Back to Journals » OncoTargets and Therapy » Volume 13
Both Methylation and Copy Number Variation Participated in the Varied Expression of PRAME in Multiple Myeloma
Authors Yang L, Dao FT, Chang Y, Wang YZ, Li LD, Chen WM, Long LY, Liu YR, Lu J, Liu KY, Qin YZ ![]()
Received 3 December 2019
Accepted for publication 3 July 2020
Published 31 July 2020 Volume 2020:13 Pages 7545—7553
DOI https://doi.org/10.2147/OTT.S240979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Lu Yang, Feng-Ting Dao, Yan Chang, Ya-Zhe Wang, Ling-Di Li, Wen-Min Chen, Ling-Yu Long, Yan-Rong Liu, Jin Lu, Kai-Yan Liu, Ya-Zhen Qin
Peking University People’s Hospital, Peking University Institute of Hematology, National Clinical Research Center for Hematologic Disease, Beijing, People’s Republic of China
Correspondence: Ya-Zhen Qin Tel/ Fax +86-10-88324672
Email [email protected]
Purpose: The cancer-testis antigen, which is a preferentially expressed antigen of melanoma (PRAME), is an ideal target for immunotherapy and cancer vaccines. Since the expression of this antigen is relevant to therapy responses, the heterogeneity in its expression and the underlying mechanism need to be investigated.
Patients and Methods: Plasma cell sorting was performed in 48 newly diagnosed multiple myeloma (MM) patients. Real-time quantitative PCR was performed to examine the PRAME transcript levels and gene copy numbers. Bisulfate clone sequencing of the PRAME promoter and exon 1b regions was performed in 4 patients. Quantitative methylation-specific PCR of the +287 CpG site was performed for all patients. The human MM cell lines RPMI8226, LP-1 and MOLP-2 were treated with 5-azacytidine.
Results: The median PRAME transcript level was 3.1% (range: 0– 298.3%) in the plasma cells sorted from the 48 MM patients. Eleven (22.9%) and 37 (77.1%) patients were individually categorized into the PRAME low- and high-expression groups according to the cut-off value of 0.05%. The methylation ratios of the promoter and the 3ʹ region of exon 1b region were both negatively related to the transcript levels. The degrees of methylation at the +287 CpG site were significantly negatively related to the transcript levels in all 48 patients (r=− 0.44, P=0.0018), and those in the high-expression group (r=− 0.69, P< 0.0001) but not those in the low-expression group (r=− 0.27, P=0.43). All 5 patients with homozygous deletions were categorized into the low-expression group. There were no significant differences in the PRAME transcript levels between the hemizygous deletion (n=8) and no deletion (n=35) groups (P=0.40). Furthermore, the PRAME transcript levels significantly increased in the MM cell lines after treatment with 5-azacytidine.
Conclusion: Both methylation and copy number variation may participate in the regulation of PRAME expression in MM; in patients with no homozygous deletion, PRAME expression is mainly controlled by methylation, and a proportion of fairly low expression is caused by homozygous deletion.
Keywords: multiple myeloma, preferentially expressed antigen of melanoma, PRAME, gene methylation, gene copy number variation
Introduction
Cancer immunotherapy has been considered a breakthrough in cancer treatment, providing durable and potent clinical efficacy in a range of cancer types.1,2 The clinical utility of cellular immunotherapy, such as adoptive T cell therapy, greatly depends on the identification of candidate target antigens. Cancer-testis antigens (CTAs) have been defined as promising targets because they are expressed at high levels in a range of human tumours, but are absent or detected at low levels in normal tissues except for the testis and placenta.3 Many clinical trials of cellular immunotherapies that target CTAs are ongoing.
As a CTA family member, preferentially expressed antigen of melanoma (PRAME) is an ideal target for immunotherapy and cancer vaccines.1,2,4,5 PRAME is expressed at very low levels in normal tissues, but is highly in solid tumours and hematological malignancies.6–17 When performing PRAME-targeted therapy, the heterogeneity of its expression should be considered because this heterogeneity may limit the therapeutic response. Therefore, studies from our laboratory and others have investigated the impact of methylation on the expression of PRAME in hematologic malignancies, such as acute myeloid leukemia (AML), myelodysplastic syndromes (MDS) and chronic myeloid leukemia (CML).18–21 Furthermore, several studies have demonstrated that treatment with the demethylating agent 5ʹ-aza-2ʹdeoxycytidine can successfully induce the expression of PRAME.20–22
Multiple myeloma (MM) is a plasma cell malignancy that remains incurable despite the significant improvement in the overall survival of MM patients in the past decade.23 We and others have shown that both the bone marrow mononuclear cells (MNCs) and sorted plasma cells of newly diagnosed MM patients exhibit greatly variation in the PRAME transcript levels.10–12,24 Furthermore, we have reported that PRAME overexpression in the bone marrow was an adverse prognostic factor of progression-free survival in MM patients treated with nonbortezomib-containing regimens.11,12 We also demonstrated that PRAME gene copy number variations (CNVs) were one mechanism that led to differences in PRAME expression.12 To date, no study has evaluated the impact of methylation on PRAME expression in MM.
In the current study, after sorting plasma cell samples from 48 MM patients at diagnosis, we simultaneously evaluated the PRAME gene methylation, copy numbers and transcript levels and found that both methylation and CNVs were involved in PRAME expression in MM. Furthermore, the PRAME transcript levels increased in MM cell lines after treatment with the demethylating agent 5-azacytidine.
Materials and Methods
Patients and Samples
A total of 48 MM patients were enrolled in the present study. These patients were diagnosed at our hospital from October 2015 to December 2018. The diagnosis was based on the International Working Group Criteria. Bone marrow (BM) samples were collected from all the patients at diagnosis, and normal BM samples were collected from healthy volunteers. Mononuclear cells (MNCs) were isolated from the BM samples by Ficoll-Hypaque gradient centrifugation. Plasma cells were sorted from MNCs using CD138 immunomagnetic beads (MiltenyiBiotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. This study was approved by the Ethics Committee of Peking University People’s Hospital. All patients and volunteers provided written informed consent in accordance with the Declaration of Helsinki.
Detection of PRAME Transcript Levels
The RNeasy Micro Kit (Qiagen, Hilden, Germany) was used to extract the RNA from the sorted plasma cells, and TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) was used to extract RNA from the cell lines according to the manufacturer’s instructions. The PRAME transcript levels were measured using complementary DNA (cDNA) samples and TaqMan-based real time quantitative RT-PCR (qRT-PCR) technology as we have described previously.12,13,17 The PRAME transcript level was calculated as PRAME transcript copies/ABL copies and is expressed as a percentage. The reproducible sensitivity of the qRT-PCR was 2 copies, and all the samples with undetectable PRAME had ≥4,000 copies of ABL, guaranteeing that ≥0.05% of the PRAME transcript levels could be detected.
Bisulfate Treatment of Genomic DNA
DNA bisulfite conversion was carried out using the EZ DNA Methylation-Lightning Kit (Zymo Research, CA, USA) according to the manufacturer’s instructions. Briefly, approximately 500 ng genomic DNA per sample was added to the Lightning Conversion Reagent and mixed. Then, the mixtures were incubated in a thermal cycler at 98°C for 8 minutes, 54°C for 1 h and 4°C for 20 h. The bisulfate-converted DNA samples were loaded in a spin column provided with the kit for desulfonation and purification.
Cloning Sequencing of Sodium Bisulfate-Treated DNA
As in our previous report, the PRAME promoter and exon 1b regions were amplified.18 As shown in Table 1, the primers for the exon 1b region reported by Ortmann et al were used, and those for the promoter region were designed by ourselves.18,20 The PCR products were cloned into the pCR2.1 plasmid vector (Invitrogen; Thermo Fisher, CA, USA), and transformed into DH5a competent cells. Eight recombinant colonies per PCR product were selected and sequenced. The methylation ratio was defined as the ratio of methylated CpG sites/all included CpG sites.
 |
Table 1 The Sequences of the Primers Used in Cloning Sequencing and qMSP |
Quantitative Methylation Specific PCR (qMSP)
Considering the negative correlation between the methylation level of individual CpG sites and the previously obtained PRAME transcript levels, the PRAME methylation pattern in the MM patients, and the efficiency and specificity of real-time quantitative PCR, +287 CpG sites located in exon 1b were chosen to perform qMSP to determine the methylation level of the PRAME gene.18 Bisulfate-treated DNA was used as a template, and KAPA SYBR® FAST qPCR Kits (kapa biosystems, Wilmington, USA) were used for amplification. The primers for the methylated PRAME +287 CpG site (abbreviated as M) are shown in Table 1, and the total DNA (abbreviated as T) was amplified with the primers for the exon 1b region that were used for clone sequencing. The amplification was performed with an ABI PRISM 7500 real-time PCR system (PE Applied Biosystems, Foster City, CA, USA) under the following conditions: incubation at 95°C for 3 minutes, followed by 40 cycles of 10 seconds at 95°C and 50 seconds at 60°C, and completed with melt curve analysis. Then, electrophoresis was performed to confirm the specificity by the amplicon size (Figure 1). Each reaction was performed in three replicates. The cycle number at which the fluorescence signal crosses a detection threshold is referred to as Ct, and the difference in the Ct values between the reactions for M and T was calculated for each sample (ΔCt=Ct (average of M replicates) – Ct (average of T replicates)). Normal plasma cell DNA served as the calibrator, and each sample’s relative PRAME methylation level was calculated using 2−ΔΔCt (ΔΔCt=ΔCt (sample) –ΔCt (calibrator)).
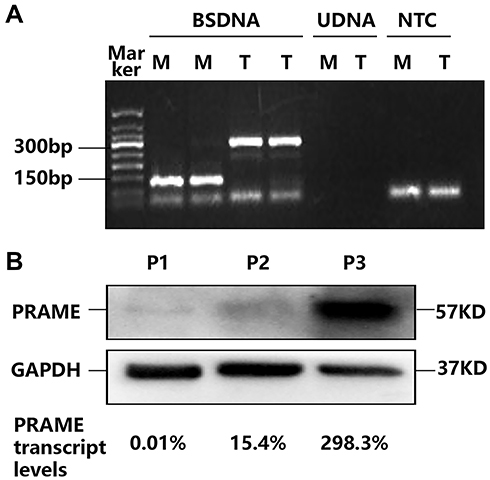 |
Figure 1 (A) The electrophoretogram of the qMSP products. The template for the amplification of bisulfate-treated DNA, bisulfate-untreated DNA and no template control are abbreviated as BSDNA, UDNA and NTC. M and T represent methylated and total DNA, respectively. (B) PRAME protein levels in the plasma cells sorted from three MM patients. |
Detection of PRAME Gene Copy Numbers
DNA was extracted from the plasma cells using the TIANamp Micro DNA Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. The PRAME gene copy numbers were measured by TaqMan-based real-time quantitative PCR (RQ-PCR) technology as we previously described.12
Cell Culture and Reagents
Human MM cell lines LP-1 and MOLP-2 were purchased from the American Type Culture Collection (ATCC). Human MM cell line RPMI8226 cell line was kindly provided by Cell Bank Shanghai Institute of Cell Biology (Shanghai, China). The cell lines were all cultured in RPMI-1640 media supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 mg/mL streptomycin, and 2 mM L-glutamine (Gibco, Life Technologies, Carlsbad, CA, USA). The cells were grown at 37°C in a humidified 5% CO2 atmosphere. 5-azacytidine was purchased from Sigma Aldrich, and the 5-azacytidine stock solution was prepared by dissolving the compound in dimethyl sulfoxide (DMSO; Sigma Aldrich, MO, USA).
Western Blot
Lysates of the sorted plasma cells from the primary patients were obtained using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China). The protein concentration was determined using the BCA Protein Assay Reagent Kit (Solarbio, Beijing, China). Equal amounts of proteins (30 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Merck Millipore, Darmstadt, Germany). The membranes were incubated with 5% nonfat skim milk in Tris-buffered Saline and Tween 20 (TBST) for 1 h, followed by hybridization at 4°C overnight with primary antibodies against PRAME (rabbit monoclonal, 1:1000; Abcam, MA, USA) and GAPDH (rabbit monoclonal, 1:1000; Cell Signaling Technology, MA, USA). After washing with TBST, the membranes were incubated with a horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:10000; Santa Cruz, USA) for 1 h. The bands were detected by enhanced chemiluminescence substrate (Thermo Fisher Scientific, USA).
Statistical Analysis
The Mann–Whitney U nonparametric test was used to compare the PRAME transcript levels in the sorted plasma cells between the two groups, and Student’s t-test was used for the comparison of the PRAME transcript levels before and after 5-azacytidine treatment. Correlations were analysed using Spearman correlation test. All P-values were obtained using two-tailed tests, and a value of P<0.05 was considered significant. The statistical analysis was performed using SPSS software version 19 (SPSS, Inc., Chicago, IL) and GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA).
Results
PRAME Transcript and Protein Expression in Plasma Cells Sorted from MM Patients
The PRAME mRNA expression varied greatly among the plasma cell samples sorted from the 48 MM patients at diagnosis, and the median PRAME transcript level was 3.1% (range: 0–298.3%). According to the sensitivity of detection of the qRT-PCR, 11 (22.9%) patients with ≤0.05% PRAME transcript levels were categorized into the PRAME low-expression group (6 had undetectable PRAME), and 37 (77.1%) patients with >0.05% PRAME transcript levels were categorized into the PRAME high-expression group. In addition, the PRAME protein could be detected in the plasma cells sorted from three patients, and the protein levels corresponded to the transcript levels (Figure 1B).
PRAME Gene Methylation Patterns
Clone sequencing was performed on the plasma cell samples sorted from 4 MM patients. P1 and P2 had low PRAME expression, and P3 and P4 had high PRAME expression. The PRAME gene CpG island methylation patterns of the promoter and exon 1b regions are shown in Figure 2. The total methylation ratios of the P1 to P4 samples were 61.3%, 59.3%, 19.4%, and 7.7%, respectively. In general, the degree of methylation decreased as the transcript levels increased. Table 2 shows the methylation ratios of the groups with low and high PRAME expression; the degree of methylation in both the promoter and exon 1b region was negatively related to the PRAME transcript levels. Furthermore, both the 5ʹ and 3ʹ regions of the promoter and exon 1b were negatively related to the PRAME transcript levels, although the 5ʹ region of the exon 1b was less methylated than the 3ʹ region in each sample (Figure 2). In addition, −130 was the only CpG site at which none of the clones was methylated (Figure 2).
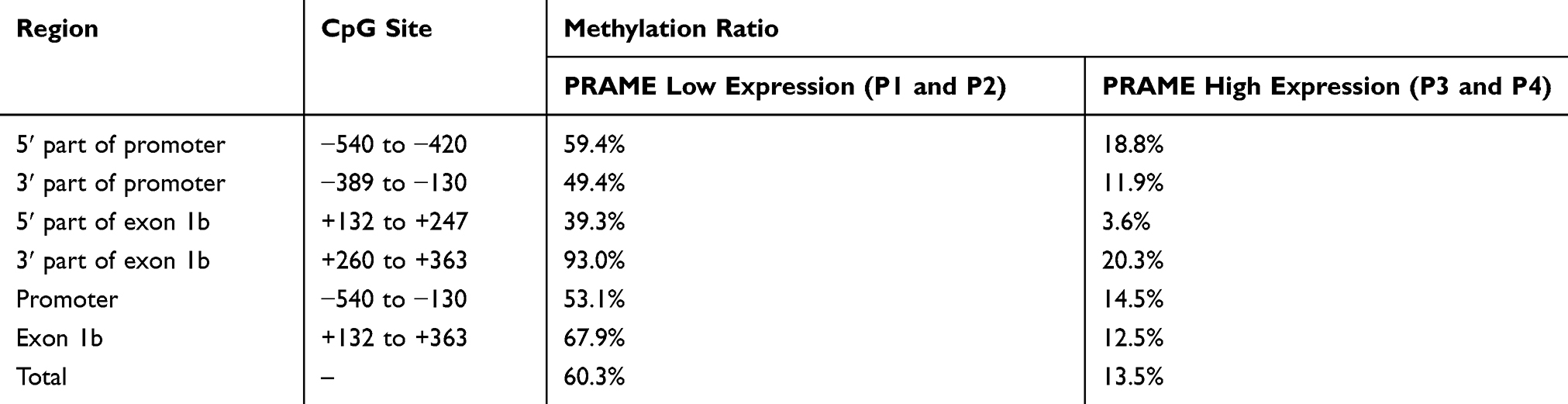 |
Table 2 The Methylation Ratios of the Individual CpG Island Regions |
 |
Figure 2 The clone sequencing results of the −540 to −130 and +132 to +363 CpG sites of the plasma cell samples sorted from 4 MM patients. The white square represents unmethylated cytosine; the black square represents methylated cytosine. |
The Relationship Between Degree of Methylation of the PRAME Gene and Its Expression
Clone sequencing showed that the methylation ratios at the PRAME +287 CpG site were 81.3% in the PRAME low-expression group and 6.25% in the high-expression group, which was similar to the total methylation ratio and negatively related to the PRAME transcript levels (Figure 2). Therefore, the methylation level at the PRAME +287 CpG site, as measured by the qMSP technique, could represent the total degree of methylation of the PRAME gene. As shown in Figure 3A, the degrees of methylation of PRAME were different among the plasma cell samples sorted from the 48 MM patients (median: 0.59, range: 0.0002–1.44), and they were significantly negatively related to the transcript levels (r=−0.44, P=0.0018). The patients with different PRAME transcript levels were further analysed separately. In the PRAME high-expression group (n=37), the degree of methylation was significantly negatively related to the transcript levels (r=−0.69, P<0.0001), whereas no relationship existed in the low-expression group (n=11, r=−0.27, P=0.43). Although the patients in the low-expression group had the range of degrees of methylation (0.13 to 1.14) that was similar to that in the high-expression group (0.00020 to 1.441), the PRAME transcripts in the low-expression group were almost undetectable. It implied that mechanisms other than methylation might be involved in low expression patients.
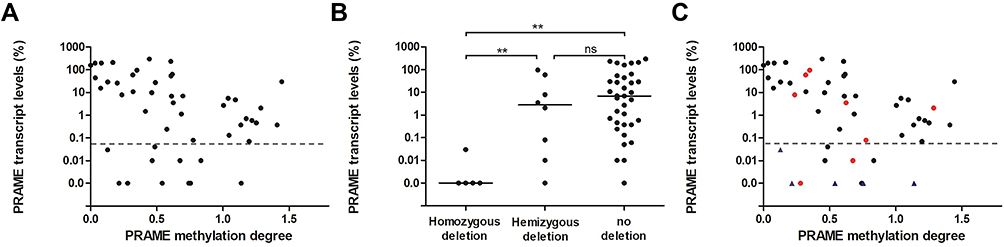 |
Figure 3 The impact of PRAME the degree of methylation and gene copy number status on PRAME expression in the plasma cell samples sorted from 48 MM patients. (A) The relationship of PRAME the degree methylation with its transcript levels. The horizontal line represents 0.05% of the PRAME transcript levels. (B) The PRAME transcript levels in samples with different gene copy number status. The horizontal lines represent the median PRAME transcript levels. ns, not significant; **P<0.01. (C) The PRAME transcript levels, degree of methylation and copy number status of each sample are simultaneously shown. The black circles, red circles, and blue triangles represent patients with no deletion, hemizygous deletion, and homozygous deletion, respectively. |
The Relationship Between PRAME Gene Copy Number and Its Expression
CNV of the PRAME gene exited among the 48 MM patients. A total of 35 (72.9%) patients had normal PRAME gene copies with a relative copy number ~1.0, whereas 5 (10.4%) and 8 (16.7%) patients individually had a homozygous deletion and hemizygous deletion, which corresponded to relative copy numbers of ~ 0 and 0.5, respectively. The PRAME transcript levels were significantly different among the three groups (homozygous deletion 0% [range: 0–0.030%] vs hemizygous deletion 2.8% [range: 0–95.9%] vs no deletion 6.7% [range: 0–298.3%], P=0.0028, Figure 3B). The patients with homozygous deletions had significantly lower PRAME transcript levels than both the patients with no deletions and patients with hemizygous deletions (P=0.0008 and 0.0020), respectively. However, there were no significant differences in the PRAME transcript levels between the patients with hemizygous deletions and those with no deletions (P=0.40). Furthermore, the frequency of hemizygous deletion in the low-expression group was similar to that in the high-expression group (18.2% vs 16.2%, P=1.0).
The Relationship Between PRAME Gene Methylation, Copy Number and Its Expression
The PRAME transcript level, degree of methylation and copy number status of each sample are simultaneously shown in Figure 3C. The degrees of methylation of the patients with no deletion, hemizygous deletion, and homozygous deletion were similar (P=0.96). The patients with hemizygous deletions dispersedly distributed in both the low-expression and high-expression groups. All 5 patients with homozygous deletions were categorized into the low-expression group despite their varied degrees of methylation; the remaining 6 patients in the low-expression group had a similar range of degrees of methylation that was similar to that of the high-expression group but almost undetectable PRAME transcript levels.
The Effect of 5-Azacytidine on PRAME Expression in MM Cell Lines
The PRAME transcript levels in the MOLP-2, LP-1 and RPMI8226 cells were 3.2%, 175.9% and 824.0%, respectively. As shown in Figure 4, the PRAME transcript levels of all 3 cell lines significantly increased after treatment with 0.25 μM and 0.5 μM 5-azacytidine for 24 h and 48 h.
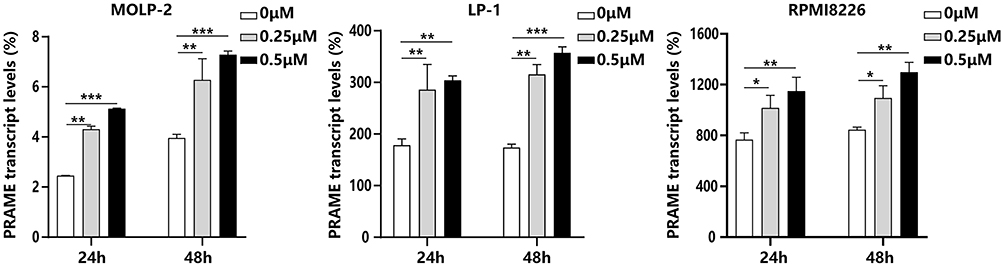 |
Figure 4 The effect of 5-azacytidine on the PRAME transcript levels in MM cell lines. *P<0.05; **P<0.01; ***P<0.001. |
Discussion
PRAME is commonly overexpressed in both solid tumours and hematologic malignancies, making it a promising target for immunotherapy.3–17 Since PRAME is relevant to therapy response, the heterogeneity in its expression and the underlying mechanism should be considered. In the current study, we investigated the impact of methylation and CNV on PRAME expression in newly diagnosed MM patients, and the impact of a demethylating agent on the PRAME transcript levels in MM cell lines.
We previously reported that normal plasma cells expressed PRAME.12 Similar to our previous report, great variation was found in the PRAME transcript levels of the plasma cells sorted from 48 MM patients was observed.12 An interesting phenomenon is that several patients had undetectable PRAME transcript levels. Because of the low percentage of plasma cells in the bone marrow of MM patients, plasma cell sorting is necessary for real evaluation. As a result, the cell numbers and the ABL copies are low for some patients. Considering the sensitivity of detection, we chose 0.05% as the cut-off value to categorize the MM patients into low- and high-PRAME expression groups.
Methylation at DNA CpG islands is an important mechanism of regulating gene expression.25,26 Previous studies have observed that aberrant hypomethylation of PRAME correlates with its expression in AML, CML and MDS.18–21 For the first time in MM plasma cells, we demonstrated a similar negative relationship between the PRAME transcript levels and the degree of methylation of the promoter and exon 1b region as well as the +287 CpG degree of methylation through clone sequencing and qMSP, respectively. Nevertheless, it seems that the methylation patterns are not identical among patients with hematologic malignancies.
We and others have demonstrated that the degree of methylation of the 3ʹ regions of the promoter and exon 1b but not the 5ʹ regions of the promoter were relevant to PRAME expression in AML/MDS patients.18–20 However, the 2 plasma samples with high PRAME expression had significantly lower degrees of methylation of both the 5ʹ and 3ʹ regions of the promoter and the exon 1b CpG region compared to the 2 plasma samples with low PRAME expression. CpG site methylation at the promoter region inhibits gene expression by directly inhibiting the binding of specific transcription factors and by indirectly recruiting methyl-CpG-binding domain (MBD) proteins, which recruit histone-modifying and chromatin-remodelling complexes to the methylated sites.27 Different methylation patterns in 5ʹ region of the promoter CpG island suggested that MM plasma cells may have unique binding transcription factors or MBD proteins that regulate their PRAME expression.
By analysing the high- and low-expression groups separately, we found a negative relationship between PRAME expression and degree of methylation in the high- but not the low-expression group. Although the low-expression group had a range of degrees of methylation that was similar to that of the high-expression group, all the plasma samples had almost undetectable PRAME levels. Therefore, methylation cannot account for the fairly low PRAME expression in MM. In addition, we found that the PRAME transcript levels of 3 MM cell lines with high PRAME expression increased after demethylated agent treatment. Thus, the combination of a demethylating agent might improve the effect of PRAME-targeted therapy.
Gene CNV is another mechanism that contributes to heterogeneous gene expression in cancer.28–31 We previously reported that both homozygous and hemizygous PRAME gene deletions occurred in MM plasma cells.12 In the current study, 5 patients had homozygous deletions, and they were all categorized into the low-expression group. It is obvious that no DNA transcript was detected. The samples with hemizygous PRAME deletions had similar PRAME transcript levels and degrees of methylation to those with no deletions. This result implied that hemizygous deletion had no significant impact on PRAME expression and that methylation mainly contributed to heterogeneous PRAME expression in MM patients with no PRAME homozygous deletion.
In addition to homozygous deletion, the mechanism that caused the low PRAME expression in the remaining 6 patients is still unclear. Genetic and other epigenetic alterations also need to be considered. Lee et al demonstrated that PRAME is upregulated by MZF1 in cooperation with DNA hypomethylation in melanoma cells.32 Whether defects in important transcription factors or MBD proteins, such as MZF1, occur in MM remains to be studied.
In conclusion, plasma cells from MM patients exhibited great variation in their PRAME transcript levels, and approximately 1/4 had fairly low PRAME expression. Through combining analysis, we demonstrated that both methylation and CNV participated in the regulation of PRAME expression; in patients with no homozygous PRAME deletion, PRAME expression was mainly controlled by methylation, and a proportion of fairly low PRAME expression was caused by homozygous deletion. It has been revealed that the demethylating agent 5-aza-2ʹ-deoxycytidine could induce PRAME expression in AML.20 We found that the PRAME transcript levels significantly increased in the MM cell lines after treatment with 5-azacytidine. The current study provides a evidence that more MM patients could be treat with PRAME-targeting immunotherapy combined with demethylating treatment. Furthermore, it should be noted that the demethylating agent is not suitable for all MM patients, and PRAME gene copies should be evaluated before treatment.
Funding
This work was supported by Peking University People’s Hospital Research and Development Funds (RDX2018-01), and the National Natural Science Foundation of China (81870125).
Disclosure
The authors declare no conflicts of interest.
References
1. Al-Khadairi G, Decock J. Cancer testis antigens and immunotherapy: where do we stand in the targeting of PRAME? Cancers. 2019;11(7):984. doi:10.3390/cancers11070984
2. van den Bulk J, Verdegaal EM, de Miranda NF. Cancer immunotherapy: broadening the scope of targetable tumours. Open Biol. 2018;8(6):180037. doi:10.1098/rsob.180037
3. Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi:10.1034/j.1600-065X.2002.18803.x
4. Fratta E, Coral S, Covre A, et al. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol Oncol. 2011;5(2):164–182. doi:10.1016/j.molonc.2011.02.001
5. Salmaninejad A, Zamani MR, Pourvahedi M, Golchehre Z, Hosseini Bereshneh A, Rezaei N. Cancer/testis antigens: expression, regulation, tumor invasion, and use in immunotherapy of cancers. Immunol Invest. 2016;45(7):619–640. doi:10.1080/08820139.2016.1197241
6. Epping MT, Bernards R. A causal role for the human tumor antigen preferentially expressed antigen of melanoma in cancer. Cancer Res. 2006;66(22):10639–10642. doi:10.1158/0008-5472.CAN-06-2522
7. Doolan P, Clynes M, Kennedy S, Mehta JP, Crown J, O’Driscoll L. Prevalence and prognostic and predictive relevance of PRAME in breast cancer. Breast Cancer Res Treat. 2008;109(2):359–365. doi:10.1007/s10549-007-9643-3
8. Bankovic J, Stojsic J, Jovanovic D, et al. Identification of genes associated with non-small-cell lung cancer promotion and progression. Lung Cancer. 2010;67(2):151–159. doi:10.1016/j.lungcan.2009.04.010
9. Neumann E, Engelsberg A, Decker J, et al. Heterogeneous expression of the tumor-associated antigens RAGE-1, PRAME, and glycoprotein 75 in human renal cell carcinoma: candidates for T-cell-based immunotherapies? Cancer Res. 1998;58(18):4090–4095.
10. Pellat-Deceunynck C, Mellerin MP, Labarriere N, et al. The cancer germ-line genes MAGE-1, MAGE-3 and PRAME are commonly expressed by human myeloma cells. Eur J Immunol. 2000;30(3):803–809. doi:10.1002/1521-4141(200003)30:3<803::AID-IMMU803>3.0.CO;2-P
11. Qin Y, Lu J, Bao L, et al. Bortezomib improves progression-free survival in multiple myeloma patients overexpressing preferentially expressed antigen of melanoma. Chin Med J. 2014;127(9):1666–1671.
12. Yang L, Wang YZ, Zhu HH, et al. PRAME gene copy number variation is related to its expression in multiple myeloma. DNA Cell Biol. 2017;36(12):1099–1107. doi:10.1089/dna.2017.3951
13. Qin Y, Zhu H, Jiang B, et al. Expression patterns of WT1 and PRAME in acute myeloid leukemia patients and their usefulness for monitoring minimal residual disease. Leuk Res. 2009;33(3):384–390. doi:10.1016/j.leukres.2008.08.026
14. van Baren N, Chambost H, Ferrant A, et al. PRAME, a gene encoding an antigen recognized on a human melanoma by cytolytic T cells, is expressed in acute leukaemia cells. Br J Haematol. 1998;102(5):1376–1379.
15. Proto-Siqueira R, Falcao RP, de Souza CA, Ismael SJ, Zago MA. The expression of PRAME in chronic lymphoproliferative disorders. Leuk Res. 2003;27(5):393–396.
16. Radich JP, Dai H, Mao M, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103(8):2794–2799. doi:10.1073/pnas.0510423103
17. Qin YZ, Zhu HH, Liu YR, et al. PRAME and WT1 transcripts constitute a good molecular marker combination for monitoring minimal residual disease in myelodysplastic syndromes. Leuk Lymphoma. 2013;54(7):1442–1449.
18. Qin YZ, Zhang YH, Qin XY, Zhu HH. Methylation pattern of preferentially expressed antigen of melanoma in acute myeloid leukemia and myelodysplastic syndromes. Oncol Lett. 2017;13(4):2823–2830. doi:10.3892/ol.2017.5790
19. Gutierrez-Cosio S, de la Rica L, Ballestar E, et al. Epigenetic regulation of PRAME in acute myeloid leukemia is different compared to CD34+ cells from healthy donors: effect of 5-AZA treatment. Leuk Res. 2012;36(7):895–899. doi:10.1016/j.leukres.2012.02.030
20. Ortmann CA, Eisele L, Nuckel H, et al. Aberrant hypomethylation of the cancer-testis antigen PRAME correlates with PRAME expression in acute myeloid leukemia. Ann Hematol. 2008;87(10):809–818. doi:10.1007/s00277-008-0514-8
21. Roman-Gomez J, Jimenez-Velasco A, Agirre X, et al. Epigenetic regulation of PRAME gene in chronic myeloid leukemia. Leuk Res. 2007;31(11):1521–1528. doi:10.1016/j.leukres.2007.02.016
22. Schenk T, Stengel S, Goellner S, Steinbach D, Saluz HP. Hypomethylation of PRAME is responsible for its aberrant overexpression in human malignancies. Genes Chromosomes Cancer. 2007;46(9):796–804.
23. Rajkumar SV, Kumar S. Multiple myeloma: diagnosis and treatment. Mayo Clin Proc. 2016;91(1):101–119. doi:10.1016/j.mayocp.2015.11.007
24. Sigalotti L, Fratta E, Coral S, et al. Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylation-regulated and functionally reverted by 5-aza-2ʹ-deoxycytidine. Cancer Res. 2004;64(24):9167–9171. doi:10.1158/0008-5472.CAN-04-1442
25. Jones PA. DNA methylation and cancer. Cancer Res. 1986;46(2):461–466.
26. Riggs AD, Jones PA. 5-methylcytosine, gene regulation, and cancer. Adv Cancer Res. 1983;40:1–30.
27. Wade PA. Methyl CpG-binding proteins and transcriptional repression. Bioessays. 2001;23(12):1131–1137. doi:10.1002/bies.10008
28. Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7(2):85–97. doi:10.1038/nrg1767
29. Largo C, Alvarez S, Saez B, et al. Identification of overexpressed genes in frequently gained/amplified chromosome regions in multiple myeloma. Haematologica. 2006;91(2):184–191.
30. Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–454. doi:10.1038/nature05329
31. Feurstein S, Rucker FG, Bullinger L, et al. Haploinsufficiency of ETV6 and CDKN1B in patients with acute myeloid leukemia and complex karyotype. BMC Genomics. 2014;15:784. doi:10.1186/1471-2164-15-784
32. Lee YK, Park UH, Kim EJ, Hwang JT, Jeong JC, Um SJ. Tumor antigen PRAME is up-regulated by MZF1 in cooperation with DNA hypomethylation in melanoma cells. Cancer Lett. 2017;403:144–151. doi:10.1016/j.canlet.2017.06.015
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.