Back to Journals » Research Reports in Clinical Cardiology » Volume 10
Brugada syndrome: updated perspectives
Authors Walia J, Steinberg C, Laksman Z
Received 15 February 2019
Accepted for publication 2 May 2019
Published 4 June 2019 Volume 2019:10 Pages 19—32
DOI https://doi.org/10.2147/RRCC.S182162
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Kones
Jagdeep Walia,1 Christian Steinberg,2 Zachary Laksman1
1Heart Rhythm Services, Division of Cardiology, Department of Medicine, University of British Columbia, Vancouver, BC, Canada; 2Cardiac Electrophysiology, Division of Cardiology, Department of Medicine, Universite Laval, Québec, Canada
Abstract: Since the first reported descriptions of Brugada syndrome, there has been a growing awareness and appreciation of the disease and its implications. From the diagnostic criteria, to risk stratification and management, there is an ongoing evolution, reclassification and re-thinking of Brugada syndrome as basic science, registry and clinical trial data shape our understanding of the pathophysiology and its clinical implications. This in-depth review sheds light on the most important literature to date, highlighting insights that shifted the global perspective on the disease. Current clinical paradigms and guidelines are presented, along with their justification, and possible opportunities for future research are explored.
Keywords: Brugada, risk stratification, sudden death, inherited arrhythmia syndrome
Introduction
Brugada syndrome (BrS) was first described in 1992 within a group of patients presenting with ST elevation in the anterior precordial leads, right bundle branch block, and recurrent aborted sudden cardiac death (SCD).1 It has since been recognized to predispose patients to malignant cardiac events leading to ventricular fibrillation (VF) and sudden cardiac arrest (SCA). Depending on the population under study, BrS has been implicated in as many as one in four sudden arrhythmogenic death syndrome cases in the setting of a structurally normal heart.2
The incidence of BrS is higher in Japan and South East Asian countries with rates reported between 0.18% and 0.27%,3,4 vs European and North American populations where BrS is less common (0.017% and 0.012% respectively).5,6 Diagnosis is often made incidentally within healthy and asymptomatic patients at a median age of 45 years.7 Most of these patients have a low risk of a future cardiac arrest3 and thus do not receive chronic medical therapy or a primary prevention defibrillator. There has been a significant effort over the past two decades to refine the diagnostic criteria, risk stratification, and medical management strategies to determine the small number of patients at significant risk. The aim of this review is to briefly summarize current knowledge of these topics in BrS.
Diagnostic criteria
Since the first expert consensus conference in the 2002,8 the diagnostic criteria for BrS have been refined in subsequent consensus reports published in 20059 and 2013,10 along with technical electrocardiogram (ECG) requirements in 201211 and management recommendations in 2017.12 The most recent expert consensus report from 201613 proposed the Shanghai Score System that awards points based on ECG features, clinical history, family history and genetic test results (Table 1). The scoring system was devised by experts using limited data, with the expectation that ongoing validation studies would be required. To date, the validity of the Shanghai Score System has been tested by one single-center retrospective study (N=393)14 that showed malignant cardiac events occurred only in patients who received scores that met diagnostic criteria.
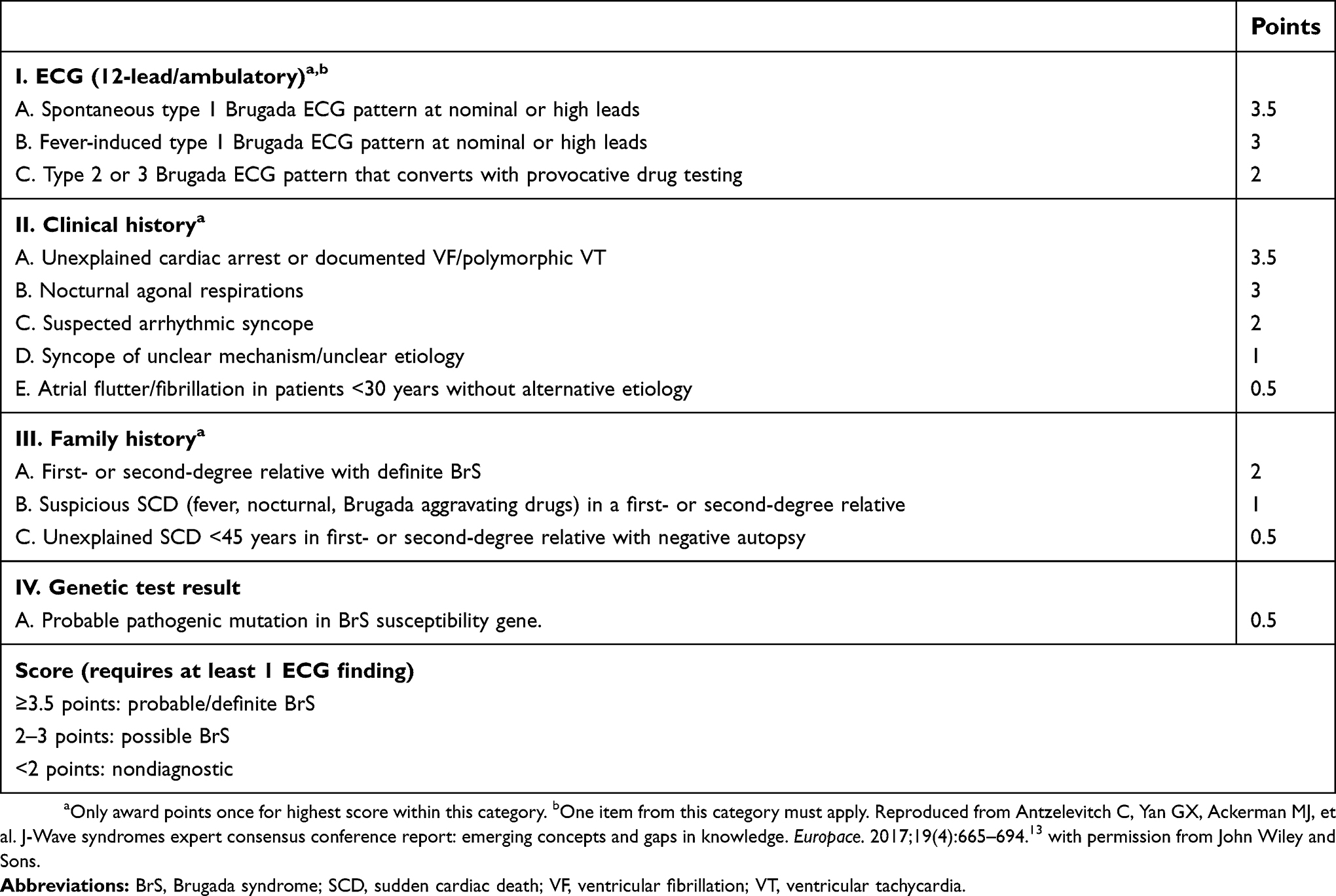 | Table 1 Proposed Shanghai Score System for diagnosis of Brugada syndrome |
Consistent with the 2013 consensus report,10 a type 1 ECG presenting spontaneously, in a febrile state or after drug challenge, in conjunction with a Shanghai Score of ≥3.5 points is required for the diagnosis of BrS. Absence of a type 1 ECG despite a Shanghai Score ≥3.5 points is nondiagnostic for BrS. However, many consider that a type 1 ECG alone is diagnostic for BrS, either occurring spontaneously or recorded during sodium channel provocation testing. There has not yet been widespread acceptance of the Shanghai Score System due to its relative complexity and lack of supporting data in its prognostic utility, and thus the majority of centers continue to diagnose BrS based on a type 1 ECG in the absence of specific reversible factors (discussed further in text). The type 1 ECG is characterized by a coved ST elevation ≥2 mm and negative T waves within ≥1 of the anterior precordial leads (V1-V3) under baseline conditions (Figure 1). A type 2 ECG is characterized by a saddleback ST elevation ≥0.5 mm within leads V1-V3 and a positive T wave in V2.
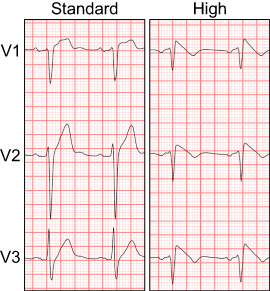 | Figure 1 12-lead electrocardiographic (ECG) recordings of a 62-year old male immediately after infusion of a 1 g procainamide bolus over 20 minutes. The diagnostic type 1 ECG for Brugada syndrome is present within leads V1–V3 when recorded in the high but not standard positions. |
Electrophysiological abnormalities of the right ventricular outflow tract (RVOT) are felt to contribute to the underlying substrate of BrS.15,16 A seminal study using cardiac magnetic resonance imaging (CMRI) on 30 patients with spontaneous or drug induced type 1 ECGs found that the RVOT was predominately superimposed with the second, third or fourth intercostal space (ICS) in 10%, 73%, and 17% of patients respectively.17 Anterior precordial lead placement within the ICS that coincided with RVOT location demonstrated the greatest degree of ST elevation. Sensitivity for detection of a spontaneous and drug induced type 1 ECG improved when ECG leads were elevated to the second and third ICS, and thus so-called high lead ECG recordings should be performed on all patients with suspected BrS.13
The type 1 ECG can be mimicked by a variety of drugs (
Drug challenge
Cerrato et al19 performed 24-hour 12-lead Holter monitoring on BrS patients diagnosed based on spontaneous type 1 ECG patterns at baseline (group A, N=75) versus type 1 ECG after provocative drug testing (group B, N=176). They assessed for type 1 ECG burden, and found persistent, intermittent and complete absence of the type 1 ECG pattern in 12%, 57% and 31% of patients within group A, and 0%, 20% and 80% of patients within group B respectively. Of note, a separate study showed that a higher type 1 ECG burden on 24-hour Holter was found in symptomatic compared to asymptomatic BrS patients.20 The necessity of a type 1 ECG pattern for BrS diagnosis is complicated by its intermittent nature, which could falsely label true BrS patients as disease-free, and redundantly expose patients to invasive provocative drug testing.
When clinically indicated, a type 1 ECG can be provoked by infusion of class I anti-arrhythmic drugs.21 Different anti-arrhythmics are employed in different countries and health regions including ajmaline, flecainide, pilsicainide and procainamide.13 The prognostic accuracy of these medications is variable, with studies reporting ajmaline to be more sensitive than flecainide,22 and IV procainamide more sensitive than oral flecainide.23 A positive drug challenge where a type 1 ECG is provoked in standard or high lead ECGs is diagnostic for BrS.13 Unfortunately without a gold standard for diagnosis, the sensitivity and specificity of drug testing is unknown.24 Drug challenge is not indicated in patients with a history of spontaneous or fever-induced type 1 ECG as it does not yield any additional diagnostic or prognostic information in these patients.
Clinical features
A number of features can raise the clinical suspicion for BrS including a personal or familial history of malignant cardiac events during a vagally enhanced state (ie, during sleep, after a large meal).25,26 Occurrence of malignant events with exercise are uncommon and would lower but not completely negate the pretest probability of BrS. If transient, ventricular arrhythmias may manifest as nocturnal agonal respirations.14 Atrial arrhythmias frequently co-present with BrS (Table 2).27–30
 | Table 2 Prevalence of atrial arrhythmias in Brugada syndrome patients |
Sex
Incidence of BrS is at least two-fold greater in males than it is in females, and tends to have a more malignant course.31 Interestingly, BrS has equal prevalence in both males and females below age 15 potentially implicating androgens in the pathogenesis of BrS.32
Age
Malignant cardiac events within BrS patients occur throughout life, and peak between the ages of 27 and 59 years.33 Many studies tend to investigate middle age adults where BrS prevalence is highest, thus data in children and the elderly are lacking, although older age at diagnosis appears to have a more benign course. The elderly, defined as aged ≥60 years, present more often with conduction disturbances manifesting as prolonged PR intervals and atrioventricular blocks.34
Within a multicenter European study investigating 106 BrS children with a mean age of 11.1 years, the presence of a spontaneous type 1 ECG and a history of prior malignant cardiac events were found to be independent predictors of future malignant cardiac events.32 In contrast, this same study reported that asymptomatic children with a drug induced type 1 ECG were at low risk of malignant cardiac events and thus did not need medical therapy beyond lifestyle measures. If a negative provocative challenge is performed in a child with a high degree of clinical suspicion for BrS, a repeat provocative test after the age of 15 could be considered.
Imaging studies
Papavassiliu et al35 was the first to show dilation of the RVOT in patients with BrS compared to controls, a finding that has been replicated in subsequent CMRI studies.36,37 Information on right ventricular (RV) dimensions have been conflicting without a clear trend observed.35,37–39 Functional abnormalities have been identified in some patients with BrS, including reduced right ventricular ejection fraction (RVEF) and right wall motion abnormalities (RWMA), however their relationship to BrS remains unclear.36–40 At this point, the role of CMRI in the diagnosis or risk stratification of patients with BrS remains unknown, and is not widely performed.
Genetic testing
The genetic substrate underlying BrS remains elusive in the majority of patients. A pathogenic variant in the SCN5A gene, encoding the pore-forming α-subunit of NaV1.5, is found in 20–25% of BrS patients, with over 300 variants identified to date.41,42 Mutations in over 20 other genes, most notably CACNA1C which has been found in as many as 5% of patients with BrS,41 have been described. A recent large-scale gene validity study by Hosseini et al43 reinterpreted the variants in the literature and suggested that only SCN5A variants could be clearly labeled as pathogenic for BrS, while a lack of supporting evidence would suggest downgrading the variant classification of the other variants previously described. Another large study examined families who had 5 or more individuals carrying the same SCN5A mutation, including the proband who was diagnosed with BrS.44 Out of the 115 mutation carriers identified, only 54 carriers (47%) presented with a type 1 ECG at baseline or after provocative flecainide or ajmaline testing. Additionally, 8 individuals from 5 families were genotyped as negative for a SCN5A mutation but still demonstrated a type 1 ECG either spontaneously or after provocative drug testing. These data indicate that the presence or absence of an SCN5A mutation does not absolutely confirm or refute the diagnosis of BrS, and may be reflective of other undiscovered genetic interactions that could explain the heritable component to their disease. It follows that genetic testing is a class IIb recommendation12 as the screening of confirmed BrS probands may be considered to identify whether there is a heritable component to their disease that would warrant cascade familial screening.7 Offering patients this testing should not be taken lightly, and pretest counselling with a genetic counsellor is strongly recommended.45
Pathophysiology
Depolarization of the cardiac sarcolemma to threshold potential evokes the rapid opening of NaV1.5 and conductance of inward Na+ current (INa) manifesting as a steep upstroke (phase 0). With sufficient NaV1.5 mediated depolarization, voltage-dependent activation of slow kinetic inward L-type Ca2+ currents (ICa-L), and outward delayed rectifier K+ currents (Ikr and Iks) occurs. Phase 1 reflects the flow of transient outward K+ current (Ito) producing early repolarization that manifests as a “notch” in the action potential (AP). As Ito inactivates, ICa-L increases and is roughly counterbalanced by Ikr and Iks producing a dome (phase 2). With eventual inactivation of ICa-L, there is net repolarization (phase 3) and return to resting membrane potential (phase 4) via Iks and Ikr with contributions from the inward K+ rectifier Ik1 at negative membrane potentials.
While INa is ubiquitously expressed throughout the heart, higher densities of Ito are located predominately in the RV, closer to the base of the heart and in the epicardium, with a predominance in the RVOT epicardium.46 The degree of Ito mediated repolarization correlates to increased action potential duration (APD) due to a longer time required for peak ICa-L recruitment.47 Through this mechanism, a transmural heterogenous “spike and dome” AP morphology develops as a result of regional differences in repolarization between the epicardium and endocardium. Within most healthy individuals, Ito prolongs the epicardial APD beyond that of the endocardium, and in this process minimizes the transmural voltage gradient between both regions, thus manifesting as a flat ST segment on the ECG.
BrS has been classically linked to loss of function mutations within SCN5A. These mutations blunt the magnitude of phase 0 depolarization throughout the heart. In the RVOT, this attenuation results in an imbalance of Ito current within the epicardium relative to the endocardium. Within the epicardium, the low membrane potential following phase 0 impairs the recruitment and conductance of ICa-L, Iks, Ikr,47 but not Ik1 which remains constitutively open at negative membrane potentials.48 Consequently, the imbalance of currents favoring repolarization during phase 2 leads to premature shortening and termination of the AP, manifesting as a heterogenous loss of AP “dome” in the epicardium vs the endocardium. On the ECG, the ensuing transmural voltage gradient produces the characteristic type 1 ECG with J-point elevation, and reversal of the normal sequence of repolarization between these two tissues resulting in inverted T waves.49 The transmural gradient can predispose patients to early depolarization of the prematurely repolarized epicardium, facilitating phase 2 re-entry by creating a substrate for circuitous electrical activity that can degenerate to VF and SCD.49
Repolarization vs depolarization hypotheses
More recently, the pathophysiology of BrS has been challenged, and has become a topic of ongoing debate. The dominant hypotheses focus on potential depolarization abnormalities, where discontinuous electrical propagation through the myocardium creates the substrate for re-entry, and repolarization abnormalities where transmural dispersion of repolarization as explained above facilitates phase 2 re-entry.
The depolarization hypothesis has been supported by the observation that at baseline, patients with BrS can have late potentials (LP) as measured by the signal averaged ECG (SAECG) (described below). This may be reflective of regions of tissue with delayed conduction, that are low voltage, delayed and fragmented within the RVOT epicardium.15 Also, at increased heart rates, augmentation of J-point elevation has been observed suggesting that conduction delay is a significant contributor.50 In addition, administration of ajmaline impairs INa and is associated with prolongation of the APD within the RVOT, and augmentation of pre-existing J-point elevation.51
Opponents of the depolarization hypothesis have argued that fragmented LPs may not reflect regions of delayed conduction, but instead are the ECG signature of masked circuitous electrical activity supportive of phase 2 re-entry and the repolarization hypothesis. Primary conduction delay is central to the depolarization hypothesis, but this was not observed within canine wedge preparations.52 Proponents of the repolarization hypothesis point out that the dynamic modulation of transmembrane currents, namely INa and Ito, can help to explain the intermittent presentation of the type 1 ECG. In addition, temperature disturbances can modulate the activity of NaV1.5, with increased temperature associated with accelerated inactivation, thus mimicking or exacerbating a loss of function SCN5A mutation.53 Testosterone can also increase Ito current, which could potentially explain the higher prevalence of BrS in males.13,54 Finally, J-point elevation has been shown to increase in the recovery phase of exercise where vagal tone is high.26,55 Vagal stimulation may increase Ito therefore explaining this ECG finding.25,26
Overlap syndromes
There is increasing recognition that BrS may not represent a single disease entity, but instead a group of disorders that produce the shared type 1 ECG phenotype. These diseases may manifest as purely electrical to purely structural in origin, but most often seem to present with features of both.
Early repolarization syndrome is an electrical disease entity in the absence of structural abnormalities that shares a number of features with BrS including the association with loss of function SCN5A variants, the observation of J-waves that are accentuated with vagal stimulation, and a male predominance among many other factors.13 There have also been patients in whom long QT syndrome, BrS, and cardiac conduction system disease can co-exist in patients and families with an associated gain of function SCN5A-E1478K variant.56
There has been significant interest in the overlap between BrS and arrhythmogenic right ventricular cardiomyopathy (ARVC) after histopathologic evidence of ARVC was identified in SCD victims who had prior recorded type 1 ECGs.57,58 Autopsy findings in patients with BrS have also revealed prominent fibrosis within the RVOT and RV epicardium with reduced expression of the gap junction protein Cx43.16
Within a more recent study, midwall left ventricular late gadolinium enhancement (LGE), a marker of pathologic myocardial fibrosis, was found in 8% (N=6) of BrS patients.36 One of these patients had T-wave inversion across all precordial leads, an ECG finding typical of ARVC, and another patient had a pathogenic desmoplakin mutation.
Risk stratification
Risk stratification for patients with BrS is a significant challenge. Definitive therapy to prevent SCD remains an implantable cardioverter defibrillator (ICD), but long-term complication rates are significant and yearly malignant event rates are low. A number of risk markers have been identified over the last few years (Table 3), with most demonstrating only modest predictive value. In addition, most arrhythmic events occur in so-called low risk Brugada patients.59 While patients with a previous cardiac arrest or history of concerning syncope are offered an ICD, most asymptomatic patients are managed with lifestyle modification alone (class I recommendation).12 Accurate identification of high-risk features within this patient population will help to offer effective therapy to the select the few that need it most.
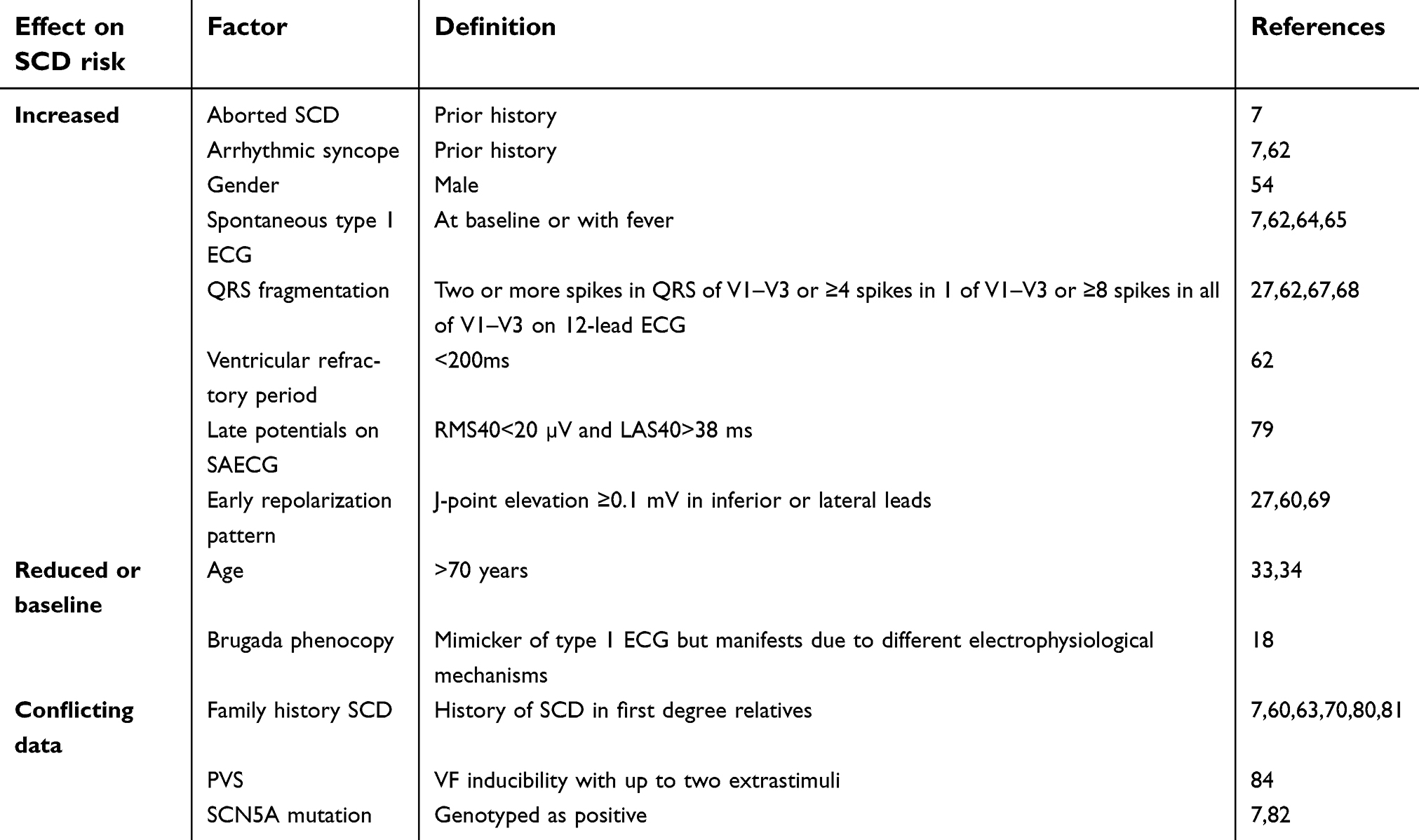 | Table 3 Effects of independent variables on SCD risk |
Symptoms
The two strongest clinical predictors for further arrhythmic events include a history of previous cardiac arrest and unexplained syncope. The yearly malignant cardiac event rates are 7.7%, 1.9% and 0.5% for previous cardiac arrest, syncope, and asymptomatic patient cohorts respectively.7 The majority of patients are asymptomatic at the time of diagnosis,31 and are usually classified as low risk. Patients with a prior history of cardiac syncope or cardiac arrest are classified as high risk for recurrence.60
Stratifying patients with a history of syncope can be difficult, and it is important to distinguish vasovagal from arrhythmic syncope based on clinical history. BrS patients can experience recurrent vasovagal syncope that is benign, and does not elevate the risk for SCD.61 Arrhythmic syncope has been consistently shown to be a predictor of future malignant cardiac events, and this effect is further strengthened when combined with the presence of a spontaneous type 1 ECG.7,62,63
A prospective study by Sacher et al61 investigated the characteristics of BrS patients with a history of syncope to determine whether syncope of arrhythmic vs nonarrhythmic origin could be readily differentiated. Patients without prodromal symptoms, that did not have an identifiable trigger, and who were unconscious for less than one minute with a rapid return to consciousness were classified as having arrhythmic syncope (N=23). Patients with loss of consciousness for greater than one minute, who had associated neurocardiogenic symptoms, and who did not endure severe physical injury were classified as having nonarrhythmic syncope (N=17). During a 5-year follow-up period, patients in the arrhythmic group experienced ventricular arrhythmias with a yearly event rate of 5.5%, and those in the nonarrhythmic group, or patients whose syncope could not otherwise be classified, did not experience any malignant cardiac events during the follow-up period.
ECG characteristics
The presence of a spontaneous type 1 ECG has been consistently shown to be associated with increased risk of malignant cardiac events,7,62,64 with a meta-analysis quantifying the increased risk as 2.3-fold,64 and patients experiencing cardiac event rates ranging from 0.5–0.8% per year.65 A greater spontaneous type 1 ECG burden on serial recordings during follow-up has also been associated with increased risk.66 In contrast, patients with a drug induced type 1 ECG have a substantially lower risk as evidenced by a lower malignant cardiac event rate and time to first event.7,62 Asymptomatic patients with fever-provoked type 1 ECGs have similar risk profiles to those patients with spontaneous patterns, with annual malignant cardiac event rates of 0.9% per year.65
A fragmented QRS has not been consistently defined, but nonetheless confers increased risk (relative risk: 3.88)67 when measured as 2 or more spikes within the QRS of V1-V3,62 or ≥4 spikes in 1 of V1–V3 or ≥8 spikes in all of V1–V3.27,68 Early repolarization pattern defined as J-point elevation ≥0.1 mV in the inferior (II, III, aVF) or lateral (I, aVL, V4–V6) leads commonly presents in BrS patients and is associated with increased risk (HR: 2.5–4.88).27,60,69 Increased risk has also been shown with augmentation of ST-elevation within leads V1–V3 within the early recovery phase after exercise testing.55
Other ECG variables that have been correlated with increased risk, but require validation due to limited datasets or conflicting data include; a prominent S-wave in lead I70 or R-wave in lead aVR,71,72 prolonged QRS duration in lead V2,73 prolonged Tpeak-Tend interval,74,75 macroscopic T-wave alternans after sodium channel blockade,76 presence of the type 1 ECG phenotype in the peripheral leads,77 and presence of first degree atrioventricular block.71
SAECG
The signal averaged ECG (SAECG) measures cardiac electrical activity using three orthogonal leads oriented in the X, Y, Z planes.78 SAECG software algorithms align each QRS complex, and average a predetermined number of complexes to eliminate noise and unmask low voltage signals.78 LPs are low voltage markers of delayed conduction, and have been correlated with increased risk for malignant cardiac events in BrS patients when the following criteria were met: root mean square voltage of the terminal 40 ms of the filtered QRS complex (RMS40) <20 µV and duration of low amplitude signals <40 µV of the QRS complex (LAS40) >38 ms.79
Age
During long-term follow-up, BrS patients aged ≥70 years did not experience any malignant cardiac events.34 This aligns with data from the multinational SABRUS study that found only 10 patients (1.5%) aged greater than 70 experienced an aborted SCA or appropriate ICD shock.33
Family history and genetics
Data describing the risk associated with a positive family history of SCD are conflicting, but the general consensus indicates that it is not a reliable independent predictor of increased risk.7,60,63,70,80,81 While there is significant evidence supporting the pathogenicity of SCN5A mutations in BrS,43 mutation status does not independently predict risk.7,82 This may be due to the identification of over 300 SCN5A variants, with each of these variants exerting different effects on INa. Due to the low prevalence of each individual variant, many larger studies do not examine the risk attributed by each variant independently, but instead segregate patients in to a simplified SCN5A positive or negative cohort.
Programmed ventricular stimulation (electrophysiology study)/pacing studies
Programmed ventricular stimulation (PVS) is an invasive electrophysiological technique that entails direct stimulation of the RV apex and RVOT, with 1–3 premature extrastimuli after a fixed number of impulses with a certain cycle length.83 Data from the multinational SABRUS study reported that VF inducibility during PVS was associated with a similar time to first arrhythmic event as those with an SCN5A mutation and family history of SCD, but longer than those with a spontaneous type 1 ECG or syncope.81 Both the PRELUDE and FINGER registries, whose patients were incorporated in the meta-analysis by Sroubek et al, discussed below, did not find inducibility to be associated with increased risk. However the PRELUDE registry did find that a ventricular refractory period <200 ms was a predictor of increased risk (HR: 3.91).7,62
A meta-analysis by Sroubek et al84 investigated the role of PVS in risk stratifying BrS patients. They pooled and analyzed data from eight prospective and observational studies, and included asymptomatic patients with a type 1 ECG and symptomatic patients with positive provocative testing. In total, 1,312 patients were analyzed with a median follow-up of 38 months. They found that inducibility of ventricular tachycardia (VT) or VF was associated with a 2–3-fold increased risk of malignant cardiac events. Higher risks were attributed to patients who were induced with a single extrastimulus versus double. A third extrastimulus did not offer any further predictive value. Patients without inducible arrhythmias, however, also experienced ventricular events in follow-up. PVS is currently classified as a class IIb recommendation12 where it may be reasonably used for risk stratification purposes by performing 1–2 extrastimuli in asymptomatic patients with a spontaneous type 1 ECG.12
Risk by combining multiple factors
Stratifying patients with multiple risk factors increases the power for prediction, for example, by combining high risk markers such as early repolarization pattern and fragmented QRS,27 or syncope and spontaneous type 1 ECG.62 Other studies have proposed score-based models that attempt to correlate higher risk with increasing score. The Shanghai Score system13 is one such model that has undergone successful initial validation.14 They found 10-year post-diagnosis lethal arrhythmic event rates to be 0%, 6.8%, 17.6% and 25.0% with scores of ≤3.0 points, 3.5 points, 4.0–5.0 points and ≥ 5.5 points respectively in patients without genetic testing. A similar score-based model proposed by Sieira et al63 incorporated spontaneous type 1 ECG, early familial SCD, inducible VT or VF with PVS, and presentation as either syncope, sinus node dysfunction, or SCD.
Management
Lifestyle
The treatment algorithm for BrS patients is summarized in Figure 2.12 Fevers should be treated aggressively with antipyretic medications. Some anti-arrhythmic, psychotropic and anesthetic medications along with substances such as cocaine and excessive alcohol should be avoided (
 | Figure 2 Management algorithm for Brugada syndrome patients. Class designations are based on the guidelines for the management of ventricular arrhythmias.12 Recommendations without class designations were derived based on expert consensus.13 Modified from Antzelevitch C, Yan GX, Ackerman MJ, et al. J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Europace. 2017;19(4):665–694.13 with permission from John Wiley and Sons.Abbreviations: ES, extrastimuli; ICD, implantable cardiac defibrillator; ILR, implantable loop recorder; NAR, nocturnal agonal respiration; RVOT, right ventricular outflow tract; SCA, sudden cardiac arrest; VT, ventricular tachycardia; VF, ventricular fibrillation. |
ICD
The young age at BrS diagnosis has the potential to translate to a high cumulative lifetime risk for ICD-related complications. A meta-analysis reported yearly appropriate ICD shock rates of 0.9% and 2.5% for primary and secondary prevention patients respectively, and yearly rates of inappropriate shocks and other ICD-related complications at 3.9% and 3.4% respectively.85 Strategies to reduce complication rates have been proposed that include strategic ICD programming and use of subcutaneous ICDs.85,86 Current class I indications for ICD therapy are reserved for patients with a spontaneous type 1 ECG with a history of malignant cardiac events or arrhythmic syncope.12
Medications
ICD candidates who experience frequent ICD shocks or those who refuse an ICD can be treated with quinidine (class I recommendation).12 Quinidine is a class 1A anti-arrhythmic drug that has Ito and IKr inhibition properties and has been shown to normalize the type 1 ECG pattern in some patients.87,88 On electrophysiological study, quinidine prevented the occurrence of PVS-induced ventricular arrhythmia.89,90 In patients whose arrhythmias were suppressed, no malignant events occurred while on chronic quinidine therapy during a 7.7–10 year follow-up period.89,90 In a nationwide Spanish study,91 patients experiencing frequent shocks or electrical storm with ICDs experienced a significant absolute reduction in shocks from a median of six shocks per patient to zero following initiation of quinidine. They noted that risk factors for recurrent shocks after initiation of quinidine were temporary discontinuation (HR: 4.6), and the number of shocks the patient experienced prior to quinidine (HR: 1.13).
Despite the effectiveness of quinidine, 76% of countries worldwide are unable to access it.92 For primary prevention, data on the use of quinidine in asymptomatic patients and those who are ICD candidates remains limited.7,93 The efficacy of quinidine versus ICD therapy has not yet been assessed. The QUIDAM study which set out to answer this question was terminated prematurely due to high patient attrition as a result of frequent side effects within the quinidine cohort. The most common side effect was diarrhea.91,93 Other side effects include drug-induced lupus, photophobia, photosensitivity, tinnitus, headaches, thrombocytopenia and esophagitis.93,94
Electrical storms are defined as three or more ventricular arrhythmias within a 24-hour period.95 Isoproterenol has demonstrated clear efficacy in treating patients with BrS who present with electrical storm.95–97 Consistent with the repolarization hypothesis, isoproterenol stimulates β-adrenergic receptors triggering an influx of ICa-L, and rebalancing inward and outward currents leading to recovery of the phase 2 epicardial dome. In theory, isoproterenol should concomitantly lead to an increase in heart rate that should worsen the storm, but this has not been observed.95–97 Alternatively, oral quinidine can be used when electrical storm is refractory to isoproterenol, as bridging from isoproterenol, on its own, or in pediatric populations.97–99
The burden of atrial arrhythmias can be high in BrS patients.27–30,71 Many of the typical treatments for atrial arrhythmias are contraindicated in BrS.18 A promising case series demonstrated the safety and efficacy of quinidine for atrial and ventricular arrhythmias in patients with BrS.100
Ablation
Patients who experience frequent ICD shocks or those who refuse an ICD can undergo catheter radiofrequency ablation (class I recommendation).12 Most ablation protocols target the arrhythmogenic substrate characterized by abnormal local electrograms with features of low voltage, fractionation and late potentials within the epicardium of the anterior and anteroseptal RVOT where there is significant Ito expression, and a minority of patients have displayed endocardial targets as well. Some patients may have a premature ventricular complex (PVC) that is inducible and can be ablated. Concomitant sodium channel blocker infusion can extend the detectable arrhythmogenic substrate. At the end of the procedure as a test of “cure”, patients may be assessed for noninducibility of previously inducible VT or VF,16,101 or the amelioration of a type 1 ECG pattern or premature triggering beats.16,101,102
Haïssaguerre et al performed endocardial ablation of inducible PVC triggers for 3 BrS patients and on follow-up over 17 months, demonstrated no recurrence of sustained ventricular arrhythmias.102 Nademanee et al15 was the first to systematically perform endocardial and epicardial mapping over the RVOT in 9 patients, and noted that the arrhythmogenic substrate localized to the anterior epicardial RVOT. Epicardial ablation in this location was associated with event-free survival over a mean of 20 months. Within a larger subsequent study (Figure 3) with epicardial voltage mapping in symptomatic BrS patients (N=135), the area encompassing the arrhythmogenic substrate within the right ventricular epicardium was greater following ajmaline infusion, and ablation of this region normalized the type 1 ECG pattern resulting in noninducibility of VT/VF.51 Over a median prospective follow-up of 10 months, all but 2 patients displayed a normal baseline and ajmaline provocation challenge. A recent systematic review has shown that endocardial mapping does not identify the arrhythmogenic substrate in 93% of cases.103 Prevention of VT/VT during long-term follow-up post ablation of PVCs, endocardial, and epicardial targets was 80%, 71% and 97% of cases respectively. Longer-term prospective follow-up is needed to corroborate the efficacy of ablation with the recurrence of any ventricular arrhythmias before its more widespread use can be adopted.
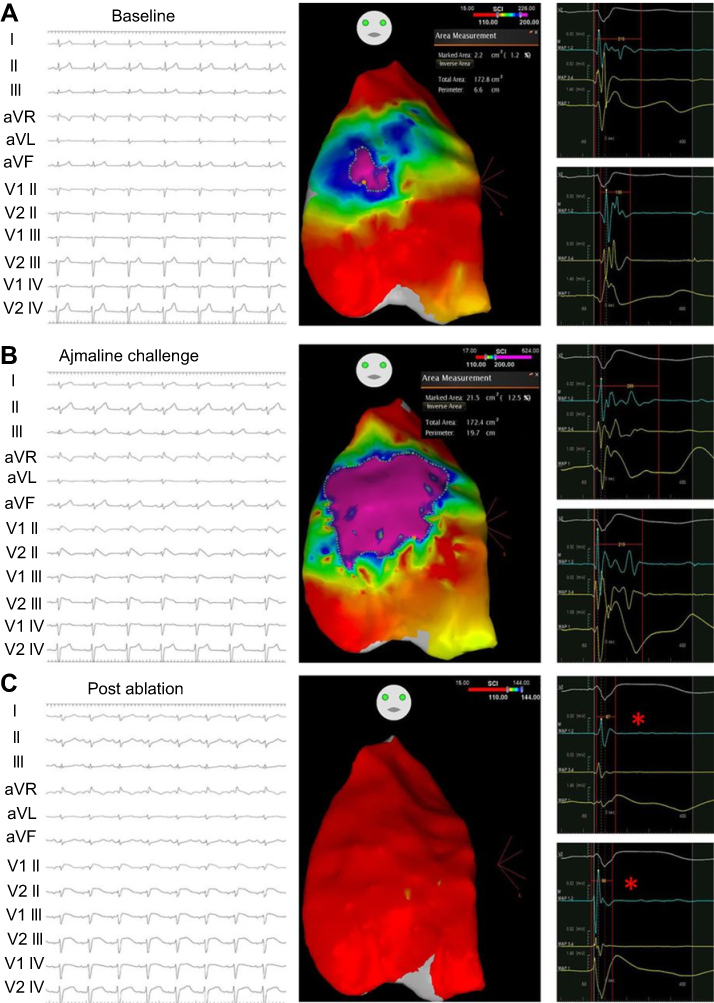 | Figure 3 Epicardial ablation of a Brugada syndrome patient. (A) Baseline type 2 ECG pattern visible with CARTO maps displaying abnormally prolonged potentials (210 ms) as purple, potentials between 110 and 200 ms as blue/green, and example electrograms on the right. (B) Expansion of tissue area displaying abnormally prolonged potentials with presentation of a type 1 ECG following ajmaline infusion. (C) Reinfusion of ajmaline after epicardial ablation has ameliorated the type 1 ECG pattern and abnormal prolonged potentials (red asterisk). Reproduced with permission from Pappone C, Brugada J, Vicedomini G, et al. Electrical substrate elimination in 135 consecutive patients with Brugada syndrome. Circ Arrhythm Electrophysiol. 2017;10(5):e005053, https://www.ahajournals.org/journal/circep. The Creative Commons license does not apply to this content. Use of the material in any format is prohibited without written permission from the publisher, Wolters Kluwer Health, Inc. Please contact [email protected] for further information.51 |
Conclusion
There is increasing recognition that a diagnosis for BrS is not binary, but instead is represented along a continuum of risk. Accurate risk stratification represents the major challenge for the management of Brugada syndrome. Conventional risk prediction models have significant limitations, and there is clearly a need for novel risk markers and models. Significant long-term complication rates from traditional transvenous ICDs have to be outweighed against the potential benefits and are rarely indicated for primary prevention. The emerging approach of ablation of the arrhythmogenic substrate shows promising preliminary results but requires longer-term studies for validation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol. 1992;20(6):1391–1396.
2. Papadakis M, Raju H, Behr Elijah R, et al. Sudden Cardiac Death With Autopsy Findings of Uncertain Significance. Circ Arrhythm Electrophysiol. 2013;6(3):588–596. doi:10.1161/CIRCEP.113.000111
3. Tsuji H, Sato T, Morisaki K, Iwasaka T. Prognosis of subjects with Brugada-type electrocardiogram in a population of middle-aged Japanese diagnosed during a health examination. Am J Cardiol. 2008;102(5):584–587. doi:10.1016/j.amjcard.2008.04.066
4. Gervacio-Domingo G, Isidro J, Tirona J, et al. The Brugada type 1 electrocardiographic pattern is common among Filipinos. J Clin Epidemiol. 2008;61(10):1067–1072. doi:10.1016/j.jclinepi.2007.11.009
5. Patel SS, Anees S, Ferrick KJ. Prevalence of a Brugada pattern electrocardiogram in an urban population in the United States. Pacing Clin Electrophysiol. 2009;32(6):704–708. doi:10.1111/j.1540-8159.2009.02354.x
6. Gallagher MM, Forleo GB, Behr ER, et al. Prevalence and significance of Brugada-type ECG in 12,012 apparently healthy European subjects. Int J Cardiol. 2008;130(1):44–48. doi:10.1016/j.ijcard.2007.07.159
7. Probst V, Veltmann C, Eckardt L, et al. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada syndrome registry. Circulation. 2010;121(5):635–643. doi:10.1161/CIRCULATIONAHA.109.887026
8. Wilde Arthur AM, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome. Circulation. 2002;106(19):2514–2519.
9. Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111(5):659–670. doi:10.1161/01.CIR.0000152479.54298.51
10. Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932–1963. doi:10.1016/j.hrthm.2013.05.014
11. Bayes de Luna A, Brugada J, Baranchuk A, et al. Current electrocardiographic criteria for diagnosis of Brugada pattern: a consensus report. J Electrocardiol. 2012;45(5):433–442. doi:10.1016/j.jelectrocard.2012.06.004
12. Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Heart Rhythm. 2018;15(10):e190–e252. doi:10.1016/j.hrthm.2017.10.035
13. Antzelevitch C, Yan GX, Ackerman MJ, et al. J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Europace. 2017;19(4):665–694. doi:10.1093/europace/euw235
14. Kawada S, Morita H, Antzelevitch C, et al. Shanghai Score System for diagnosis of Brugada syndrome: validation of the score system and system and reclassification of the patients. JACC Clin Electrophysiol. 2018;4(6):724–730. doi:10.1016/j.jacep.2018.02.009
15. Nademanee K, Veerakul G, Chandanamattha P, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123(12):1270–1279. doi:10.1161/CIRCULATIONAHA.110.972612
16. Nademanee K, Raju H, de Noronha SV, et al. Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome. J Am Coll Cardiol. 2015;66(18):1976–1986. doi:10.1016/j.jacc.2015.08.862
17. Veltmann C, Papavassiliu T, Konrad T, et al. Insights into the location of type I ECG in patients with Brugada syndrome: correlation of ECG and cardiovascular magnetic resonance imaging. Heart Rhythm. 2012;9(3):414–421. doi:10.1016/j.hrthm.2011.10.032
18. Baranchuk A, Nguyen T, Ryu MH, et al. Brugada phenocopy: new terminology and proposed classification. Ann Noninvasive Electrocardio. 2012;17(4):299–314. doi:10.1111/j.1542-474X.2012.00525.x
19. Cerrato N, Giustetto C, Gribaudo E, et al. Prevalence of type 1 brugada electrocardiographic pattern evaluated by twelve-lead twenty-four-hour holter monitoring. Am J Cardiol. 2015;115(1):52–56. doi:10.1016/j.amjcard.2014.10.007
20. Gray B, Kirby A, Kabunga P, et al. Twelve-lead ambulatory electrocardiographic monitoring in Brugada syndrome: potential diagnostic and prognostic implications. Heart Rhythm. 2017;14(6):866–874. doi:10.1016/j.hrthm.2017.02.026
21. Antzelevitch C, Patocskai B. Ajmaline-induced slowing of conduction in the right ventricular outflow tract cannot account for ST elevation in patients with type I Brugada ECG. Circ Arrhythm Electrophysiol. 2017;10(10):e005775. doi:10.1161/CIRCEP.117.005775
22. Wolpert C, Echternach C, Veltmann C, et al. Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm. 2005;2(3):254–260. doi:10.1016/j.hrthm.2004.11.025
23. Hashemi A, Shahrzad S, Shahrzad S, et al. Positive Brugada challenge test in V1R–V3R as a predictor of malignant prognosis in Brugada patients. Ann Noninvas Electrocardiol. 2013;18(5):421–426. doi:10.1111/anec.12055
24. Viskin S, Rosso R. Read my lips: a positive Ajmaline test does not always mean you have Brugada syndrome∗. JACC Clin Electrophysiol. 2017;3(12):1409–1411. doi:10.1016/j.jacep.2017.05.016
25. Ikeda T, Abe A, Yusu S, et al. The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17(6):602–607. doi:10.1111/j.1540-8167.2006.00424.x
26. Mizumaki K, Fujiki A, Tsuneda T, et al. Vagal activity modulates spontaneous augmentation of ST elevation in the daily life of patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2004;15(6):667–673. doi:10.1046/j.1540-8167.2004.03601.x
27. Tokioka K, Kusano KF, Morita H, et al. Electrocardiographic parameters and fatal arrhythmic events in patients with Brugada syndrome: combination of depolarization and repolarization abnormalities. J Am Coll Cardiol. 2014;63(20):2131–2138. doi:10.1016/j.jacc.2014.01.072
28. Kusano KF, Taniyama M, Nakamura K, et al. Atrial fibrillation in patients with Brugada syndrome: relationships of gene mutation, electrophysiology, and clinical backgrounds. J Am Coll Cardiol. 2008;51(12):1169–1175. doi:10.1016/j.jacc.2007.10.060
29. Giustetto C, Cerrato N, Gribaudo E, et al. Atrial fibrillation in a large population with Brugada electrocardiographic pattern: prevalence, management, and correlation with prognosis. Heart Rhythm. 2014;11(2):259–265. doi:10.1016/j.hrthm.2013.10.043
30. Schimpf R, Giustetto C, Eckardt L, et al. Prevalence of supraventricular tachyarrhythmias in a cohort of 115 patients with Brugada syndrome. Ann Noninvas Electrocardiol. 2008;13(3):266–269. doi:10.1111/j.1542-474X.2008.00230.x
31. Benito B, Sarkozy A, Mont L, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol. 2008;52(19):1567–1573. doi:10.1016/j.jacc.2008.07.052
32. Andorin A, Behr ER, Denjoy I, et al. Impact of clinical and genetic findings on the management of young patients with Brugada syndrome. Heart Rhythm. 2016;13(6):1274–1282. doi:10.1016/j.hrthm.2016.02.013
33. Milman A, Andorin A, Gourraud JB, et al. Age of first arrhythmic event in Brugada syndrome: data from the SABRUS (survey on arrhythmic events in Brugada syndrome) in 678 patients. Circ Arrhythm Electrophysiol. 2017;10:12. doi:10.1161/CIRCEP.117.005222
34. Conte G, De Asmundis C, Sieira J, et al. Clinical characteristics, management, and prognosis of elderly patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2014;25(5):514–519. doi:10.1111/jce.12359
35. Papavassiliu T, Wolpert C, FlÜChter S, et al. Magnetic resonance imaging findings in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2004;15(10):1133–1138. doi:10.1046/j.1540-8167.2004.03681.x
36. Gray B, Gnanappa GK, Bagnall RD, et al. Relations between right ventricular morphology and clinical, electrical and genetic parameters in Brugada Syndrome. PLoS One. 2018;13(4):e0195594. doi:10.1371/journal.pone.0195594
37. Papavassiliu T, Veltmann C, Doesch C, et al. Spontaneous type 1 electrocardiographic pattern is associated with cardiovascular magnetic resonance imaging changes in Brugada syndrome. Heart Rhythm. 2010;7(12):1790–1796. doi:10.1016/j.hrthm.2010.09.004
38. Catalano O, Antonaci S, Moro G, et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur Heart J. 2009;30(18):2241–2248. doi:10.1093/eurheartj/ehp252
39. Bastiaenen R, Cox AT, Castelletti S, et al. Late gadolinium enhancement in Brugada syndrome: a marker for subtle underlying cardiomyopathy? Heart Rhythm. 2017;14(4):583–589. doi:10.1016/j.hrthm.2016.12.004
40. Tessa C, Del Meglio J, Ghidini Ottonelli A, et al. Evaluation of Brugada syndrome by cardiac magnetic resonance. Int J Cardiovasc Imaging. 2012;28(8):1961–1970. doi:10.1007/s10554-012-0009-5
41. Curcio A, Santarpia G, Indolfi C. The Brugada syndrome- from gene to therapy. Circ J. 2017;81(3):290–297. doi:10.1253/circj.CJ-16-0971
42. Kapplinger JD, Tester DJ, Alders M, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33–46. doi:10.1016/j.hrthm.2009.09.069
43. Hosseini SM, Kim R, Udupa S, et al. Reappraisal of reported genes for sudden arrhythmic death. Circulation. 2018;138(12):1195–1205. doi:10.1161/CIRCULATIONAHA.118.035070
44. Probst V, Wilde Arthur AM, Barc J, et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet. 2009;2(6):552–557. doi:10.1161/CIRCGENETICS.109.853374
45. Spoonamore KG, Ware SM. Genetic testing and genetic counseling in patients with sudden death risk due to heritable arrhythmias. Heart Rhythm. 2016;13(3):789–797. doi:10.1016/j.hrthm.2015.11.013
46. Boukens BJ, Christoffels VM, Coronel R, Moorman AF. Developmental basis for electrophysiological heterogeneity in the ventricular and outflow tract myocardium as a substrate for life-threatening ventricular arrhythmias. Circ Res. 2009;104(1):19–31. doi:10.1161/CIRCRESAHA.108.188698
47. Morita H, Zipes DP, Wu J. Brugada syndrome: insights of ST elevation, arrhythmogenicity, and risk stratification from experimental observations. Heart Rhythm. 2009;6(11,Supplement):S34–S43. doi:10.1016/j.hrthm.2009.07.018
48. Kurata HT, Phillips LR, Rose T, et al. Molecular basis of inward rectification: polyamine interaction sites located by combined channel and ligand mutagenesis. J Gen Physiol. 2004;124(5):541–554. doi:10.1085/jgp.200409159
49. Naccarelli GV, Antzelevitch C. The Brugada syndrome: clinical, genetic, cellular, and molecular abnormalities. Am J Med. 2001;110(7):573–581.
50. Amin Ahmad S, de Groot Elisabeth AA, Ruijter Jan M, Wilde Arthur AM, Tan Hanno L. Exercise-induced ECG changes in Brugada syndrome. Circ Arrhythm Electrophysiol. 2009;2(5):531–539. doi:10.1161/CIRCEP.109.862441
51. Pappone C, Brugada J, Vicedomini G, et al. Electrical substrate elimination in 135 consecutive patients with Brugada syndrome. Circ Arrhythm Electrophysiol. 2017;10(5):e005053. doi:10.1161/CIRCEP.117.005053
52. Szel T, Antzelevitch C. Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J Am Coll Cardiol. 2014;63(19):2037–2045. doi:10.1016/j.jacc.2014.01.067
53. Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85(9):803–809.
54. Shimizu W, Matsuo K, Kokubo Y, et al. Sex hormone and gender difference—role of testosterone on male predominance in Brugada syndrome. J Cardiovasc Electrophysiol. 2007;18(4):415–421. doi:10.1111/j.1540-8167.2006.00743.x
55. Makimoto H, Nakagawa E, Takaki H, et al. Augmented ST-segment elevation during recovery from exercise predicts cardiac events in patients with Brugada syndrome. J Am Coll Cardiol. 2010;56(19):1576–1584. doi:10.1016/j.jacc.2010.06.033
56. Veltmann C, Barajas-Martinez H, Wolpert C, et al. Further insights in the most common SCN5A mutation causing overlapping phenotype of long QT syndrome, Brugada syndrome, and conduction defect. J Am Heart Assoc. 2016;5:7. doi:10.1161/JAHA.116.003379
57. Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G. Right bundle branch block, right precordial st-segment elevation, and sudden death in young people. Circulation. 2001;103(5):710–717.
58. Coronel R, Casini S, Koopmann TT, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation. 2005;112(18):2769–2777. doi:10.1161/CIRCULATIONAHA.105.532614
59. Raju H, Papadakis M, Govindan M, et al. Low prevalence of risk markers in cases of sudden death due to Brugada syndrome relevance to risk stratification in Brugada syndrome. J Am Coll Cardiol. 2011;57(23):2340–2345. doi:10.1016/j.jacc.2010.11.067
60. Kamakura S, Ohe T, Nakazawa K, et al. Long-term prognosis of probands with Brugada-pattern ST-elevation in leads V1-V3. Circ Arrhythm Electrophysiol. 2009;2(5):495–503. doi:10.1161/CIRCEP.108.816892
61. Sacher F, Arsac F, Wilton SB, et al. Syncope in Brugada syndrome patients: prevalence, characteristics, and outcome. Heart Rhythm. 2012;9(8):1272–1279. doi:10.1016/j.hrthm.2012.04.013
62. Priori SG, Gasparini M, Napolitano C, et al. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol. 2012;59(1):37–45. doi:10.1016/j.jacc.2011.08.064
63. Sieira J, Conte G, Ciconte G, et al. A score model to predict risk of events in patients with Brugada syndrome. Eur Heart J. 2017;38(22):1756–1763. doi:10.1093/eurheartj/ehx119
64. Bayoumy A, Gong M-Q, Christien LKH, et al. Spontaneous type 1 pattern, ventricular arrhythmias and sudden cardiac death in Brugada syndrome: an updated systematic review and meta-analysis. J Geriatr Cardiol. 2017;14(10):639–643. doi:10.11909/j.issn.1671-5411.2017.10.010
65. Mizusawa Y, Morita H, Adler A, et al. Prognostic significance of fever-induced Brugada syndrome. Heart Rhythm. 2016;13(7):1515–1520. doi:10.1016/j.hrthm.2016.03.044
66. Castro Hevia J, Dorantes Sanchez M, Martinez Lopez F, et al. Multiple serial ECGs aid with the diagnosis and prognosis of Brugada syndrome. Int J Cardiol. 2019;277:130–135. doi:10.1016/j.ijcard.2018.08.089
67. Meng L, Letsas KP, Baranchuk A, et al. Meta-analysis of fragmented QRS as an electrocardiographic predictor for arrhythmic events in patients with Brugada syndrome. Front Physiol. 2017;8:678. doi:10.3389/fphys.2017.00678
68. Morita H, Kusano KF, Miura D, et al. Fragmented QRS as a marker of conduction abnormality and a predictor of prognosis of Brugada syndrome. Circulation. 2008;118(17):1697–1704. doi:10.1161/CIRCULATIONAHA.108.770917
69. Kawata H, Morita H, Yamada Y, et al. Prognostic significance of early repolarization in inferolateral leads in Brugada patients with documented ventricular fibrillation: a novel risk factor for Brugada syndrome with ventricular fibrillation. Heart Rhythm. 2013;10(8):1161–1168. doi:10.1016/j.hrthm.2013.04.009
70. Calo L, Giustetto C, Martino A, et al. A new electrocardiographic marker of sudden death in Brugada syndrome: the S-wave in lead I. J Am Coll Cardiol. 2016;67(12):1427–1440. doi:10.1016/j.jacc.2016.01.024
71. Maury P, Rollin A, Sacher F, et al. Prevalence and prognostic role of various conduction disturbances in patients with the Brugada syndrome. Am J Cardiol. 2013;112(9):1384–1389. doi:10.1016/j.amjcard.2013.06.033
72. Babai Bigi MA, Aslani A, Shahrzad S. aVR sign as a risk factor for life-threatening arrhythmic events in patients with Brugada syndrome. Heart Rhythm. 2007;4(8):1009–1012. doi:10.1016/j.hrthm.2007.04.017
73. Ohkubo K, Watanabe I, Okumura Y, et al. Prolonged QRS duration in lead V2 and risk of life-threatening ventricular arrhythmia in patients with Brugada syndrome. Int Heart J. 2011;52(2):98–102.
74. Maury P, Sacher F, Gourraud JB, et al. Increased Tpeak-Tend interval is highly and independently related to arrhythmic events in Brugada syndrome. Heart Rhythm. 2015;12(12):2469–2476. doi:10.1016/j.hrthm.2015.07.029
75. Tse G, Gong M, Li CKH, et al. T(peak)-T(end), T(peak)-T(end)/QT ratio and T(peak)-T(end) dispersion for risk stratification in Brugada Syndrome: a systematic review and meta-analysis. J Arrhythmia. 2018;34(6):587–597. doi:10.1002/joa3.12118
76. Tada T, Kusano KF, Nagase S, et al. Clinical significance of macroscopic T-wave alternans after sodium channel blocker administration in patients with Brugada syndrome. J Cardiovasc Electrophsiol. 2008;19:56–61.
77. Rollin A, Sacher F, Gourraud JB, et al. Prevalence, characteristics, and prognosis role of type 1 ST elevation in the peripheral ECG leads in patients with Brugada syndrome. Heart Rhythm. 2013;10(7):1012–1018. doi:10.1016/j.hrthm.2013.03.001
78. Steinberg JS, Berbari EJ. The signal-averaged electrocardiogram: update on clinical applications. J Cardiovasc Electrophysiol. 1996;7(10):972–988.
79. Huang Z, Patel C, Li W, et al. Role of signal-averaged electrocardiograms in arrhythmic risk stratification of patients with Brugada syndrome: a prospective study. Heart Rhythm. 2009;6(8):1156–1162. doi:10.1016/j.hrthm.2009.05.007
80. Sarkozy A, Sorgente A, Boussy T, et al. The value of a family history of sudden death in patients with diagnostic type I Brugada ECG pattern. Eur Heart J. 2011;32(17):2153–2160. doi:10.1093/eurheartj/ehr129
81. Milman A, Hochstadt A, Andorin A, et al. Time-to-first appropriate shock in patients implanted prophylactically with an implantable cardioverter-defibrillator: data from the survey on arrhythmic events in Brugada Syndrome (SABRUS). EP Europace. 2018;21(5):796–802.
82. Raharjo SB, Maulana R, Maghfirah I, et al. SCN5A gene mutations and the risk of ventricular fibrillation and syncope in Brugada syndrome patients: a meta-analysis. J Arrhythmia. 2018;34(5):473–477. doi:10.1002/joa3.12097
83. Kossaify A, Refaat M. Programmed ventricular stimulation–indications and limitations: a comprehensive update and review. Hellenic J Cardiol. 2013;54(1):39–46.
84. Sroubek J, Probst V, Mazzanti A, et al. Programmed ventricular stimulation for risk stratification in the brugada syndrome: a pooled analysis. Circulation. 2016;133(7):622–630. doi:10.1161/CIRCULATIONAHA.115.017885
85. Olde Nordkamp LR, Postema PG, Knops RE, et al. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: a systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. 2016;13(2):443–454. doi:10.1016/j.hrthm.2015.09.010
86. Rudic B, Tulumen E, Berlin V, et al. Low prevalence of inappropriate shocks in patients with inherited arrhythmia syndromes with the subcutaneous implantable defibrillator single center experience and long-term follow-up. J Am Heart Assoc. 2017;6:10. doi:10.1161/JAHA.117.006265
87. Alings M, Dekker L, Sadee A, Wilde A. Quinidine induced electrocardiographic normalization in two patients with Brugada syndrome. Pacing Clin Electrophysiol. 2001;24(9 Pt 1):1420–1422.
88. Zhou P, Yang X, Li C, Gao Y, Hu D. Quinidine depresses the transmural electrical heterogeneity of transient outward potassium current of the right ventricular outflow tract free wall. J Cardiovasc Dis Res. 2010;1(1):12–18. doi:10.4103/0975-3583.59979
89. Belhassen B, Rahkovich M, Michowitz Y, Glick A, Viskin S. Management of Brugada syndrome: thirty-three-year experience using electrophysiologically guided therapy with class 1A antiarrhythmic drugs. Circ Arrhythm Electrophysiol. 2015;8(6):1393–1402. doi:10.1161/CIRCEP.115.003109
90. Bouzeman A, Traulle S, Messali A, et al. Long-term follow-up of asymptomatic Brugada patients with inducible ventricular fibrillation under hydroquinidine. Europace. 2014;16(4):572–577. doi:10.1093/europace/eut279
91. Anguera I, García-Alberola A, Dallaglio P, et al. Shock reduction with long-term quinidine in patients with Brugada syndrome and malignant ventricular arrhythmia episodes. J Am Coll Cardiol. 2016;67(13):1653–1654. doi:10.1016/j.jacc.2016.01.042
92. Viskin S, Wilde AAM, Guevara-Valdivia ME, et al. Quinidine, a life-saving medication for Brugada syndrome, is inaccessible in many countries. J Am Coll Cardiol. 2013;61(23):2383–2387. doi:10.1016/j.jacc.2013.02.077
93. Andorin A, Gourraud J-B, Mansourati J, et al. The QUIDAM study: hydroquinidine therapy for the management of Brugada syndrome patients at high arrhythmic risk. Heart Rhythm. 2017;14(8):1147–1154. doi:10.1016/j.hrthm.2017.04.019
94. Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110(13):1731–1737. doi:10.1161/01.CIR.0000143159.30585.90
95. Maury P, Couderc P, Delay M, Boveda S, Brugada J. Electrical storm in Brugada syndrome successfully treated using isoprenaline. EP Europace. 2004;6(2):130–133. doi:10.1016/j.eupc.2003.11.009
96. Jongman JK, Jepkes-Bruin N, Ramdat Misier AR, et al. Electrical storms in Brugada syndrome successfully treated with isoproterenol infusion and quinidine orally. Neth Heart J. 2007;15(4):151–155.
97. Watanabe A, Fukushima Kusano K, Morita H, et al. Low-dose isoproterenol for repetitive ventricular arrhythmia in patients with Brugada syndrome. Eur Heart J. 2006;27(13):1579–1583. doi:10.1093/eurheartj/ehl060
98. Bettiol K, Gianfranchi L, Scarfo S, Pacchioni F, Pedaci M, Alboni P. Successful treatment of electrical storm with oral quinidine in Brugada syndrome. Ital Heart J Suppl. 2005;6(7):601–602.
99. Mok NS, Chan NY, Chiu AC. Successful use of quinidine in treatment of electrical storm in Brugada syndrome. Pacing Clin Electrophysiol. 2004;27(6 Pt 1):821–823. doi:10.1111/j.1540-8159.2004.00537.x
100. Halperin L, Mellor G, Talajic M, Krahn A, Tadros R, Laksman Z. Quinidine effective for the management of ventricular and atrial arrhythmias associated with Brugada syndrome. HeartRhythm Case Rep. 2018;4(7):270–272. doi:10.1016/j.hrcr.2018.01.008
101. Shelke A, Tachil A, Saggu D, Jesuraj ML, Yalagudri S, Narasimhan C. Catheter ablation for electrical storm in Brugada syndrome: results of substrate based ablation. Indian Heart J. 2018;70(2):296–302. doi:10.1016/j.ihj.2017.07.019
102. Haïssaguerre M, Extramiana F, Hocini M, et al. Mapping and ablation of ventricular fibrillation associated with long-QT and Brugada syndromes. Circulation. 2003;108(8):925–928. doi:10.1161/01.CIR.0000088781.99943.95
103. Fernandes GC, Fernandes A, Cardoso R, et al. Ablation strategies for the management of symptomatic Brugada syndrome: a systematic review. Heart Rhythm. 2018;15(8):1140–1147. doi:10.1016/j.hrthm.2018.03.019
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.