Back to Journals » Biologics: Targets and Therapy » Volume 18
Biophysical Analysis of Therapeutic Antibodies in the Early Development Pipeline
Authors Willis LF ![]() , Kapur N
, Kapur N ![]() , Radford SE, Brockwell DJ
, Radford SE, Brockwell DJ ![]()
Received 12 October 2024
Accepted for publication 10 December 2024
Published 21 December 2024 Volume 2024:18 Pages 413—432
DOI https://doi.org/10.2147/BTT.S486345
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Shein-Chung Chow
Video abstract presented by Willis.
Views: 165
Leon F Willis,1 Nikil Kapur,2 Sheena E Radford,1 David J Brockwell1
1School of Molecular and Cellular Biology, Astbury Centre for Structural Molecular Biology, Faculty of Biological Sciences, University of Leeds, Leeds, LS2 9JT, UK; 2School of Mechanical Engineering, Faculty of Engineering and Physical Sciences, University of Leeds, Leeds, LS2 9JT, UK
Correspondence: David J Brockwell, Email [email protected]
Abstract: The successful progression of therapeutic antibodies and other biologics from the laboratory to the clinic depends on their possession of “drug-like” biophysical properties. The techniques and the resultant biophysical and biochemical parameters used to characterize their ease of manufacture can be broadly defined as developability. Focusing on antibodies, this review firstly discusses established and emerging biophysical techniques used to probe the early-stage developability of biologics, aimed towards those new to the field. Secondly, we describe the inter-relationships and redundancies amongst developability assays and how in silico methods aid the efficient deployment of developability to bring a new generation of cost-effective therapeutic proteins from bench to bedside more quickly and sustainably.
Keywords: developability, biophysics, protein, antibody, analytical techniques
Introduction: The “Biologics Era” and the Drug Discovery Pipeline
Therapeutic proteins, or biologics, interact with their targets with a different mechanism of action and often greater specificity than their small-molecule counterparts, giving rise to fewer side-effects.1 These features have thus made the development of proteins as therapies desirable over the past few decades. The advent of recombinant DNA technology in the 1970’s and 1980’s brought the biopharmaceutical industry into its modern era.2–5 Prior to this, therapeutic peptides and proteins derived from animal6 or donor sources (eg insulin7 and blood factors8) were used to treat patients deficient in these vital macromolecules.5 A further vital breakthrough was the development of hybridoma technology by Köhler and Milstein,9 and using this approach, the first monoclonal antibody (mAb) therapy (Orthoclone®) was approved by the Food and Drug Administration (FDA) in 1986.10 Since then, many technological breakthroughs have been made,11 including a variety of display methods, (such as phage-4 and ribosome display12) that now allow the routine and efficient identification of highly avid mAbs.13,14 Additionally, mAbs have additional modes of action (effector function) afforded to them through their crystallizable fraction (Fc) region; the isotype of the antibody can modulate its interaction with different receptors, eg, neonatal receptor allows for “recycling” of the molecule from the serum giving a long half-life15 or interaction with Fcγ receptor IIIa to trigger antibody-dependent cellular cytotoxicity.15,16 By contrast, peptide therapeutic agents lack such diverse modes of action nor a means of extending their half-life; unnatural amino acids or chemical modifications are necessary to improve this. For example, the blockbuster type-2 diabetes and anti-obesity drug, Ozempic® has two amino acid substitutions/modifications (Ala8 to α-aminoisobutyric acid and Lys34 to Arg) in addition to conjugating a C18 fatty acid via a hydrophilic linker to the ε-amino group of Lys26 in the semaglutide peptide. This increases its half-life by several days compared to the wild-type peptide.17,18
High target affinity, however, is only one feature that is required for a successful mAb therapeutic to transition from “bench to bedside” (Figure 1).19 These additional properties, identified over the last forty years include specificity, potency and manufacturability, and are probed using a “toolkit” of different techniques.20 The process of identifying whether any initial “hit molecules” possess the critical quality attributes required for the economic manufacture of an efficacious and safe medicine (Figure 1b) is known as “developability”.21–23 The varied physicochemical profile of a molecule needs to be evaluated using multiple different methods, with each method probing (mainly) one specific feature, eg, hydrophobicity Figure 1. Technological advancements in instrumentation, computation and fundamental science have meant that this toolkit is always changing in terms of size and contents.20
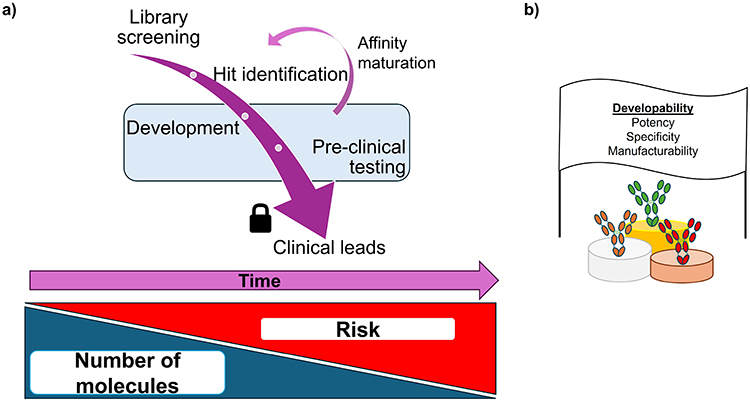 |
Figure 1 Overview of the drug development pipeline. (a) Screening libraries of molecules to identify hits against the target(s) of interest will whittle the number of molecules down from several thousand to tens of molecules. An array of developability assays will be employed to interrogate these hits, with the most successful undergoing further tests to ascertain if they will progress to the clinic. These clinical leads have their sequence locked at this point, with late-stage formulation deciding the dosage and delivery method. Low quantities of material are produced (tens of milligrams from tens–hundreds of mL of cell culture) for early development, increasing to gram and kilogram scale (from thousands of liters of cells) at the clinical end. The cost of clinical trials (both financially and in terms of patient benefit) means that increased risk is attached to molecules that reach the clinic, thus developability assays and the data they generate are so important. The blue region represents the scope of this review. (b) Ultimately, one aims to use developability assays to find potent molecules, with no off-target interactions that can be made easily, at scale and administered safely into patients. |
In this review, we survey the array of biophysical techniques used in early-phase drug discovery to aid the selection of developable biopharmaceuticals. Focusing on antibody-based therapies, commonly used experimental and in silico methods are discussed, prefaced with a simple overview of how they work. New techniques are then highlighted together with the potential of machine learning and artificial intelligence to exploit the ever-increasing quantities of training data, which together may rationalize the developability framework. This is of increasing importance as a wide range of next-generation therapeutics based on mAb scaffolds with more challenging biophysical properties for manufacturing are entering development.24 This is a challenging yet exciting time, with biophysical characterization fundamental in the development of the next-generation of therapeutic proteins.
Folding and Functionality: From Conformational to Colloidal Stability
Before we look at the methods used to analyze therapeutic proteins, it is important to consider the species one needs to analyze in term of their size, abundance and stability (thermodynamic and kinetic); all of which can be modulated by their physico-chemical environment. We define the native state as the correctly folded three-dimensional structure of protein through which desirable biological function is achieved (Figure 2). Given its functional importance within the context of the biologics sphere, the characterization of this state, excursions from it and the parameters that affect it are a key part of drug developability screening.25,26 It should be noted that these principles apply to all globular proteins, not just therapeutic mAbs.
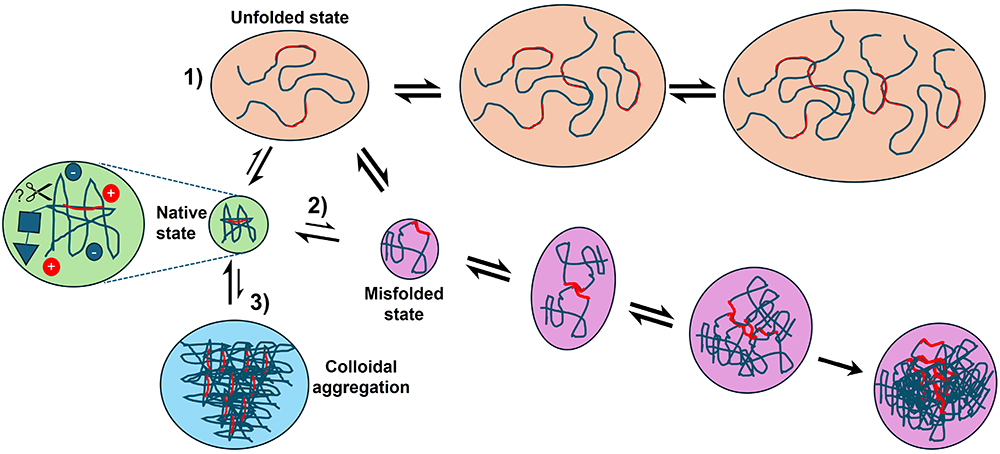 |
Figure 2 Overview of the aggregation pathways of biopharmaceuticals. Most therapeutic proteins are folded into a defined 3D-structure (green circles). The native protein’s surface charge, dynamics, amenability to cleavage by proteases (for example) are all important for it to maintain this structure. Extremes of pH or temperature may bring about the complete unfolding of a protein, leading to aggregation via its exposed aggregation-prone regions (in red) via pathway 1. Along pathway 2 (purple route), partial unfolding of the native state (or misfolding from 1) results in an aggregation-prone misfolded state, with these species able to multimerize and eventually precipitate from solution. Finally, it is increasingly clear that some proteins can reversibly self-associate into crystalline precipitates or a liquid-like phase (colloidal aggregation, blue circle). The length- and timescales of all of the above mean a wide range of assays are necessary to probe the states above and their interconversion. |
Relatively harsh conditions (eg extremes of pH,27–29 temperature30–32 or in the presence of chaotropic denaturants such as urea,33,34 guanidine hydrochloride34,35 or organic solvents)36,37 will bring about the global unfolding of a protein (Figure 2). Unfolded proteins can self-associate and aggregate due to exposure of previously buried hydrophobic residues to the solvent. There is a consensus in the literature25,38,39 that for biopharmaceuticals, most aggregation takes place from a misfolded state, brought about by perturbations to the native state by physical, chemical or mechanical means. For this review, we define an aggregate as a proteinaceous entity comprising two or more mis- or un-folded species.40
Colloidal stability refers to the dispersity of the sample, ie, a low dispersity sample contains species of mainly one size, eg, monomers, whereas a highly disperse sample would contain monomers and aggregates over a broad range of size/length scales.41,42 While it is established that under certain conditions, proteins can self-associate into a solid-state crystal lattice (ie native state aggregation),43 many have shown that proteins can self-associate and undergo liquid–liquid phase separation (Figure 2).44–46 While crystallization is being explored as an alternative means of purifying proteins at scale,47 both of these aggregation phenomena are generally deleterious for mAbs during production.
In-Silico Analysis of Therapeutic Protein Sequences: Identifying Risks and Liabilities
A plethora of computational methods have been developed to aid the development of therapeutic proteins (Figure 3).48–50 Generally, these algorithms use a sliding window of primary sequence to compute an output score describing a certain biophysical property such as net charge or hydrophobicity on a “per residue” basis or averaged over the entire protein.51 Experimentally relevant conditions can often be fed into the algorithms to tune the output, eg, changing pH or ionic strength, which are known to influence aggregation kinetics by affecting electrostatic interactions between proteins.25 These algorithms can be applied to either the primary sequence or higher-order protein structures, allowing the structural context of problematic sequences to be taken into account with structures derived either experimentally or from homology models using programs such as MODELLER,52 and ABodyBuilder.53 Sequences or regions of protein, which promote aggregation or have low solubility, for example, can thus be identified, aiding the redesign of protein sequences or the selection of more developable candidates. Most algorithms developed to probe this have been trained on intrinsically disordered amyloidogenic peptides, eg, TANGO54 and AGGRESCAN;55 however, some of these have been adapted or developed specifically to look at therapeutic proteins, including antibodies.56–60
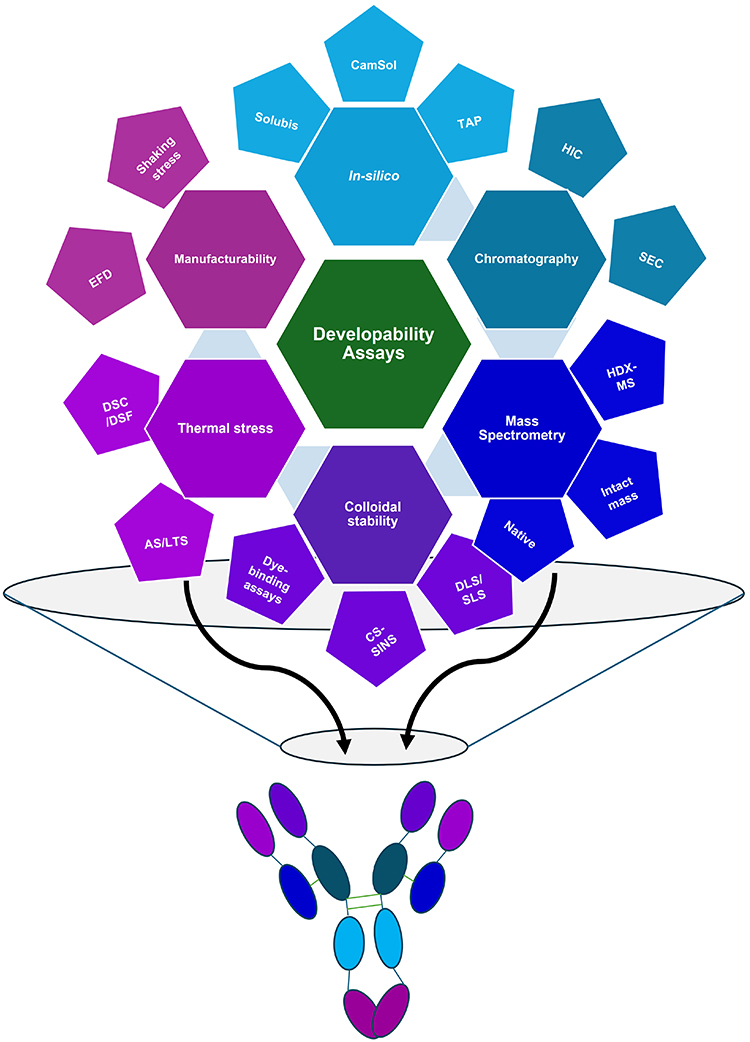 |
Figure 3 Schematic depicting the “family tree” of developability assays, discussed in this review. The assay groups radiating from the center (going clockwise from the top) are discussed in this review: In-silico assays, Chromatography-based assays, Mass spectrometry-based approaches, Methods to probe colloidal stability, thermal stress assays and finally, assays to probe manufacturability. As developability assays from each branch probe different physicochemical features of a molecule, by “harvesting fruit” from different branches of this tree, one can gain a holistic understanding of a molecule’s developability profile, represented by the mAb colored by the branches above. |
The TANGO algorithm was developed twenty years ago and uses a statistical mechanics approach to predict the β-sheet aggregation propensity of a given protein/peptide sequence.54 While amyloid aggregates consist of a cross-β architecture,61,62 thus the excellent prediction of amyloidogenic sequences with this algorithm, non-amyloidogenic aggregates can still contain β-sheet rich aggregates (binding dyes such as Thioflavin T).63 Antibodies contain multiple immunoglobulin domains, comprising two β-sheets cross-linked via a conserved disulfide bond.64,65 The algorithm can thus predict aggregation for a broad range of input sequences. As shown in Figure 2, aggregation of biopharmaceuticals is thought to mainly take place through the misfolded state,25 by exposing aggregation-prone regions (APRs) buried in the natively folded core, to the solvent. Here, APRs are defined as a window of five or more amino-acids with a high average TANGO score and while ordinarily buried in the native state, these APRs may become solvent-exposed upon environmental perturbation (eg38). The Solubis approach66,67 calculates both the aggregation propensity (using TANGO) and the contribution of a given APR towards a protein’s folding stability (ΔGcontrib) using the FoldX algorithm.68 Using this approach, variants with reduced aggregation propensity can then be designed which suppress the TANGO score but do not thermodynamically destabilize the protein. For example, placing a lysine residue, a known “gatekeeper” which disfavors β-sheet formation,69 into an APR would decrease its TANGO score, but would adversely affect the conformational stability of a protein if the APR in question was found in the hydrophobic core of a protein.
Other algorithms have been developed to identify APRs formed from spatially proximal residues that may be distal in primary sequence. The Spatial Aggregation Propensity (SAP) algorithm has been developed to identify solvent-exposed APRs in antibodies using a molecular dynamics-based approach.70 Dynamics in the protein, an inherent property of monoclonal antibodies for their function,71 could expose these APRs to the solvent over time. This algorithm has been used to identify problematic APRs in antibodies, for example, the MEDI1912 protein, whose “WFL” residue patch causes notable developability issues such as self-association72 and poor manufacturability.73 This approach was expanded to create a “developability index” which also computes an antibody’s net charge,74 which is increasingly recognized as an important physicochemical parameter to consider for a developable biopharmaceutical.75–78
Solubility can be defined as the maximum quantity of a solute that can be dissolved in a given volume of solvent at equilibrium.79 When proteins aggregate, solvent-solute interactions between water and the protein are disrupted, allowing the APRs to self-associate (to yield a favorable protein–protein interaction).80 Solubility, while related to aggregation, can be treated slightly differently with respect to its prediction, as charge will greatly affect protein solubility in water (a polar solvent). Hydrophobicity is of course another key parameter related to solubility.77,81 A notable tool to predict protein solubility is the CamSol algorithm, developed by Sormanni et al.82 Based on physicochemical properties of amino acids in a protein, a seven-residue sliding window is used to predict the local solubility, which can then be averaged over the length of the sequence. As described above, CamSol scores can also be calculated to account for structural context (ie buried or solvent exposed). Similar to Solubis, rational design of mutants, which favor solubility can be performed with this program,82 as well as the identification of hotspots which disfavor solubility, such as the WFL example above.56
When employing these algorithms for antibody engineering, it is important that any amino-acid substitutions designed to reduce aggregation and or increase solubility do not affect binding affinity to the target,49,83 which is especially challenging as residues in the complementarity determining regions (CDRs) are the regions usually subjected to mutational scanning during discovery.84 To minimize this, the Therapeutic Antibody Profiler (TAP) has been developed to interrogate the CDRs for five developability liabilities including charge density and proximity, length and hydrophobicity.85 An ever-growing database of therapeutic antibodies is used to benchmark amber (warning) and red (danger) thresholds below/above which a given TAP parameter is deemed problematic. Recently, TAP has been expanded to account for differences in both the modelling of antibody structures (aided by machine-learning algorithms (eg ImmuneBuilder)),86 to distinguish between kappa and lambda light chains in therapeutic mAbs, with the latter trending towards poorer developability outcomes.87 Interestingly this observation concurs with the observation that lambda light chains are found relatively more commonly in patients with light-chain amyloidosis.88–90
Efforts have also been made to predict aberrant post-translational modifications in proteins (eg deamidation) based on known sequence motifs, which are linked to these.91 Recently, these sequence features have been identified in germline and therapeutic antibodies, based on mining large sequence databases to generate the Liability Antibody Profiler (LAP).92 Based on their location in the protein (eg solvent exposure) as well as the frequency of these liabilities in germline (ie natural) and therapeutic proteins, a particular liability is ranked in terms of its real risk. For example, identification of a solvent-exposed Met residue which does not exist in the same position in any marketed mAbs may indicate that this is a prime candidate for substitution to minimize the risk of oxidation.92
In summary, computation characterization of the physicochemical characteristics of stretches of amino acids in proteins, corrected based on their structural context, has allowed various algorithms to be developed which identify APRs, regions of poor solubility and other developability liabilities in proteins. The prevalence of these tools in the development of therapeutics will continue to increase as the use of artificial intelligence/machine learning models and the availability of large volumes of sequence data becomes more ubiquitous in the 21st century.93
Chromatography-Based Methods: The Analytical Workhorses of the Biopharmaceutical Industry
The high-throughput (HTP), robust and versatile nature of high-performance liquid chromatography (HPLC) methods means they are employed routinely in the analysis of biopharmaceuticals.94 The chemistry of the stationary phase (the column resin) and the mobile phase (the solvent) work in tandem to resolve different analyte(s) within a sample that can be quantified by a variety of (usually) spectroscopic methods. Proteins and peptides can either be analyzed in their native state using aqueous buffers at near neutral pH or in a denatured form by using non-polar resin and polar solvents at low pH in reversed-phase methods95 with the latter method often prior to analysis by mass spectrometry (MS, see next section). As the characterization of the native state is often the principal aim of developability assessment (see above), this section will focus on normal-phase chromatographic methods.
Size-exclusion chromatography (SEC) has been described as the workhorse method to characterize therapeutic proteins.96,97 Molecules are fractionated by the resin according to their mass and shape.98 Detection of the species as they elute from the column, most frequently by in-line UV-Vis absorbance spectroscopy, allows for the separation and simultaneous quantification of aggregates, monomeric protein, and fragments.97 By measuring samples stored over a period of time, this method is routinely employed to track changes in monomer/fragment/aggregate content in a sample over time at a defined temperature, setting its shelf-life.99,100 The guidelines from the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) mandate this method for therapeutic proteins, based on its power in this regard.101,102 However, SEC does have some caveats. Samples are often diluted prior to analysis or dilute during resolution on the column, which may favor the dissociation of some aggregates back to monomer (thus masking the true aggregate content of the original sample).11,103
Furthermore, some proteins will interact with the column resin (often silica-based for HP-SEC), leading to longer retention times72 erroneously suggesting,98 a much smaller apparent molecular weight. In the absence of changes in mass (verified by MS, for example) longer retention times are usually indicative of “sticky” molecules.72,104 This property has been shown to correlate with other indicators of poor developability, such as poor growth in the presence of beta-lactam antibiotics (indicative of high aggregation propensity)105,106 in an in-vivo tripartite beta-lactamase assay (TPBLA) of antibody variants.83 Chromatography column vendors have developed SEC resins with a stand-up monolayer, which mimic the exterior of proteins.107 For these resins, long retention times (caused by aberrant adsorption to the resin) have been shown to correlate with unfavorable developability parameters, such as colloidal stability as measured using cross-interaction chromatography (CIC), a method, which functionalizes SEC resin with a different IgG sequence to that of the analyte.108
As both hydrophobicity and overall charge can dictate the aggregation and solubility of protein. Hydrophobicity can be probed directly using hydrophobic interaction chromatography (HIC), where a high ionic strength mobile phase (eg 2 M ammonium sulfate) is used to “salt” proteins onto a hydrophobic resin. Dilution of the salt in the mobile phase results in the elution of proteins in order of increasing hydrophobicity (thus long retention times correlate with increased molecular hydrophobicity).104,109 Whether the adsorption process leads to increased exposure of previously buried hydrophobic residues (eg110–112) is a potential caveat of the technique.
Resins of both negative or positive charge (for cation- and anion-exchange chromatography, respectively) are used in ion-exchange chromatography (IEX) to separate analytes based on their charge. The pH of the mobile phase can be used to tune adsorption to the resin, exploiting the isoelectric point (pI) of the analyte of interest, with high ionic strength typically used to elute species from the column.113 While generally employed during bioprocessing in polishing steps to remove nucleic acids or charge variants from the desired drug substance,114 both cation-115,116 and to a lesser-extent, anion-exchange chromatography117 have been used to identify molecules with aberrant charge profiles, often coupled to mass spectrometry113,116 (see below). So-called “mixed mode” resins have been developed which have characteristics encompassing both hydrophobic and charged (ie polar) character.118 Such resins have been used to separate different heavy- and light-chain combinations of bispecific antibodies when used in a SEC-resin, with multi-angle static light scattering used to corroborate the molecular weights of the species separated by these.119
Whilst not a chromatographic method per se, capillary electrophoresis uses an electric field in a functionalized capillary120 or microfluidic chip121,122 to separate molecules based on their charge and mass (ie charge density). By varying the pH in the capillary, the device can be used to perform capillary isoelectric focusing (cIEF). This has been used to look at charge variants of antibodies, with such deviations stemming from aberrant post-translational modifications (PTMs) caused by deamidation, glycosylation and other chemical changes to the higher order structure of the protein.122–124
The varied chemistry of chromatography resins, together with the precision of a HPLC autosampler, mean that HPLC-based methods have become routine for the analysis of therapeutic proteins. Aberrant biophysical behavior (namely fragmentation, aggregation, unwanted surface adsorption and charge variation) can be detected and quantified, depending on the combination of stationary and mobile phases employed. As alluded to above, MS is often employed downstream of these methods.
Mass Spectrometry (MS)- The Main MAM in Developability Assessment
Multi-attribute methods (or MAMs) are analytical methods capable of discerning multiple critical quality attributes (such as low aggregation propensity) from one sample, as described under the Quality-by-Design framework.125,126 MS is one such MAM which has found itself well-placed in the development of therapeutic proteins, due to the rapid advancement and commercialization of these instruments over the past decade,127 and in the software used to analyze the complex data these instruments generate.128 In its simplest application (the accurate mass determination of proteins or their peptide derivatives), most MS measurements of therapeutic proteins are prefaced by a HPLC-based separation of the analyte, often by reversed-phase (RP) liquid chromatography. An ion-pairing agent, such as formic acid or trifluoroacetic acid (TFA), is typically added to the mobile phase, making protein analytes more hydrophobic than usual, thus increasing their resolution from an RP column.95,129 The protein is eluted in a volatile organic solvent (typically acetonitrile), facilitating the transmission of the analyte from solution phase into the gas phase. For an in-depth discussion of different MS configurations, we direct the reader to more specialist reviews.130,131
Measured masses, obtained with a high degree of precision using MS (typically ± 0.01% of the protein’s molecular weight) that are different to the calculated mass of the primary sequence can indicate proteolysis of the sample or incorrect disulfide bonding.132 To look for residue-level evidence of PTMs, a “bottom-up” approach is often employed, whereby the protein will first be enzymatically digested by a protease (typically trypsin or a mixture of other proteases); if disulfide bonds are present, the sample is reduced and alkylated using standard protocols,133 with salts then removed prior to the LC-MS experiment.134 It is known that certain PTMs can adversely affect the chemical stability of a protein, namely: deamidation,135 incorrect disulfide bonding132,136 or glycosylation137,138 and oxidation.139–141 The primary sequence of a protein, particularly if this is an mAb, can be analyzed using algorithms to identify motifs, which are liable to these aberrant PTMs. For example, it is known that the dipeptide motifs of Asn followed by either Gly or Ser are liable to undergo deamidation at the Asn residue,135,142 decreasing the chemical and conformational stability of the mAb. These can be readily engineered out of the sequence (identified using the LAP algorithm described above)92 if they lie outside of functional regions of the mAb, eg, the paratope.
Glycosylation is another key attribute of antibodies, as N-linked glycosylation of a conserved Asn residue in the CH2 domain of an IgG is important for both the thermal stability of an antibody137,138,143 and Fc-receptor binding.144 The so-called effector functions of an antibody can dictate half-life in vivo (due to binding to the neo-natal Fc receptor) or aid killing of target cells through, eg, antibody-dependent cellular cytotoxicity.15 Cell line engineering,145 as well as modifications to residues in the hinge,146,147 can affect the type of glycans formed in the endoplasmic reticulum (ER) during expression. mAbs can be deglycosylated to analyze the intact mass (of both mAb and glycan, the latter particularly important for the detection of different glycoforms) using enzymes such as PNGase F,148 using the HPLC to aid their separation.
Due to the development of commercially available automated systems,149 Hydrogen-Deuterium exchange (HDX) coupled to MS is being increasingly used to identify particularly dynamic regions in therapeutic proteins.150 Typically, experiments are performed where the (protiated) protein of interest is incubated in a D2O-based buffer. Under these conditions (a vast excess of deuterons versus protons) backbone amide protons exchange with deuterons,151 the rate of which being dependent on pH, temperature, solvent accessibility and amino acid side chain.152 After incubation for variable time periods, exchange is quenched (through low pH and cold temperature), and the protein digested (typically with immobilized pepsin, which is most active at pH 2.5), then performing LC-MS. The peptide ions are often fragmented in the gas-phase to facilitate their sequencing (see134 for more detail). One can then piece together the peptide information and map this back onto the 3D structure (or model) of the protein, to identify dynamic regions from protected regions. This has been done to identify regions of proteins/amino acid substitutions which modulate aggregation.150,153–155
The structural mass spectrometry toolkit allows one to probe the HOS of a protein, with a growing body of literature linking these data to favorable/aberrant developability parameters.156–159 By introducing proteins (or other analytes) into the gas phase under much gentler conditions, using volatile additives such as ammonium acetate, native mass spectrometry proteins and their higher-order species can analyzed in conditions that maintain tertiary and quaternary structure.160,161 A more novel use of MS for protein developability has recently been the use of collision-induced unfolding to probe the gas-phase stability of different therapeutic proteins.162,163 This involves incrementally increasing the energy of ions in the ion trap of mass spectrometer, then measuring their collisional cross-sectional (CCS) area using an ion-mobility cell. This acts like a “gas-phase chromatography column”, with compact ions interacting less with the buffer gas in the ion mobility cell than more expanded ones, thus the former arrive at the detector first.164,165 Increased collision energy thus results in the unfolding of a protein in the gas phase, detected by a later arrival time. This has been shown to be able to identify different glycoforms based on their gas-phase stability.166 Another approach to “gently” introduce proteins into the gas phase is to use capillary electrophoresis, as described above. This can be applied to both intact molecules (ie in a “top-down” fashion), as well as on peptides, in a bottom-up fashion167 analogous to the LC-MS/MS approach above.
From the perspective of early-stage developability assessment, while confirmation of the correct intact mass of a protein is routinely employed for hits, more in-depth analysis would only typically be performed as molecules progress through the development pipeline, due to the increased amount of analytical effort (in terms of time) required to perform these latter experiments. We envisage this becoming more routine over time, as suggested by the increasing amount of literature in this area (with over 200 papers a year published during this decade).131
Colloidal Stability Assessment: Probing Self- and Cross-Interactions in Therapeutic Protein Samples
The word “colloid” stems from the Greek, meaning “glue-like”. Protein solutions can be thought of as colloids based on their properties.168,169 In Figure 2, self-association between monomers, through native state aggregation, or non-native aggregation (leading to the formation of oligomers and eventually precipitates) pertains to the colloidal stability of a therapeutic protein solution. In addition to self-interaction, therapeutic proteins will encounter other biological macromolecules throughout their lifetime (expression, purification and after administration), where cross-interaction with other antibodies, antigens or other off-target biomolecules can lead to undesired manufacturing and therapeutic outcomes (see Aussewöger et al for more detail).170 A variety of methods have been developed to probe colloidal stability in its broadest sense.
Dynamic light scattering (DLS), which correlates the time-dependent fluctuations in the intensity of light scattered by solutes within a solution, is commonly employed to characterize colloidal stability.171 This allows one to infer the diffusion coefficient of solutes and by extension, their hydrodynamic radii (RH).42 Further analysis (using the regularization algorithm for example)172 allows the size-distribution of species within the sample to be determined. As the intensity of scattered light is proportional to RH6 aggregates will scatter far more light than the native monomer.171 This makes DLS a particularly sensitive means of detecting aggregation within a sample but conversely makes precise quantification of the populations within polydisperse solutions extremely challenging.171,173 Nanoparticle tracking analysis (which visualizes and counts aggregates larger than 50 nm in diameter),173,174 mass photometry (an imaging-based method where the scattering footprint of a protein landing on the imaging surface is proportional to its mass)175,176 and microfluidic-based technologies (reviewed by Otzen et al recently177) can be used to circumvent these issues.
DLS can now be performed on multiple samples in a plate-reader format.178 By measuring the DLS of protein samples across a concentration range (typically from sub-milligram/mL to 5 mg/mL), the diffusion coefficient can be determined and plotted as a function of concentration. The intercept of a straight-line graph fitted to these data (which will have either a negative (attractive), flat or positive (repulsive) gradient, yields the diffusion coefficient at infinite dilution for the sample. The gradient of the straight-line graph relates to the diffusion interaction parameter (kD, units mL/g).179 This parameter can give an indication of whether or not proteins interact (negative kD) or repel (positive kD) one another as a function of concentration, which is useful when screening molecules179 or different formulations.72 Kingsbury et al’s determination of the kD values for a large panel of mAbs and correlation of these against 22 other properties showed that repulsive behavior between the molecules correlated with favorable pharmacokinetic properties, highlighting the importance of kD as a developability parameter.179 It should be noted that the sign of the kD value is a rule of thumb, as recently the opposite trend has been tied to unfavorable viscosity at elevated concentration.180
In contrast to DLS, static light scattering (SLS) measures the time-independent scattering of light by solutes in a sample.42,181 The intensity of the scattering is dependent on the concentration and mass of the sample.181 Using more sophisticated equipment, one can measure the SLS of protein solutions as a function of concentration in the same fashion as above. The second osmotic virial coefficient (commonly called B22 or A22) describes attractive (positive values) or repulsive interactions (negative values) between proteins as a function of concentration.182,183 It can be seen therefore that kD and B22 values are generally inversely correlated;182 (but note180 is an important exception). Both parameters are important as attractive interactions as a function of concentration are likely to impact the final drug product, which is increasingly formulated at concentrations exceeding 100 mg/mL if administered by sub-cutaneous injection via a pre-filled syringe.184–186
In addition to these sophisticated measurements, simple turbidity assays (such as the optical density of a protein sample at 350 nm) can be performed but is very sensitive to the size range and distribution of aggregates within the sample.187 The sizing and counting of subvisible and visible particles (spanning 10’s–1000’s microns in size) is critical,188 as aggregates in this range are linked to adverse immunogenic effects in vivo.189,190 Light obscuration and microflow imaging191,192 have traditionally been used to achieve this aim, with machine learning approaches being developed to aid discrimination of proteinaceous particles from other adventitious contaminants.193 More recently, background membrane imaging (which uses image processing to improve signal to noise) has been developed to look for these particles, using sub-mL volumes for analysis.194
Molecular crowding (see195 for physical basis) has been a commonly employed method to indirectly probe the colloidal stability of therapeutic proteins. Polyethylene glycol (PEG) has been used as a crowding agent to force proteins closer to one another (akin to their environment under high concentrations).196 By determining the concentration of protein remaining in solution (using a constant initial protein concentration) over a range of PEG concentrations, one can determine the colloidal stability of the protein,56,72 including the inference of its “concentratability” (ie a protein’s theoretical solubility limit).197
More sophisticated HTP methods have been developed to monitor self-association of proteins. Affinity-capture self-association nanoparticle spectroscopy (AC-SINS) was developed by the Tessier group, conjugating polyclonal human IgG-targeting antibodies onto gold nanoparticles (AuNPs).198,199 If a target mAb exhibits low dispersity (ie was monomeric), the AuNPs remain disperse in solution. Conversely, self-association of the target mAbs results in the agglomeration of the AuNPs, with the plasmon wavelength (absorbance) of the particles red-shifting as a result. This has been used to screen panels of antibody sequences,104,199 with trends being identified between net-positively charged CDRs and self-association by AC-SINS.200 Modification to the AuNP preparation protocol increases the diversity of formulation buffers compatible with the method.201,202
Cross-interaction (ie off-target binding to bio-molecules other than the epitope) is an important developability parameter as increased retention times on this resin have been linked to poor solubility at elevated protein concentrations.108 Methods have consequently been developed which conjugate or adsorb other proteins, including antibodies {Clonal self-interaction Biolayer Interferometry (CSI-BLI)203 and Cross-interaction chromatography108,204} and either specific antigens {Baculovirus particle (BVP) enzyme-linked immunosorbent assay (ELISA),205 or a broad range of antigens (Poly-specificity reagent (PSR))}206 to different stationary phases (chips, chromatography resin and well-plates). Test proteins are then incubated with these functionalized species, with interaction yielding longer retention times/increased ELISA signals with rapid clearance of drugs in animal models when BVP scores are high.109 The reader is directed to Jain et al,104 Ausserwöger et al170 and Norden et al207 for more details of these methods.
In summary, the examination of the colloidal stability of a therapeutic protein is important with respect to its pharmacology and eventual formulation as a medicine. Aggregates have also been shown to cause illicit immunological effects in vivo, including neutralization of drugs (Anti-drug antibodies)208 or anaphylaxis. Thus, the characterization and mitigation of aggregation and off-target interaction is pivotal in early-phase drug development.
Thermal Stress: Turning Up the Heat on Biopharmaceuticals
It is well-known that heating proteins can trigger their unfolding and aggregtion. From the perspective of biopharmaceutical development, the final drug product is often formulated to be kinetically stable at refrigerated temperatures for typically two years or more.101,186,209 It would be impractical to subject all early-stage candidates to a real-time stability screen based on both the long timescales and shallow amplitude of the degradation kinetics under real-time storage conditions.99,100,210–212 Instead, samples are often incubated at elevated temperatures to increase their rate of degradation. Typical “accelerated stability” (AS) conditions for early-phase biologics are incubation at 40°C for 2–4 weeks.210,213–215 The regulatory agencies mandate such testing as part of the development of new drugs.216
Can one use the rates of degradation at elevated temperatures to infer what happens at lower (refrigerator) temperature? Classically, the temperature dependence of reaction rate follows Arrhenius kinetics, whereby the rate of a reaction increases as a function of temperature (a 10 K increase in temperature typically doubles the observed reaction rate). Recently, several Arrhenius-based kinetic models have been developed which can exploit protein-degradation kinetics at elevated temperatures (and short time frames) and use the data to directly predict the long-term storage stability of molecules.99,100,212,217,218 Others in the field, however, have shown that not all proteins obey this kinetic regime,210,211,219,220 meaning AS serves as a “rule of thumb” test to identify particularly thermally labile molecules or formulations from a panel.104,209
Reasons for deviation from an Arrhenius regime may depend on the distinct aggregation mechanism of the protein in question. AS conditions are typically below the apparent melting temperature (Tm,app) of a protein (the temperature at which 50% of the molecules are unfolded). Tm,app can be deduced in a HTP fashion using differential scanning fluorimetry (DSF). If a protein contains a Trp residue (conveniently, Ab-based therapies contain several Trp residues as they are conserved as part of the immunoglobulin fold),221 then the intrinsic fluorescence can be followed as a function of temperature. Protein unfolding will result in a red-shift in the fluorescence emission maximum of the Trp residues. Plotting the ratio of the emission at 350 nm (Trp exposed suggestive of an unfolded protein) versus 330 nm (Trp buried and suggestive of a folded protein) allows a sigmoidal curve to be fitted to the data and the Tm,app extracted from the mid-point.222–224 Furthermore, the inflection point of this curve represents Tonset. As well as setting the upper limit for an AS study (typically 10 K below the Tm,app to prevent aggregation taking place predominantly from the unfolded state, not the misfolded state), this method can identify thermally labile molecules and formulations.213 Some instruments which monitor intrinsic fluorescence can also use SLS simultaneously to infer the apparent temperature of aggregation (Tagg) based on an increase in scattering intensity as a function of temperature.224,225 Such instruments fit into the MAM framework by allowing one to obtain multiple developability parameters from one sample, maximizing characterization from the smaller quantities of material available in early drug development.
The “gold standard” method for obtaining Tm,app is differential scanning calorimetry (DSC). This is because cooperativity of unfolding observed by DSF can often mean that the unfolding transitions of different domains in the protein are poorly resolved.226 DSC involves heating up a sample and measuring the difference in temperature between the reference cell and sample cell upon unfolding of different domains in the protein (ie release of heat, due to bond breaking, being exothermic). The transition midpoint yields the Tm,app, whereas the peak area would yield change in heat capacity.227 The sensitivity of the method means multiple transitions can be observed for Ab domains.227 Blech et al have recently shown that multiple thermodynamic parameters can be obtained from DSC measurements (eg enthalpy change between folded and unfolded states), which can then be linked to the kinetic aggregation mechanism for a protein in question.228 However, larger quantities of material (>100 μL at protein concentrations ~1 mg/mL, compared to tens of microliters at a similar concentration for DSF) are needed to perform these measurements. DSF is thus likely to be performed in the first instance when quantities of material are lower, albeit with the caveats mentioned above.
Assessing Manufacturability: Shaking Up the Developability Pipeline
Of the various stresses biologics experience as part of their manufacturing, hydrodynamic forces (namely shear and extensional flows, as well as interaction with both solid-liquid and air-liquid interfaces) are ubiquitous.229–231 Such mechanical forces are thought to be able to initiate protein aggregation, mainly through the adsorption of proteins to interfaces. Subsequent partial unfolding and aggregation can take place on the surface, with flow then able to dislodge these species into bulk solution to regenerating these interfaces.110,231,232 The bulk flow may also induce formation of the aggregation-prone state,233–236 though this is controversial.231,237. Although poorly defined, orbital shaking studies appear to generate many of the hydrodynamic stresses described above and have been used to identify aggregation-prone molecules,187,238 as well as the type and concentration of surfactant excipients, which protect against these interfacial stresses.239,240
However, the mechanistic drivers of aggregation in this mixed type of stress can be difficult to define. Devices have been developed to subject proteins to defined shear flows, often in the presence of a solid–liquid interface of known chemistry,241–243 with this latter factor playing the dominant role in any subsequent aggregation. We have developed a small-volume extensional flow device (EFD), which subjects proteins to a predominantly elongational flow in the presence of solid (glass)-liquid interfaces.73,187,236,244 Others have built similar devices,232,237,245 including a recent example that can subject later-stage formulations to realistic hydrodynamic forces encountered during syringe actions.246 Though this latter application falls slightly outside the scope of this review, one could envisage the utilization of these devices for the rapid small-scale assessment of proteins to manufacturing stresses, something which appears to be a unique developability feature not described by the other assays mentioned in this review.244
How Do We Make Sense of Developability Data?
The many critical quality attributes required by a candidate therapeutic has driven the deployment of a wide array of methods to assess developability, but how these can be integrated is unclear. For example, if multiple methods are available to measure the same developability attribute (eg HIC and SMAC chromatography measuring molecular “stickiness”), then will one method provide adequate insight? If these assays predict conflicting developability outcomes, which do you trust? Once the data have been generated, how can one integrate the outputs to make an informed holistic decision on candidate selection?
Firstly, it is important to understand how assays relate to one another. Jain et al used Spearman’s rank correlation to look at the pairwise interaction between the behavior of 137 clinically relevant mAb sequences in 12 different developability assays.104 Hierarchical clustering was used to group the assays based on the responses of the mAbs; for example, HIC and SMAC chromatography yielded similar results.104 Both we244 and others228,247–251 have subsequently used similar approaches (sometimes using Pearson’s correlation instead of Spearman) to understand how novel assays/analyses compare to the established methods discussed herein, as well as infer (numerically) relationships between developability assays. There is an argument that applying multiple methods that probe the same biophysical property of a molecule is not the best use of resources, as one ends up with degenerate data. Using one technique from each branch of the “family tree” of developability assays (outlined in Figure 3) may be a better approach.
Secondly, statistical (and in a more simplistic sense, anecdotal) evidence can aid the decision to remove a molecule from a development campaign based on its behavior in a given developability assay. While liabilities (such as primary sequence motifs, which could lead to poor chemical stability252 or aggregation propensity60) can be engineered out in early-stage development, or by the addition of surfactant during formulation253,254 this becomes increasingly difficult the further the molecule progresses through the clinic. Acceptable thresholds are often placed on particular developability assays either from the regulator (eg particle levels255), scientific reasoning (based on long-term stability data, for example), or other more arbitrary cut-offs.99,100 This latter approach would allow one to discard (for example) the worst molecules in a panel if they score a “red flag” in a particular assay.104 This becomes less clear cut if a molecule performs poorly in one DA but well in other unrelated assays.
Machine learning/artificial intelligence approaches have gained much traction over the past five years in the biophysical analysis of proteins.93,202,256,257 For example, Makowski et al used CS (charge stabilised)-SINS and PSR to screen the self-association and polyspecificity, respectively, of a panel of 80 clinically-relevant antibody sequences. A machine-learning model was trained on these data (plus others) to effectively predict which mutations would ameliorate aberrant behavior in these assays, which was validated experimentally.202 Most recently, clinical and natural sequence databases have been mined to compute different sequence and structural liabilities across antibodies.92,257 As the biopharmaceutical industry embraces Industry 4.0, the leveraging of large datasets in this fashion will only aid the identification of the rules of developability for both current and next-generation molecules.
The biopharmaceutical pipeline has seen an increase in diversity in terms of both the pharmacological targets and the molecular architecture of the molecules used to interact with these.24 The number of modalities is only set to increase over the next few years.24 How the relatively established rules used to develop mAbs applies to these modalities in the future, remains to be seen, though efforts to this aim have begun.251 Some rules could be the same (for example, a molecule obeying Arrhenius kinetics will allow prediction of long-term stability).100 Others could change in light of allosteric effects, or due to the availability of a richer and larger dataset as technical challenges in analyzing these modalities are overcome . The field will inevitably rise to the challenge to bring about the successful development of the next-generation of biopharmaceutical proteins.
In conclusion, therapeutic proteins, in particular, monoclonal antibodies, have emerged as powerful medicines over the past forty years due to their potency, specificity and multiple modes of action. However, the multiple mechanisms by which they can degrade means an array of “developability” assays have been developed to identify defective molecules and select only those with drug-like properties. These analyses probe the varied molecular features of these complex molecules. The integration of computational and statistical workflows with experimental methods, especially as the field continues to grow, will aid the development of the therapeutic proteins of the future.
Funding
All the authors acknowledge funding from the EPSRC (EP/Z533063/1). NK is supported by a Royal Academy of Engineering Chair. SER is also funded by a Royal Society Professorial Fellowship (RSRP\R1\211057).
Disclosure
The authors declare no competing financial interests arising from this work.
References
1. Mcgonigle P. How biologics have changed the drug discovery landscape. Annu Rev Pharmacol Toxicol. 2024;207:20. doi:10.1146/annurev-pharmtox-061724
2. Goeddel DV, Kleid DG, Bolivar F, et al. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc Natl Acad Sci USA. 1979;76(1):106–110. doi:10.1073/pnas.76.1.106
3. Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12(1):433–455. doi:10.1146/annurev.iy.12.040194.002245
4. Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization: human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222(3):581–597. doi:10.1016/0022-2836(91)90498-U
5. Walsh G. Biopharmaceuticals: Biochemistry and Biotechnology.
6. Redwan ERM. Animal-derived pharmaceutical proteins. J Immunoassay Immunochem. 2009;30(3):262–290. doi:10.1080/15321810903084400
7. Mathieu C, Martens PJ, Vangoitsenhoven R. One hundred years of insulin therapy. Nat Rev Endocrinol. 2021;17(12):715–725. doi:10.1038/s41574-021-00542-w
8. Schramm W. The history of haemophilia - A short review. Thromb Res. 2014;134:S4–S9. doi:10.1016/j.thromres.2013.10.020
9. Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–497. doi:10.1038/256495a0
10. Group* OMTS. A randomized clinical trial of OKT3 monoclonal antibody for acute rejection of cadaveric renal transplants. New Engl J Med. 1985;313(6):337–342. doi:10.1056/NEJM198508083130601
11. Mahler H-C, Friess W, Grauschopf U, Kiese S. Protein aggregation: pathways, induction factors and analysis. J Pharm Sci. 2009;98(9):2909–2934. doi:10.1002/jps.21566
12. Hanes J, Schaffitzel C, Knappik A, Plückthun A. Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display. Nat Biotechnol. 2000;18:1287–1292. doi:10.1038/82407
13. Szlezák N, Evers M, Wang J, Pérez L. The role of big data and advanced analytics in drug discovery, development, and commercialization. Clin Pharmacol Ther. 2014;95(5):492–495. doi:10.1038/clpt.2014.29
14. Crescioli S, Kaplon H, Chenoweth A, Wang L, Visweswaraiah J, Reichert JM. Antibodies to watch in 2024. MAbs. 2024;16(1). doi:10.1080/19420862.2023.2297450
15. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5(OCT):1–17. doi:10.3389/fimmu.2014.00520
16. Weiner LM, Murray JC, Shuptrine CW. Antibody-based immunotherapy of cancer. Cell. 2012;148(6):1081–1084. doi:10.1016/j.cell.2012.02.034
17. Knudsen LB, Lau J. The discovery and development of liraglutide and semaglutide. Front Endocrinol. 2019;10(APR). doi:10.3389/fendo.2019.00155
18. Lau J, Bloch P, Schäffer L, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. 2015;58(18):7370–7380. doi:10.1021/acs.jmedchem.5b00726
19. Mieczkowski C, Zhang X, Lee D, et al. Blueprint for antibody biologics developability. MAbs. 2023;15(1):2185924. doi:10.1080/19420862.2023.2185924
20. Svilenov HL, Arosio P, Menzen T, Tessier P, Sormanni P. Approaches to expand the conventional toolbox for discovery and selection of antibodies with drug-like physicochemical properties. MAbs. 2023;15(1):2164459. doi:10.1080/19420862.2022.2164459
21. Fernández-Quintero ML, Ljungars A, Waibl F, et al. Assessing developability early in the discovery process for novel biologics. MAbs. 2023;15(1). doi:10.1080/19420862.2023.2171248
22. Liu Y, Caffry I, Wu J, et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs. 2014;6(2):483–492.
23. Shukla AA, Thömmes J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010;28(March):253–261. doi:10.1016/j.tibtech.2010.02.001
24. Wilkinson I, Hale G. Systematic analysis of the varied designs of 819 therapeutic antibodies and Fc fusion proteins assigned international nonproprietary names. MAbs. 2022;14(1):2123299. doi:10.1080/19420862.2022.2123299
25. Wang W, Roberts CJ. Protein aggregation – mechanisms, detection, and control. Int J Pharm. 2018;550(1–2):251–268. doi:10.1016/j.ijpharm.2018.08.043
26. Roberts CJ. Protein aggregation and its impact on product quality. Curr Opin Biotechnol. 2014;30C:211–217. doi:10.1016/j.copbio.2014.08.001
27. Le WP, Yan SX, Zhang YX, Zhou HM. Acid-induced folding of yeast alcohol dehydrogenase under low pH conditions. J Biochem. 1996;119(4):674–679. doi:10.1093/oxfordjournals.jbchem.a021295
28. Mazzer AR, Perraud X, Halley J, O’Hara J, Bracewell DG. Protein A chromatography increases monoclonal antibody aggregation rate during subsequent low pH virus inactivation hold. J Chromatogr A. 2015;1415:83–90. doi:10.1016/j.chroma.2015.08.068
29. Wei-Ping LE, Yan SX, Sen LI, Zhong HN, Zhou HM. Alkaline unfolding and salt-induced folding of yeast alcohol dehydrogenase under high pH conditions. Int J Pept Protein Res. 1996;47(6):484–490. doi:10.1111/j.1399-3011.1996.tb01099.x
30. Menzen T, Friess W, Niessner R, Haisch C. Laser-induced breakdown detection of temperature-ramp generated aggregates of therapeutic monoclonal antibody. Eur J Pharm Biopharm. 2015;94:463–467. doi:10.1016/j.ejpb.2015.07.001
31. Correia C, Tavares E, Lopes C, et al. Stability of protein formulations at subzero temperatures by isochoric cooling. J Pharm Sci. 2020;109(1):316–322. doi:10.1016/j.xphs.2019.06.017
32. Rosa M, Roberts CJ, Rodrigues MA. Connecting high-temperature and low temperature protein stability and aggregation. PLoS One. 2017;12(5):1–12. doi:10.1371/journal.pone.0176748
33. Tezuka-Kawakami T, Gell C, Brockwell DJ, Radford SE, Smith DA. Urea-induced unfolding of the immunity protein Im9 monitored by spFRET. Biophys J. 2006;91(5):L42–L44. doi:10.1529/biophysj.106.088344
34. Emadi S, Behzadi M. A comparative study on the aggregating effects of guanidine thiocyanate, guanidine hydrochloride and urea on lysozyme aggregation. Biochem Biophys Res Commun. 2014;450(4):1339–1344. doi:10.1016/j.bbrc.2014.06.133
35. Heyda J, Okur HI, Hladílková J, et al. Guanidinium can both cause and prevent the hydrophobic collapse of biomacromolecules. J Am Chem Soc. 2017;139(2):863–870. doi:10.1021/Jacs.6b11082
36. Tamura H, Nakama T, Rossen A, Ishikita H, Fujita M. Organic solvent-induced structural changes in a protein confined in a giant coordination cage. Chem Lett. 2024;53(5):upae101. doi:10.1093/chemle/upae101
37. Uversky VN, Narizhneva NV, Kirschstein SO, Winter S, Löber G. Conformational transitions provoked by organic solvents in β-lactoglobulin: can a molten globule like intermediate be induced by the decrease in dielectric constant? Fold Des. 1997;2(3):163–172. doi:10.1016/S1359-0278(97)00023-0
38. Codina N, Hilton D, Zhang C, et al. An expanded conformation of an antibody fab region by X-ray scattering, molecular dynamics, and smFRET identifies an aggregation mechanism. J Mol Biol. 2019;431(7):1409–1425. doi:10.1016/j.jmb.2019.02.009
39. Banks DD, Zhang J, Siska CC. Relationship between native-state solubility and non-native aggregation of recombinant human granulocyte colony stimulating factor: practical implications for protein therapeutic development. Mol Pharm. 2014;11(10):3431–3442. doi:10.1021/mp500165j
40. Willis LF. The Effects of Flow on Therapeutic Protein Aggregation. University of Leeds; 2018. https://etheses.whiterose.ac.uk/21963/.
41. Hofmann M, Gieseler H. Predictive screening tools used in high-concentration protein formulation development. J Pharm Sci. 2018;107(3):772–777. doi:10.1016/j.xphs.2017.10.036
42. Minton AP. Recent applications of light scattering measurement in the biological and biopharmaceutical sciences. Anal Biochem. 2016;501:4–22. doi:10.1016/j.ab.2016.02.007
43. Yang H, Peczulis P, Inguva P, Li X, Heng JYY. Continuous protein crystallisation platform and process: case of lysozyme. Chem Eng Res Des. 2018;136:529–535. doi:10.1016/j.cherd.2018.05.031
44. Lilyestrom WG, Yadav S, Shire SJ, Scherer TM. Monoclonal antibody self-association, cluster formation, and rheology at high concentrations. J Phys Chem B. 2013;117(21):6373–6384. doi:10.1021/jp4008152
45. Sarangapani PS, Weaver J, Parupudi A, et al. Both reversible self-association and structural changes underpin molecular viscoelasticity of mab solutions. J Pharm Sci. 2016;105(12):3496–3506. doi:10.1016/j.xphs.2016.08.020
46. Rekhi S, Garcia CG, Barai M, et al. Expanding the molecular language of protein liquid–liquid phase separation. Nat Chem. 2024;16(7):1113–1124. doi:10.1038/s41557-024-01489-x
47. Shah UV, Jahn NH, Huang S, Yang Z, Williams DR, Heng JYY. Crystallisation via novel 3D nanotemplates as a tool for protein purification and bio-separation. J Cryst Growth. 2017;469:42–47. doi:10.1016/j.jcrysgro.2016.09.029
48. Smiatek J, Jung A, Bluhmki E. Towards a digital bioprocess replica: computational approaches in biopharmaceutical development and manufacturing. Trends Biotechnol. 2020;1–13. doi:10.1016/j.tibtech.2020.05.008
49. Baran D, Pszolla MG, Lapidoth GD, et al. Principles for computational design of binding antibodies. Proc Natl Acad Sci U S A. 2017;114(41):10900–10905. doi:10.1073/pnas.1707171114
50. Norman RA, Ambrosetti F, Bonvin AMJJ, et al. Computational approaches to therapeutic antibody design: established methods and emerging trends. Brief Bioinform. 2019;(October):1–19. doi:10.1093/bib/bbz095
51. Prabakaran R, Rawat P, Thangakani AM, Kumar S, Gromiha MM. Protein aggregation: in silico algorithms and applications. Biophys Rev. 2021;13(1):71–89. doi:10.1007/s12551-021-00778-w
52. Fiser A, Šali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi:10.1016/S0076-6879(03)74020-8
53. Leem J, Dunbar J, Georges G, Shi J, Deane CM. ABodyBuilder: automated antibody structure prediction with data–driven accuracy estimation. MAbs. 2016;8(7):1259–1268. doi:10.1080/19420862.2016.1205773
54. Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22(10):1302–1306. doi:10.1038/nbt1012
55. Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics. 2007;8(1):65. doi:10.1186/1471-2105-8-65
56. Sormanni P, Amery L, Ekizoglou S, Vendruscolo M, Popovic B. Rapid and accurate in silico solubility screening of a monoclonal antibody library. Sci Rep. 2017;7(1):8200. doi:10.1038/s41598-017-07800-w
57. Sormanni P, Aprile FA, Vendruscolo M. Third generation antibody discovery methods: in silico rational design. Chem Soc Rev. 2018;47(24):9137–9157. doi:10.1039/C8CS00523K
58. Waight AB, Prihoda D, Shrestha R, et al. A machine learning strategy for the identification of key in silico descriptors and prediction models for IgG monoclonal antibody developability properties. MAbs. 2023;15(1). doi:10.1080/19420862.2023.2248671
59. Kuriata A, Iglesias V, Pujols J, Kurcinski M, Kmiecik S, Ventura S. Aggrescan3D (A3D) 2.0: prediction and engineering of protein solubility. Nucleic Acids Res. 2019;47(W1):W300–W307. doi:10.1093/nar/gkz321
60. Gil-Garcia M, Banó-Polo M, Varejao N, et al. Combining structural aggregation propensity and stability predictions to redesign protein solubility. Mol Pharm. 2018;15(9):3846–3859. doi:10.1021/acs.molpharmaceut.8b00341
61. Geddes AJ, Parker KD, Atkins EDT, Beighton E. “Cross-β” conformation in proteins. J Mol Biol. 1968;32(2):343–358.
62. Wilkinson M, Xu Y, Thacker D, et al. Structural evolution of fibril polymorphs during amyloid assembly. Cell. 2023;186(26):5798–5811.e26. doi:10.1016/j.cell.2023.11.025
63. Batzli KM, Love BJ. Agitation of amyloid proteins to speed aggregation measured by ThT fluorescence: a call for standardization. Mat Sci Eng. 2015;48:359–364. doi:10.1016/j.msec.2014.09.015
64. Feige MJ, Buchner J. Principles and engineering of antibody folding and assembly. Biochim Biophys Acta Proteins Proteom. 2014;1844(11):2024–2031. doi:10.1016/j.bbapap.2014.06.004
65. Feige MJ, Groscurth S, Marcinowski M, et al. An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol Cell. 2009;34(5):569–579. doi:10.1016/j.molcel.2009.04.028
66. Van Durme J, De Baets G, Van Der Kant R, et al. Solubis: a webserver to reduce protein aggregation through mutation. Protein Eng Des Sel. 2016;29(8):285–289. doi:10.1093/protein/gzw019
67. van der Kant R, Karow-Zwick AR, Van Durme J, et al. Prediction and reduction of the aggregation of monoclonal antibodies. J Mol Biol. 2017;429(8):1244–1261. doi:10.1016/j.jmb.2017.03.014
68. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33(Web Server):W382–W388. doi:10.1093/nar/gki387
69. Khodaparast L, Khodaparast L, Gallardo R, et al. Aggregating sequences that occur in many proteins constitute weak spots of bacterial proteostasis. Nat Commun. 2018;9(1):866. doi:10.1038/s41467-018-03131-0
70. Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci USA. 2009;106(29):11937–11942. doi:10.1073/pnas.0904191106
71. Yang D, Kroe-Barrett R, Singh S, Roberts CJ, Laue TM. IgG cooperativity–is there allostery? Implications for antibody functions and therapeutic antibody development. MAbs. 2017;9(8):1231–1252. doi:10.1080/19420862.2017.1367074
72. Dobson CL, Devine PWA, Phillips JJ, et al. Engineering the surface properties of a human monoclonal antibody prevents self-association and rapid clearance in vivo. Sci Rep. 2016;6(November):38644. doi:10.1038/srep38644
73. Willis LF, Kumar A, Dobson J, et al. Using extensional flow to reveal diverse aggregation landscapes for three IgG1 molecules. Biotechnol Bioeng. 2018;115(5):1216–1225. doi:10.1002/bit.26543
74. Lauer TM, Agrawal NJ, Chennamsetty N, Egodage K, Helk B, Trout BL. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci. 2012;101(1):102–115. doi:10.1002/jps.22758
75. Lai PK, Gallegos A, Mody N, Sathish HA, Trout BL. Machine learning prediction of antibody aggregation and viscosity for high concentration formulation development of protein therapeutics. MAbs. 2022;14(1). doi:10.1080/19420862.2022.2026208
76. Austerberry JI, Dajani R, Panova S, et al. The effect of charge mutations on the stability and aggregation of a human single chain Fv fragment. Eur J Pharm Biopharm. 2017;115:18–30. doi:10.1016/j.ejpb.2017.01.019
77. Hebditch M, Warwicker J. Charge and hydrophobicity are key features in sequence-trained machine learning models for predicting the biophysical properties of clinical-stage antibodies. PeerJ. 2019;2019(12). doi:10.7717/peerj.8199
78. Filoti DI, Shire SJ, Yadav S, Laue TM. Comparative study of analytical techniques for determining protein charge. J Pharm Sci. 2015;104(7):2123–2131. doi:10.1002/jps.24454
79. Gamsjäger H, Lorimer JW, Scharlin P, Shaw DG. Glossary of terms related to solubility: (IUPAC recommendations 2008). Pure and Appl Chem. 2008;80(2):233–276. doi:10.1351/pac200880020233
80. Bremer A, Farag M, Borcherds WM, et al. Deciphering how naturally occurring sequence features impact the phase behaviours of disordered prion-like domains. Nat Chem. 2021. doi:10.1038/s41557-021-00840-w
81. Hebditch M, Warwicker J. Web-based display of protein surface and pH-dependent properties for assessing the developability of biotherapeutics. Sci Rep. 2019;9(1):1969. doi:10.1038/s41598-018-36950-8
82. Sormanni P, Aprile FA, Vendruscolo M. The CamSol method of rational design of protein mutants with enhanced solubility. J Mol Biol. 2015;427(2):478–490. doi:10.1016/j.jmb.2014.09.026
83. Ebo JS, Saunders JC, Devine PWA, et al. An in vivo platform to select and evolve aggregation-resistant proteins. Nat Commun. 2020;11(1):1816. doi:10.1038/s41467-020-15667-1
84. Hanning KR, Minot M, Warrender AK, Kelton W, Reddy ST. Deep mutational scanning for therapeutic antibody engineering. Trends Pharmacol Sci. 2022;43(2):123–135. doi:10.1016/j.tips.2021.11.010
85. Raybould MIJ, Marks C, Krawczyk K, et al. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci USA. 2019;116(10):4025–4030. doi:10.1073/pnas.1810576116
86. Abanades B, Wong WK, Boyles F, Georges G, Bujotzek A, Deane CM. ImmuneBuilder: deep-learning models for predicting the structures of immune proteins. Commun Biol. 2023;6(1):575. doi:10.1038/s42003-023-04927-7
87. Raybould MIJ, Turnbull OM, Suter A, Guloglu B, Deane CM. Contextualising the developability risk of antibodies with lambda light chains using enhanced therapeutic antibody profiling. Commun Biol. 2024;7(1). doi:10.1038/s42003-023-05744-8
88. Weber B, Hora M, Kazman P, et al. The antibody light-chain linker regulates domain orientation and amyloidogenicity. J Mol Biol. 2018;430(24):4925–4940. doi:10.1016/j.jmb.2018.10.024
89. Morgan GJ, Usher GA, Kelly JW. Incomplete refolding of antibody light chains to non-native, protease-sensitive conformations leads to aggregation: a mechanism of amyloidogenesis in patients? Biochemistry. 2017;56(50):6597–6614. doi:10.1021/acs.biochem.7b00579
90. Huang X, Wang Q, Jiang S, Chen W, Zeng C, Liu Z. The clinical features and outcomes of systemic AL amyloidosis: a cohort of 231 Chinese patients. Clin Kidney J. 2015;8(1):120–126. doi:10.1093/ckj/sfu117
91. Xu Y, Wang D, Mason B, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11(2):239–264. doi:10.1080/19420862.2018.1553476
92. Satława T, Tarkowski M, Wróbel S, et al. LAP: liability antibody profiler by sequence & structural mapping of natural and therapeutic antibodies. PLoS Comput Biol. 2024;20(3). doi:10.1371/journal.pcbi.1011881
93. Rathore AS, Nikita S, Thakur G, Mishra S. Artificial intelligence and machine learning applications in biopharmaceutical manufacturing. Trends Biotechnol. 2023;41(4):497–510. doi:10.1016/j.tibtech.2022.08.007
94. Magil SG. Biopharmaceutical characterization techniques for early phase development of proteins. Biopharm Int. 2005;2005(3).
95. Carr D. HPLC Columns A guide to the analysis and purification of proteins and peptides by reversed-phase HPLC. Available from: www.hplc.eu/Downloads/ACE_Guide_Peptides.pdf.
96. Uliyanchenko E. Size-exclusion chromatography - from high-performance to ultra-performance. Anal Bioanal Chem. 2014;406(25):6087–6094. doi:10.1007/s00216-014-8041-z
97. Hong P, Koza S, Bouvier ESP. A review size-exclusion chromatography for the analysis of protein biotherapeutics and their aggregates. J Liq Chromatogr Relat Technol. 2012;35(20):2923–2950. doi:10.1080/10826076.2012.743724
98. Cytiva. Size exclusion chromatography 2; 2020. Available from: https://cdn.cytivalifesciences.com/api/public/content/digi-11639-pdf?_gl=1*gacgsq*_gcl_au*MTYxMzUxOTIyNy4xNzI2MjI0NjY0.
99. Bunc M, Hadži S, Graf C, Bončina M, Lah J. Aggregation time machine: a platform for the prediction and optimization of long-term antibody stability using short-term kinetic analysis. J Med Chem. 2022;65(3):2623–2632. doi:10.1021/acs.jmedchem.1c02010
100. Dillon M, Xu J, Thiagarajan G, Skomski D, Procopio A. Predicting the long-term stability of biologics with short-term data. Mol Pharm. 2024;21(9):4673–4687. doi:10.1021/acs.molpharmaceut.4c00609
101. ICH. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Ich Harmonised Tripartite Guideline Stability Testing of New Drug Substances and Products q1a(r2); 2003.
102. Pharmacopeia US. Analytical Procedures for Recombinant Therapeutic Monoclonal Antibodies; 2024. doi:10.31003/USPNF_M6297_02_01
103. Murphy RM, Roberts CJ. Protein misfolding and aggregation research: some thoughts on improving quality and utility. Biotechnol Prog. 2013;29(5):1109–1115. doi:10.1002/btpr.1812
104. Jain T, Sun T, Durand S, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci USA. 2017;114(5):944–949. doi:10.1073/pnas.1616408114
105. Foit L, Morgan GJ, Kern MJ, et al. Optimizing protein stability in vivo. Mol Cell. 2009;36(5):861–871. doi:10.1016/j.molcel.2009.11.022
106. Saunders JC, Young LM, Mahood RA, et al. An in vivo platform for identifying inhibitors of protein aggregation. Nat Chem Biol. 2016;12(2):94–101. doi:10.1038/nchembio.1988
107. Kohli N, Jain N, Geddie ML, Razlog M, Xu L, Lugovskoy AA. A novel screening method to assess developability of antibody-like molecules. MAbs. 2015;7(4):752–758. doi:10.1080/19420862.2015.1048410
108. Jacobs SA, Wu S-J, Feng Y, Bethea D, O’Neil KT. Cross-interaction chromatography: a rapid method to identify highly soluble monoclonal antibody candidates. Pharm Res. 2010;27(1):65–71. doi:10.1007/s11095-009-0007-z
109. Estep P, Caffry I, Yu Y, et al. An alternative assay to hydrophobic interaction chromatography for high-throughput characterization of monoclonal antibodies. MAbs. 2015;7(3):553–561. doi:10.1080/19420862.2015.1016694
110. Kopp MRG, Grigolato F, Zürcher D, et al. Surface-induced protein aggregation and particle formation in biologics: current understanding of mechanisms, detection and mitigation strategies. J Pharm Sci. 2023;112(2):377–385. doi:10.1016/j.xphs.2022.10.009
111. Alemie MN, Bright R, Nguyen NH, et al. Surface chemistry induced igg unfolding and modulation of immune responses. ACS Appl Mater Interfaces. 2024;16(38):50507–50523. doi:10.1021/acsami.4c12883
112. Wang X, Wang J, Han Y, et al. Utilizing a hydrophobic primary container surface to reduce the formation of subvisible particles in monoclonal antibody solution caused by fluid shear. Eur J Pharm Biopharm. 2024;204:114502. doi:10.1016/j.ejpb.2024.114502
113. Muneeruddin K, Nazzaro M, Kaltashov IA. Characterization of intact protein conjugates and biopharmaceuticals using ion-exchange chromatography with online detection by native electrospray ionization mass spectrometry and top-down tandem mass spectrometry. Anal Chem. 2015;87(19):10138–10145. doi:10.1021/acs.analchem.5b02982
114. Zydney AL. New developments in membranes for bioprocessing – a review. J Memb Sci. 2021;620. doi:10.1016/j.memsci.2020.118804
115. Sule SV, Fernandez JE, Mecozzi VJ, et al. Assessing the impact of charge variants on stability and viscosity of a high concentration antibody formulation. J Pharm Sci. 2017;106(12):3507–3514. doi:10.1016/j.xphs.2017.08.016
116. Duivelshof BL, Bouvarel T, Pirner S, et al. Enhancing selectivity of protein biopharmaceuticals in ion exchange chromatography through addition of organic modifiers. Int J Mol Sci. 2023;24(23):16623. doi:10.3390/ijms242316623
117. Neill A, Nowak C, Patel R, et al. Characterization of recombinant monoclonal antibody charge variants using OFFGEL fractionation, weak anion exchange chromatography, and mass spectrometry. Anal Chem. 2015;87(12):6204–6211. doi:10.1021/acs.analchem.5b01452
118. Arakawa T, Ponce S, Young G. Isoform separation of proteins by mixed-mode chromatography. Protein Expr Purif. 2015;116:144–151. doi:10.1016/j.pep.2015.08.013
119. Jiang H, Xu W, Liu R, et al. Characterization of bispecific antibody production in cell cultures by unique mixed mode size exclusion chromatography. Anal Chem. 2020;92(13):9312–9321. doi:10.1021/acs.analchem.0c01641
120. Tagliaro F, Manetto G, Crivellente F, Smith FP. A brief introduction to capillary electrophoresis. Forensic Sci Int. 1998;92(2–3):75–88. doi:10.1016/S0379-0738(98)00010-3
121. Redman EA, Batz NG, Mellors JS, Ramsey JM. Integrated microfluidic capillary electrophoresis-electrospray ionization devices with online MS detection for the separation and characterization of intact monoclonal antibody variants. Anal Chem. 2015;87:2264–2272. doi:10.1021/ac503964j
122. Redman EA, Mellors JS, Starkey JA, Ramsey JM. Characterization of intact antibody drug conjugate variants using microfluidic capillary electrophoresis-mass spectrometry. Anal Chem. 2016;88(4):2220–2226. doi:10.1021/acs.analchem.5b03866
123. Dai J, Lamp J, Xia Q, Zhang Y. Capillary isoelectric focusing-mass spectrometry method for the separation and online characterization of intact monoclonal antibody charge variants. Anal Chem. 2018;90(3):2246–2254. doi:10.1021/acs.analchem.7b04608
124. Herr AE, Molho JI, Drouvalakis KA, et al. On-chip coupling of isoelectric focusing and free solution electrophoresis for multidimensional separations. Anal Chem. 2003;75(5):1180–1187. doi:10.1021/ac026239a
125. Auclair JR, Rathore AS. The multi-attribute method (MAM) for the characterization of biopharmaceuticals. LC-GC North America. 2021;39(1):28–32. doi:10.56530/lcgc.na.gi5577l2
126. ICH. Pharmaceutical development Q8. ICH Harmonised Tripartite Guideline. 2009;8(August):1–28.
127. Yang F, Zhang J, Buettner A, et al. Mass spectrometry-based multi-attribute method in protein therapeutics product quality monitoring and quality control. MAbs. 2023;15(1). doi:10.1080/19420862.2023.2197668
128. Chen C, Hou J, Tanner JJ, Cheng J. Bioinformatics methods for mass spectrometry-based proteomics data analysis. Int J Mol Sci. 2020;21(8). doi:10.3390/ijms21082873
129. Hancock WS, Bishop CA, Prestidge RL, Harding DRK, Hearn MTW. Reversed-phase, high-pressure liquid chromatography of peptides and proteins with ion-pairing reagents. Science. 1978;200(4346):1168–1170. doi:10.1126/science.206966
130. Britt HM, Beveridge R, Calabrese AN. A special issue of essays in biochemistry on structural mass spectrometry. Essays Biochem. 2023;67(2):147–149. doi:10.1042/EBC20230006
131. Li X. Recent applications of quantitative mass spectrometry in biopharmaceutical process development and manufacturing. J Pharm Biomed Anal. 2023;234. doi:10.1016/j.jpba.2023.115581
132. Cramer CN, Haselmann KF, Olsen JV, Nielsen PK. Disulfide linkage characterization of disulfide bond-containing proteins and peptides by reducing electrochemistry and mass spectrometry. Anal Chem. 2016;88(3):1585–1592. doi:10.1021/acs.analchem.5b03148
133. Suttapitugsakul S, Xiao H, Smeekens J, Wu R. Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics. Mol Biosyst. 2017;13(12):2574–2582. doi:10.1039/c7mb00393e
134. Leitner A. Cross-linking and other structural proteomics techniques: how chemistry is enabling mass spectrometry applications in structural biology. Chem Sci. 2016;7(8):4792–4803. doi:10.1039/c5sc04196a
135. Phillips JJ, Buchanan A, Andrews J, et al. Rate of asparagine deamidation in a monoclonal antibody correlating with hydrogen exchange rate at adjacent downstream residues. Anal Chem. 2017;89(4):2361–2368. doi:10.1021/acs.analchem.6b04158
136. Yang M, Dutta C, Tiwari A. Disulfide-bond scrambling promotes amorphous aggregates in lysozyme and bovine serum albumin. J Phys Chem B. 2015;119:3969–3981. doi:10.1021/acs.jpcb.5b00144
137. Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 2011;3(6):568–576. doi:10.4161/mabs.3.6.17922
138. Zhang Y. Understanding the impact of Fc glycosylation on its conformational changes by molecular dynamics simulations and bioinformatics. Mol BioSyst. 2015;11(12):3415–3424. doi:10.1039/C5MB00602C
139. Venkatesh S, Tomer KB, Sharp JS. Rapid identification of oxidation-induced conformational changes by kinetic analysis. Rapid Commun Mass Spectrom. 2007;21(23):3927–3936. doi:10.1002/rcm.3291
140. Larson NR, Wei Y, Prajapati I, et al. Comparison of polysorbate 80 hydrolysis and oxidation on the aggregation of a monoclonal antibody. J Pharm Sci. 2020;109(1):633–639. doi:10.1016/j.xphs.2019.10.069
141. Yang R, Jain T, Lynaugh H, et al. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. MAbs. 2017;9(4):646–653. doi:10.1080/19420862.2017.1290753
142. Lu X, Nobrega RP, Lynaugh H, et al. Deamidation and isomerization liability analysis of 131 clinical-stage antibodies. MAbs. 2019;11(1):45–57. doi:10.1080/19420862.2018.1548233
143. Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol. 2003;325(5):979–989. doi:10.1016/S0022-2836(02)01250-0
144. Lin CW, Tsai MH, Li ST, et al. A common glycan structure on immunoglobulin G for enhancement of effector functions. Proc Natl Acad Sci U S A. 2015;112(22):201513456. doi:10.1073/pnas.1513456112
145. Nguyen NTB, Leung HW, Pang KT, et al. Optimizing effector functions of monoclonal antibodies via tailored N-glycan engineering using a dual landing pad CHO targeted integration platform. Sci Rep. 2023;13(1). doi:10.1038/s41598-023-42925-1
146. Edgeworth MJ, Phillips JJ, Lowe DC, Kippen AD, Higazi DR, Scrivens JH. Global and local conformation of human IgG antibody variants rationalizes loss of thermodynamic stability. Angewandte Chemie Intl Ed. 2015;54(50):15156–15159. doi:10.1002/anie.201507223
147. Hale G, De Vos J, Davy AD, Sandra K, Wilkinson I. Systematic analysis of Fc mutations designed to reduce binding to Fc-gamma receptors. MAbs. 2024;16(1):2402701. doi:10.1080/19420862.2024.2402701
148. van de Bovenkamp FS, Derksen NIL, Ooijevaar-de Heer P, Rispens T. The enzymatic removal of immunoglobulin variable domain glycans by different glycosidases. J Immunol Methods. 2019;467:58–62. doi:10.1016/j.jim.2019.02.005
149. Groves K, Ashcroft AE, Cryar A, et al. Reference protocol to assess analytical performance of higher order structural analysis measurements: results from an interlaboratory comparison. Anal Chem. 2021;93(26):9041–9048. doi:10.1021/acs.analchem.0c04625
150. Majumdar R, Middaugh CR, Weis DD, Volkin DB. Hydrogen-deuterium exchange mass spectrometry as an emerging analytical tool for stabilization and formulation development of therapeutic monoclonal antibodies. J Pharm Sci. 2015;104:327–345. doi:10.1002/jps.24224
151. Miranker A, Robinson CV, Radford SE, Aplin RT, Dobson CM. Detection of transient protein folding populations by mass spectrometry. Science. 1993;262(5135):896–900. doi:10.1126/science.8235611
152. Clarke J, Itzhakit LS. Hydrogen exchange and protein folding. Curr Opin Struct Biol. 1998;8(1):112–118.
153. Moorthy BS, Zarraga IE, Kumar L, et al. Solid-state hydrogen-deuterium exchange mass spectrometry: correlation of deuterium uptake and long-term stability of lyophilized monoclonal antibody formulations. Mol Pharm. 2018;15(1):1–11. doi:10.1021/acs.molpharmaceut.7b00504
154. Wood VE, Groves K, Cryar A, Quaglia M, Matejtschuk P, Dalby PA. HDX and in silico docking reveal that excipients stabilize G-CSF via a combination of preferential exclusion and specific hotspot interactions. Mol Pharm. 2020;17(12):4637–4651. doi:10.1021/acs.molpharmaceut.0c00877
155. Wood VE, Groves K, Wong LM, et al. Protein engineering and HDX identify structural regions of G-CSF critical to its stability and aggregation. Mol Pharm. 2022;19(2):616–629. doi:10.1021/acs.molpharmaceut.1c00754
156. Jiang Y, Li C, Li J, Gabrielson JP, Wen J. Technical decision making with higher order structure data: higher order structure characterization during protein therapeutic candidate screening. J Pharm Sci. 2015;104:1533–1538. doi:10.1002/jps.24406
157. Wei H, Mo J, Tao L, et al. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discov Today. 2014;19(1):95–102. doi:10.1016/j.drudis.2013.07.019
158. Kerr RA, Keire DA, Ye H. The impact of standard accelerated stability conditions on antibody higher order structure as assessed by mass spectrometry. MAbs. 2019;11(5):930–941. doi:10.1080/19420862.2019.1599632
159. Degueldre M, Wielant A, Girot E, et al. Native peptide mapping–A simple method to routinely monitor higher order structure changes and relation to functional activity. MAbs. 2019;11(8):1391–1401. doi:10.1080/19420862.2019.1634460
160. Konermann L, Vahidi S, Sowole MA. Mass spectrometry methods for studying structure and dynamics of biological macromolecules. Anal Chem. 2014;86(1):213–232. doi:10.1021/ac4039306
161. Konermann L. Addressing a common misconception: ammonium acetate as neutral ph “buffer” for native electrospray mass spectrometry. J Am Soc Mass Spectrom. 2017;28(9):1827–1835. doi:10.1007/s13361-017-1739-3
162. Tian Y, Ruotolo BT. The growing role of structural mass spectrometry in the discovery and development of therapeutic antibodies. Analyst. 2018;143(11):2459–2468. doi:10.1039/c8an00295a
163. Tian Y, Han L, Buckner AC, Ruotolo BT. Collision induced unfolding of intact antibodies: rapid characterization of disulfide bonding patterns, glycosylation, and structures. Anal Chem. 2015;87(22):11509–11515. doi:10.1021/acs.analchem.5b03291
164. Ruotolo BT, Benesch JLP, Sandercock AM, Hyung SJ, Robinson CV. Ion mobility-mass spectrometry analysis of large protein complexes. Nat Protoc. 2008;3(7):1139–1152. doi:10.1038/nprot.2008.78
165. Ferguson CN, Gucinski-Ruth AC. Evaluation of ion mobility-mass spectrometry for comparative analysis of monoclonal antibodies. J Am Soc Mass Spectrom. 2016;27(5):822–833. doi:10.1007/s13361-016-1369-1
166. Tian Y, Ruotolo BT. Collision induced unfolding detects subtle differences in intact antibody glycoforms and associated fragments. Int J Mass Spectrom. 2018;425:1–9. doi:10.1016/j.ijms.2017.12.005
167. Giorgetti J, Beck A, Leize-Wagner E, François YN. Combination of intact, middle-up and bottom-up levels to characterize 7 therapeutic monoclonal antibodies by capillary electrophoresis – mass spectrometry. J Pharm Biomed Anal. 2020;182. doi:10.1016/j.jpba.2020.113107
168. Nicoud L, Owczarz M, Arosio P, Morbidelli M. A multiscale view of therapeutic protein aggregation: a colloid science perspective. Biotechnol J. 2015;10(3):367–378. doi:10.1002/biot.201400858
169. Skar-Gislinge N, Ronti M, Garting T, et al. A colloid approach to self-assembling antibodies. Mol Pharm. 2019;16(6):2394–2404. doi:10.1021/acs.molpharmaceut.9b00019
170. Ausserwöger H, Schneider MM, Herling TW, et al. Non-specificity as the sticky problem in therapeutic antibody development. Nat Rev Chem. 2022;6(12):844–861. doi:10.1038/s41570-022-00438-x
171. Hassan PA, Rana S, Verma G. Making sense of brownian motion: colloid characterization by dynamic light scattering. Langmuir. 2014;31(1):3–12. doi:10.1021/la501789z
172. Provencher SW. CONTIN: a general purpose constrained regularisation program for inverting noisy linear algebraic and integral equations. Comput Phys Commun. 1982;27:229–242.
173. Filipe V, Hawe A, Jiskoot W. Critical evaluation of nanoparticle tracking analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates. Pharm Res. 2010;27(5):796–810. doi:10.1007/s11095-010-0073-2
174. Krueger AB, Carnell P, Carpenter JF. Characterization of factors affecting nanoparticle tracking analysis results with synthetic and protein nanoparticles. J Pharm Sci. 2016;105(4):1434–1443. doi:10.1016/j.xphs.2016.02.005
175. Hundt N, Cole D, Hantke MF, Miller JJ, Struwe WB, Kukura P. Direct observation of the molecular mechanism underlying protein polymerization. Sci Adv. 2022;8(35):31. doi:10.1126/sciadv.abm7935
176. Young G, Hundt N, Cole D, et al. Quantitative mass imaging of single biological macromolecules. Science. 2018;360(6387):423–427. doi:10.1126/science.aar5839
177. Otzen DE, Buell AK, Jensen H. Microfluidics and the quantification of biomolecular interactions. Curr Opin Struct Biol. 2021;70:8–15. doi:10.1016/j.sbi.2021.02.006
178. Some D. WP5003-automated-dynamic-and-static-light-scattering-in-microwell-plates. Available from: https://www.wyatt.com/library/application-notes/wp5003-dynapro-plate-reader-iii-automated-dynamic-and-static-light-scattering-in-microwell-plates-for-fast-productive-development-of-biologics.html.
179. Kingsbury JS, Saini A, Auclair SM, et al. A single molecular descriptor to predict solution behavior of therapeutic antibodies. Sci Adv. 2020;6(32):eabb0372. doi:10.1126/sciadv.abb0372
180. Yuan G, Salipante PF, Hudson SD, et al. Flow activation energy of high-concentration monoclonal antibody solutions and protein–protein interactions influenced by NaCl and sucrose. Mol Pharm. 2024;21(9):4553–4564. doi:10.1021/acs.molpharmaceut.4c00460
181. Wyatt PJ. Light scattering and the solution properties of macromolecules. Anal Chim Acta. 1993;272:1–40.
182. Ghosh R, Calero-Rubio C, Saluja A, Roberts CJ. relating protein-protein interactions and aggregation rates from low to high concentrations. J Pharm Sci. 2016;105(3):1086–1096. doi:10.1016/j.xphs.2016.01.004
183. Svilenov HL, Winter G. formulations that suppress aggregation during long-term storage of a bispecific antibody are characterized by high refoldability and colloidal stability. J Pharm Sci. 2020;109(6):2048–2058. doi:10.1016/j.xphs.2020.03.011
184. Jiskoot W, Hawe A, Menzen T, Volkin DB, Crommelin DJA. Ongoing challenges to develop high concentration monoclonal antibody-based formulations for subcutaneous administration: Quo Vadis? J Pharm Sci. 2022;111(4):861–867. doi:10.1016/j.xphs.2021.11.008
185. Garidel P, Kuhn AB, Schäfer LV, Karow-Zwick AR, Blech M. High-concentration protein formulations: how high is high? Eur J Pharm Biopharm. 2017;119:353–360. doi:10.1016/j.ejpb.2017.06.029
186. Strickley RG, Lambert WJ. A review of formulations of commercially available antibodies. J Pharm Sci. 2021;110(7):2590–2608.e56. doi:10.1016/j.xphs.2021.03.017
187. Willis LF, Toprani V, Wijetunge S, et al. Exploring a role for flow-induced aggregation assays in platform formulation optimisation for antibody-based proteins. J Pharm Sci. 2024;113(3):625–636. doi:10.1016/j.xphs.2023.10.031
188. Pharmacopeia US. General chapter particulate matter in injections | USP-NF. US Pharmacopeia. Available from: http://www.pharmacopeia.cn/v29240/usp29nf24s0_c788.html.
189. Moussa EM, Panchal JP, Moorthy BS, et al. Immunogenicity of therapeutic protein aggregates. J Pharm Sci. 2016;105(2):417–430. doi:10.1016/j.xphs.2015.11.002
190. Pham NB, Meng WS. Protein aggregation and immunogenicity of biotherapeutics. Int J Pharm. 2020;585(June). doi:10.1016/j.ijpharm.2020.119523
191. Zölls S, Tantipolphan R, Wiggenhorn M, et al. Particles in therapeutic protein formulations, part 1: overview of analytical methods. J Pharm Sci. 2012;101(3):914–935. doi:10.1002/jps.23001
192. Zölls S, Weinbuch D, Wiggenhorn M, et al. Flow imaging microscopy for protein particle analysis-a comparative evaluation of four different analytical instruments. AAPS J. 2013;15(4):1200–1211. doi:10.1208/s12248-013-9522-2
193. Daniels AL, Calderon CP, Randolph TW. Machine learning and statistical analyses for extracting and characterizing “fingerprints” of antibody aggregation at container interfaces from flow microscopy images. Biotechnol Bioeng. 2020;117(11):3322–3335. doi:10.1002/bit.27501
194. Murphy MI, Bruque M, Hanford A, et al. Qualitative high-throughput analysis of subvisible particles in biological formulations using backgrounded membrane imaging. J Pharm Sci. 2022;111(6):1605–1613. doi:10.1016/j.xphs.2022.03.010
195. Rivas G, Minton AP. Macromolecular crowding in vitro, in vivo, and in between. Trends Biochem Sci. 2016;41(11):970–981. doi:10.1016/j.tibs.2016.08.013
196. Wälchli R, Fanizzi F, Massant J, Arosio P. Relationship of PEG-induced precipitation with protein-protein interactions and aggregation rates of high concentration mAb formulations at 5 °C. Eur J Pharm Biopharm. 2020;151(January):53–60. doi:10.1016/j.ejpb.2020.03.011
197. Toprani VM, Joshi SB, Kueltzo LA, Schwartz RM, Middaugh CR, Volkin DB. A micro–polyethylene glycol precipitation assay as a relative solubility screening tool for monoclonal antibody design and formulation development. J Pharm Sci. 2016;105(8):2319–2327. doi:10.1016/j.xphs.2016.05.021
198. Sule SV, Dickinson CD, Lu J, Chow CK, Tessier PM. Rapid analysis of antibody self-association in complex mixtures using immunogold conjugates. Mol Pharm. 2013;10(4):1322–1331. doi:10.1021/mp300524x
199. Alam ME, Geng SB, Bender C, et al. Biophysical and sequence-based methods for identifying monovalent and bivalent antibodies with high colloidal stability. Mol Pharm. 2018;15(1):150–163. doi:10.1021/acs.molpharmaceut.7b00779
200. Rabia LA, Zhang Y, Ludwig SD, Julian MC, Tessier PM. Net charge of antibody complementarity-determining regions is a key predictor of specificity. Protein Eng Des Sel. 2018;31(11):409–418. doi:10.1093/protein/gzz002
201. Starr CG, Makowski EK, Wu L, et al. Ultradilute measurements of self-association for the identification of antibodies with favorable high-concentration solution properties. Mol Pharm. 2021;18(7):2744–2753. doi:10.1021/acs.molpharmaceut.1c00280
202. Makowski EK, Wang T, Zupancic JM, et al. Optimization of therapeutic antibodies for reduced self-association and non-specific binding via interpretable machine learning. Nat Biomed Eng. 2024;8(1):45–56. doi:10.1038/s41551-023-01074-6
203. Sun T, Reid F, Liu Y, et al. High throughput detection of antibody self-interaction by bio-layer interferometry. MAbs. 2013;5(6):838–841.
204. Hedberg SHM, Rapley J, Haigh JM, Williams DR. Cross-interaction chromatography as a rapid screening technique to identify the stability of new antibody therapeutics. Eur J Pharm Biopharm. 2018;133(October):131–137. doi:10.1016/j.ejpb.2018.10.009
205. Hötzel I, Theil FP, Bernstein LJ, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs. 2012;4(6):753–760. doi:10.4161/mabs.22189
206. Xu Y, Roach W, Sun T, et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Prot Eng Des Sel. 2013;26(10):663–670.
207. Norden DM, Navia CT, Sullivan JT, Doranz BJ. The emergence of cell-based protein arrays to test for polyspecific off-target binding of antibody therapeutics. MAbs. 2024;16(1). doi:10.1080/19420862.2024.2393785
208. Van schouwenburg PA, Kruithof S, Votsmeier C, et al. Functional analysis of the anti-adalimumab response using patient-derived monoclonal antibodies. J Biol Chem. 2014;289(50):34482–34488. doi:10.1074/jbc.M114.615500
209. Krause ME, Sahin E. Chemical and physical instabilities in manufacturing and storage of therapeutic proteins. Curr Opin Biotechnol. 2019;60:159–167. doi:10.1016/j.copbio.2019.01.014
210. Wälchli R, Vermeire PJ, Massant J, Arosio P. Accelerated aggregation studies of monoclonal antibodies: considerations for storage stability. J Pharm Sci. 2020;109(1):595–602. doi:10.1016/j.xphs.2019.10.048
211. Goldberg DS, Lewus RA, Esfandiary R, et al. Utility of high throughput screening techniques to predict stability of monoclonal antibody formulations during early stage development. J Pharm Sci. 2017;106(8):1971–1977. doi:10.1016/j.xphs.2017.04.039
212. Kuzman D, Bunc M, Ravnik M, Reiter F, Žagar L, Bončina M. Long-term stability predictions of therapeutic monoclonal antibodies in solution using Arrhenius-based kinetics. Sci Rep. 2021;11(1):20534. doi:10.1038/s41598-021-99875-9
213. Nowak C, Cheung K, J M, et al. Forced degradation of recombinant monoclonal antibodies: a practical guide. MAbs. 2017;9(8):1217–1230. doi:10.1080/19420862.2017.1368602
214. Tamizi E, Jouyban A. Forced degradation studies of biopharmaceuticals: selection of stress conditions. Eur J Pharm Biopharm. 2016;98:26–46. doi:10.1016/j.ejpb.2015.10.016
215. Halley J, Chou YR, Cicchino C, et al. An industry perspective on forced degradation studies of biopharmaceuticals: survey outcome and recommendations. J Pharm Sci. 2020;109(1):6–21. doi:10.1016/j.xphs.2019.09.018
216. ICH. ICH topic Q 5 C quality of biotechnological products: stability testing of biotechnological/biological products step 5 note for guidance on quality of biotechnolo-gical products: stability testing of biotechnological/biological products (cpmp/ich/138/95) proposed date for coming into operation; 1996. Available from: http://www.emea.eu.int.
217. Dai L, Davis J, Nagapudi K, et al. Predicting long-term stability of an oral delivered antibody drug product with accelerated stability assessment program modeling. Mol Pharm. 2024;21(1):325–332. doi:10.1021/acs.molpharmaceut.3c00877
218. Klijn ME, Hubbuch J. Correlating multidimensional short-term empirical protein properties to long-term protein physical stability data via empirical phase diagrams. Int J Pharm. 2019;560(November 2018):166–174. doi:10.1016/j.ijpharm.2019.02.006
219. Wang W, Roberts CJ. Non-Arrhenius protein aggregation. AAPS J. 2013;15(3):840–851. doi:10.1208/s12248-013-9485-3
220. Sarkar D, Kang P, Nielsen SO, Qin Z. Non-Arrhenius reaction-diffusion kinetics for protein inactivation over a large temperature range. ACS Nano. 2019;13(8):8669–8679. doi:10.1021/acsnano.9b00068
221. Halaby DM, Poupon A, Mornon JP. The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein Eng Des Select. 1999;12(7):563–571. doi:10.1093/protein/12.7.563
222. He F, Woods CE, Becker GW, Narhi LO, Razinkov VI. High‐throughput assessment of thermal and colloidal stability parameters for monoclonal antibody formulations. J Pharm Sci. 2011;100(12):5126–5141.
223. Brader ML, Estey T, Bai S, et al. Examination of thermal unfolding and aggregation profiles of a series of developable therapeutic monoclonal antibodies. Mol Pharm. 2015;12(4):1005–1017. doi:10.1021/mp400666b
224. Svilenov H, Markoja U, Winter G. Isothermal chemical denaturation as a complementary tool to overcome limitations of thermal differential scanning fluorimetry in predicting physical stability of protein formulations. Eur J Pharm Biopharm. 2018;125(January):106–113. doi:10.1016/j.ejpb.2018.01.004
225. Robinson MJ, Matejtschuk P, Bristow AF, Dalby PA. Tm-values and unfolded fraction can predict aggregation rates for granulocyte colony stimulating factor variant formulations but not under predominantly native conditions. Mol Pharm. 2018;15(1):256–267. doi:10.1021/acs.molpharmaceut.7b00876
226. Augustijn D, Mahapatra S, Streicher W, et al. Novel non-linear curve fitting to resolve protein unfolding transitions in intrinsic fluorescence differential scanning fluorimetry. Eur J Pharm Biopharm. 2019;142(March):506–517. doi:10.1016/j.ejpb.2019.06.001
227. Nemergut M, Žoldák G, Schaefer JV, et al. Analysis of IgG kinetic stability by differential scanning calorimetry, probe fluorescence and light scattering. Protein Science. 2017;26(11):2229–2239. doi:10.1002/pro.3278
228. Blech M, Melien R, Tschammer N, Presser B, Hinderberger D, Garidel P. Expanding the toolbox for predictive parameters describing antibody stability considering thermodynamic and kinetic determinants. Pharm Res. 2021;38(12):2065–2089. doi:10.1007/s11095-021-03120-x
229. Cromwell MEM, Hilario E, Jacobson F. Protein aggregation and bioprocessing. AAPS J. 2006;8(3):E572–9. doi:10.1208/aapsj080366
230. Li J, Krause ME, Chen X, et al. Interfacial stress in the development of biologics: fundamental understanding, current practice, and future perspective. AAPS J. 2019;21(3). doi:10.1208/s12248-019-0312-3
231. Willis LF, Radford SE, Kapur N, Brockwell DJ. Does flow play a role in protein aggregation during bioprocessing? Available from: https://www.bioprocessonline.com/doc/does-flow-play-a-role-in-protein-aggregation-during-bioprocessing-0001.
232. Thite NG, Ghazvini S, Wallace N, Feldman N, Calderon CP, Randolph TW. Interfacial adsorption controls particle formation in antibody formulations subjected to extensional flows and hydrodynamic shear. J Pharm Sci. 2023;112(11):2766–2777. doi:10.1016/j.xphs.2023.07.010
233. Simon S, Krause HJ, Weber C, Peukert W. Physical degradation of proteins in well-defined fluid flows studied within a four-roll apparatus. Biotechnol Bioeng. 2011;108(12):2914–2922. doi:10.1002/bit.23257
234. Bekard IB, Dunstan DE. Shear-induced deformation of bovine insulin in Couette flow. J Phys Chem B. 2009;113(25):8453–8457. doi:10.1021/jp903522e
235. Sing CE, Alexander-Katz A. Elongational flow induces the unfolding of von Willebrand factor at physiological flow rates. Biophys J. 2010;98(9):L35–L37. doi:10.1016/j.bpj.2010.01.032
236. Dobson J, Kumar A, Willis LF, et al. Inducing protein aggregation by extensional flow. Proc Natl Acad Sci USA. 2017;114(18):4673–4678. doi:10.1073/pnas.1702724114
237. Grigolato F, Arosio P. Synergistic effects of flow and interfaces on antibody aggregation. Biotechnol Bioeng. 2020;117(2):417–428. doi:10.1002/bit.27212
238. Kiese S, Papppenberger A, Friess W, Mahler HC. Shaken, not stirred: mechanical stress testing of an IgG1 antibody. J Pharm Sci. 2008;97(10):4347–4366. doi:10.1002/jps.21328
239. Agarkhed M, O’Dell C, Hsieh MC, Zhang J, Goldstein J, Srivastava A. Effect of surfactants on mechanical, thermal, and photostability of a monoclonal antibody. AAPS PharmSciTech. 2017;19:79–92. doi:10.1208/s12249-017-0845-7
240. Hanson MG, Katz JS, Ma H, et al. Effects of hydrophobic tail length variation on surfactant-mediated protein stabilization. Mol Pharm. 2020;17(11):4302–4311. doi:10.1021/acs.molpharmaceut.0c00737
241. Biddlecombe JG, Smith G, Uddin S, et al. Factors influencing antibody stability at solid-liquid interfaces in a high shear environment. Biotechnol Prog. 2009;25:1499–1507. doi:10.1002/btpr.211
242. Defante AP, Kalonia CK, Keegan E, et al. The impact of the metal interface on the stability and quality of a therapeutic fusion protein. Mol Pharm. 2020;17(2):569–578. doi:10.1021/acs.molpharmaceut.9b01000
243. Gerlt MS, Meier EM, Dingfelder F, Zürcher D, Müller M, Arosio P. microfluidic stress device to decouple the synergistic effect of shear and interfaces on antibody aggregation. J Pharm Sci. 2024;113(8):2161–2169. doi:10.1016/j.xphs.2024.05.024
244. Willis LF, Kumar A, Jain T, et al. The uniqueness of flow in probing the aggregation behavior of clinically relevant antibodies. Eng Rep. 2020;2(5). doi:10.1002/eng2.12147
245. Grabarek AD, Bozic U, Rousel J, et al. What makes polysorbate functional? Impact of polysorbate 80 grade and quality on IgG stability during mechanical stress. J Pharm Sci. 2020;109(1):871–880. doi:10.1016/j.xphs.2019.10.015
246. Du Y, Song J, Lu L, et al. Design of a reciprocal injection device for stability studies of parenteral biological drug products. J Pharm Sci. 2024;113(5):1330–1338. doi:10.1016/j.xphs.2023.12.014
247. Gentiluomo L, Svilenov HL, Augustijn D, et al. Advancing therapeutic protein discovery and development through comprehensive computational and biophysical characterization. Mol Pharm. 2020;17(2):426–440. doi:10.1021/acs.molpharmaceut.9b00852
248. Kopp MRG, Wolf Pérez AM, Zucca MV, et al. An accelerated surface-mediated stress assay of antibody instability for developability studies. MAbs. 2020;12(1):1815995. doi:10.1080/19420862.2020.1815995
249. Wolf Pérez AM, Sormanni P, Andersen JS, et al. In vitro and in silico assessment of the developability of a designed monoclonal antibody library. MAbs. 2019;11(2):388–400. doi:10.1080/19420862.2018.1556082
250. Jain T, Prinz B, Marker A, et al. Assessment and incorporation of in vitro correlates to pharmacokinetic outcomes in antibody developability workflows. MAbs. 2024;16(1):2384104. doi:10.1080/19420862.2024.2384104
251. Condado-Morales I, Dingfelder F, Waibel I, et al. A comparative study of the developability of full-length antibodies, fragments, and bispecific formats reveals higher stability risks for engineered constructs. MAbs. 2024;16(1). doi:10.1080/19420862.2024.2403156
252. Sharma VK, Patapoff TW, Kabakoff B, et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014;111(52):18601–18616. doi:10.1073/pnas.1421779112
253. Katz JS, Chou DK, Christian TR, et al. Emerging challenges and innovations in surfactant-mediated stabilization of biologic formulations. J Pharm Sci. 2022;111(4):919–932. doi:10.1016/j.xphs.2021.12.002
254. Brosig S, Cucuzza S, Serno T, et al. Not the usual suspects: alternative surfactants for biopharmaceuticals. ACS Appl Mater Interfaces. 2023;15(29):34540–34553. doi:10.1021/acsami.3c05610
255. Ripple DC, Dimitrova MN. Protein particles: what we know and what we do not know. J Pharm Sci. 2012;101(10):3568–3579. doi:10.1002/jps.23242
256. Narayanan H, Dingfelder F, Condado Morales I, et al. Design of biopharmaceutical formulations accelerated by machine learning. Mol Pharm. 2021;18(10):3843–3853. doi:10.1021/acs.molpharmaceut.1c00469
257. Bashour H, Smorodina E, Pariset M, et al. Biophysical cartography of the native and human-engineered antibody landscapes quantifies the plasticity of antibody developability. Commun Biol. 2024;7(1):922. doi:10.1038/s42003-024-06561-3
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.