Back to Journals » OncoTargets and Therapy » Volume 13
Bufalin Induces Glioma Cell Death by Apoptosis or Necroptosis
Authors LingHu HR, Luo H, Gang L
Received 20 December 2019
Accepted for publication 4 May 2020
Published 27 May 2020 Volume 2020:13 Pages 4767—4778
DOI https://doi.org/10.2147/OTT.S242567
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Hai Rui LingHu,1,* Hui Luo,2,* Lin Gang2,*
1Department of Neurosurgery and Neurocritical Care, Beijing Chaoyang Integrative Medicine Emergency Medical Center, Beijing 100022, People’s Republic of China; 2Characteristic Medical Center of Chinese People’s Armed Police Force (PAP), Tianjin 300162, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hai Rui LingHu
Department of Neurosurgery and Neurocritical Care, Beijing Chaoyang Integrative Medicine Emergency Medical Center, Chaoyang District, Beijing 100022, People’s Republic of China
Tel +8613622098542
Email [email protected]
Introduction: Bufalin is a component of Chinese traditional medicine, Chansu, which is reported to induce cell death among various kinds of tumors. Apoptosis evasion is a common problem of cancer treatment.
Materials and Methods: The proliferation of U-87 and U-373 treated by bufalin combined with or without apoptosis inhibitor was detected by MTT assay. The protein levels related to apoptosis and necroptosis were measured by Western blotting. Immunoprecipitation (IP) was applied for monitoring the formation of necrosome. The gene knockdown by CRISPR/Cas9 was applied to determine the roles of the proteins in apoptosis and necroptosis.
Results: In this study, we found that bufalin could induce apoptosis or necroptosis when U-87 and U-373 escaped from apoptosis. Bufalin triggered cell death by upregulating tumor necrosis factor (TNF) -α, TNF receptor 1 (TNFR1) and receptor-interacting protein 1 (RIPK1). Antagonizing cellular inhibitor of apoptosis 1 (cIAP1) and cIAP2 were also contributory. Caspase-8 activation led to apoptosis. When caspase-8 was functionally lost, necrosome consisted of RIPK1, receptor-interacting protein 3 (RIPK3) and mixed lineage kinase domain-like protein (MLKL) formed and necroptosis happened. The knockdown of above genes or the drug treatment confirmed the mechanism of bufalin-induced cell death. Cytotoxicity of bufalin to caspase-8 knockdown cell lines made control cell lines more sensitive to bufalin in their mixture.
Discussion: The cytotoxicity of bufalin to U-87 and U-373 was by inducing apoptosis or necroptosis when they were sensitive to apoptosis or not. The results indicated that seeking for treatments that could induce apoptosis and necroptosis was a good solution for the tumor evasion of apoptosis.
Keywords: glioma, bufalin, RIPK1, apoptosis, necroptosis
Introduction
Glioma is the most common brain tumor.1,2 Although surgical resection is a valid therapy, its high recurrence rate is a hidden danger.3 In terms of chemotherapy, the well-known temozolomide is only effective in a little portion of glioma patients with high rate of drug resistance.1
In antitumor treatments, numbers of drugs have been tested. Escaping from apoptosis is one of the most impressive problems during treatment administration.4–6 So new targets triggering the alternative cell death for apoptosis are in urgent need. Necroptosis is seemed to be a choice because it only happens when the activity of caspase-8 (cysteinyl aspartate-specific proteinase-8), one of the key initiators of apoptosis, is blocked naturally or manually.7,8
Necroptosis is characterized by the formation of necrosome that is made up of RIPK1 (receptor-interacting serine/threonine-protein kinase 1), RIPK3 and MLKL (mixed lineage kinase domain-like protein). RIPK1 is a node diverging into NF-κB, apoptosis or necroptosis under different conditions. The destination depends on the modification states switching between ubiquitination and phosphorylation.9 On the condition that TNFR1 is activated by TNF-α, ubiquitination of RIPK1 by cIAPs and LUBAC results in cell alive through NF-κB signal pathway,10 and deubiquitination of RIPK1 leads to apoptosis11 or necroptosis which is determined by its phosphorylation states.12 Once phosphorylated, RIPK1 then catalyzes the phosphorylation of RIPK3, which launches necroptosis and can be inhibited by necrostatin-1 (Nec-1), the specific inhibitor of RIPK1. The phosphorylated RIPK3 further phosphorylates MLKL, the main executor of necroptosis killing cells by punching on the membranes.13
Bufalin is a component of Chinese traditional medicine, Chansu14 that has been reported to show antitumor activity, containing inducing apoptosis15 or necroptosis.16 But how bufalin determines to go through apoptosis or necroptosis is still unclear. As to glioma, the antitumor activity of bufalin is yet barely reported.
In this study, we found that bufalin could induce either apoptosis or necroptosis in U-87 and U-373 when caspase-8 was active or inactive. Besides, bufalin-induced necroptosis in caspase-8 activity absent cells could enhance bufalin-induced apoptosis in caspase-8 activity present cells. This study suggested that bufalin might be a potential drug for apoptosis insensitive gliomas.
Materials and Methods
Reagents
Bufalin was purchased from Sigma-Aldrich (B0261). Caspase-8 inhibitor, zVAD.fmk (S7023), was purchased from Selleck. Nec-1 was from MedChemExpress (HY-15760). Anti-RIPK1 (ab72139), anti-MLKL (ab184718), anti-cIAP1 (ab108361), anti-cIAP2 (ab32059), anti-caspase-8 (ab32397) antibodies were purchased from Abcam. Anti-β-actin antibody was obtained from Proteintech (20536-1-AP). Anti-RIPK3 antibody (NBP1-77299) was purchased from Novus.
Cell Culture
Cell lines of human glioma, U-87 and U-373 (the Cell Bank of the Shanghai Chinese Academy of Sciences) were cultured with 10% fetal bovine serum (Gibco, 10099) in DMEM high glucose medium at 37°C, 5% CO2.
Gene Knockdown
Gene knockdown was performed by CRISPR/Cas9 with lentiCRISPRv2. Virus particles were collected by transfected lentiCRISPRv2 combined with psPAX2, pMD2G to 293T for 48 h. All cell lines were infected by virus particles for 24 hs and cultured with 2 ng/μL puromycin after another 48 hs. The gene-modified cell lines were applied for further studies after cultured with 2 ng/μL puromycin for 7 days. The knockdown sequences were as following: TNF-α, 5ʹ-TATCTCGACTTTGCCGAGTC-3ʹ; TNFR1, 5ʹ-ATATACCCCTCAGGGGTTAT-3ʹ; RIPK1, 5ʹ-CGGCTTTCAGCACGTGCATC-3ʹ; RIPK3, 5ʹ-CAGTGTTCCGGGCGCAACAT-3ʹ; MLKL, 5ʹ-TTGAAGCATATTATCACCCT-3ʹ; caspase-8, 5ʹ-TCCTTTGCGGAATGTAGTCC-3ʹ; cIAP1, 5ʹ-GAGTTCTTGATACGAATGAA-3ʹ; cIAP2, 5ʹ-TTACCCTCATCTACTTGAAC-3ʹ.
Cell Viability
Each well was seeded with 2×103 cells in 100 μL medium. Twenty-four hours later, drugs were added into each well according to different concentrations indicated. Another 24 h later, cell viabilities were measured by the CellTiter 96® AQueous One Solution Cell Proliferation Assay according to the manual after treatments. The absorbance at 490 nm was measured as the cell viabilities.
Western Blot Analysis and Immunoprecipitation
Cells were scratched in lysis buffer (1% NP-40, 150 mM NaCl, 10 mM Tris, pH 8.0), with cocktail (HY-K0010, MedChemExpress, America) on the ice. Bradford was applied for the concentration determination. As for immunoprecipitation, antibody for RIPK3 was added at ratio of 1:100 and incubated at 4°C for 4 h. Then, protein A/G magnetic beads (88802, Thermo Scientific, America) were added and incubated overnight.
In terms of Western blot, equal amount of protein was separated by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis). Nitrocellulose membrane was used for immunoblotting. All primary antibodies were incubated overnight at 4°C and secondary antibodies were incubated for 1 h at room temperature.
Statistical Analysis
Comparison between two groups was analyzed by Student’s t-test. P ≤ 0.05 was considered significant. All results were written as mean ± SD.
Results
Bufalin Could Trigger Either Necroptosis or Apoptosis Both in U-87 and U-373
To investigate the possible death forms of apoptosis and necroptosis triggered by bufalin in glioma, a series of 4 times diluted concentrations of bufalin combined with zVAD.fmk or Nec-1 were administrated to U-87 and U-373. And, the survival rates after 24 h are shown in Figure 1A and B. As shown, the survival rates were decreased sharply along with the bufalin concentration increasing. And the lowest survival rates of U-87 and U-373 were 49.1% and 45.5%, which located at the concentration of 1,000 nM. However, when apoptosis was blocked by zVAD.fmk, the cell death induced by bufalin was exacerbated. Details were as follows. Bufalin combined with zVAD.fmk reduced survival rate to 84.7% and 66.9% at the concentration as low as 15.6 nM in U-87 and U-373, respectively. But little effect was observed with bufalin alone at that concentration. Furthermore, ranging from 15.6 nM to the extreme concentration in this study (1000nM), the administration of zVAD.fmk also exaggerated cell death induced by bufalin. Although Nec-1 showed little help to bufalin alone induced cell survival rate, it completely reversed the cell death induced by the combination of zVAD.fmk with all concentrations of bufalin.
 |
Figure 1 Bufalin induced apoptosis or necroptosis when caspase-8 was absent in U-87 and U-373. Survival rates of U-87 (A) and U-373 (B) treated with different concentrations of bufalin combined with Nec-1or zVAD. TNFα/TNFR1-based cell death-related universal protein (cIAP1, cIAP2, RIPK1), apoptosis specific protein (caspase-8) and necroptosis specific protein (RIPK3 and MLKL) expression in U-87 (C) and U-373 (D) when treated with different combinations of bufalin, Nec-1 and zVAD. Immunoprecipitation analyses of necrosome formation when U-87 (E) and U-373 (F) was treated with bufalin or bufalin/zVAD. **P < 0.001. |
The different responses to Nec-1 when U-87 and U-373 incubated by bufalin with or without zVAD.fmk indicated the diverse cell death forms. The failure blockage to cell death by Nec-1 indicated apoptosis but not necroptosis happened in the context of bufalin alone. The aggravation of cell death triggered by bufalin with zVAD.fmk was a character of apoptosis switching to necroptosis.17 Moreover, the blockage of cell death by Nec-1 proved the occurrence of necroptosis when U-87 and U-373 were incubated with bufalin and zVAD.fmk synergetically.
Above all, we could speculate that bufalin alone induced apoptosis in U-87 and U-373 and that zVAD.fmk switched bufalin-induced apoptosis to necroptosis. To verify the conjecture, we determined the apoptosis and necroptosis related protein levels on various conditions of drug administration in U-87 and U-373 (Figure 1C and D).
As the ligand and receptor, TNF-α and TNFR1 increased sharply when bufalin showed up. As the intracellular inhibitors of cell death, cIAP1 and cIAP2 decreased sharply in both cell lines when cell death signal showed up under the stimulation of bufalin. The alteration of cIAP1 and cIAP2 always determined the destiny of RIPK1. By measuring the protein level of RIPK1, we found that bufalin promoted the expression of RIPK1. As previous prediction, bufalin-induced U-87 and U-373 apoptosis. So, we tested the caspase-8, a key apoptosis-related molecular downstream of RIPK1. The expression, especially the mature state of caspase-8 was upregulated, which proved the apoptosis induced by bufalin.
As mentioned above, bufalin-induced apoptosis in U-87 and U-373 (Figure 1C and D) and apoptosis inhibitor promoted cell death other than cell survival (Figure 1A and B), which was in accordance with the feature of necroptosis. To verify the phenomenon, we conducted the necroptosis-related detection by measuring necroptosis pathway-related protein levels. Like apoptosis, as the cell death signal, TNF-α and TNFR1 raised up and cIAP1 and cIAP2 reduced after the administration of bufalin combined with zVAD.fmk. Necrosome was the landmark of necroptosis. We found its components, RIPK1, RIPK3 and MLKL, all increased under the treatment of bufalin combined with zVAD.fmk. However, the protein levels could not distinguish the two death forms induced by bufalin with or without zVAD.fmk (Figure 1C and D). Therefore, we detected the formation of necrosome in U-87 and U-373 (Figure 1E and F). Although bufalin induced the upregulation of RIPK1, RIPK3 and MLKL, the interaction of RIPK3 with RIPK1 and with MLKL was not seen. However, RIPK3 interacted with RIPK1 and MLKL tightly in U-87 and U-373 incubated by the combination of bufalin and zVAD.fmk, which confirmed the occurrence of necroptosis.
TNF-α and TNFR1 Were Required for Cytotoxicity of Bufalin
The above results had shown the possible pathways induced by bufalin with or without zVAD.fmk. However, whether the above pathways contributed to the cell death was not sure. To evaluate the potential mechanism, we first knocked down TNF-α and TNFR1, respectively. As ratiocinated, TNF-α knockdown by CRISPR/Cas9 eased the cytotoxicity of bufalin whatever zVAD.fmk present or not in U-87 (Figure 2A and B) and U-373 (Figure 2C and D). As the receptor of TNF-α, TNFR1 worked as the transmitter9,18 and showed the similar trends, in which its knockdown significantly blocked the apoptosis and necroptosis when bufalin or bufalin/zVAD was treated with U-87 (Figure 2E and F) and U-373 (Figure 2G and H). These findings indicated that upregulation of TNF-α and TNFR1 activation was necessary for bufalin stirred apoptosis or necroptosis in U-87 and U-373.
 |
Figure 2 TNFα and TNFR1 were required for bufalin-induced apoptosis and necroptosis. TNFα (A) and TNFR1 (E) were knocked down by CRISPR/Cas9 in U-87 and detected by Western blot. Cells were treated with bufalin (1000 nM) or bufalin/zVAD (50 μM) (B, F) for 24 h, and cell survival rates were measured by MTS. **P < 0.001. TNFα (C) and TNFR1 (G) were knocked down by CRISPR/Cas9 in U-373 and detected by Western blot. Cells were treated with bufalin (1000 nM) or bufalin/zVAD (50 μM) (D, H) for 24 h, and cell survival rates were measured by MTS. **P < 0.001. |
cIAP1 or cIAP2 Knockdown Amplified Cytotoxicity of Bufalin in U-87 and U-373
Downstream of TNF-α/TNFR1 signaling, the modification state of RIPK1 determined whether the cell went through death or not. During the process, cIAP1 and cIAP2 were two important molecules that ubiquitinated RIPK1 and prevented cell from death. Here, we knocked down the both. We found that cIAP1 knockdown reduced cell survival of U-87 (Figure 3A and B) and U-373 (Figure 3C and D). But the inhibition rate of bufalin to them reduced heavily. For example, cIAP1 knockdown of U-87 reduced survival rate to 65.3%. However, compared to the inhibition rate in control U-87 treated with bufalin [51.2%, inhibition rate=1- (survival rate of U-87 treated with bufalin/survival rate of U-87 treated with vehicle)], inhibition rate of bufalin in the cIAP1 knockdown U-87 reduced to 35.0% [inhibition rate=1- (survival rate of cIAP1 knockdown U-87 treated with bufalin/survival rate of cIAP1 knockdown U-87 treated with vehicle)]. The inhibition rate of cIAP1 knockdown U-87 treated with bufalin/zVAD.fmk also reduced (83.6% vs 80.2%) (Figure 3B).
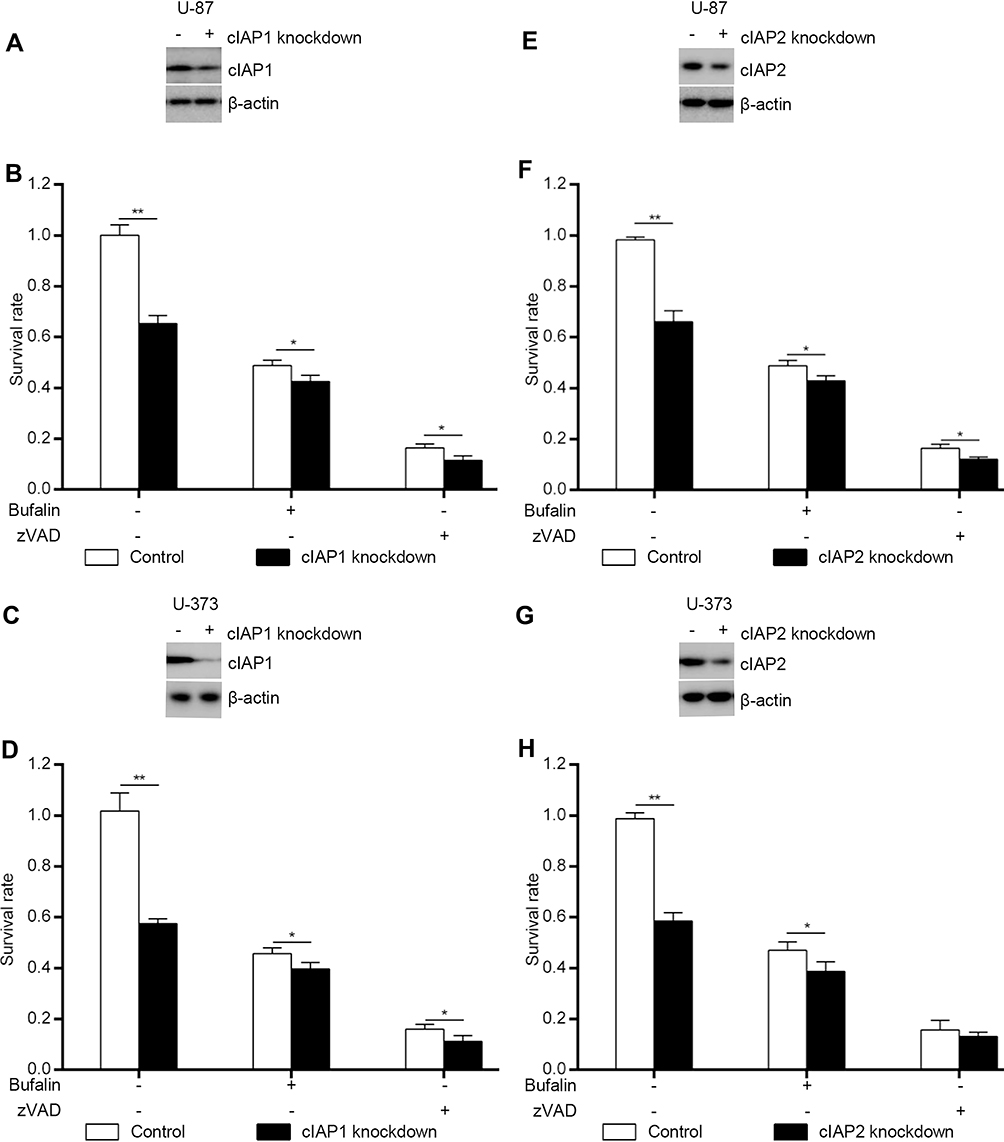 |
Figure 3 cIAP1 and cIAP2 were required for bufalin-induced apoptosis and necroptosis. cIAP1 (A) and IAP2 (E) were knocked down by CRISPR/Cas9 in U-87 and detected by Western blot. Cells were treated with bufalin (1000 nM) or bufalin/zVAD (50 μM) (B, F) for 24 h, and cell survival rates were measured by MTS. *P < 0.05; **P < 0.001. cIAP1 (C) and cIAP2 (G) were knocked down by CRISPR/Cas9 in U-373 and detected by Western blot. Cells were treated with bufalin (1000 nM) or bufalin/zVAD (50 μM) (D, H) for 24 h, and cell survival rates were measured by MTS. *P < 0.05; **P < 0.001. |
Similar phenomenon was observed in U-373 (Figure 3D). Knockdown of cIAP1 in U-373 resulted in inhibition rates dropping down from 55.2% to 37.6% or from 85.3% to 80.0% when treated with bufalin or bufalin/zVAD.fmk.
Likewise, cIAP2 knockdown also increased cell death. But the inhibition rates when treated with bufalin or bufalin/zVAD.fmk also reduced in U-87 (Figure 3E and F) and U-373 (Figure 3G and H).
Giving the results of cIAP1 and cIAP2 knockdown, we could infer that cIAP1 and cIAP2 inhibition was also one of the reasonable mechanisms of bufalin cytotoxicity.
RIPK1 Contributed to Both Necroptosis and Apoptosis
RIPK1 was a multifunction molecular contributed to NF-κB signaling pathway-dependent cell alive, caspase-8 dependent apoptosis or necrosome dependent necroptosis. In this work, bufalin led to RIPK1 dependent apoptosis or necroptosis. So, we knocked down RIPK1 to reveal its role in bufalin cytotoxicity. In U-87, RIPK1 knockdown (Figure 4A) raised cell survival rate from 48.8% up to 85.5% in the aspect of bufalin mediated apoptosis and remedied cell survival rate from 16.3% to 93.5% as to bufalin/zVAD.fmk caused necroptosis (Figure 4B). Considering results above, it seemed that RIPK1 was more indispensable to necroptosis than apoptosis.
 |
Figure 4 RIPK1 was required for bufalin-induced apoptosis and necroptosis. RIPK1 was knocked down by CRISPR/Cas9 in U-87 (A) and in U-373 (C), then detected by Western blot. Cells were treated with bufalin (1000 nM) or bufalin/zVAD (50 μM) (B, D) for 24 h, and cell survival rates were measured by MTS. **P < 0.001. |
When it came to U-373, RIPK1 knockdown (Figure 4C) almost blocked bufalin and bufalin/zVAD.fmk aroused cell death completely with survival rate of 94.1% and 92.7% (Figure 4D), respectively.
The RIPK1 knockdown in U-87 and U-373 demonstrated that RIPK1 was required for both bufalin and bufalin/zVAD.fmk induced apoptosis and necroptosis.
The State of Caspase-8 Determined the Death Form Induced by Bufalin Both in U-87 and U-373
Along with the TNF-α/TNFR1/RIPK1 axis, the caspase-8 was the referee that determined whether the cell went through apoptosis or necroptosis.19 Caspase-8 might play important role in the shifting between bufalin and bufalin/zVAD.fmk induced cell death. Thus, we conducted caspase-8 knockdown. Agreeing with zVAD.fmk administration, caspase-8 knockdown enhanced bufalin-induced cell death in U-87 (Figure 5A and B) and U-373 (Figure 5C and D).
 |
Figure 5 Caspase-8 determined destination of apoptosis or necroptosis when U-87 and U-373 were treated with bufalin. Caspase-8 was knocked down by CRISPR/Cas9 in U-87 (A) and in U-373 (C), then detected by Western blot. Cells were treated with bufalin (1000 nM) or bufalin/zVAD (50 μM) (B, D) for 24 h, and cell survival rates were measured by MTS. *P < 0.05; **P < 0.001. |
Judging from the data above, we could know that caspase-8 was necessary for the bufalin-caused apoptosis and its deletion could switch apoptosis to the drastic cell death, necroptosis.
RIPK3 and MLKL Only Contributed to Bufalin-Induced Necroptosis when zVAD.fmk Was Present
Necroptosis and apoptosis divided into two forks downstream of RIPK1. Although caspase-8 impacted necroptosis, the real executors were RIPK3 and MLKL.13,17 Theoretically, the knockdown of RIPK3 and MLKL only surrendered to necroptosis but not apoptosis. To verify the assumption, we knocked down RIPK3 and MLKL in U-87 and U-373, severally. In accordance with our hypothesis, RIPK3 knockdown nearly kept U-87 (Figure 6A and B) and U-373 (Figure 6C and D) from death completely in the presence of bufalin/zVAD.fmk. Compared with control groups, RIPK3 knockdown made no sense to bufalin-induced apoptosis both in U-87and U-373.
 |
Figure 6 RIPK3 and MLKL were prerequisite for bufalin/zVAD induced necroptosis. RIPK3 (A) and MLKL (E) were knocked down by CRISPR/Cas9 in U-87 and detected by Western blot. Cells were treated with bufalin (1000 nM) or bufalin/zVAD (50 μM) (B, F) for 24 h, and cell survival rates were measured by MTS. **P < 0.001. RIPK3 (C) and MLKL (G) were knocked down by CRISPR/Cas9 in U-373 and detected by Western blot. Cells were treated with bufalin (1000 nM) or bufalin/zVAD (50 μM) (D, H) for 24 h, and cell survival rates were measured by MTS. **P < 0.001. |
Without any exception, MLKL knockdown just shielded necroptosis induced by bufalin/zVAD.fmk but not apoptosis induced by bufalin in U-87 (Figure 6E and F) and U-373 (Figure 6G and H).
The results demonstrated that bufalin/zVAD.fmk induced cell death depended on RIPK3 and MLK.
Bufalin Mediated Necroptosis in Caspase-8 Deletion U-87 and U-373 Promoted Bufalin-Induced Apoptosis in Normal Groups
As was stated above, bufalin could kill U-87 and U-373 whatever they were sensitive to apoptosis or not. In fact, tumors always gained mutations during growing. And apoptosis loss was one of the escaping approaches from death. Sometimes the tumor tissues were made up of cancer cells both sensitive or insensitive to apoptosis. We mixed caspase-8 knockdown U-87 and U-373 with control cell lines at ratios of 1:3, 2:2 and 3:1. The survival rates at 1 μM at 24 h were measured (Figure 7A and B). As shown, the survival rates were all less than predicted in U-87 and U-373.
 |
Figure 7 Bufalin mediated necroptosis in caspase-8 absent cell lines made bufalin more cytotoxic to induce apoptosis in normal cell lines. U-87 (A) and U-373 (B) with caspase-8 knockdown were mixed with the control cell lines at different ratios and treated with bufalin (1000 nM) for 24 h. Survival rates were measured with MTS. |
Giving the survival rates above, we could tell that the bufalin-induced cell death in caspase-8 knockdown U-87 and U-373 made the control cells easier to die in the circumstance of bufalin.
Discussion
Apoptosis is one of the main forms of cancer cell death for drug treatment now. In the past decades, plenty of drugs or targets have been tested to inducing tumor apoptosis. During the treatment, resistance to apoptosis is one of the main reasons for treatment failure. The apoptosis failure becomes a challenge to the treatment of cancer. Searching for the succedaneum of apoptosis is in urgent need.
Necroptosis just happens on the condition that the activity of caspase-8 is blocked, which is complementary to apoptosis. Although necroptosis needs the inactivation of caspase-8, not all caspase-8 inactivation goes to necroptosis.20–22 Therefore, discovering the key regulators switching apoptosis to necroptosis in tumors is of great concern.
Recently, a few kinds of drugs have been reported to lead to necroptosis,16,23–26 which provides the possibility for drug-induced necroptosis in tumors.
RIPK3 and MLKL are two key molecules for necroptosis downstream of RIPK1. Necrosome consists of RIPK1, RIPK3 and MLKL is the dominating executor and landmark of necroptosis.26
In this study, we found that bufalin could induce apoptosis in U-87 and U-373, spontaneously. And this process went along with TNF-α, TNFR1, RIPK1 and caspase-8 activity increasing and cIAP1, cIAP2 decreasing. The knockdown of TNF-α, TNFR1, RIPK1 blocked bufalin-induced apoptosis, effectively. That meant that TNF-α, TNFR1 and RIPK1 were necessary for bufalin-induced apoptosis. The knockdown of cIAP1 and cIAP2 reduced the inhibition rates, which meant that they were also responsible for the cell death.
When it comes to necroptosis, the similar phenomena were observed when TNF-α, TNFR1 and RIPK1 were knocked down. Different with bufalin-induced apoptosis, cIAP1 and cIAP2 knockdown made a little difference to the cell survival rates of bufalin/zVAD.fmk induced necroptosis.
Besides the common regulators of apoptosis and necroptosis, necroptosis characterized by RIPK3 and MLKL regulation. The knockdown of them really reduced the bufalin/zVAD.fmk induced necroptosis. Although bufalin could induce the upregulation of RIPK3 and MLKL (Figure 1C and D), the knockdown did no help to the induced apoptosis in U-87 (Figure 6B and F) and U-373 (Figure 6D and H).
Giving apoptosis and necroptosis mentioned in the above cell models, the knockdown of cIAP1 and cIAP2 affected cell death or bufalin sensitivity weakly. In fact, downstream of TNFR1, except cIAP1 and cIAP2, linear ubiquitin chain assembly complex (LUBAC),27 A2010 and CYLD28,29 were also in charge of RIPK1 modification. So cIAP1 and cIAP2 might not be the only molecules responsible for the modification of RIPK1 in the models. More focus on the other molecules was helpful for understanding the mechanism comprehensively.
As the results above, caspase-8 knockdown turned bufalin-induced apoptosis to necroptosis. That meant that bufalin could inhibit U-87 and U-373 no matter they were sensitive to apoptosis or not, which figured out the problem of apoptosis escaping without extra treatment. What was more, caspase-8 knockdown made normal cell lines more sensitive to bufalin in U-87 (Figure 7A) and U-373 (Figure 7B). Whether the damage-associated molecular pattern (DAMP) aroused from necroptosis or other reasons contributed to the extra cell death was unknown. However, that was benefit for the antitumor activity of bufalin.
While lots of details of bufalin-induced cell death with or without zVAD.fmk were vague, the switching to necroptosis when cell escaped from apoptosis showed a new tumor treatment design. This was the first work, as we have known, to explore the drug induced both apoptosis and necroptosis, concurrently. To explore more treatments that could inhibit cancer cell proliferation by initiating apoptosis or necroptosis like bufalin was a potential direction.
Conclusion
Our study reported multifunction of bufalin in necroptosis and apoptosis mediated by RIPK1 on glioma, which resolved the problem of apoptosis resistance during treatment. By this, bufalin might be a potential antitumor drug for more widely use.
Acknowledgments
This study was funded by National Natural Science Foundation Major Program of China (81891003), National High-tech R&D Program of China (2016YFC1101502), Science & Technology Program of Tianjin (15ZXLCSY00040), and Technology Research Projects of PAL (AWS15J001).
Disclosure
The authors report no conflicts of interest in this work.
References
1. J Dabrowski M, Wojtas B. Global DNA methylation patterns in human gliomas and their interplay with other epigenetic modifications. Int J Mol Sci. 2019;20(14):3478. doi:10.3390/ijms20143478
2. Vigneswaran K, Neill S, Hadjipanayis CG. Beyond the World Health Organization grading of infiltrating gliomas: advances in the molecular genetics of glioma classification. Ann Transl Med. 2015;3(7):95. doi:10.3978/j.issn.2305-5839.2015.03.57
3. Altieri R, Zenga F, Fontanella MM, et al. Glioma surgery: technological advances to achieve a maximal safe resection. Surg Technol Int. 2015;27:297–302.
4. Tamtaji OR, Mirzaei H, Shamshirian A, et al. New trends in glioma cancer therapy: targeting Na(+)/H (+) exchangers. J Cell Physiol. 2019. doi:10.1002/jcp.29014
5. Saggioro FP, Neder L, Stavale JN, et al. Fas, FasL, and cleaved caspases 8 and 3 in glioblastomas: a tissue microarray-based study. Pathol Res Pract. 2014;210(5):267–273. doi:10.1016/j.prp.2013.12.012
6. Hirose Y, Berger MS, Pieper RO. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001;61(5):1957–1963.
7. Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16(6):329–344. doi:10.1038/nrm3999
8. Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138(2):229–232. doi:10.1016/j.cell.2009.07.006
9. Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16(7):689–697. doi:10.1038/ni.3206
10. Gerlach B, Cordier SM, Schmukle AC, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471(7340):591–596. doi:10.1038/nature09816
11. O’Donnell MA, Hase H, Legarda D, Ting AT. NEMO inhibits programmed necrosis in an NFκB-independent manner by restraining RIP1. PLoS One. 2012;7(7):e41238. doi:10.1371/journal.pone.0041238
12. Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. doi:10.1038/nchembio.83
13. Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–227. doi:10.1016/j.cell.2011.11.031
14. Panesar NS. Bufalin and unidentified substance(s) in traditional Chinese medicine cross-react in commercial digoxin assay. Clin Chem. 1992;38(10):2155–2156. doi:10.1093/clinchem/38.10.2155
15. Li D, Qu X, Hou K, et al. PI3K/Akt is involved in bufalin-induced apoptosis in gastric cancer cells. Anticancer Drugs. 2009;20(1):59–64. doi:10.1097/CAD.0b013e3283160fd6
16. Li Y, Tian X, Liu X, Gong P. Bufalin inhibits human breast cancer tumorigenesis by inducing cell death through the ROS-mediated RIP1/RIP3/PARP-1 pathways. Carcinogenesis. 2018;39(5):700–707. doi:10.1093/carcin/bgy039
17. He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell. 2009;137(6):1100–1111. doi:10.1016/j.cell.2009.05.021
18. Vasilikos L, Spilgies LM, Knop J, Wong WW. Regulating the balance between necroptosis, apoptosis and inflammation by inhibitors of apoptosis proteins. Immunol Cell Biol. 2017;95(2):160–165. doi:10.1038/icb.2016.118
19. Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 2013;27(15):1640–1649. doi:10.1101/gad.223321.113
20. Varfolomeev E, Blankenship JW, Wayson SM, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell. 2007;131(4):669–681. doi:10.1016/j.cell.2007.10.030
21. Vince JE, Wong WW, Khan N, et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell. 2007;131(4):682–693. doi:10.1016/j.cell.2007.10.037
22. Petersen SL, Wang L, Yalcin-Chin A, et al. Autocrine TNFα signaling renders human cancer cells susceptible to smac-mimetic-induced apoptosis. Cancer Cell. 2007;12(5):445–456. doi:10.1016/j.ccr.2007.08.029
23. Gentilin E, Simoni E, Candito M, Cazzador D, Astolfi L. Cisplatin-induced ototoxicity: updates on molecular targets. Trends Mol Med. 2019;25(12):1123–1132. doi:10.1016/j.molmed.2019.08.002
24. Wang JN, Liu MM, Wang F, et al. RIPK1 inhibitor Cpd-71 attenuates renal dysfunction in cisplatin-treated mice via attenuating necroptosis, inflammation and oxidative stress. Clin Sci. 2019;133(14):1609–1627. doi:10.1042/CS20190599
25. Yu WN, Lai YJ, Ma JW, et al. Citronellol induces necroptosis of human lung cancer cells via TNF-alpha pathway and reactive oxygen species accumulation. In Vivo. 2019;33(4):1193–1201. doi:10.21873/invivo.11590
26. Huang C, Luo Y, Zhao J, et al. Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One. 2013;8(6):e66326. doi:10.1371/journal.pone.0066326
27. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–320. doi:10.1038/nature14191
28. Onizawa M, Oshima S, Schulze-Topphoff U, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol. 2015;16(6):618–627. doi:10.1038/ni.3172
29. Wang L, Du F, Wang X. TNF-α induces two distinct caspase-8 activation pathways. Cell. 2008;133(4):693–703. doi:10.1016/j.cell.2008.03.036
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.