU-loaded VES-CSO (BU@VeC) micelles, VES-CSO/TPGS mixed micelles (BU@VeC/T MM) and VES-CSO/TPGS-RGD mixed micelles (BU@VeC/T-RGD MM) were prepared by organic solvent emulsification–evaporation as described,30,31 except that BU (1 mg) was separately contained in 200 μL of chloroform. Excess unentrapped BU was removed using 0.45 μm filters.
The average particle size and zeta potential of the micelles were determined using a Malvern Zetasizer Nano ZS90 DLS (Malvern Instruments, Malvern, UK) instrument. TEM (JEM-2100; JEOL, Tokyo, Japan) was used to examine the surface morphology of micelles.
The DL capability of the micelles was determined using HPLC. BU-loaded MM solution (300 μL) was transferred into a 1.5 mL volumetric flask and diluted with methanol. After the samples were vortexed and sonicated, 20 μL of supernatant was injected into the HPLC system to analyze BU concentration. The HPLC system (Agilent 1260 Infinity, Agilent, Japan) was equipped with a PLATISIL™ ODS column (250×4.6 mm, 5 μm) with a mobile phase of acetonitrile and water (70:30), with the flow rate and column temperature set at 1.0 mL/min and 30°C, respectively. The signals were recorded by UV detector at 296 nm. A calibration line was conducted to determine the BU concentration in the range of 1–50 μg/mL. The r2-value of peak area against BU concentration was at 0.999.
The DL content and encapsulation efficiency were calculated using the following equations.
Drug release in vitro
The release profile of BU from micelles was analyzed by dialysis using a membrane with molecular weight cut off 10,000 Da. In total, 2 mL of different micelles was dialyzed against 18 mL of PBS (0.02 mol/L, pH 7.4) at 37°C under shaking at 100 rpm/min. At predetermined periods (0.5, 1, 2, 3, 4, 6, 8, 10, 12 hours), 0.5 mL aliquots were collected and replaced by 0.5 mL fresh PBS. The BU content in each samples was determined by HPLC. AR was calculated as the following formula:
|
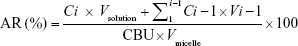
where i is from 0 to n, C is the determined concentration, V is the volume of aliquots taken out of the medium, and Vsolution is the volume of the total medium.
Cell lines and cell cytotoxicity
The HCT116 human colon cancer cells were purchased from the Cell Bank of the Chinese Academy of Sciences. The human MDR colon cancer cell lines (HCT116/L-OHP) were established by our laboratory. The HCT116/L-OHP cells were cultured in the medium with 5 μg/mL of L-OHP to maintain the resistance phenotype. LoVo/ADR cell lines were obtained from Shanghai Yan Sheng Industrial Co., Ltd. DOX (8 μg/mL) was added to the medium of LoVo/ADR cells to preserve resistance. Both cell lines were cultured in RPMI-1640 containing 10% FBS at 37°C under an atmosphere of 5% CO2 and incubated for minimum 1 week in drug-free medium before use.
The cytotoxicity of various MM against HCT116/L-OHP and LoVo/ADR cells was assessed using the CCK-8 assay. Cells were seeded into 96-well plates at a density of 104 cell/well and cultured overnight at 37°C, which was replaced by fresh media containing free BU, BU@VeC, BU@VeC/T, and BU@VeC/T-RGD MM at different concentrations for 48 hours incubation. Finally, each well was added with 10 μL of CCK-8 and further cultured for 1–4 hours. The absorbance was determined by a microplate reader (Thermo Fisher Scientific) at 450 nm.
Cellular uptake
The intracellular uptake of VeC/T and VeC/T-RGD MM was conducted by loading C6 (500 μg/mL) as a fluorescent dye instead of BU, which was examined by a CLSM (Leica, Heidelberg, Germany). LoVo/ADR cells at a density of 105 cell per well were seeded and cultured in 24-well plates for 24 hours. Then, the medium was replaced with fresh 1640 media containing free C6, C6-labeled VeC, C6-labeled VeC/T MM, and C6-labeled VeC/T-RGD MM for 0.5 or 2 hours at 37°C. In addition, in order to explore the targeting effect of RGD, cells were incubated with free c(RGDfK) peptide (0.3 μg/mL) solution and empty VeC/T-RGD MM for subsequent processing.
Cell apoptosis
The apoptotic effect of different BU-loaded micelles on HCT116/LOHP and LoVo/ADR cells was analyzed by FACScan low cytometry using the Annexin V/PI Apoptosis Detection Kit. Cells were seeded in six-well plates (1×105 cells/mL) and cultured with free BU, BU@VeC, BU@VeC/T, and BU@VeC/T-RGD MM (1×10−3 μg BU/mL) for 48 hours. Then, the attached and detached cells were collected, double-stained with 5 μL PI and 5 μL Annexin V-FITC, and were analyzed (FL-1:Ex=488 nm, Em=530 nm; FL-2:Ex=488 nm, Em=620 nm) by FACS (BD Biosciences, San Jose, CA, USA).
Rho123 accumulation assay by flow cytometry
After treating with different formulations for 48 hours, LoVo/ADR cells were cultured with Rho123 (1 μg/mL) for 90 minutes, respectively. Finally, cells were collected and washed three times with PBS, and determined by flow cytometry as described.35
Western blot analysis
LoVo/ADR cells were treated by BU-loaded micelles as described above, and cells were lyzed in radio immunoprecipitation assay buffer on the ice. Equivalent amounts of proteins (40 μg/lane) were separated on 8%–15% SDS-PAGE and transferred to poly(vinylidene fluoride) membranes. The membranes were blocked with 5% albumin from bovine serum, for 2 h, and treated with the primary antibodies overnight and then incubated with corresponding horseradish peroxidase-labeled secondary antibodies. Finally, proteins were visualized by chemiluminescence.
In vivo imaging
To study the tumor-targeting capability of VeC/T-RGD MM in vivo, IR775 (62.5 μg/mL) instead of BU was encapsulated as a fluorescence probe in VeC/T and VeC/T-RGD MM. Free IR775, IR775+VeC/T-RGD MM, IR775@VeC/T MM, and IR775@VeC/T-RGD MM (200 μL) were injected through the vena caudalis (tail vein) into the tumor-bearing LoVo/ADR xenografted mice. Fluorescence imaging of the mice was performed at 2, 12, 24, and 48 hours after injection by using LIVIS Lumina series III (PerkinElmer, Waltham, MA, USA).
In vivo antitumor therapy
A total of 6–8 weeks athymic nude mice (BALB/c-nu/nu) were used to establish LoVo/ADR xenograft model, and all procedures were performed under the approval of the Administrative Panel on Laboratory Animal Care of the Putuo District Center Hospital. All animals received care following the guidelines outlined in the “Laboratory Animal-Guideline for ethical review for animal welfare.” LoVo/ADR cells at a density of 1×107 cells/mouse were subcutaneously injected into the shoulder in the nude mice. When the tumor volumes reached 150 mm3, the mice were randomized into six groups (six/group) and intravenously administered by different BU-loaded micelles, including BU@VeC, BU@VeC/T, and BU@VeC/T-RGD MM at BU dose of 2.0 mg/kg, and saline as a negative control every 2 day for 3 weeks (seven-time injection). The body weight and tumor sizes were measured at regular intervals. At the end of the treatment, the mice were euthanized, and organs (heart, liver, lung, spleen, and kidney) were harvested for histological analysis using the hematoxylin and eosin (H&E) staining.
Endocytic pathway of VeC/T-RGD MM
LoVo/ADR cells were preincubated in serum-free RPMI 1640 medium containing 100 mg/mL amiloride, 10 μg/mL chlorpromazine, and 15 mg/mL nystatin for 30 minutes, respectively. The media were then changed to fresh serum-free medium containing the inhibitors and C6 @VeC/T-RGD MM at 3 μg/mL C6 concentration and incubated for another 2 hours. The cells were washed with PBS, harvested, and then intracellular fluorescence was determined by flow cytometry.
Pharmacokinetic studies
Two groups (n=3) of male Sprague Dawley rats (250±10 g) purchased from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, People’s Republic of China) were intraperitoneal injected with BU or BU@VeC/T-RGD MM (1 mg/kg), and blood samples were collected from postorbital venous plexus veins at selected time points (5, 15, 30, 45, 60, 90, 120, 240, and 360 minutes). All blood samples were centrifuged (3,500 rpm/min) and the supernatant was stored at -80°C until analysis. In total, 300 μL acetonitrile was added to 100 μL of supernatant sample. The mixture was vortexed for 30 seconds and 1.0 mL ethylene acetate was added and centrifuged at 3,500 rpm/min for 10 minutes. Then, 2 mL of the supernatant was collected and evaporated to dryness and the residue was then redissolved in mobile phase (100 μL). Then, a 10 μL aliquot of supernatant was injected into the HPLC system.
Statistics
Statistical analysis was performed using GraphPad Prism 5.0 software. Differences between groups were analyzed by two-tailed Student’s t-test. P-values below 0.05 were considered significant.
Results
Preparation and characterization of mixed micelles
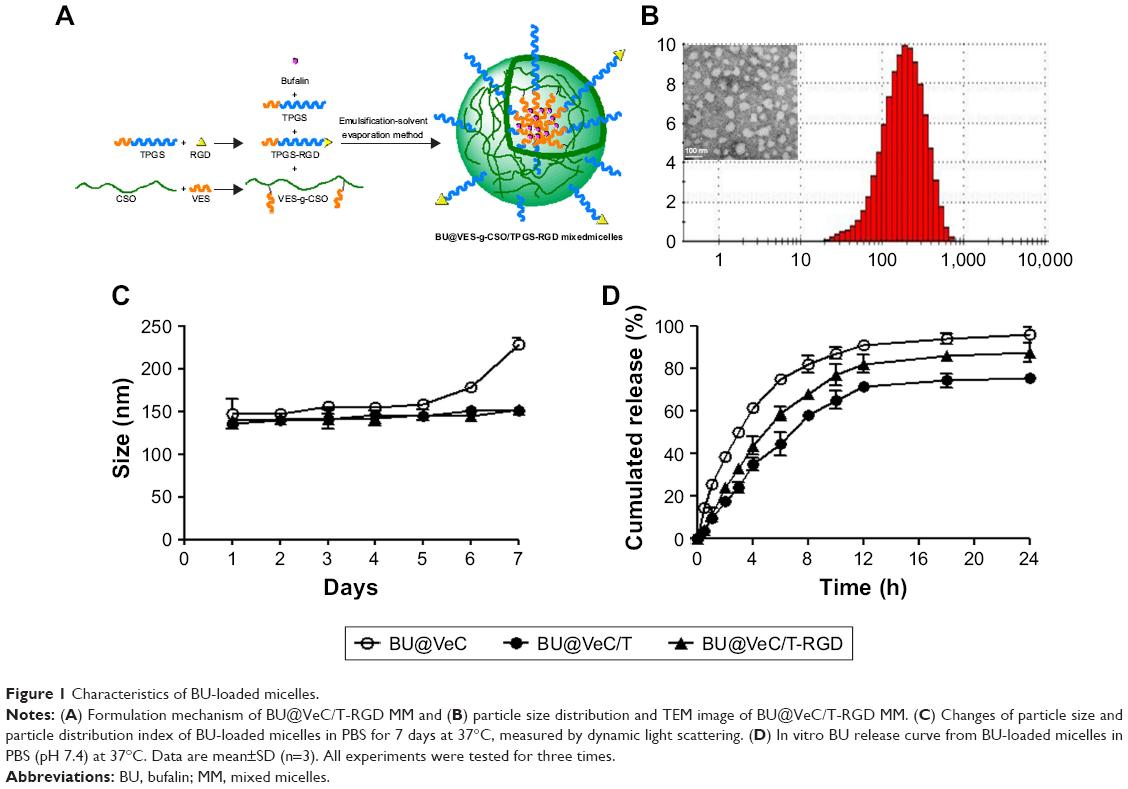
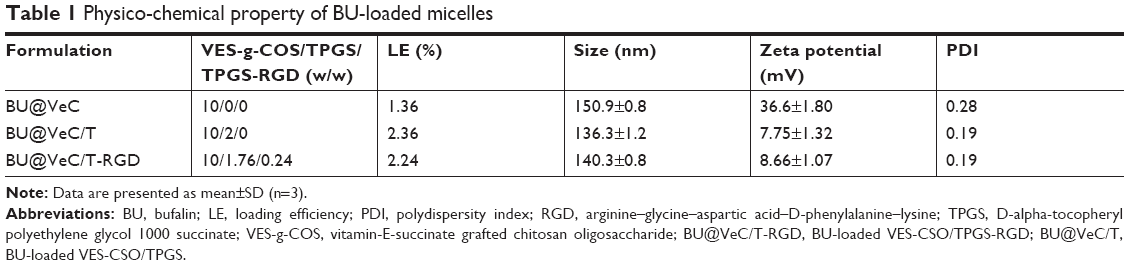
The BU-loaded mixed micelles were prepared by emulsion solvent evaporation method (Figure 1A). The particle size and zeta potential were measured by DLS, and the data are summarized in Table 1. The mean particle size of BU-loaded VES-CSO (BU@VeC) micelles was 150.9 nm with ξ potential of 36.6 mV, while the size and ξ potential of BU-loaded VES-CSO/TPGS mixed micelles (BU@VeC/T MM) decreased to 136.3 nm and 7.75 mV, which may be attributed to the insertion of TPGS (DS% of VES=3.64%). The particle size of the mixed micelles was smaller than mono-micelles. This was because TPGS and VES had the same hydrophobic fragments, so they could be embedded in the core of the spherical structure of the micelles and tightly encapsulate drugs, thus making the mixed micelles more stable. In addition, TPGS has a negative charge, which might reduce the zeta potential of the micelles. The LE of BU@VeC/T MM was 2.36%, which was higher than that of BU@VeC micelles, due to the solubility enhancement of TPGS. In contrast, after conjugation of RGD on the surface of the particles, the size of BU-loaded VES-CSO/TPGS-RGD mixed micelles (BU@VeC/T-RGD MM) showed a slight increase to 140.3 nm, while the ξ potential increased to 8.66 mV and the LE% had a slight decrease of 2.24%. The reason might be that the total content of TPGS was slightly lower, because a part of TPGS was in conjunction with RGD as the target head, resulting in slightly larger particle size and potential. As shown in Table S1, the particle size of the BU-loaded micelle was smaller than the blank micelle, because the drug molecules had intermolecular forces such as van der Waals force, hydrophobic bond, and hydrogen bond with the VES fragment of the hydrophobic core of the micelle. The combined effect of these forces could make the micelle core shrink and make the micelle contract more tightly, which led to BU being tightly wrapped in the hydrophobic core and a reduction in the particle size. The TEM image showed that BU-loaded MM were spherical with a uniform dispersion (Figure 1B). All the micelles showed a relatively narrow PDI (<0.3). These results were in accordance with our previous studies.
 | Figure 1 Characteristics of BU-loaded micelles. |
 | Table 1 Physico-chemical property of BU-loaded micelles |
During the stability experiment, micelles were stored at 4°C for 1 week. The size of BU@VeC micelles showed a remarkable change over 5 days. Comparatively, BU@VeC/T and BU@VeC/T-RGD MM maintained their original sizes, suggesting that the mixed micelles were of satisfactory stability due to the compacted inner core, composed of VES-g-CSO and TPGS (Figure 1C).
As shown in Figure 1D, in vitro drug release from different BU-loaded MM was evaluated in pH 7.4 PBS at 37°C. During the initial 1 hour, BU was rapidly released from BU@VeC micelles, BU@VeC/T MM, and BU@VeC/T-RGD MM at 25.2%, 17.6%, and 11.1%, respectively. The release rate of the three micelles gradually slowed down after 2 hours, which was followed by steady release patterns. Accumulative BU release over 24 hours followed a descending order of VeC micelles > VeC/T-RGD MM > VeC/T MM. In addition, the release kinetic behavior of BU from these formulations was also studied under acidic conditions (pH 5.0) for 24 hours. The trends were in agreement with the results in pH 7.4 (Figure S1). It is speculated that BU might be partially adsorbed on the surface of the micelle, and it might get quickly desorbed and released from the surface of the carrier at the beginning of the release experiment. As the hydrophobic core constructed by VES was not close enough, the structure of BU@VeC micelles was relatively loose. In contrast, due to the role of TPGS in BU@VeC/T-RGD and BU@VeC/T MM, the hydrophobic core was close, and the drug was relatively stable and not easy to release, resulting in relative sustained release. The diffusion manner was correlated with the characteristics of the micelles.
Cellular uptake study
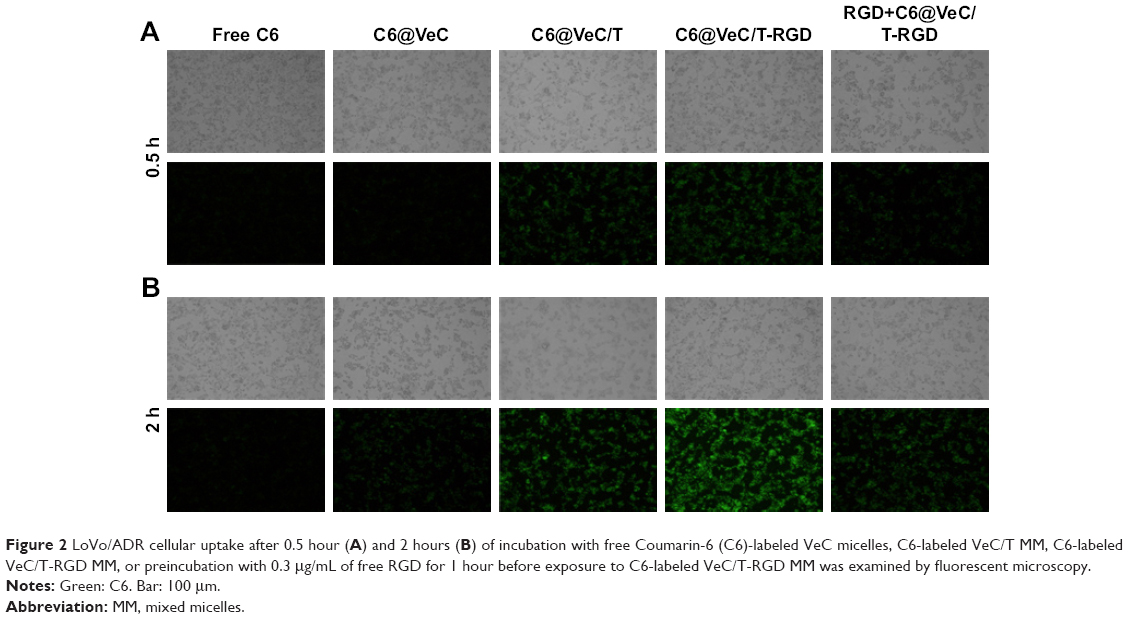
Cellular uptake of micelles was observed by replacing BU with C6, which was evaluated in LoVo/ADR cells by CLSM (Figure 2). The results showed that after 2 hours of incubation, the uptake of C6@VeC/T-RGD MM was significantly higher compared with the other formulations. This could have resulted from the addition of TPGS and because of RGD-mediated active targeting, which increased the cellar uptake of micelles. In addition, TPGS overcame MDR by suppressing P-gp, which may further enhance the cellular uptake of C6. After preincubation with free RGD peptide, the cellular uptake of C6-labeled VeC/T-RGD MM was reduced, which might be due to the competitive binding of integrin to free ligands. Furthermore, the results of uptake mechanism of VeC/T-RGD MM on LoVo/ADR cells demonstrated that the uptake of VeC/T-RGD MM was dependent on clathrin- and caveolin-mediated endocytosis process (Figure S2). These results indicated that RGD-targeted VeC/T MM could improve cell uptake ability and enhance the cancer cell targeting of cargos, most likely due to the high expression of integrin αvβ3 in LoVo/ADR cells.
 | Figure 2 LoVo/ADR cellular uptake after 0.5 hour (A) and 2 hours (B) of incubation with free Coumarin-6 (C6)-labeled VeC micelles, C6-labeled VeC/T MM, C6-labeled VeC/T-RGD MM, or preincubation with 0.3 μg/mL of free RGD for 1 hour before exposure to C6-labeled VeC/T-RGD MM was examined by fluorescent microscopy. |
In vitro cell cytotoxicity
Cytotoxicity of BU, BU@VeC micelles, BU@VeC/T, and BU@VeC/T-RGD MM against drug-resistant colon cancer HCT116/LOHP and LoVo/ADR cells at various concentrations was evaluated by CCK-8 assay. As shown in Figure 3, the cytotoxicity in drug-resistant cells was in the following order: BU<BU@VeC micelles<BU@VeC/T MM<BU@VeC/T-RGD MM. When BU was loaded into micelles, its solubility was increased, and it was more easily taken up by cells in the form of micelles. Thus, more BU entered the cells and greater toxicity was achieved. The increased cytotoxicity against LoVo/ADR cells observed upon the addition of TPGS might be attributed to the potentiated cellular uptake of TPGS-inserted mixed micelles. BU@VeC/T-RGD MM could further enhance cytotoxicity by promoting intracellular drug concentration through integrin-mediated endocytosis. Accordingly, it was observed that the combination of increased solubility, enhanced cellular uptake by efficient intracellular delivery, active targeting effect of RGD, and P-gp inhibition by TPGS led to significant enhancement of BU cytotoxicity by BU@VeC/T-RGD MM.
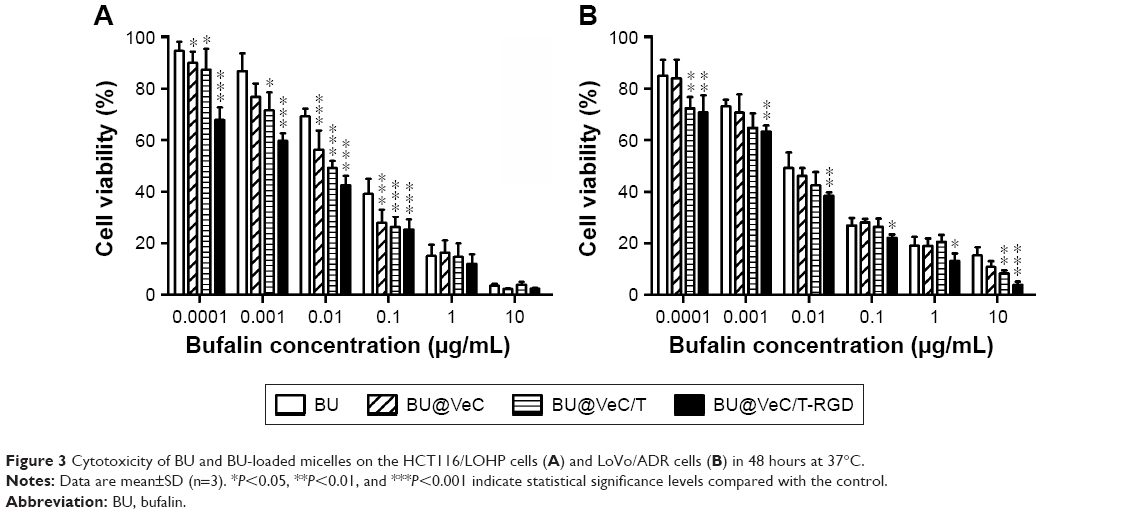 | Figure 3 Cytotoxicity of BU and BU-loaded micelles on the HCT116/LOHP cells (A) and LoVo/ADR cells (B) in 48 hours at 37°C. |
These results further agreed with the cellular uptake test, which confirmed that the enhanced inhibitory effect might be due to the higher intracellular uptake and overcoming of MDR by inhibition of P-gp.
Apoptosis assay and expression of apoptosis-related proteins
First, the effect of apoptosis induction in LoVo/ADR cells by different formulations was quantified through counting of apoptotic cells in early and late stages using flow cytometry analysis. As shown in Figure 4A and B, the percentages of apoptotic cells for BU, BU@VeC micelles, BU@VeC/T, and BU@VeC/T-RGD MM were 7.5%, 10.5%, 14.8%, 18.4%, and 20.5%, respectively. Due to the increased solubility and cellular uptake of BU upon encapsulation in micelles, the apoptotic ratio of BU-loaded MM was higher than free BU. Compared with mono-micelles, the mixed micelles could increase the apoptotic ratio, which may be attributed to the insertion of TPGS. RGD peptide could target tumor cells through receptor-mediated endocytosis, thus increasing the uptake of drugs as well as their intracellular concentration, resulting in an increased apoptotic ratio.
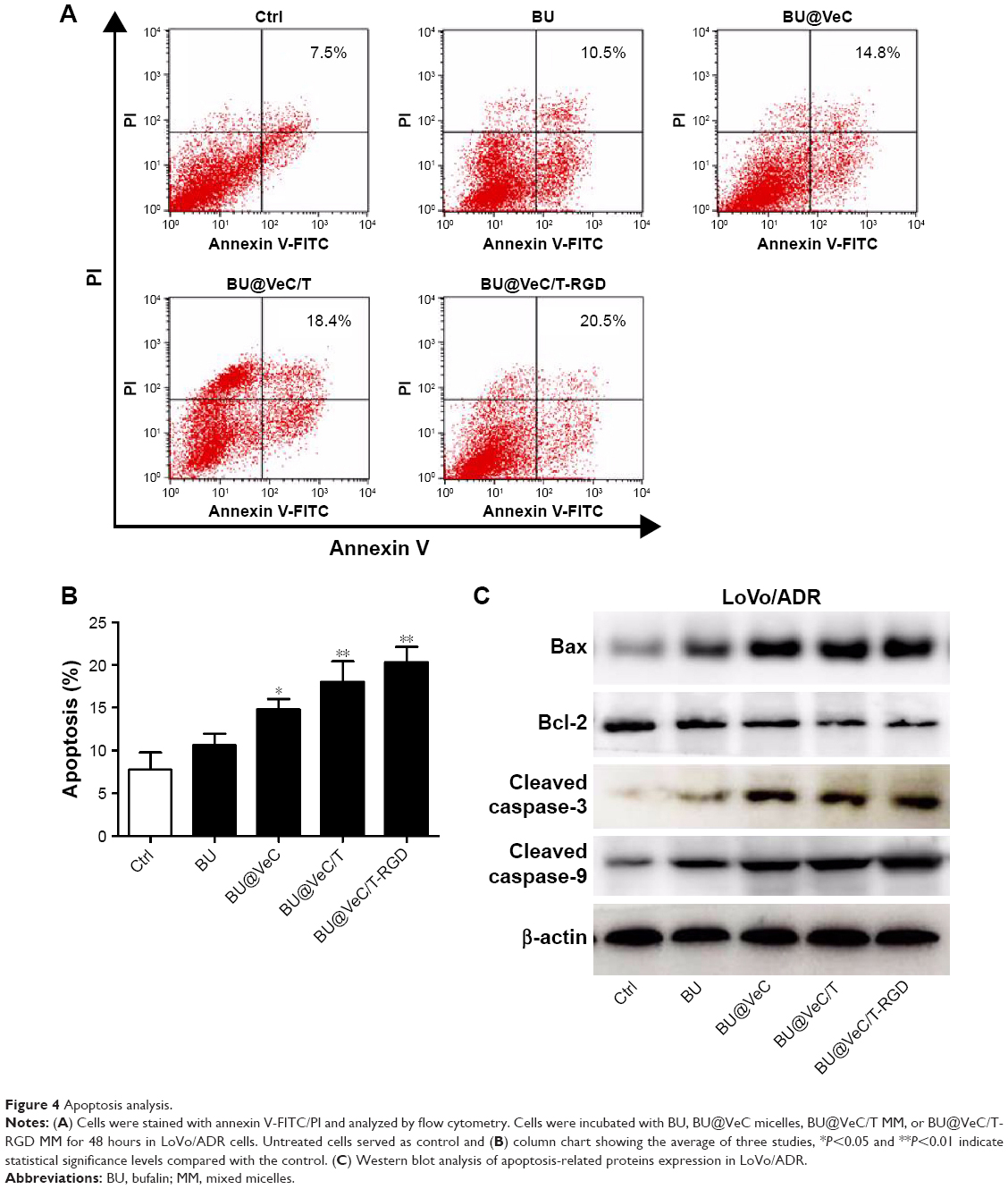 | Figure 4 Apoptosis analysis. |
To explore the molecular mechanism of apoptosis induction, the expression of apoptosis-related proteins (Bcl-2, Bax, and cleaved caspase-3, -9) was examined using Western blot analysis. We found that, compared with free BU, BU-loaded micelles further enhanced the levels of pro-apoptotic molecules Bax and cleaved caspase-3, -9, and decreased the expressions of anti-apoptotic molecules Bcl-2 in LoVo/ADR cells (Figures 4C and S3). Our results supported the previously reported antitumor mechanisms of BU, which was involved in induction of apoptosis by the mitochondrial pathway.36–40 The action of regulatory proteins in BU-loaded micelles was more significant than free BU.
Together, these results demonstrated that mixed micelles increase the pro-apoptotic effect of BU.
Intracellular accumulation of Rho123 and protein expression of P-gp
The intracellular accumulation of Rho123, a P-gp substrate, by different formulations was observed using flow cytometry in LoVo/ADR cells. As shown in Figure 5A, compared with free Rho123, strong MFI was observed after exposure to a mixture of Rho123 and TPGS, which confirmed TPGS as a P-gp inhibitor for reversal of MDR. Furthermore, we encapsulated Rho123 into different micelles to acquire Rho123@VeC micelles, Rho123@VeC/T, and Rho123@VeC/T-RGD MM, and examined their cellular uptake of Rho123. As shown in Figure 5B, compared with free Rho123, Rho123@VeC micelles group did not show any reinforcement of the MFI. However, both Rho123@VeC/T and Rho123@VeC/T-RGD MM groups showed a great capacity for promoting the intracellular uptake of Rho123. These results confirmed the P-gp-inhibiting effect of TPGS-incorporated mixed micelles.
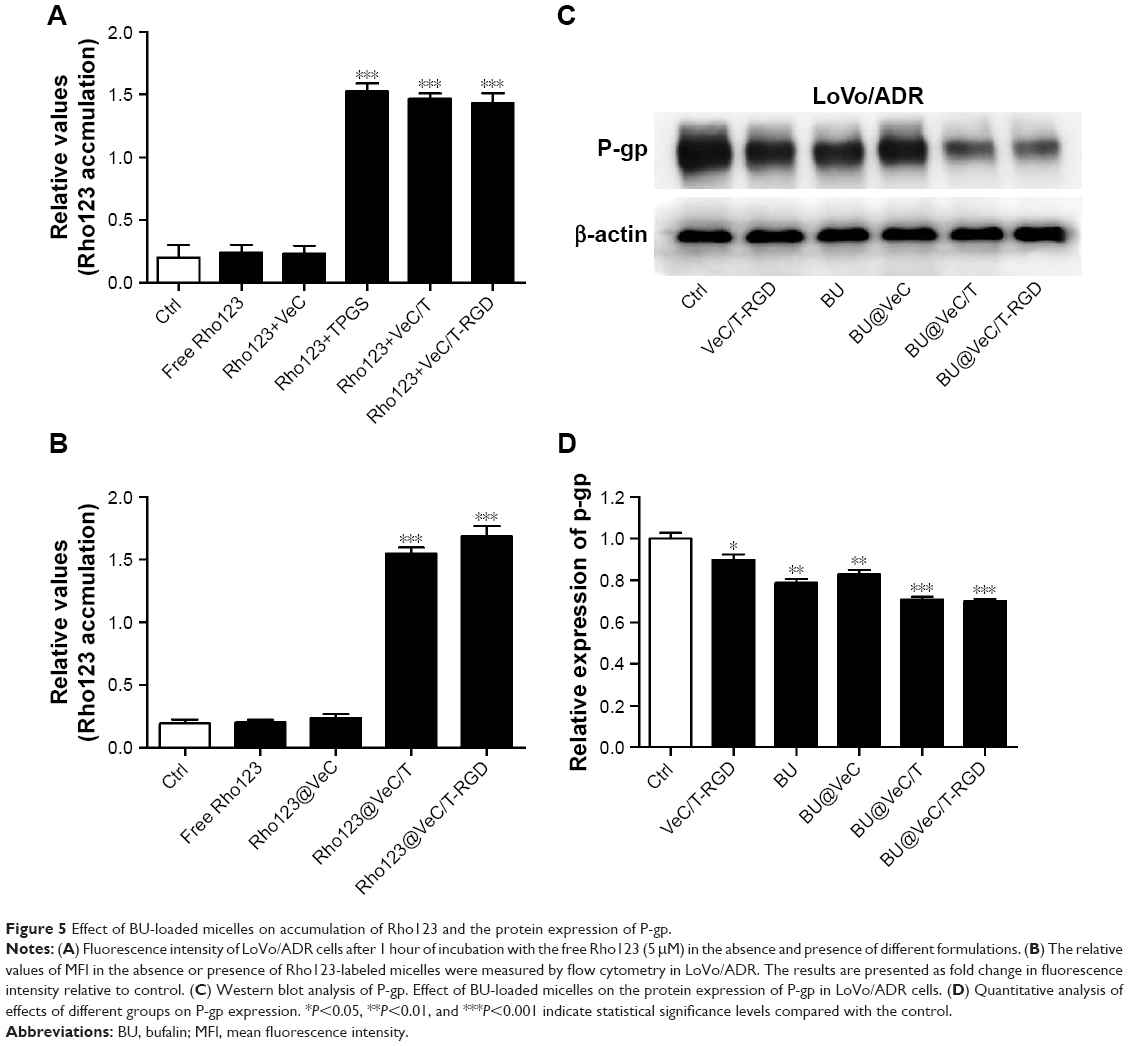 | Figure 5 Effect of BU-loaded micelles on accumulation of Rho123 and the protein expression of P-gp. |
To further investigate the effect of BU-loaded micelles on reversing MDR, we determined the effect of different formulations on P-gp protein in LoVo/ADR cells using Western blot assay. As shown in Figure 5C and D, treatment with empty VeC/T-RGD, free BU, BU@VeC micelles, BU@VeC/T, and BU@VeC/T-RGD MM reduced the P-gp expression by 10%, 21%, 18%, 29%, and 30%, respectively. Consistent with above results for Rho123, empty mixed micelles could inhibit P-gp, likely due to the role of TPGS. In addition, our group had confirmed that free BU could effectively inhibit P-gp expression. Thus, BU@VeC/T-RGD MM combined the double-inhibiting effect of BU and mixed micelles, which led to a significant reduction in P-gp expression.
Therefore, these results indicate that not only BU-loaded VeC/T-RGD MM but also empty VeC/T-RGD MM could serve as nano-inhibitors to decrease the expression of P-gp in resistant colon cancer cells.
Our previous studies showed that BU can effectively inhibit P-gp-mediated MDR in colon cancer. The mechanism involves downregulation of P-gp expression and inhibition of P-gp ATP enzyme activity. However, because of its low water solubility and high toxicity, its application was limited to combination with chemotherapeutic drugs at low dose. In this paper, the development of nano-micelles addressed the above shortcomings of BU, which widens the scope for its clinical application in MDR cancer therapy.
In vivo imaging
The whole-body fluorescence images of mice are shown in Figure 6. At 12 hours post-injection, strongest fluorescence signal was observed, especially in the liver. The fluorescence intensity at 24 hours was slightly higher than that at 48 hours. With time, the fluorescence signal showed a sharp decline due to clearance in the groups treated with free IR775, nontargeted VeC/T MM, and a mixture of VeC/T-RGD MM plus free IR775. Comparatively, VeC/T-RGD MM displayed a strong fluorescence in the tumor as early as 12 hours post-injection, which also showed a longer residence time and was still retained in the tumor tissue even at 48 hours. Our results indicated that the tumor targeting and accumulation of VeC/T-RGD MM can be attributed to a combination of the EPR effect by PEGylation and the RGD peptide, which confers the ability of active targeting to micelles.
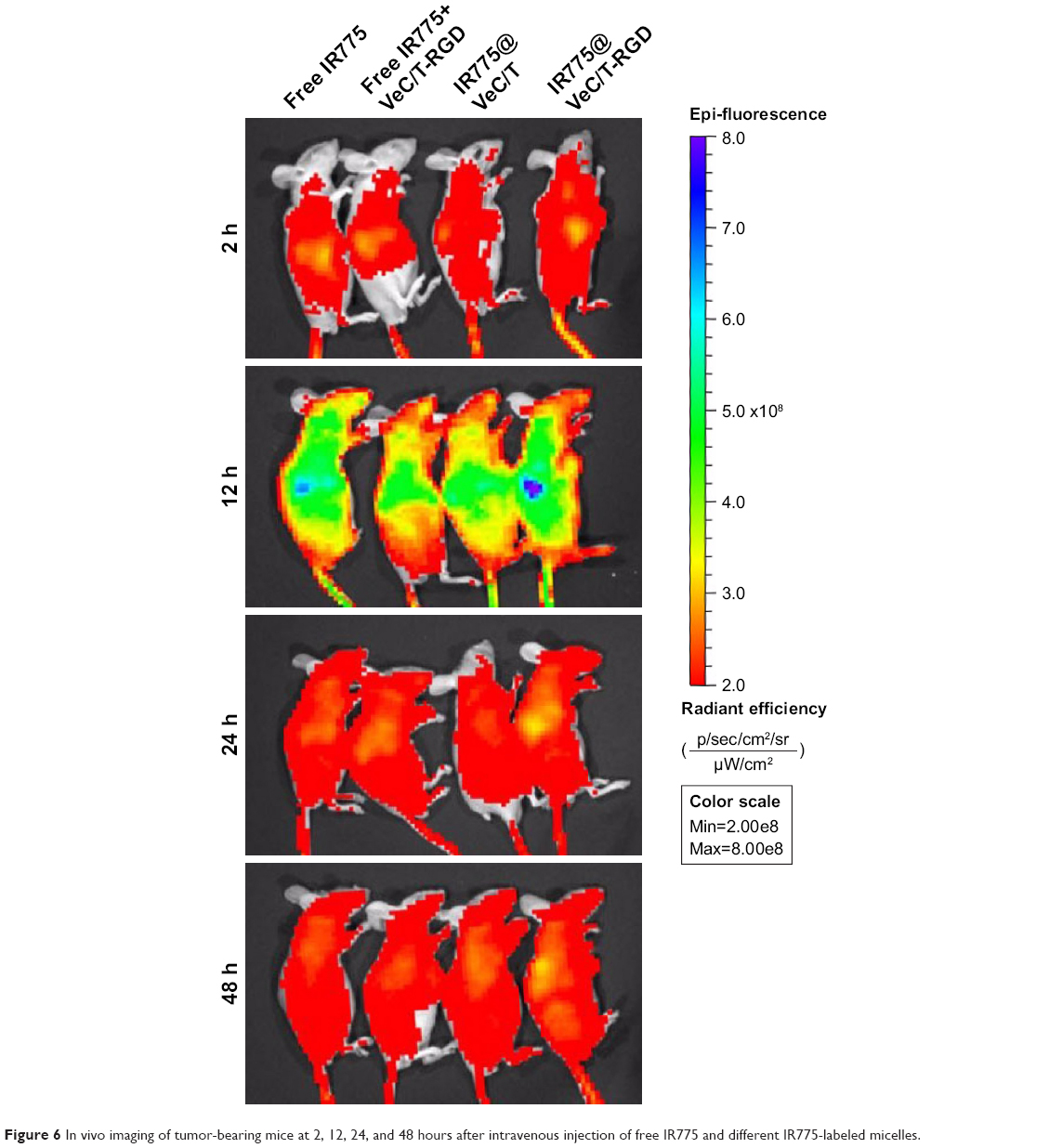 | Figure 6 In vivo imaging of tumor-bearing mice at 2, 12, 24, and 48 hours after intravenous injection of free IR775 and different IR775-labeled micelles. |
In vivo antitumor effect
The in vivo antitumor efficacy of BU-loaded micelles against drug-resistant tumor growth was evaluated in LoVo/ADR tumor-bearing nude mice. Compared with BU alone, all the BU-loaded micelles presented a greater efficacy of tumor growth suppression. Specifically, BU@VeC/T-RGD MM inhibited the tumor growth by 65%, which was significantly higher than the 22% inhibition by free BU (Figure 7A and B). This was mainly associated with the improved pharmacokinetic behavior (Figure S4; Table S2) and tumor-targeting capability of the mixed micelles functionalized by RGD and TPGS. After treatment, the weight of four groups (empty VeC/T-RGD MM, BU@VeC micelles, BU@VeC/T MM, and BU@VeC/T-RGD MM) and saline group showed no remarkable change, whereas free-BU group reduced the mice weight by 15%, likely due to BU toxicity (Figure 7C).
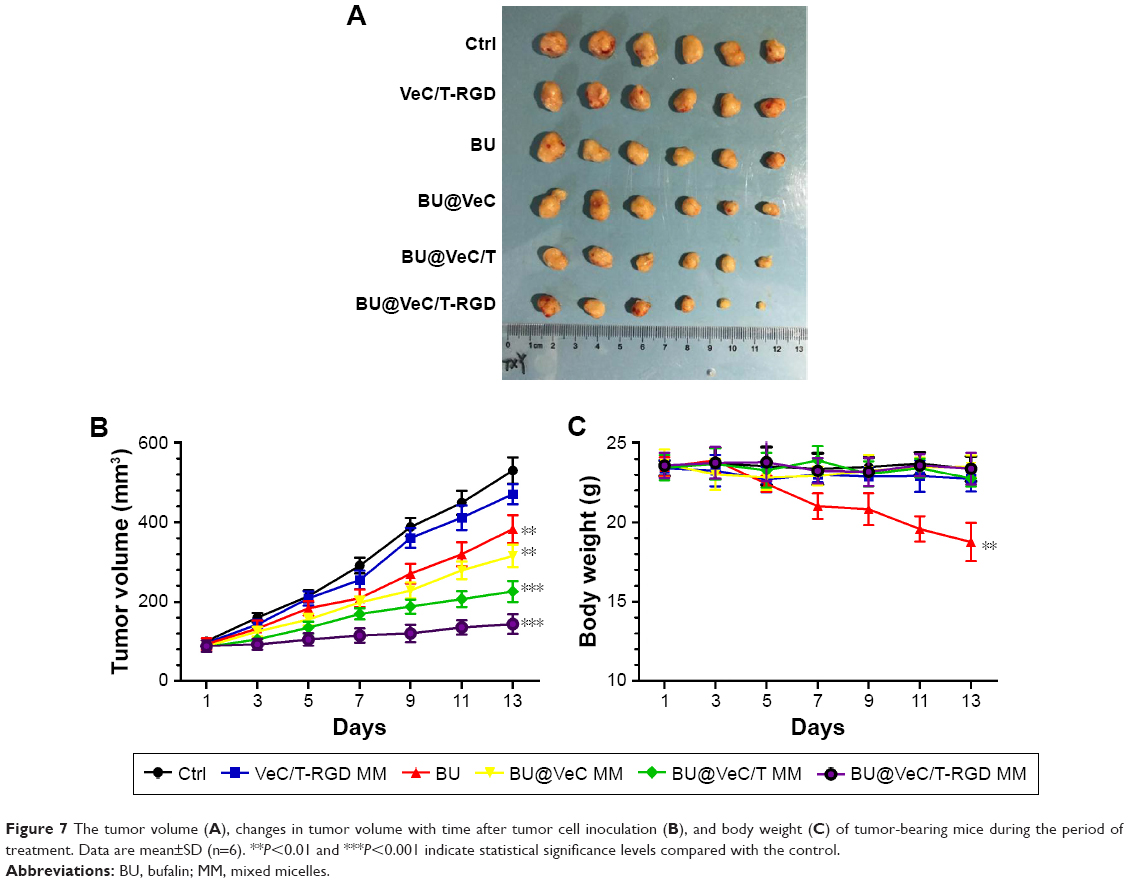 | Figure 7 The tumor volume (A), changes in tumor volume with time after tumor cell inoculation (B), and body weight (C) of tumor-bearing mice during the period of treatment. Data are mean±SD (n=6). **P<0.01 and ***P<0.001 indicate statistical significance levels compared with the control. |
In addition, we analyzed histopathological changes in the heart, liver, spleen, lung, and kidney. The major organs had different degrees of pathological damages in free-BU-treated mice group, including cardiomyocyte necrosis, connective tissue proliferation, neutrophil infiltration, liver cell necrosis, renal tubular necrosis, and apoptosis, whereas no obvious damages in major organs were detected in the micelles group (Figure 8).
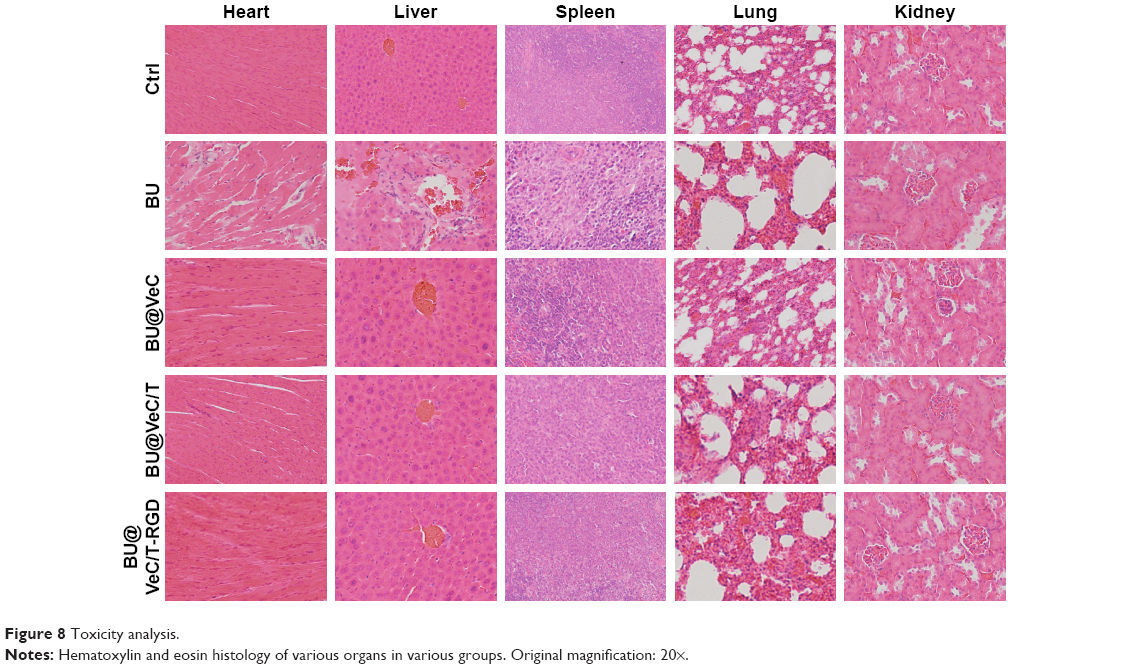 | Figure 8 Toxicity analysis. |
These data demonstrated that VeC/T-RGD MM not only enhanced the antitumor effects of BU but also reduced its toxicity in vivo, suggesting the potential value of VeC/T-RGD MM for clinical application.
Taken together, the present in vivo results clearly demonstrate the potential of BU@VeC/T-RGD MM as a novel antitumor therapy. The advantages of BU@VeC/T-RGD MM compared with native BU, including higher solubility, EPR effect, intracellular uptake, improved pharmacokinetic behavior, tumor targeting, and P-gp inhibiting, strongly strengthen its anticancer effect against drug-resistant colon cancer. Further investigations should focus on its mechanisms of in vivo antitumor actions, and its reversing effect of MDR should be explored in different tumor models.
Conclusion
In this study, we prepared a multifunctional mixed micelles composed of VES-CSO and TPGS-RGD for tumor-targeted delivery of BU by emulsion solvent evaporation method. In addition to improved water solubility and stability, VeC/T-RGD MM exhibited sustained-release manner of BU, higher intracellular uptake, and enhanced cytotoxicity in LoVo/ADR cells. Moreover, VeC/T-RGD MM were shown to increase apoptosis rate and inhibit P-gp expression than free BU in LoVo/ADR cells. In LoVo/ADR drug-resistant tumor-bearing nude mice model, VeC/T-RGD MM demonstrated significantly improved antitumor activity, with less toxicity compared with BU alone, owing to the ERP effect and RGD-mediated tumor targeting, apoptosis inducing, and P-gp inhibition, leading to MDR-inhibiting effect in vivo. In consequence, BU-loaded VeC/T-RGD MM is a promising multifunctional drug delivery system with stronger antitumor effect against drug-resistant colon cancer.
Abbreviations
ABC, ATP-binding cassette; AR, accumulative release rate; BU, bufalin; CLSM, confocal laser scanning microscopy; C6, Coumarin-6; CSO, chitosan oligosaccharide; DOX, Doxorubicin; DL, drug-loading; DLS, dynamic light scattering; EPR, enhanced permeation and retention; FBS, fetal bovine serum; HPLC, high-performance liquid chromatography; LE, loading efficiency; MDR, multidrug resistance; MFI, mean fluorescence intensity; MW, molecular weight; PEG, polyethylene glycol; P-gp, P-glycoprotein; PDI, polydispersity index; RGD, arginine-glycine-aspartic acid peptide; TEM, transmission electron microscopy; TPGS, D-alpha-tocopheryl polyethylene glycol 1000 succinate; TPGS-RGD, RGD-conjugated D-alpha-tocopheryl polyethylene glycol 1000 succinate; VES, vitamin E succinate; VES-g-CSO, vitamin E succinate-grafted chitosan oligosaccharide; VeC, VES-grafted-CSO micelles; VeC/T MM, VES-grafted-CSO/TPGS mixed micelles; VeC/T-RGD MM, VES-grafted-CSO/TPGS-RGD mixed micelles; BU@VeC, BU-loaded VES-grafted-CSO micelles; BU@VeC/T MM, BU-loaded VES-grafted-CSO/TPGS mixed micelles; BU@VeC/T-RGD MM, BU-loaded VES-grafted CSO/TPGS-RGD mixed micelles.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant nos. 81473482 and 81603502).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Lage H. An overview of cancer multidrug resistance: a still unsolved problem. Cell Mol Life Sci. 2008;65(20):3145–3167. | ||
Li W, Zhang H, Assaraf YG, et al. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat. 2016;27:14–29. | ||
Mohammad IS, He W, Yin L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed Pharmacother. 2018;100:335–348. | ||
Breier A, Gibalova L, Seres M, Barancik M, Sulova Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med Chem. 2013;13(1):159–170. | ||
Amin ML. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights. 2013;7:DTI.S12519–DTI.S12534. | ||
Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets. 2013;13(3):11–346. | ||
Wang J, Seebacher N, Shi H, Kan Q, Duan Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget. 2017;8(48):84559–84571. | ||
Sun J, Xu K, Qiu Y, et al. Bufalin reverses acquired drug resistance by inhibiting stemness in colorectal cancer cells. Oncol Rep. 2017;38(3):1420–1430. | ||
Tsai SC, Yang JS, Peng SF, et al. Bufalin increases sensitivity to AKT/mTOR-induced autophagic cell death in SK-HEP-1 human hepatocellular carcinoma cells. Int J Oncol. 2012;41(4):1431–1442. | ||
Yin PH, Liu X, Qiu YY, et al. Anti-tumor activity and apoptosis-regulation mechanisms of bufalin in various cancers: new hope for cancer patients. Asian Pac J Cancer Prev. 2012;13(11):5339–5343. | ||
Zhang ZJ, Yang YK, Wu WZ. Bufalin attenuates the stage and metastatic potential of hepatocellular carcinoma in nude mice. J Transl Med. 2014;12:57. | ||
Yuan ZT, Shi XJ, Yuan YX, et al. Bufalin reverses ABCB1-mediated drug resistance in colorectal cancer. Oncotarget. 2017;8(29):48012–48026. | ||
Yuan Z, Ye Y, Gao F, et al. Chitosan-graft-β-cyclodextrin nanoparticles as a carrier for controlled drug release. Int J Pharm. 2013;446(1–2):191–198. | ||
Xue X, Liang XJ. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin J Cancer. 2012;31(2):100–109. | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. | ||
Ramasamy T, Ruttala HB, Gupta B, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: A comprehensive review. J Control Release. 2017;258(258):226–253. | ||
Ramasamy T, Ruttala HB, Chitrapriya N, et al. Engineering of cell microenvironment-responsive polypeptide nanovehicle co-encapsulating a synergistic combination of small molecules for effective chemotherapy in solid tumors. Acta Biomater. 2017;48(48):131–143. | ||
Huang Y, Zhao Y, Liu F, Liu S. Nano Traditional Chinese Medicine: Current Progresses and Future Challenges. Curr Drug Targets. 2015;16(13):1548–1562. | ||
Hu Q, Liang B, Sun Y, et al. Preparation of bufalin-loaded pluronic polyetherimide nanoparticles, cellular uptake, distribution, and effect on colorectal cancer. Int J Nanomedicine. 2014;9:4035–4041. | ||
Liu T, Huang Q. Biodegradable brush-type copolymer modified with targeting peptide as a nanoscopic platform for targeting drug delivery to treat castration-resistant prostate cancer. Int J Pharm. 2016;511(2):1002–1011. | ||
Liu Y, Wang P, Sun C, et al. Bioadhesion and enhanced bioavailability by wheat germ agglutinin-grafted lipid nanoparticles for oral delivery of poorly water-soluble drug bufalin. Int J Pharm. 2011;419(1–2):260–265. | ||
Shi XJ, Qiu YY, Yu H, et al. Increasing the anticancer performance of bufalin (BUF) by introducing an endosome-escaping polymer and tumor-targeting peptide in the design of a polymeric prodrug. Colloids Surf B Biointerfaces. 2018;166:224–234. | ||
Tian X, Yin H, Zhang S, et al. Bufalin loaded biotinylated chitosan nanoparticles: an efficient drug delivery system for targeted chemotherapy against breast carcinoma. Eur J Pharm Biopharm. 2014;87(3):445–453. | ||
Yin P, Wang Y, Qiu Y, et al. Bufalin-loaded mPEG-PLGA-PLL-cRGD nanoparticles: preparation, cellular uptake, tissue distribution, and anticancer activity. Int J Nanomedicine. 2012;7:3961–3969. | ||
Zhang H, Huang N, Yang G, Lin Q, Su Y. Bufalin-loaded bovine serum albumin nanoparticles demonstrated improved anti-tumor activity against hepatocellular carcinoma: preparation, characterization, pharmacokinetics and tissue distribution. Oncotarget. 2017;8(38):13. | ||
Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res. 2010;27(12):2569–2589. | ||
Song Y, Cai H, Yin T, et al. Paclitaxel-loaded redox-sensitive nanoparticles based on hyaluronic acid-vitamin E succinate conjugates for improved lung cancer treatment. Int J Nanomed. 2018;13:1585–1600. | ||
Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49(2):175–186. | ||
Song J, Huang H, Xia Z, et al. TPGS/Phospholipids Mixed Micelles for Delivery of Icariside II to Multidrug-Resistant Breast Cancer. Integr Cancer Ther. 2016;15(3):390–399. | ||
Assanhou AG, Li W, Zhang L, et al. Reversal of multidrug resistance by co-delivery of paclitaxel and lonidamine using a TPGS and hyaluronic acid dual-functionalized liposome for cancer treatment. Biomaterials. 2015;73:284–295. | ||
Chen W, Zou Y, Zhong Z, Haag R. Cyclo(RGD)-Decorated Reduction-Responsive Nanogels Mediate Targeted Chemotherapy of Integrin Overexpressing Human Glioblastoma In Vivo. Small. 2017;13(6):1601997. | ||
Wu J, Feng S, Liu W, Gao F, Chen Y. Targeting integrin-rich tumors with temoporfin-loaded vitamin-E-succinate-grafted chitosan oligosaccharide/d-α-tocopheryl polyethylene glycol 1000 succinate nanoparticles to enhance photodynamic therapy efficiency. Int J Pharm. 2017;528(1–2):287–298. | ||
Chen Y, Feng S, Liu W, Yuan Z, Yin P, Gao F. Vitamin E Succinate-Grafted-Chitosan Oligosaccharide/RGD-Conjugated TPGS Mixed Micelles Loaded with Paclitaxel for U87MG Tumor Therapy. Mol Pharm. 2017;14(4):1190–1203. | ||
Yuan Z, Shi X, Qiu Y, et al. Reversal of P-gp-mediated multidrug resistance in colon cancer by cinobufagin. Oncol Rep. 2017;37(3):1815–1825. | ||
Ding DW, Zhang YH, Huang XE, An Q, Zhang X. Bufalin induces mitochondrial pathway-mediated apoptosis in lung adenocarcinoma cells. Asian Pac J Cancer Prev. 2014;15(23):10495–10500. | ||
Hsu CM, Tsai Y, Wan L, Tsai FJ. Bufalin induces G2/M phase arrest and triggers autophagy via the TNF, JNK, BECN-1 and ATG8 pathway in human hepatoma cells. Int J Oncol. 2013;43(1):338–348. | ||
Liu F, Tong D, Li H, et al. Bufalin enhances antitumor effect of paclitaxel on cervical tumorigenesis via inhibiting the integrin α2/β5/FAK signaling pathway. Oncotarget. 2016;7(8):8896–8907. | ||
Miao Q, Bi LL, Li X, et al. Anticancer effects of bufalin on human hepatocellular carcinoma HepG2 cells: roles of apoptosis and autophagy. Int J Mol Sci. 2013;14(1):1370–1382. | ||
Tian X, Dai S, Sun J, et al. Bufalin Induces Mitochondria-Dependent Apoptosis in Pancreatic and Oral Cancer Cells by Downregulating hTERT Expression via Activation of the JNK/p38 Pathway. Evid Based Complement Alternat Med. 2015;2015:1–12. |
Supplementary materials
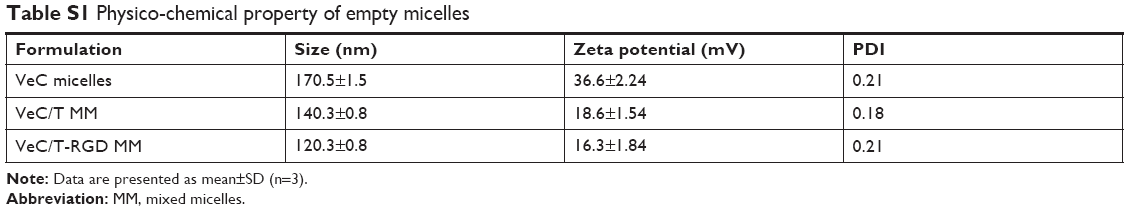 | Table S1 Physico-chemical property of empty micelles |
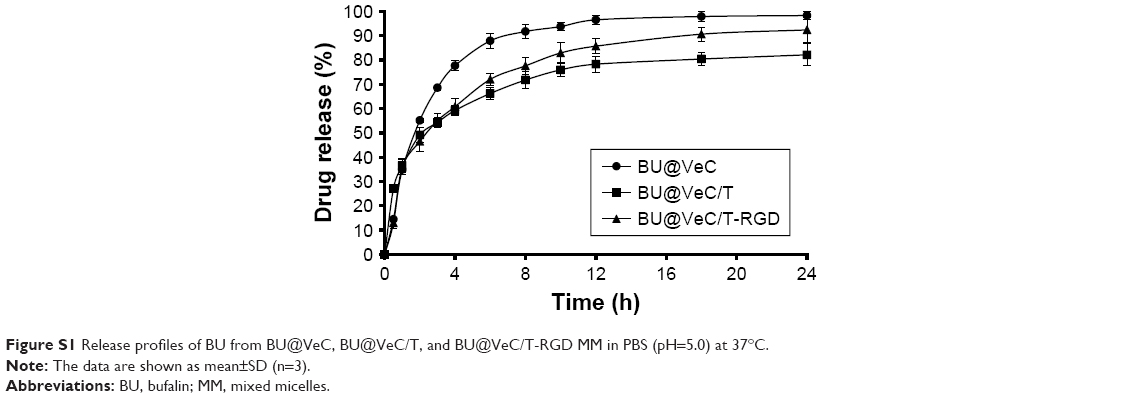 | Figure S1 Release profiles of BU from BU@VeC, BU@VeC/T, and BU@VeC/T-RGD MM in PBS (pH=5.0) at 37°C. |
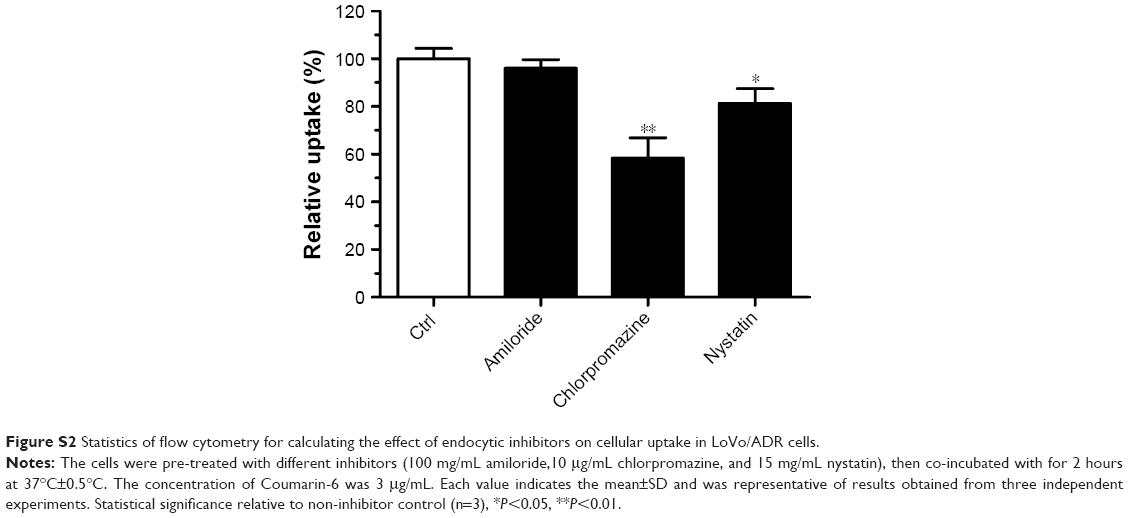 | Figure S2 Statistics of flow cytometry for calculating the effect of endocytic inhibitors on cellular uptake in LoVo/ADR cells. |
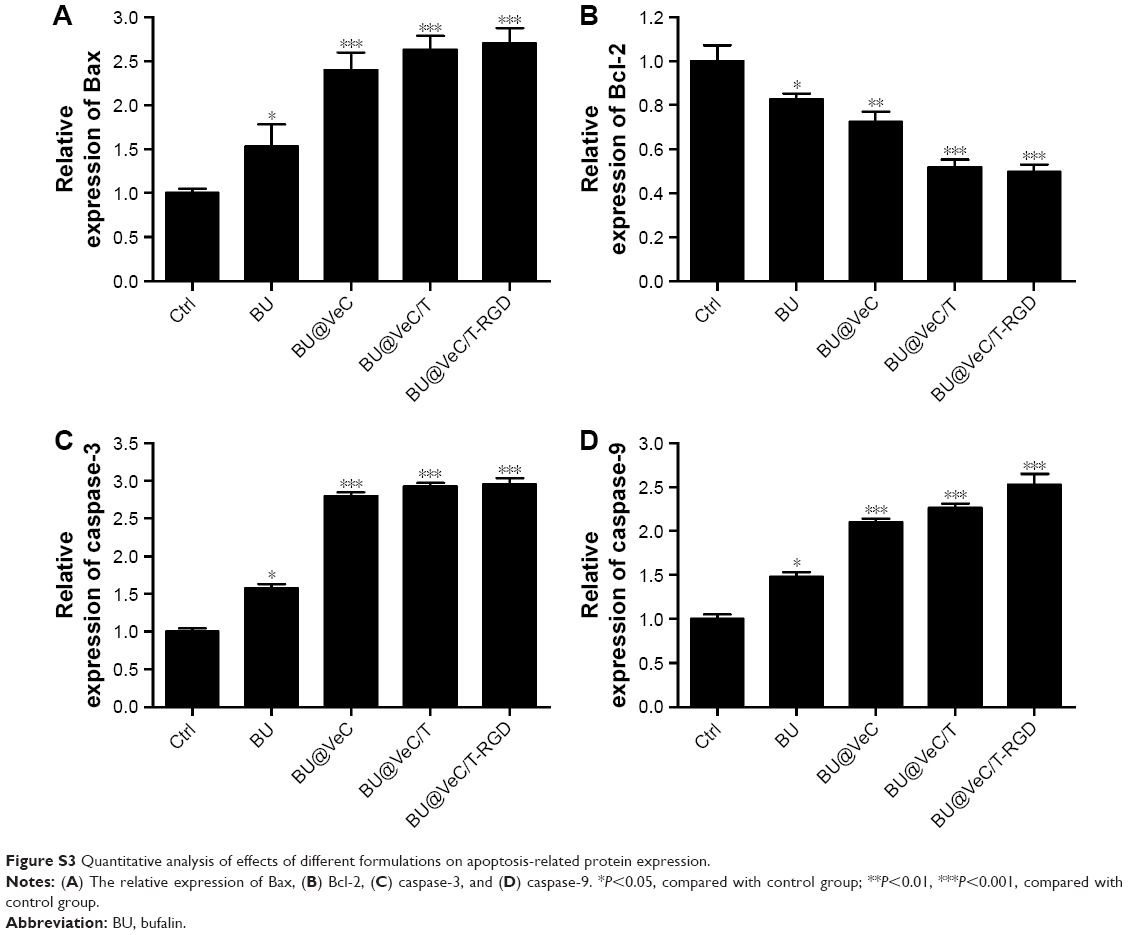 | Figure S3 Quantitative analysis of effects of different formulations on apoptosis-related protein expression. |
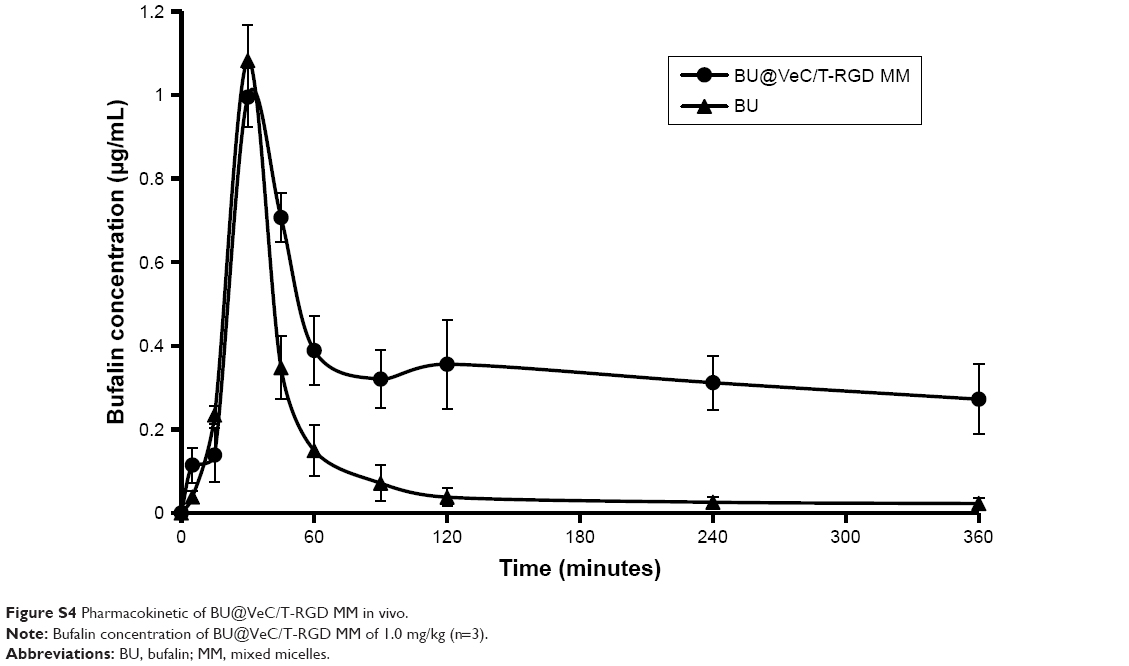 | Figure S4 Pharmacokinetic of BU@VeC/T-RGD MM in vivo. |
 | Table S2 Pharmacokinetic parameters of BU and BU@VeC/T-RGD MM |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.