Back to Journals » International Journal of Nanomedicine » Volume 15
Both Intracranial and Intravenous Administration of Functionalized Carbon Nanotubes Protect Dopaminergic Neuronal Death from 6-Hydroxydopamine
Authors Kim OH ![]() , Park JH, Son JI, Kim KY, Lee HJ
, Park JH, Son JI, Kim KY, Lee HJ
Received 11 August 2020
Accepted for publication 10 September 2020
Published 8 October 2020 Volume 2020:15 Pages 7615—7626
DOI https://doi.org/10.2147/IJN.S276380
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel Rubinstein
Ok-Hyeon Kim,1 Jun Hyung Park,2 Jong In Son,1 Kyung-Yong Kim,1 Hyun Jung Lee1,2
1Department of Anatomy and Cell Biology, College of Medicine, Chung-Ang University, Seoul, Republic of Korea; 2Department of Global Innovative Drugs, Graduate School of Chung-Ang University, Seoul, Republic of Korea
Correspondence: Hyun Jung Lee
Chung-Ang University, Rm615 Bd105, 84 Heuksuk-Ro, Dongjak-gu, Seoul 06974, Republic of Korea
Tel +82-2-820-5434
Email [email protected]
Purpose: Although single-walled nanotubes (SWNTs) with functional groups have been suggested as a potential nanomedicine to treat neuronal disorders, effective routes to administer SWNTs have not been compared thus far. The blood–brain barrier is a considerable challenge for the development of brain-targeting drugs, and therefore functionalized SWNT routes of administration have been needed for testing Parkinson’s disease (PD) treatment. Here, effective administration routes of functionalized SWNTs were evaluated in PD mouse model.
Methods: Three different administration routes were tested in PD mouse model. Functionalized SWNTs were injected directly into the lateral ventricle three days before (Method 1) or after (Method 2) 6-hydroxydopamine (6-OHDA) injection to compare the protective effects of SWNTs against dopaminergic neuronal death or functionalized SWNTs were injected intravenously at three and four days after 6-OHDA injection (Method 3). Asymmetric behaviors and histological assessment from all animals were performed at two weeks after 6-OHDA injection.
Results: Ventricular injections of SWNTs both before or after 6-OHDA exposure protected dopaminergic neurons both in the substantia nigra and striatum and alleviated rotational asymmetry behavior in PD mice. Moreover, intravenous administration of SWNTs three and four days after 6-OHDA injection also prevented neuronal death and PD mice behavioral impairment without apparent cytotoxicity after six months post-treatment.
Conclusion: Our study demonstrates that functionalized SWNTs could effectively protect dopaminergic neurons through all administration routes examined herein. Therefore, SWNTs are promising nanomedicine agents by themselves or as therapeutic carriers to treat neuronal disorders such as PD.
Keywords: single-walled nanotubes, Parkinson’s disease, intracranial, intravenous, nanomedicine
Introduction
Parkinson’s disease (PD) is the second-most common neurodegenerative disorder after Alzheimer’s disease, and leads to various impairments in the patients’ motor abilities and system.1 PD is known to induce the malfunction or death of neurons in the brain, particularly those in the substantia nigra (SN) region.2 In turn, this neuronal degeneration leads to a reduction in dopamine (DA) production, which induces ataxia and tremors in the hands, as well as balance and coordination difficulties.3 Despite the ongoing progress on PD treatment research, a cure for this condition has not yet been developed. Symptoms of PD can be alleviated with a DA precursor (ie, levodopa) or DA agonist; however, many side effects including nausea or orthostatic hypotension have been reported as a result of their chronic administration.
Over the past decades, nanotechnology and nanomedicine have been considered promising therapeutic tools for neurodegenerative diseases, including PD. Among them, carbon nanotubes (CNTs) have been shown to possess a wide applicability range for the relief of neural disorder symptoms via neural interfaces,4 neuronal differentiation,5 and neural stimulation.6 In the aforementioned studies, single-walled nanotubes (SWNTs) or multi-walled nanotubes (MWNTs) were functionalized with a chemical group or copolymer to enhance biocompatibility and reduce cytotoxicity. Additionally, CNT functionalization has not been linked to harmful effects such as reductions in agglomeration, chirality, and impurities, all of which contribute to CNT toxicity.7 Many in vitro studies have shown improvements in the growth and differentiation of neural stem cells by creating a conductive environment with functionalized CNTs.8 Although the outstanding conductivity and favorable neural properties of CNTs have been shown in many in vitro studies,9 only a few in vivo applications of CNTs have been reported so far regarding the neuroprotective effects of CNTs in the brain tissue. Moon et al reported the ability of hydrophobic CNTs impregnated with subventricular-zone neural progenitor cells to repair damaged neural tissues following stroke.10 Moreover, SWNTs functionalized with an amine group ameliorated damage and sped up the recovery of post-stroke brain tissue compared to other groups.11 Among these studies, CNTs were administered in vivo directly into the ventricle or parenchyma of the brain via intracranial injection, a procedure that is both invasive and painful. However, given that one of the largest limitations for brain-targeting drug delivery systems or treatment methods is the restricted entry of active compounds to the central nervous system via the blood–brain barrier, nanomedical approaches have been limited to directly target brain tissue through intracranial injections. Thus, minimally invasive approaches need to be developed to enhance nanoparticle delivery, including nanomedicine.
In this study, the effects of different administration routes on the neuroprotective effects of SWNTs were estimated and analyzed in PD mice. Routes of administration have a direct impact on drug bioavailability. Here, SWNTs were functionalized with polyethylene glycol (PEG) to improve biocompatibility, and their function was compared based on exposure route (intracranial or intravenous injections). We found that intracranial injection of SWNTs before or after 6-hydroxydopamine (6-OHDA) administration in PD mice brains could prevent dopaminergic neuron death and behavioral asymmetry. Moreover, intravenous injection of SWNTs after 6-OHDA administration also alleviated neuronal damage of the SN and striatum (ST) of PD mice brains and reduced abnormal behaviors in PD mice, suggesting that functionalized SWNTs possess PD therapeutic potential both via intracranial and intravenous administration. Therefore, functionalized SWNTs could be applied as a nanomedicine using a less invasive approach.
Materials and Methods
FE-TEM Specimen Preparation and Imaging PEG-SWNTs
Commercially available SWNTs functionalized with polyethylene glycol (PEG-SWNTs) were purchased from Sigma Aldrich (St. Louis, MO, USA). The morphology and dispersibility of PEG-SWNTs in a water suspension were analyzed via field-emission transmission electron microscopy (FE-TEM; JEM-F200, JEOL); 300 mesh copper TEM grids (TED PELLA, Inc., Redding, CA, USA) were used for TEM experiments. For TEM specimen preparation, the PEG-SWNTs were suspended in distilled water (D.W.). The vials were bath sonicated for 24 hours or until agglomerates broke apart. Afterward, the PEG-SWNT suspensions were drop-casted onto the TEM grids and dried in a 70°C dry oven for at least 24 hours.
Animal Preparation and Surgical Procedures
Male ICR mice (25–30 g) were purchased from Samtako (Osan-si, Gyeonggi-do, Korea). All animals were caged at 5 mice per cage density for least 1 week prior to the start of the experiments and maintained on a normal laboratory diet and tap water ad libitum in an air-conditioned room (21 ± 2°C) with a 12 hours light cycle. All experimental procedures were approved by the Institutional Animal Care Use Committee (IACUC) of Chung-Ang University (Approval ID:11–0012) and carried out according to the ‘Guide for the Care and Use of Laboratory Animals’ published by the National Academy Press (NIH Publication No. 85–23, revised 1996). For the PD model, desipramine (25 mg/kg; Sigma-Aldrich) was intraperitoneally injected 1 hour before 6-OHDA (Sigma-Aldrich) administration. Animals were anesthetized using intraperitoneal injections of zoletil (30 mg/kg, Virbac, Carros, France) and rompun (10 mg/kg, Bayer, Leverkusen, Germany) and then placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) using a 10 μL Hamilton syringe fitted with a steel cannula. 6-OHDA (10 μg; 4 μg/μL, 2.5 μL) dissolved in 0.2 mg/mL L-ascorbic acid was injected into the right ST (AP: 0.8 mm, ML: −2.0 mm, DV: −3.3 mm from bregma and dura). Applied dose was chosen as 2 μg per mouse for intracranial injection and 400 μg per mouse for intravenous injection according to previous studies.12–14 PEG-SWNTs (2 μg; 1mg/mL, 2 μL) dissolved in DW were injected into the right lateral ventricle (AP: −0.22 mm, ML: −1.1 mm, DV: −2.2 mm from the bregma and dura) at an injection rate of 0.5 μL/min. After injection, the cannula remained in situ for an additional 5 min before being withdrawn. Control treatments were conducted by injecting 2 μL of DW as described above. In sham-operated mice, only the scalps were dissected under anesthetization (n=10). The experiment was conducted with different PEG-SWNT infusion routes. 1) Either PEG-SWNTs (n=16) or DW (n=18) were injected into the ventricle 3 days prior to 6-OHDA injection into the ST; 2) PEG-SWNTs (n=16) or DW (n=16) were injected into the ventricle 3 days after 6-OHDA administration; 3) PEG-SWNT (n=20, 200 μg, 1 mg/mL, 200 μL) or DW (n=18) were injected into the tail vein once a day (200 μL/day) on days 3 and 4 after 6-OHDA administration. Animals
Behavioral Tests
Rotation was measured using modified automated “Rotometer” bowls.15 Drug-induced asymmetric rotational behaviors were tested using apomorphine (0.5 mg/kg, subcutaneous injection) 2 weeks after 6-OHDA administration. Ten minutes after apomorphine injection, animal activity was recorded for 60 minutes. Net rotations (contralateral turns-ipsilateral turns) were counted over a 60-min period beginning 10 min after the administration of apomorphine. 6-OHDA-exposed mice developed a preference for spontaneous turning toward the side contralateral to 6-OHDA injection. Results were expressed as net turns/60 min.
Cell Culture and Differentiation of SH-SY5Y Cells
The human neuroblastoma SH-SY5Y cell line was obtained from American Type Culture Collection (ATCC, CRL-2266, Rockville, MD, USA). Cells were maintained in DMEM (GE Healthcare, Chicago, IL, USA) supplemented with 10% FBS (GE Healthcare) and 1% penicillin/streptomycin (ThermoFisher Scientific, Waltham, MA, USA) in a 5% CO2 humidified atmosphere at 37°C. Upon reaching 80% confluence, the cells were disaggregated by trypsinization and seeded onto glass-bottomed 6-well plates at a 105 cells per glass density. SH-SY5Y cells were differentiated using retinoic acid (RA, Sigma-Aldrich) with 2.5% serum at a final concentration of 10 μM. The cells were differentiated for 7 days, and the medium was replenished every 2 days. Neurite length was assessed from five microscopic images taken at random locations using the ImageJ software.
Cell Viability
Cell viability was determined with the alamarBlue cell viability assay kit (Molecular Probes, Eugene, Oregon, USA) following the manufacturer’s instructions. Briefly, SH-SY5Y cells were seeded onto 96-well culture plates at a 4 × 104 cell per well density and allowed to adhere overnight. Various concentrations (0, 5, 10 and 20 μg/mL) of PEG-SWNT were added into cells with or without 50 μM of 6-OHDA, and cells were allowed to incubate for 24 hours. PEG-SWNTs were pretreated 1 hour prior to 6-OHDA treatment or treated with 6-OHDA simultaneously. At the end of the experiment, 10% v/v alamarBlue (10 μL) was added into each well and fluorescent intensity was measured (excitation 530 nm, emission 590 nm) using a fluorescence plate reader (Glomax, Promega; Madison, Wisconsin, USA).
Reactive Oxygen Species Measurement
A 2ʹ,7-dichlorofluorescein diacetate (DCF-DA) assay (DCFDA/H2DCFDA-Cellular ROS Assay Kit, Abcam; Cambridge, England) was performed to characterize intracellular ROS (reactive oxygen species) levels. The cells were plated onto a 96-well plate and exposed to different doses of PEG-SWNT and 6-OHDA. Three hours after treatment, the cells were stained with 20 μM H2DCFDA and incubated at 37°C for 45 min in the dark. The fluorescence intensity of the cells was detected at 485 nm excitation and 535 nm emission with a fluorescence plate reader (Glomax, Promega); images were captured using a fluorescence microscope (Eclipse Ti2, Nikon, Minato-ku, Tokyo, Japan).
Measurement of Mitochondrial Membrane Potential
Changes in mitochondrial membrane potential were estimated using tetramethylrhodamine ethyl ester (TMRE), which is a cationic potentiometric dye that accumulates preferentially in energized mitochondria driven by the membrane potential. The cells were incubated with 100 nM TMRE for 15 min at 37°C and the TMRE fluorescent intensity was then measured at a 549 nm excitation and 574 nm emission using a fluorometer (Glomax, Promega); images were captured using a fluorescence microscope as described above.
Immunohistochemistry
After 2 weeks of 6-OHDA injection, the animals were deeply anesthetized and transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde in PBS. Harvested brains were stored in 4% paraformaldehyde for 24 hours and then sequentially transferred to 15%, 20%, 25%, and 30% sucrose solutions until they sank. After overnight storage in 30% sucrose, the brains were removed from the solution and stored at −80°C until needed. Sections (20-μm thick) were prepared and then processed for immunohistochemical detections. Free-floating sections were incubated with 0.3% H2O2 for 30 min, placed in a blocking buffer for 1 hour at 37°C, and then incubated with anti-tyrosine hydroxylase (TH) (1:200, Merck Millipore, Burlington, MA, USA) overnight at room temperature. After washing, the sections were incubated with appropriate biotinylated secondary antibodies (1:200, Vector Laboratories Inc., San Francisco, CA, USA), followed by avidin-biotin-peroxidase (1:200, Vector Laboratories) for 1 hour at 37°C. Immunoreactivity was visualized with 0.05% diaminobenzidine tetrahydrochloride, and sections were imaged with an inverted microscope (Leica microscope system, Wetzlar, Hesse, Germany) equipped with a Leica camera (Leica).
Western Blot Analyses
For Western blot analyses, animals were sacrificed 2 weeks after 6-OHDA injection. Brains were rapidly removed from mice, and the ST was dissected immediately on ice. Each tissue was homogenized with a lysis buffer (137 mM NaCl, 20 mM Tris, pH 8.0, 1% NP40, 10% glycerol, 1 mM PMSF, 10 mg/mL aprotinin, 1 mg/mL leupeptin, and 0.5 mM sodium vanadate). The homogenates were centrifuged at 13,000 rpm at 4°C for 15 min, and supernatants were collected for Western blot analysis. Protein content was determined using the Bradford method. Equal amounts of protein extracts from brain tissue were separated by SDS-PAGE and then transferred to a nitrocellulose membrane; blots were probed with antibodies specific for TH (1:1000, Thermo Fisher Scientific), N-Cadherin (1:1000, Santa Cruz Biotechnology, Dallas, TX, USA), and β-actin (1:1000, Santa Cruz Biotechnology). The blots were then probed with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies, and immunoreactive proteins were detected using an ECL detection system (Thermo Fisher Scientific). The intensities of protein bands were quantified using ImageJ. Quantification of relative protein levels was performed by comparing each protein to β-actin or its non-phosphorylated isoform. The value was expressed as a percentage by normalizing the ipsilateral region to the contralateral region.
GOT and GPT Assay
Blood was collected for analysis via a cardiac puncture at the time of sacrifice. Afterward, glutamate oxalate transaminase (GOT) and glutamate pyruvate transaminase (GPT) assays were used to measure hepatotoxicity via serum analysis using commercially available kits (Asan Pharmaceutical, Seoul, Korea) according to the manufacturer’s instructions.
Statistical Analysis
Data were expressed as the mean ± SEM. Group results were analyzed using a t-test or one-way analysis of variance (ANOVA) and Holm–Sidak’s method for multiple comparisons using the GraphPad Prism 8 software (GraphPad, Inc., CA, USA). P-values of <0.05, <0.01, and <0.0001 were considered to be significant.
Results
Characterization of PEG-SWNTs
Commercially available PEG-SWNTs were acquired to conduct the experiments described herein. The PEGs were linked to the surface of the SWNTs at a 20 to 80 weight ratio (Supplementary Figure 1). Prior to its administration, 1 mg/mL PEG-SWNT solution was prepared in DW and dispersed by ultrasonication for 24 h. After dispersion, PEG-SWNTs were scanned by TEM. Figure 1 shows that the PEG-SWNTs were well separated and evenly distributed.
 |
Figure 1 Characterization of PEG-SWNTs. PEG-SWNTs were dissolved in distilled water at a 1 mg/mL concentration and dispersed by ultrasonication for 24 hours. (A) Transmission electron microscope (TEM) images of dispersed PEG-SWNTs. (B and C) are magnified versions of the colored box in (A). |
PEG-SWNTs Ameliorate Abnormal Behavior of PD Mice
Unilateral 6-OHDA-induced lesions lead to behavioral abnormalities in apomorphine-injected mice, which exhibited ipsilateral loss of TH+ neurons in the SN pars compacta (SNpc) and ST, which is the golden standard for PD mimicking.16 To investigate the protective effect of PEG-SWNTs and compare their effectiveness as a function of administration route, three different experiments were conducted, as shown in Figure 2A: method 1) intracranial injection of PEG-SWNTs 3 days before 6-OHDA injection; method 2) intracranial injection 3 days after 6-OHDA injection, or method 3) intravenous injection at days 3 and 4 after 6-OHDA injection. All mice from each method were assessed for behavior asymmetry and sacrificed for histological analyses 14 days after 6-OHDA injection. For behavior tests, apomorphine was subcutaneously injected into each mouse at a 0.5 mg/kg dose, and asymmetrical rotation of mice to their contralateral side was monitored for 1 hour. In method 1, the 6-OHDA-injected group showed 334.6 net turns per hour, whereas the PEG-SWNT pre-administered group showed a significant reduction in abnormal rotation induced by 6-OHDA (334.6±13.6 for 6-OHDA only vs 186.2±12.2 for PEG-SWNT pre-injection before 6-OHDA, *P<0.05). In method 2, the 6-OHDA group exhibited 418.2 net turns per hour, whereas the post-injection of PEG-SWNTs resulted in 168.2 net turns per hour (418.2±21.5 for 6-OHDA only vs 168.2±49.6 for post-injection of PEG-SWNTs after 6-OHDA; ***P<0.001). Moreover, intravenous injection of PEG-SWNTs after 6-OHDA treatment resulted in a significant reduction of abnormal rotation compared to the 6-OHDA-only group (408.2±59.7 for 6-OHDA-only vs 253.8±22.2 for the intravenous injection of PEG-SWNTs after 6-OHDA; *P<0.05). In summary, all PEG-SWNT-injected groups exhibited a reduced rotation number regardless of the administration route (Figure 2B).
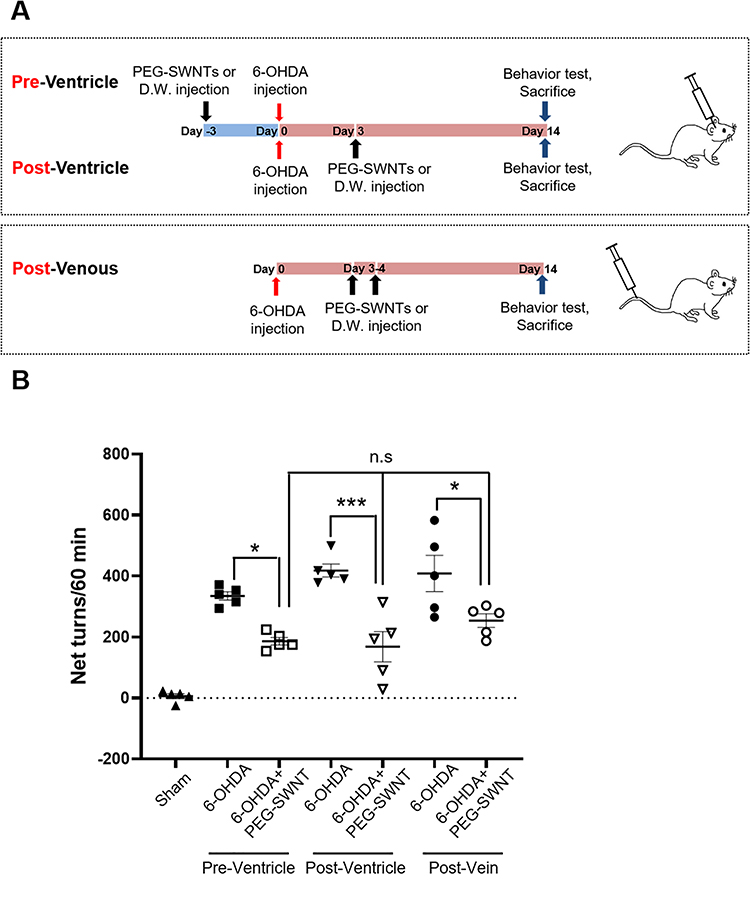 |
Figure 2 Experimental design and assessment for Parkinson’s disease model. (A) The PD model was administered with PEG-SWNTs via different routes. In Method 1, PEG-SWNTs (2 µL; 2 µg) or DW (2 µL) were injected into the lateral ventricle of each mouse three days before 6-OHDA (10 µg) was injected into the ipsilateral ST. In Method 2, PEG-SWNTs or DW were injected three days after 6-OHDA administration. In Method 3, PEG-SWNTs (200 µL; 200 µg) or DW (200 µL) were injected into the tail vein once per day at the third and fourth day after 6-OHDA administration. At two weeks after 6-OHDA injection, apomorphine-induced rotation tests and histological assessment were performed. (B) Apomorphine was subcutaneously injected into mice, after which rotation behavior was evaluated. The number of rotations (net turns = contralateral turns-ipsilateral turns) was recorded for an hour. All data were expressed as mean ± S.E.M. (One way ANOVA, all pair-wise multiple comparisons were performed with Holm–Sidak’s method, *P < 0.05, ***P < 0.0001; n = 5 for each group). |
TH+ Neurons are Rescued by PEG-SWNTs
Given that PEG-SWNTs were found to prevent abnormal behavior in PD mice, histological analysis was performed to determine whether dopaminergic neurons in ST and SNpc could also be protected by PEG-SWNTs. As shown in Figure 3A, the ipsilateral loss of tyrosine hydroxylase (TH)-positive neurons was observed in 6-OHDA-induced PD mice brains compared to the contralateral side of each group or the sham group. However, the PEG-SWNT-injected group showed a higher population of TH+ neurons in the ST regardless of the method of administration (Figure 3A). Interestingly, intracranial injection of PEG-SWNTs (both pre- and post-) resulted in a greater population of TH-positive neurons than the intravenous-injected PEG-SWNTs group. The TH+ neurons in the SNpc were also protected significantly in the PEG-SWNTs group compared to the 6-OHDA-only group (Figure 3B).
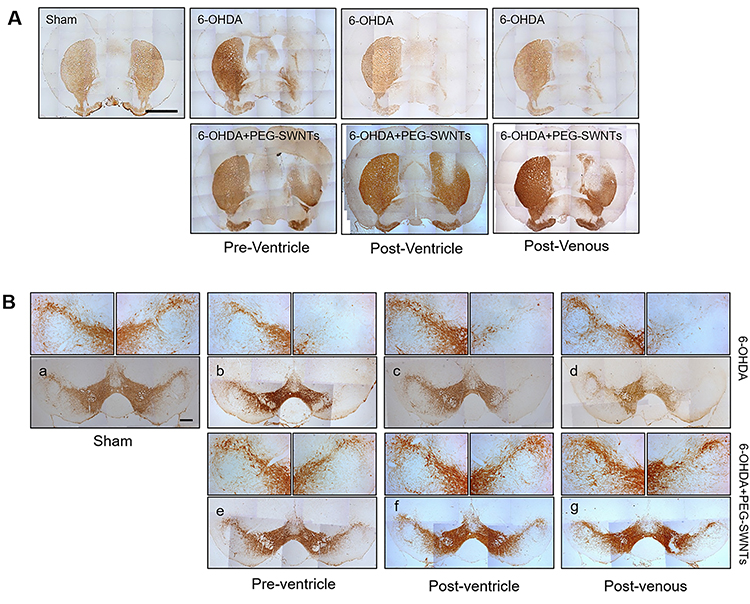 |
Figure 3 Histological assessment of PEG-SWNTs injected PD animals. (A) Immunohistochemistry staining for TH+ neurons in the ST two weeks after 6-OHDA injection. Scale bar = 1 mm. (B) Immunohistochemistry staining for TH+ neurons in the SN pars compacta (SNpc) two weeks after 6-OHDA injection. Scale bar = 200 µm. |
Although TH protein expression levels in the ST in the ipsilateral hemisphere were also markedly reduced by 6-OHDA injection, pre- and post-PEG-SWNTs-injected mice exhibited further less reductions in TH protein in the ipsilateral hemisphere under all three administration methods (Figure 4). Moreover, pre-intracranial injection of PEG-SWNTs significantly protected TH-positive neurons in 6-OHDA-exposed mice, which showed similar levels of TH expression to those of the sham group (Figure 4B).
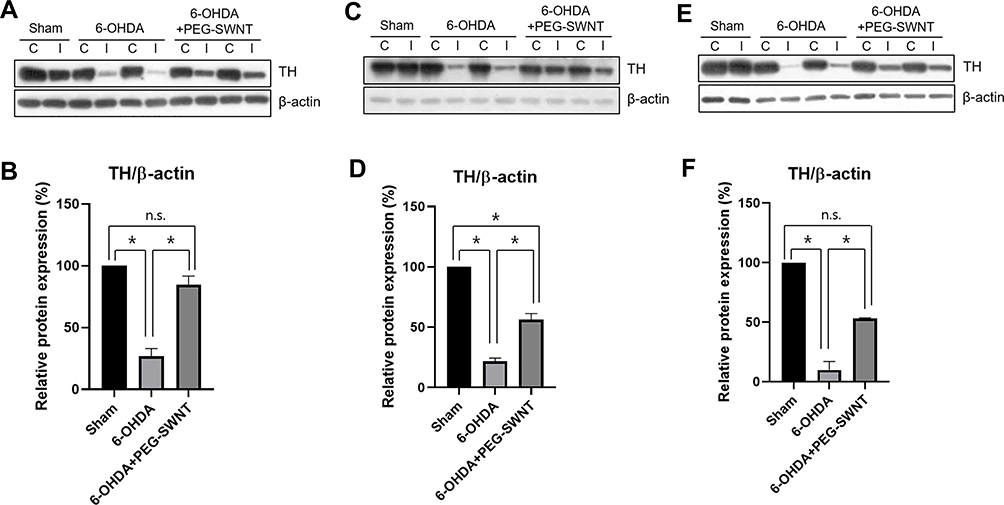 |
Figure 4 Western blot analysis of TH protein level in mice ST by PEG-SWNTs. The effect of PEG-SWNT administration route on TH protein expression was evaluated in PD mice ST. (A and B) TH expression in Method 1; (C and D) TH expression in Method 2; (E and F) TH expression in Method 3. (One-way ANOVA, all pair-wise multiple comparisons were performed with Holm–Sidak’s method, *P < 0.05; n = 4 for each group). |
PEG-SWNTs Promote Neural Differentiation and Neurite Outgrowth
Since neural cadherin (N-cadherin) is a known regulator of neural polarity and neuron development,17 we confirmed N-cadherin expression in the PD mouse model. In our previous study, N-cadherin expression was significantly enhanced in cases of SWNT-induced neuroprotection against brain damage by stroke.11 As shown in Figure 5, N-cadherin expression was significantly decreased in the ipsilateral hemisphere of the PD mouse model compared to the contralateral hemisphere. Interestingly, the ipsilateral side in the PEG-SWNTs-injected PD mouse model exhibited a highly enhanced expression of N-cadherin compared to the contralateral side. Chen et al reported that N-cadherin is an important regulator of neurite outgrowth.18 To investigate PEG-SWNT-induced neurite outgrowth enhancement, human neuroblastoma SH-SY5Y cells were differentiated into neurons on the surface of a PEG-SWNT-treated or control glass coverslip, after which the length of each neurite was estimated. After seven days of differentiating the SH-SY5Y cells with 10 µM of retinoic acid (RA), cellular morphology was captured and the neurite length of each cell was quantified with the ImageJ software. At day 7 post-differentiation, PEG-SWNT-treated SH-SY5Y cells showed higher MAP2 expression than the cells on the control glass coverslip (lower panel in Figure 6A). Additionally, as shown in Figure 6A (upper panel) and B, the neurites of the PEG-SWNT-treated SH-SY5Y cells were also longer and more complexed than the control cells.
 |
Figure 5 Western blot analysis of N-cadherin expression in the mouse ST by PEG-SWNTs. The effect of PEG-SWNT administration route on N-cadherin expression was evaluated in PD mice ST. (A and B) N-cadherin expression in Method 1; (C and D) N-cadherin expression in Method 2; (E and F) N-cadherin expression in Method 3. (One-way ANOVA, *P < 0.05; n = 4 for each group). |
 |
Figure 6 Effect of PEG-SWNTs on SH-SY5Y cell neurite outgrowth. SH-SY5Y cells were differentiated with 10 µM of retinoic acid for seven days on either a PEG-SWNT-treated surface or a control glass coverslip. (A) The upper panel shows bright field images for each group and the lower panel indicates microtubule-associated protein 2 (MAP2) stained cells on each surface. (B) The neurite length of differentiated SH-SY5Y cells on each surface was quantified with the ImageJ 1.52a software. (unpaired t-test, *P < 0.05; n = 5 for each group). |
PEG-SWNTs Attenuate 6-OHDA-Induced Oxidative Stress
To investigate how PEG-SWNTs prevent neuronal death induced by 6-OHDA, the oxidative stress level of SH-SY5Y cells with and without PEG-SWNTs was estimated. As shown in Figure 7, exposure to 50 μM of 6-OHDA resulted in high levels of reactive oxygen species (ROS) after 3 hours in SH-SY5Y cells cultured on glass coverslips, whereas cells grown on a PEG-SWNT coated surface exhibited only small increases in ROS (Figure 7A). When the cells were directly co-treated with PEG-SWNTs and 6-OHDA (Figure 7B) or pre-treated with PEG-SWNTs 1 hour prior to 6-OHDA exposure (Figure 7C), ROS levels were also dramatically reduced. Given that mitochondrial impairment and oxidative damage in the brain play a key role in PD pathogenesis,19,20 mitochondrial membrane potential was also evaluated. TMRE, a cationic potentiometric dye, is used as an indicator of mitochondrial membrane potential integrity. As shown in Figure 7D, vehicle-treated cells showed bright red fluorescence, whereas 6-OHDA treatment significantly reduced the mitochondrial membrane potential of SH-SY5Y cells cultured on a glass surface. In contrast, the cells grown on the PEG-SWNT coated surface exhibited relatively intact mitochondrial membrane potentials. 100 μM of Vitamin C (L-ascorbic acid) was used as a positive control in this experiment, as it is known to act as a free radical scavenger and possesses antioxidant effects. When the cells were directly co-treated with PEG-SWNTs and 6-OHDA (Figure 7E) or pre-treated with PEG-SWNTs 1 hour prior to 6-OHDA exposure (Figure 7F), collapsed mitochondrial membrane potential was significantly recovered. Additionally, the alamarBlue assay demonstrated that PEG-SWNTs prevented neuronal cell death caused by a 24 h exposure to 6-OHDA (Supplementary Figure 2A and B). PEG-SWNT co-treatment or pre-treatment attenuated neuronal cell death caused by 50 μM 6-OHDA exposure, and PEG-SWNTs alone did not induce cellular damage (Supplementary Figure 2C).
 |
Figure 7 Antioxidant effect of PEG-SWNTs on SH-SY5Y cells. (A) SH-SY5Y cells were differentiated with 10 μM retinoic acid for seven days on either a PEG-SWNT-treated surface or a control glass coverslip. Each group was treated with a vehicle solution or 50 μM of 6-OHDA for 3 hours, after which DCF-DA was added and allowed to incubate for 45 min at 37°C; representative images were the captured. (B and C) The cells were seeded in 96-well plates and treated with different PEG-SWNT concentrations either simultaneously with 6-OHDA (B) or 1 hour prior to 6-OHDA treatment (C). Three hours after 6-OHDA treatment, DCF-DA was added and allowed to incubate, after which fluorescence intensity was detected with a fluorescence plate reader (See Materials and Methods for detail) (n=3 independent experiments; One-way ANOVA, #P<0.05 Con vs 6-OHDA; *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 6-OHDA vs 6-OHDA+PEG-SWNTs groups). (D) The differentiated cells were then seeded on either PEG-SWNT-coated surfaces or control glass coverslips. Each group was treated with vehicle or 50 μM of 6-OHDA for 6 hours, after which TMRE was added and allowed to incubate for an additional 15 min at 37°C. Fluorescent images were chosen randomly per each group. (E and F) The cells were seeded onto 96-well plates and treated with different PEG-SWNTs concentrations either simultaneously with 6-OHDA (E) or 1 hour prior to 6-OHDA treatment (F). Six hours after 6-OHDA treatment, 100 nM of TMRE was added and allowed to incubate for 15 additional minutes, after which the fluorescence intensity of each well was measured with a fluorescence plate reader (see the Materials and Methods for more details) (n=3 independent experiments; one-way ANOVA, #P<0.05, ##P<0.001 Con vs 6-OHDA; **P<0.01, ****P<0.0001 6-OHDA vs 6-OHDA+PEG-SWNTs). |
Discussion
This study demonstrated the neuroprotective effect of functionalized SWNTs on a 6-OHDA-induced PD model. Additionally, various SWNT routes of administration into the brain tissue were compared to assess their effectiveness. Pre-intracranial injection of SWNTs resulted in the most effective protection of TH-positive neurons in the ST compared to the other administration methods. Moreover, PEG-SWNT pre-administration through the skull of PD mice enhanced TH protein expression to levels as high as those of the sham group (Figure 4A and B). Nonetheless, all SWNT-injected groups showed significant brain tissue neuroprotection and SWNT treatment also alleviated asymmetric behavior in PD mice compared to the 6-OHDA-only group.
CNTs have been previously applied to reduce neuronal damage or improve neural function in PD models.13,21 Vitale et al utilized CNT fiber as a microelectrode to enhance neural activity and reduce inflammatory responses in PD mice due to its great biocompatibility.21 For PD treatment, Guo et al utilized PEG-SWNTs as a drug carrier and linked DA onto the surface of PEG-SWNTs to deliver DA into the parkinsonian brain.13 Although CNTs have been proposed as devices or vehicles to deliver bioactive compounds to treat neurodegenerative diseases, we demonstrate here that functionalized SWNTs per se have the potential to prevent ST and SNpc neuronal damage in the pre-injected group, as well as protect TH-positive neurons in the post-administered group. Additionally, this study monitored the long-term toxicity of PEG-SWNTs for treated all animals over six months (Supplementary Figures 3 and 4). Animal body weights were only affected by 6-OHDA injection for acute monitoring, whereas PD mice and PEG-SWNT-treated PD mice showed similar body weights at the end of long-term tracking. Moreover, we found that systemic intravenous injection of PEG-SWNTs did not increase GOT or GPT blood levels at two weeks, three months, and six months after SWNT injection, suggesting that functionalized SWNTs do not pose significant long-term cytotoxicity. A few previous studies regarding the in vivo application of CNT have also reported that intravenously injected CNTs were rapidly cleared through renal excretion22 or opsonization through reticuloendothelial system (RES) capture,23 resulting in low toxicity. Moreover, Nunes et al reported that intracranial-injected CNTs could be partially biodegraded or uptaken by microglia,24 thus supporting the safety of direct CNT application as a nanomedicine.
Early studies on CNTs revealed that they raised cytotoxicity and inflammation in the lung and liver.25,26 CNT length and surface modifications (ie, functionalization) may result in different CNT toxicities after in vivo application. Long fibers and large CNT aggregates typically induce asbestos-like toxicity, which is characterized by chronic inflammation and the formation of fibrosis in the lungs.25 In contrast, short and functionalized CNTs tend to be more easily cleared by phagocytosis or excreted through other routes.27 Kolosnjaj-Tabi et al exposed Swiss mice to SWNTs of different lengths and reported that well-functionalized SWNTs (<300 nm) were eliminated via the kidneys and bile ducts.28 However, large aggregates of SWNTs (>10 µm) induced severe granuloma formation, whereas smaller aggregates (<10 µm) could be engulfed by phagocytes but were not cleared for several months. The PEG-SWNTs utilized in this study were 0.5–0.6 µm long; thus, we assume that our SWNTs could be cleared rapidly, thereby delivering protective effects without cytotoxicity or inflammation.
Moreover, our results demonstrated that pre- and post-administration of SWNTs protects dopaminergic neurons in PD mice. Moreover, the in vitro results demonstrated that the neurite outgrowth of SH-SY5Y cells was elongated and more branched on the PEG-SWNT-treated surface than on glass coverslips. Additionally, PEG-SWNTs could visibly reduce oxidative stress caused by 6-OHDA exposure by preventing the collapse of mitochondrial membrane potential in SH-SY5Y cells, which indicated that PEG-SWNTs act as potent antioxidants. A few studies have also reported the antioxidant activity of SWNTs due to their high electron affinity.29,30 It is assumed that the antioxidant properties of SWNTs might potentiate the protective effect of PEG-SWNTs on dopaminergic neurons. Nevertheless, the molecular mechanisms underlying these protective effects must be further elucidated in future studies.
This study demonstrated that intravenous injection of SWNTs protected mice from dopaminergic neuronal death and reduced abnormal behavior induced by 6-OHDA. Kafa et al demonstrated that functionalized MWNTs were able to cross the blood–brain barrier (BBB) by using an in vitro co-culture system and in vivo systemic injection.31 In their study, CNTs were proven to effectively penetrate the BBB via receptor-mediated transcytosis. Moreover, 6-OHDA-induced brain lesions may alter BBB permeability,32 meaning that intravenously injected CNTs can cross the BBB more easily. In the present study, intravenously injecting PEG-SWNTs after 6-OHDA treatment attenuated abnormal behavior and neuronal loss in PD mice; thus, it was assumed that injected PEG-SWNTs could penetrate the BBB and act as a nanomedicine. In summary, PEG-SWNTs have great potential to protect dopaminergic neurons in a PD mouse model by either intracranial or intravenous injection, indicating that PEG-SWNTs could become promising materials as nanomedicine either alone or in combination with other drugs.
Conclusion
In this study, PEG-SWNTs were administered for treating PD mouse model through different routes. Both pre- and post-intracranial injection of PEG-SWNTs in PD mice significantly protected dopaminergic neurons from 6-OHDA induced brain damage and reduced abnormal behavior of PD mice. Moreover, intravenous injection of PEG-SWNTs also reduced neuronal damage and behavioral abnormality by 6-OHDA in PD mice, indicating PEG-SWNTs are promising materials to treat Parkinson’s disease. Further, less invasive route of PEG-SWNTs, intravenous injection also showed significant protection of ST and SNpc in PC mice, suggesting PEG-SWNTs have a great potential as nanomedicine agents combined with or without therapeutic drugs.
Abbreviations
SWNT, single-walled nanotube; MWNT, multi-walled nanotube; CNT, carbon nanotube; PD, Parkinson’s disease; 6-OHDA, 6-hydroxydopamine; SN, substantia nigra; SNpc, substantia nigra pars compacta; DA, dopamine; PEG, polyethylene glycol; ST, striatum; RA, retinoic acid; BBB, blood–brain barrier.
Funding
This study was supported by a Chung-Ang University Research Grant in 2019 and by a National Research Foundation of Korea (NRF) grant funded by the Korean government [No. 2020R1A2C2011617].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nikalje AP. Nanotechnology and its applications in medicine. Med Chem. 2015;5:2. doi:10.4172/2161-0444.1000247
2. De Lau LM, Breteler PMP. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi:10.1016/S1474-4422(06)70471-9
3. Mazzoni P, Shabbott B, Cortés JC. Motor control abnormalities in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009282. doi:10.1101/cshperspect.a009282
4. Wang L, Zhang L, Xue X, Ge G, Liang X. Enhanced dispersibility and cellular transmembrane capability of single-wall carbon nanotubes by polycyclic organic compounds as chaperon. Nanoscale. 2012;4:3983–3989.
5. Malarkey EB, Fisher KA, Bekyarova E, Liu W, Haddon RC, Parpura V. Conductive single-walled carbon nanotube substrates modulate neuronal growth. Nano Lett. 2009;9:264–268. doi:10.1021/nl802855c
6. Wang K, Fishman HA, Dai H, Harris JS. Neural stimulation with a carbon nanotube microelectrode array. Nano Lett. 2006;6:2043–2048. doi:10.1021/nl061241t
7. Rodriguez-Yañez Y, Muñoz B, Albores A. Mechanisms of toxicity by carbon nanotubes. Toxicol Mech Methods. 2013;23:178–195. doi:10.3109/15376516.2012.754534
8. Kam NW, Jan E, Kotov NA. Electrical stimulation of neural stem cells mediated by humanized carbon nanotube composite made with extracellular matrix protein. Nano Lett. 2009;9:273–278. doi:10.1021/nl802859a
9. Lovat V, Pantarotto D, Lagostena L, et al. Carbon nanotube substrates boost neuronal electrical signaling. Nano Lett. 2005;5:1107–1110.
10. Moon SU, Kim J, Bokara KK, et al. Carbon nanotubes impregnated with subventricular zone neural progenitor cells promotes recovery from stroke. Int J Nanomed. 2012;7:2751–2765.
11. Lee HJ, Park J, Yoon OJ, et al. Amine-modified single-walled carbon nanotubes protect neurons from injury in a rat stroke model. Nat Nanotechnol. 2011;6:121–125. doi:10.1038/nnano.2010.281
12. Godin AG, Varela JA, Gao Z, et al. Single-nanotube tracking reveals the nanoscale organization of the extracellular space in the live brain. Nat Nanotechnol. 2016;12:238–243. doi:10.1038/nnano.2016.248
13. Guo Q, You H, Yang X, et al. Functional single-walled carbon nanotubes ‘CAR’ for targeting dopamine delivery into the brain of parkinsonian mice. Nanoscale. 2017;9:10832. doi:10.1039/C7NR02682J
14. Ma X, Zhong L, Guo H, et al. Multiwalled carbon nanotubes induced hypotension by regulating the central nervous system. Adv Funct Mater. 2018;28:1705479. doi:10.1002/adfm.201705479
15. Ungerstedt U, Arbuthnott GW. Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res. 1970;24(3):485–493. doi:10.1016/0006-8993(70)90187-3
16. Tieu KA. Guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb Perspect Med. 2011;1:a009316. doi:10.1101/cshperspect.a009316
17. Rajwar YC, Jain N, Bhatia G, Sikka N, Garg B, Walia E. Expression and significance of cadherins and its subtypes in development and progression of oral cancers: a review. J Clin Diagnostic Res. 2015;9:ZE05–ZE07.
18. Chen Q, Chen TJ, Letourneau PC, Costa LDF, Schubert D. Modifier of cell adhesion regulates n-cadherin-mediated cell-cell adhesion and neurite outgrowth. J Neurosci. 2005;25:281–290. doi:10.1523/JNEUROSCI.3692-04.2005
19. Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:120–131. doi:10.1111/j.1749-6632.2003.tb07470.x
20. Jenner P, Olanow CW. Understanding cell death in Parkinson’s disease. Ann Neurol. 1998;44:S7284. doi:10.1002/ana.410440712
21. Vitale F, Summerson SR, Aazhang B, Kemere C, Pasquali M. Neural stimulation and recording with bidirectional, soft carbon nanotube fiber microelectrodes. ACS Nano. 2015;9:4465–4474. doi:10.1021/acsnano.5b01060
22. Singh R, Pantarotto D, Lacerda L, et al. Tissue biodistribution and blood clearance rates of intravenously administered carbon nanotube radiotracers. Proc Natl Acad Sci. 2006;103:3357–3362. doi:10.1073/pnas.0509009103
23. Owens III DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;397:93–102. doi:10.1016/j.ijpharm.2005.10.010
24. Nunes A, Bussy C, Gherardini L, et al. In vivo degradation of functionalized carbon nanotubes after stereotactic administration in the brain cortex. Nanomedicine. 2012;7:1485–1494. doi:10.2217/nnm.12.33
25. Poland CA, Duffin R, Kinloch I, et al. Carbon nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat Nanotechnol. 2008;3:423–428. doi:10.1038/nnano.2008.111
26. Lam CW, James JT, McCluskey R, Hunter RL. Pulmonary toxicity of single-wall carbon nanotubes in mice 7 and 90 days after intratracheal instillation. Toxicol Sci. 2004;77:126–134. doi:10.1093/toxsci/kfg243
27. Murphy FA, Poland CA, Duffin R, et al. Length-dependent retention of carbon nanotubes in the pleural space of mice initiates sustained inflammation and progressive fibrosis on the parietal pleura. Am J Pathol. 2011;178:2587–2600. doi:10.1016/j.ajpath.2011.02.040
28. Kolosnjaj-Tabi J, Hartman KB, Boudjemaa S, et al. In vivo behavior of large doses of ultrashort and full-length single-walled carbon nanotubes after oral and intraperitoneal administration to Swiss mice. ACS Nano. 2010;4:1481–1492. doi:10.1021/nn901573w
29. Lucente-Schultz RM, Moore VC, Leonard AD, et al. Antioxidant single-walled carbon nanotubes. J Am Chem Soc. 2009;131:3934–3941. doi:10.1021/ja805721p
30. Watts PCP, Fearon PK, Hsu WK, Billingham NC, Kroto HW, Walton DRM. Carbon nanotubes as polymer antioxidants. J Mater Chem. 2003;13:491–495. doi:10.1039/b211328g
31. Kafa H, Wang JTW, Rubio N, et al. The interaction of carbon nanotubes with an in vitro blood-brain barrier model and mouse brain in vivo. Biomaterials. 2015;53:437–452. doi:10.1016/j.biomaterials.2015.02.083
32. Carvey PM, Zhao CH, Hendey B, et al. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur J Neurosci. 2005;22:1158–1168. doi:10.1111/j.1460-9568.2005.04281.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.