Back to Journals » International Journal of Nanomedicine » Volume 13
Biomimetic quantum dot-labeled B16F10 murine melanoma cells as a tool to monitor early steps of lung metastasis by in vivo imaging
Authors Díaz-García VM ![]() , Guerrero S, Díaz-Valdivia N, Lobos-González L, Kogan M
, Guerrero S, Díaz-Valdivia N, Lobos-González L, Kogan M ![]() , Pérez-Donoso JM
, Pérez-Donoso JM ![]() , Quest AFG
, Quest AFG
Received 15 February 2018
Accepted for publication 30 May 2018
Published 16 October 2018 Volume 2018:13 Pages 6391—6412
DOI https://doi.org/10.2147/IJN.S165565
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Víctor Manuel Díaz-García,1–4 Simón Guerrero,1,2,5 Natalia Díaz-Valdivia,1,2 Lorena Lobos-González,2,6,7 Marcelo Kogan,2,5 José Manuel Pérez-Donoso,3 Andrew FG Quest1,2
1Cellular Communication Laboratory, Center for Studies on Exercise, Metabolism and Cancer (CEMC), Faculty of Medicine, Universidad de Chile, Santiago, Chile; 2Advanced Center for Chronic Diseases (ACCDiS), Faculty of Medicine, Universidad de Chile, Santiago, Chile; 3BioNanotechnology and Microbiology Laboratory, Center for Bioinformatics and Integrative Biology (CBIB), Faculty of life Sciences, Universidad Andres Bello, Santiago, Chile; 4Facultad de Ingeniería y Tecnología, Universidad San Sebastián, Concepción 4080871, Chile; 5Department of Pharmacological and Toxicological Chemistry, Faculty of Chemical and Pharmaceutical Sciences, Universidad de Chile, Santiago, Chile; 6Fundación Ciencia y Vida, Santiago, Chile; 7Centro de Medicina Regenerativa, Facultad de Medicina, Clínica Alemana, Universidad del Desarrollo , Santiago, Chile
Background: Numerous studies have proposed the use of fluorescent semiconductor nanoparticles or quantum dots (QDs) as novel tools to label cells and tumors. However, QD applications are limited by their toxicity in biological systems and little is known about whether QDs affect the capacity of cancer cells to metastasize. Previously, we described the “biomimetic” synthesis of CdTe-QDs (QDs-glutathione [GSH]) with increased biocompatibility and the potential utility in labeling cells.
Purpose: In order to determine the feasibility of using QDs-GSH as a tool for tracking tumor cells during early metastasis, we characterized here for the first time, the in vitro and in vivo effects of the incorporation of green or red biomimetic QDs-GSH into B16F10 cells, a syngeneic mouse melanoma line for metastasis assays in C57BL/6 mice.
Methods: B16F10 cells were labeled with green or red biomimetic QDs-GSH in the presence or absence of n-acetylcysteine. Then, migration, invasion and proliferation of labeled B16F10 were evaluated in vitro. Finally, the B16F10 cells labeled with red QDs-GSH were used to monitor in vivo lung metastasis at early time points (5 minutes to 24 hours) or after 21 days in C57BL/6 mice.
Results: We developed a methodology that allows obtaining QDs-GSH-labeled B16F10 cells (nearly 100% viable labeled cells), which remained viable for at least 5 days and migrated similarly to control cells. However, proliferation, invasion, and the capacity to form metastatic nodules in the lungs were severely attenuated. Fluorescence imaging revealed that distribution/accumulation of QDs-GSH-labeled B16F10 cells could be tracked following injection into C57BL/6 mice (syngeneic preclinical metastasis model) and that these cells preferentially accumulated in the perialveolar area in lungs as early as 5 minutes post-injection.
Conclusion: The methodology described here represents a useful alternative for monitoring initial events during tumor cell metastasis.
Keywords: cell tracking, biomimetic, invasion, proliferation, migration, cancer
Introduction
Cancer is a leading cause of death world-wide1 and frequently attributable to tumor cell dissemination to distant organs, a process referred to as metastasis.2,3 The evolution of metastasis in vivo is difficult to follow, mainly because only a few tumor cells that enter the blood stream successfully colonize distant organs in a clinically detectable fashion.3 Standard in vivo experimental approaches to study metastasis involve the injection of metastatic cells into the blood stream of a mouse and subsequent evaluation of distant organ invasion/colonization as metastatic foci after sacrificing the animal, generally several weeks post-inoculation of the tumor cells. Accordingly, these traditional methods are considered “black box” experiments, given that the events occurring between initial administration of cells and metastatic tumor formation in the target organ cannot be directly observed.4
For in vivo imaging studies, fluorescence stands out over other techniques due to its low cost, high signal as well as spatial and temporal resolution, and the possibility of simultaneously evaluating multiple labels.4,5 The main disadvantages of fluorescence-based techniques include the instability of fluorescent probes and autofluorescence of biomolecules.4 In the last decade, sensitivity, versatility and stability of fluorescence imaging applications have increased notably, thanks to the development of semiconductor nanoparticles (NPs) or quantum dots (QDs).4–6
QDs are inorganic, semiconducting, and fluorescent NPs, ranging in size from 1 to 20 nm approximately.7–9 The QD size determines their fluorescence emission wavelength, which may vary from 450 nm (blue-green) to 650 nm (red).7,8,10,11 QDs possess a number of spectroscopic properties that make them particularly interesting for studies in vivo: 1) elevated resistance to chemical degradation and photobleaching; 2) narrow and almost symmetric fluorescence peaks; 3) a broad light absorption spectrum that allows fine-tuning for processing of optical images and multispectral analysis of multiple objects simultaneously;7,10,11 and 4) a sensitivity that may be 2–3-fold higher than other organic fluorophores commonly used for detection in tissues.10,11 The aforementioned characteristics of QDs make them a promising tool for tumor and/or cell labeling, raising the specter of being able to track essentially real-time tumor cell migration, and their accumulation in distant organs.12
A major disadvantage of using QDs in biological systems is their toxicity associated with the formation of reactive oxygen species (ROS) and the release of heavy metal ions from the QDs core (ie, Cd+2 and Pb+2, among others).13–16 These ions interact with intracellular thiolated proteins and glutathione (GSH), thereby altering the cellular redox environment16–19 and these changes likely affect the behavior of QDs-labeled metastatic cells.20 More recently, in an attempt to bypass these limitations, a new generation of cadmium-free QDs was developed, with essentially the same spectroscopic advantages but lacking detrimental effects linked to the presence of cadmium. Nonetheless, some toxic effects attributable to physical and surface properties of these QDs remain.21
In this perspective, a few years ago, we developed a protocol to synthesize, in an ecofriendly and biocompatible manner, cadmium-telluride QDs8 (Patent No US 20130284979).22 This method, referred to as “Biomimetic,” permits obtaining rapidly and easily, hydrophilic QDs, stabilized with GSH on their surface (QDs-GSH), which emit fluorescence between 480 nm (green) and 650 nm (red), depending upon their size.8,9 QDs-GSH fluorescence was shown to remain stable for months when kept at room temperature. Moreover, QDs-GSH do not precipitate in buffers, cells, or bacterial culture media, in contrast to classic QDs.8 It is noteworthy that for QDs-GSH, no significant detrimental effects on human gastric cancer cell (MKN45) viability and morphology were detected after QDS-GSH internalization, even when incubated at concentrations up to 200 μg/mL.8,9,23 Nevertheless, it still remained to be determined how QDs-GSH affect migration, invasion, and metastasis of cancer cells.
In the current study, we extensively characterized the effects of QDs-GSH internalization on the viability, proliferation, and invasion in vitro, as well as lung metastasis in vivo of B16F10 murine melanoma cells in syngeneic C57BL/6 mice, a preclinical melanoma model.24,25 QDs-GSH-labeled B16F10 cells were used to track the early accumulation/distribution of these metastatic cells in C57BL/6 mice. Although QDs-GSH internalization severely reduced the ability of B16F10 cells to form visible metastatic nodules 21 days post-injection, the QDs-GSH-labeled B16F10 cells could be successfully tracked during initial colonization of distant organs including the lung.
Materials and methods
QDs-GSH chemical synthesis
QDs-GSH were synthesized by protocols previously described by Pérez-Donoso et al.8,22 Citrate/NaBH4 15 mM (pH=9.4) solution was used as the synthesis buffer, to which CdCl2 40 μM, K2TeO310 μM, and GSH 10 mM were added. The synthesis solution was incubated at 90°C shaking vigorously for 2 (green QDs-GSH [gQDs-GSH]) or 6 hours (red QDs-GSH [rQDs-GSH]) and the synthesis reaction was stopped by cooling to 4°C. QDs-GSH were dialyzed in Citrate/NaBH4 15 mM at pH 9.4, during 2 hours. Afterwards, QDs-GSH were precipitated using 2 volumes of ethanol for 10 minutes at −20°C. Solutions were then centrifuged at 24,000× g at 4°C for 20 minutes. Pellets were purified and vacuum dried for 24 hours at room temperature. Finally, QDs were weighed and dissolved in nanopure water.
QDs-GSH characterization
Solutions of different QDs-GSH (10 mg/mL) were characterized by spectroscopy. Characterization was performed in 96-well plates, using a multimodal multiplate reader (fluorescence and absorbance) (Sinergy-H1M; Biotek). Size and zeta potential were determined by dynamic light scattering and laser electrophoresis Doppler, respectively (Zetasizer nano S90 light; Malvern Instruments Limited, Malvern, UK). The core size of rQDs-GSH was determined by using a JEOL/JEM 1200 EX II transmission electron microscope (Figure S1).
Cell culture and QDs-GSH incubation
B16F10 cells (provided originally by Dr Laurence Zitvogel, Institut Gustave Roussy, Villejuif, France) were cultured for 24 hours in Roswell Park Memorial Institute medium (RPMI medium) (Gibco™; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Bioind) and antibiotics (penicillin 100 UI/mL, streptomycin 100 μg/mL), at 37°C in 5% CO2. All procedures and the use of these cells were approved by the unit of risk prevention and biosafety, medical school, University of Chile and the institutional committee of care and use of animals (CICUA) of the University of Chile. Afterwards, QDs-GSH treatments were performed with the previously described culture medium supplemented with lipofectamine 4 μL/mL (Invitrogen), plus 0, 50, 100, 200, or 400 μg/mL of each QDs-GSH. N-acetylcysteine (NAC) treatments of 0, 2, 4, 8, or 10 mM were performed using the same conditions, but only for rQDs-GSH.
Fluorescence-activated cell sorting (FACS)
Cell viability, labeled viable cells, mean fluorescence intensity (MFI) and ROS levels were determined by FACS flow cytometry. B16F10 cells treated with red and green QDs-GSH were washed twice with PBS, detached with trypsin-EDTA 0.5%, and centrifuged at 150× g for 5 minutes. Then, cell pellets were washed with Flow Buffer (BD Biosciences, San José, CA, USA) and centrifuged as mentioned earlier. B16F10 cells treated with gQDs-GSH were incubated for 10 minutes with propidium iodide (PI) 10 μg/mL (Thermo Fisher Scientific) for death cell differentiation. On the other hand, B16F10 cells treated with rQDs-GSH were tested for green live cells-labeling (Calcein), with LIVE/DEAD® Viability/Cytotoxicity Kit, for mammalian cells (Thermo Fisher) dilution 1:100,000 to determine viability and with dihydrodichlorofluorescein (H2DCFDA) (Invitrogen) dilution 1:10,000 to determine ROS levels. Five thousand events were analyzed to determine size (forward scatter [FSC]) and granularity parameters (side scatter [SSC]), excluding cell debris. Cellular gQDs-GSH-fluorescence, cellsH2DCFDA and cellsCalcein were determined using the FITC channel (green fluorescence); rQDs-GSH and PI mark were tracked in the PerCP-Cy5 channel (red fluorescence). Both emissions were obtained by laser excitation at 488 nm. The percentage of viable labeled cells was obtained, for either red- or green-labeled cells, with the following equation:
|

MFI was determined for viable QDs-labeled cells and control cells by FACs. Data were analyzed using the FCS Express four plus research edition software (De Novo Software, Los Angeles, CA, USA).
Confocal microscopy
Viable cells labeled or not with QDs-GSH were evaluated using a Zeiss Microscope model LSM 700. The excitation laser was set at 488 nm. Green and red fluorescence were tracked at 510 and 600 nm, respectively. All optical sections obtained for co-localization studies were processed with ImageJ 1.47 v software (National Institutes of Health, USA).
For invasion assays, fixed cells attached to the lower membrane surface were visualized using a Zeiss Microscope, model LSM 700. Wavelength excitation laser was set at 405 nm. DAPI and rQDs-GSH fluorescence were tracked at 460 and 580 nm, respectively. Images of either B16F10 or B16F10QDs-GSH-10NAC cells attached to the lower side of the well membrane were obtained. Both types of cells were labeled with DAPI 1:1,000 for 5 minutes. Afterwards, membranes were washed twice and fixed in DACO fluorescent solution.
Fluorescence microscopy
Membrane-attached cells after migration were visualized using a CKX41 inverted microscope and a fluorescence light source U-LH100HG (Olympus, Japan). Images were processed with ImageJ 1,47 v software (National Institutes of Health, USA).
Quantification of reduced thiols
B16F10 and MKN45 cells were cultured until they reached ~90% confluence in 100 mm plates. Afterwards, cells were collected, washed with PBS and resuspended in 200 μL sulfosalicylic acid 5% (SSA). Samples were divided into two equivalent portions, one for total protein measurement (BCA assay) and the other for the determination of intracellular thiols. Cells were lyzed using 20 freeze/thaw cycles and subsequently centrifuged at 32,000× g for 20 minutes. Supernatants were collected and reduced thiols (RSH) were quantified with Ellman’s reagent. Following incubation for 5 minutes at 37°C, the absorbance of supernatants at 412 nm was determined.26,27 Calibration curves were obtained using GSH solutions of known concentration. Intracellular RSH levels were normalized to cellular protein content in milligrams.
Migration assays
Assays were performed using 8 μm pore polycarbonate membrane transwells (diameter 6.5 mm) (Corning, NY, USA). After coating the lower membrane surface with 100 μL of fibronectin 2 μg/mL, membranes were washed with PBS. Afterwards, transwells were immersed in RPMI medium supplemented with 10% FBS. Suspensions of 1.5×105 cells (B16F10, B16F10QDs-GSH-4NAC or B16F10QDs-GSH-10NAC) prepared in serum-free RPMI medium were seeded into the upper chamber of transwells. After 2 hours, inserts were collected and the cells present in the upper chamber were removed. Cells attached to the lower membrane surface after migration were fixed and stained with a 0.1% crystal violet solution in methanol 20% for 2 hours at 37°C. Membranes were then gently washed, dried at room temperature, mounted in Mowiol, and observed under a light microscope. At least seven fields were evaluated per experiment to determine the number of transmigrated cells.
Invasion assays
Assays were performed using Matrigel Invasion Chambers (8 μm; Corning, NY, USA), which were rehydrated with RPMI media supplemented with 10% FBS. Subsequently, suspensions of either B16F10 or B16F10QDs-GSH-10NAC cells (2×105) prepared in serum-free RPMI medium were seeded into the upper transwell chamber. After 24 hours, inserts were collected and cells remaining in the upper chamber were removed. Cells that invaded the matrigel and then attached to the lower membrane surface were fixed in cold methanol for 2 minutes and then stained with 1% Toluidine Blue for 2 hours. Membranes were then gently washed, dried at room temperature, mounted in Mowiol, and observed under a light microscope. At least seven fields were evaluated per experiment to determine the number of cells that invaded the matrigel.
rQDs-GSH cell labeling persistence and proliferation assay
This assay was performed using 1×105 viable cells (B16F10 cells and B16F10QDs-GSH-10NAC). Cells were seeded in 60 mm plates and cultured with RPMI-supplemented media. Afterwards, cells were detached with trypsin-EDTA 0.5% and centrifuged at 150× g for 5 minutes. Cell pellets were washed in PBS and centrifuged at 150× g for 5 minutes; the supernatants were discarded and cells were resuspended in 200 μL of FBS. Then, the total number of cells 1–5 days post-inoculation was quantified in a Neubauer chamber (Marienfeld Superior, Lauda-Königshofen, Germany) after trypan blue staining 0.5% (biological industries). Percentage of viability, labeled viable cells, and MFI were determined by FACS, as previously described.
In vivo metastasis assays
Experiments were done essentially following previously described protocols.25 Briefly, C57BL/6 mice were injected intravenously with 2×105 B16F10 or B16F10QDs-GSH-10NAC cells in 500 μL of physiological saline solution. Then, 21 days post-injection, mice were sacrificed. Lungs were fixed in Fekete’s solution. Black lung tumor was separated from the rest of the lung tissue and weighed. Metastasis was expressed as black tissue mass/total lung mass percentage (%) post-fixation.
Mouse imaging
C57BL/6 mice were injected intravenously with 2×105 B16F10 or B16F10QDs-GSH-10NAC cells, in 500 μL of physiological saline solution. Mice were observed for 6 hours using the in vivo imaging system (IVIS; In-Vivo FX PRO; Bruker, Billerica, MA, USA). Excitation and emission wavelengths were 410 and 600 nm, respectively. Afterwards, mice were euthanized and lungs were collected for ex-vivo imaging.
In vivo imaging
C57BL/6 mice were injected intravenously with 2×105 B16F10Control and B16F10QDs-GSH-10NAC cells in 500 μL of physiological saline solution. Mice were sacrificed at 5 minutes, 30 minutes, 2 hours, 6 hours, and 24 hours post-injection. Lungs, heart, spleen, thymus, liver, and kidneys were harvested and fixed in Fekete’s solution for 1 week. Fluorescence present at the different time points was quantified using IVIS, and are shown as the average of relative optical intensity (ROI). Fluorescence data are presented as percentages, considering the baseline fluorescence of unlabeled organs as 100%.
Immunohistochemical analysis
The organs obtained from C57BL6 mice 6 and 24 hours after injection of B16F10QDs-GSH-10NAC, B16F10Calcein, or B16F10Control cells were fixed for 24 hours in pH7-buffered formalin (10%) and then paraffin-embedded. For tissue samples, 5 μm-thick serial paraffin sections were collected on silanized slides (DAKO) and deparaffinized in two consecutive 5-minute incubations with xylene. One section was then stained with hematoxylin and eosin (H&E). The others were rehydrated in two consecutive 3-minute incubations with 98% and 90% ethanol each, followed by a single incubation in Diethyl pyrocarbonate (DEPC)-treated distilled water for 5 minutes. Sections were then incubated in 2.5 μg/mL of Proteinase K (Invitrogen) at room temperature for 20 minutes and then rinsed twice for 3 minutes in DEPC-treated water. Nuclei were stained with DAPI for 5 minutes and washed. The samples were then mounted on cover slips and embedded with fluorescence mounting medium (DAKO). The images were captured using a fluorescence microscope (Olympus, Tokyo, Japan).
Proof-of-concept
B16F10 cells stably transfected with either the empty pLacIOP vector [B16F10(Mock)] or the vector containing the Caveolin-1 (CAV1)-encoding insert [B16F10(CAV-1)] have been described previously.25,28 These cells were then labeled with rQDs-GSH as described earlier (B16F10QDs-GSH-10NAC). C57BL/6 mice were injected intravenously with 2×105 of the respective cells in 500 μL of physiological saline solution. Mice were sacrificed 6 hours post-injection of cells and lungs were fixed in Fekete’s solution for 1 week. Then, fluorescence was quantified (average ROI) using the IVIS system.
Statistical analysis
Data shown are the average±SEM of at least three independent experiments. Statistical significance was determined at a 95% CI using the nonparametric Mann–Whitney test, for the comparison of two groups.
All in vivo assays were performed as blind studies (in vivo metastasis assays and in vivo images) or double-blind studies (mice and lung images, immunohistochemistry assay, and proof-of-concept). All in vivo experiments were performed following the protocols of management and care of animals established by the CICUA of the University of Chile. CICUA approval code: CBA # 0837 FMUCH.
Results and discussion
Synthesis and characterization of QDs-GSH
In this study, green and red QDs-GSH were synthesized using the biomimetic method developed by our laboratory.8,9,22 Data summarizing the size distribution, zeta potential, and emission peak wavelengths of QDs-GSH are shown in Table 1. For gQDs-GSH, zeta potential values were greater than those of rQDs-GSH. GSH is a tripeptide, with a thiol and two carboxyl-terminal groups, which confer negative charges to the molecule.17 Thus, the elevated zeta potential is indicative of a higher GSH content in the gQDs-GSH. We previously characterized by X-ray photoelectron spectroscopy (XPS) the GSH-QD capping and reported a lower GSH content in rQDs-GSH than gQDs-GSH.23 Importantly, differences in QDs size (gQDs-GSH vs rQDs-GSH) and charge have previously been shown to affect cell viability.7,9,15,21,29 In addition, we have previously reported that green biomimetic QDs-GSH contain less Cd2+ than rQDs-GSH (18% and 30%, respectively).8,9 Therefore, in vitro assays were performed to evaluate cell behavior after incorporation of gQDs-GSH and rQDs-GSH.
 | Table 1 Size (hydrodynamic diameter), Z potential, and emission peaks of green and rQDs-GSH |
Characterization of QDs-GSH internalization and effects in B16F10 cells
B16F10 murine melanomas are cancer cells with an elevated metastatic potential that specifically accumulate in the lungs following tail vein injection of syngeneic C57BL/6 recipient animals, where they form readily detectable, pigmented metastatic nodules 15–21 days post-injection.24,25 This is a well-established preclinical model that is employed to evaluate the metastatic potential of cancer cells.24,25 To label the B16F10 cells, they were incubated with gQDs-GSH or rQDs-GSH at concentrations ranging from 0 (control) to 50, 100, 200, or 400 μg/mL, in the presence of 4 μL/mL of lipofectamine, for 24 hours. Lipofectamine was used to promote cell internalization of QDs-GSH, following a previously described protocol8 (Patent No US 20130284979).22 Then, cell viability, percentage of viable labeled cells, and the MFI of cells transfected with different concentrations of QDs-GSH were determined by flow cytometry (Figure 1A). For gQDs-GSH, cell viability in general and viability of labeled cells were substantially reduced at concentrations of 50 μg/mL or higher (Figure 1B). In this respect, rQDs-GSH were significantly less toxic and also the MFI for labeled cells was higher in comparison to those obtained with gQDs-GSH (Figure 1C and D). Note that rQDs-GSH only induced significant decreases in cell viability when used at higher concentrations (400 μg/mL). These observations are consistent with reports by Lovrić et al,30 who showed that smaller sized QDs more readily crossed intracellular membranes to enter the nucleus and other subcellular compartments, like mitochondria.8,31 The nuclear pore complex permits entry of small molecules up to ~9 nm in diameter by passive diffusion.32 Our results show that gQDs-GSH and rQDs-GSH possess a hydrodynamic diameter of ~1.8 and ~5.1 nm (Table 1), respectively, indicating that both QDs-GSH probably can cross the nuclear membrane, although gQDs-GSH are likely do so more efficiently. However, QDs toxicity may also relate to the differences in zeta potential between gQDs-GSH and rQDs-GSH, given that greater negative zeta potential values, as is the case for gQDs-GSH, are known to reduce cell survival31 (Table 1).
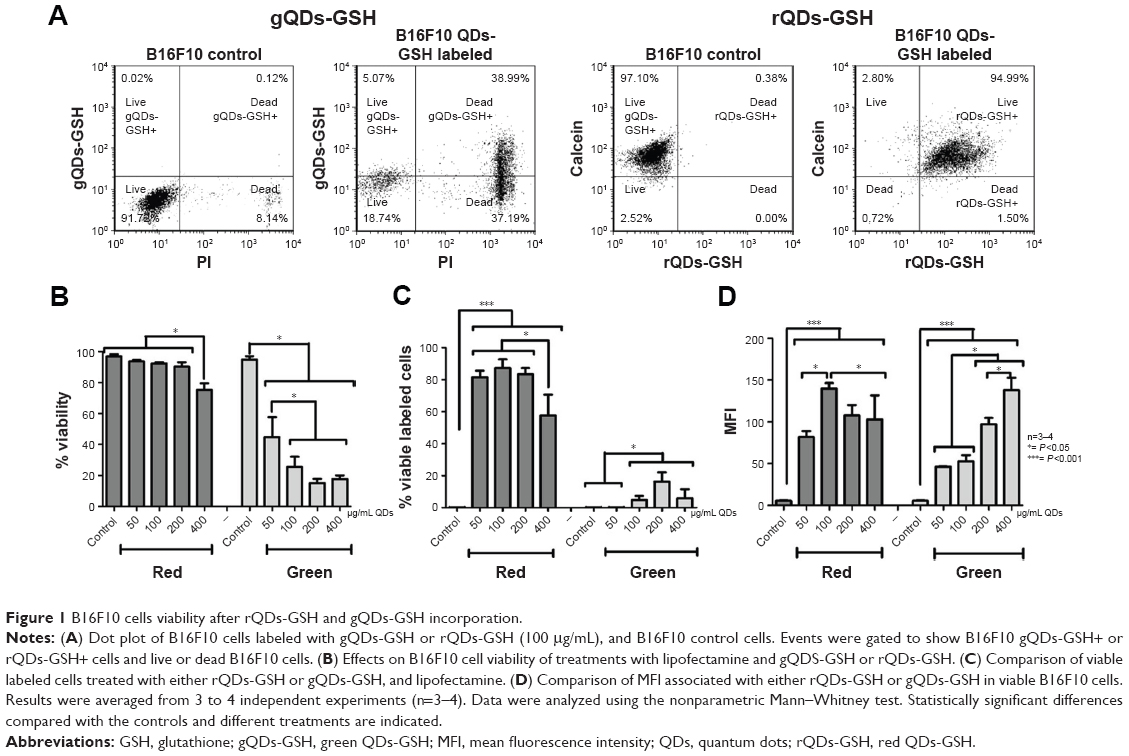 | Figure 1 B16F10 cells viability after rQDs-GSH and gQDs-GSH incorporation. |
Of note, the detrimental effects observed in B16F10 cells incubated with gQDs-GSH were significantly elevated in comparison to the toxic effects of biomimetic QDs previously reported for MKN45 cells.8,9,23 This may simply reflect cell-specific parameters of predisposition to QDs-GSH toxicity, as has been reported previously in different cell lines exposed to inorganic NPs.33 Alternatively, it is well established that the generation of ROS represents one of the main mechanisms by which QDs induce damage to biological systems.13–15,19 Intracellular thiols (RSH) play an important role as mediators of tolerance to intracellular ROS.17,18 Thus, we hypothesized that the differences in sensitivity to QDs-GSH observed for B16F10 and MKN45 cells may be due to different levels of RSH. To evaluate this possibility, intracellular RSH levels were determined in each cell type. As suspected, significantly lower RSH levels were observed in B16F10 cells than in MKN45 cells, suggesting that B16F10 cells may, for this reason, tolerate to a lesser extent QD-induced oxidative damage than MKN45 cells (Figure S2).
NAC treatment reduces rQDs-GSH effects in B16F10 cells and increases cell labeling
QDs increase ROS in biological systems,14 and ROS, on the other hand, affects migration and invasion of metastatic cells.34 Therefore, we hypothesized that NAC should ameliorate detrimental QDs-GSH effects on metastatic cells, given that NAC increases intracellular RSH levels.17 Moreover, NAC is generally beneficial and protects cells against organic stress induced by inorganic NPs,33 as well as provides greater protection against QDs damage than other non-thiol anti-oxidants, such as trolox.30 Given our results showing that incubation of cells with 100 μg/mL of rQDs-GSH resulted in >80% viable labeled cells, similar to the percentage of viable labeled cells reported by Voura et al,12 with the best mean MFI values (Figure 1C and D), as well as the fact that fluorescence emission at longer wavelengths is absorbed to a lesser extent by tissues,4 we focused on characterizing the behavior of B16F10 cells after labeling with 100 μg/mL rQDs-GSH.
To assess the benefits of NAC, B16F10 cells were cultured as previously mentioned, but this time different concentrations of NAC were added in both culture steps (Figure 2A). Afterwards, cell viability, ROS levels, viable labeled cells, and MFI associated with QDs-GSH presence in viable cells were detected by flow cytometry (Figure 2B). rQDs-GSH were incorporated by B16F10 cells and intracellular distribution was similar (Figure 2C) to that described in previous studies.8,9,23 ROS levels were lower when cells were co-incubated with rQDs-GSH in the presence of NAC (Figure 2D). Nearly 100% viable labeled B16F10 cells were obtained when incubated with 10 mM NAC (Figure 2E). Also, a general increase in MFI values after exposure to 8 and 10 mM NAC was observed (Figure 2F). Because fluorescence is proportional to the QDs-GSH concentration,8 this suggests that addition of NAC to cell cultures promotes QDs-GSH internalization, possibly by reducing ROS formation upon QDs uptake.30 Finally, NAC treatments allowed obtaining nearly 100% viable labeled cells with increased fluorescence signal and ROS levels similar to unlabeled cells.
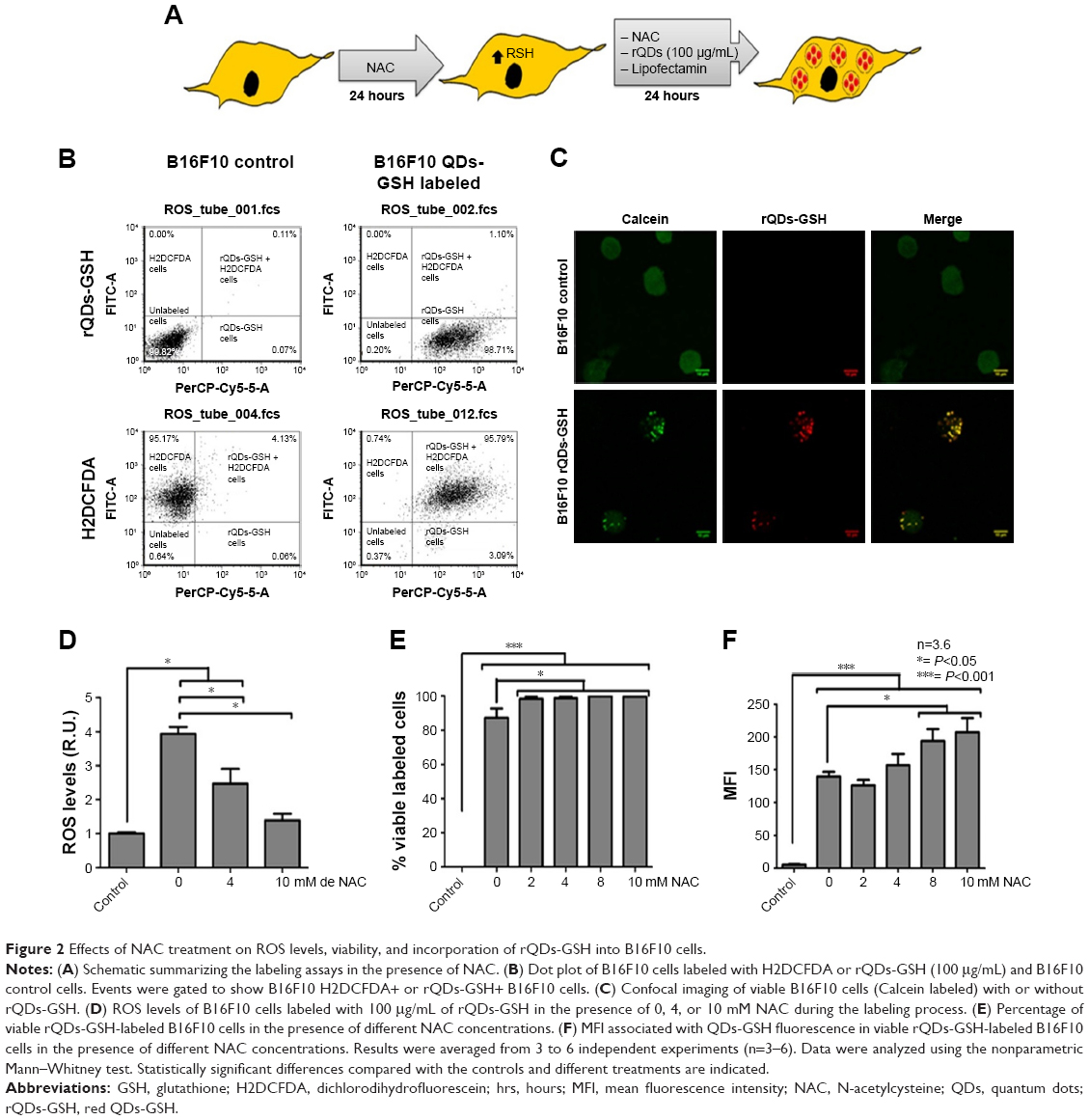 | Figure 2 Effects of NAC treatment on ROS levels, viability, and incorporation of rQDs-GSH into B16F10 cells. |
Effects of rQDs-GSH and NAC treatment on functional parameters of B16F10 cells
To compare NAC effects, cells were incubated with 100 μg/mL of rQDs-GSH in the presence of either 4 (maximum NAC concentration that did not significantly increase the MFI) or 10 mM NAC (upper limit NAC concentration used for treatment) to generate B16F10QDs-GSH-4NAC and B16F10QDs-GSH-10NAC, respectively. Subsequently, cells were evaluated in vitro in migration and invasion assays.
Migration was 2.8 times higher for B16F10QDs-GSH-4NAC than control cells (Figure 3A and B). On the other hand, no differences in migration were observed when B16F10QDs-GSH-10NAC cells were compared with control cells. This suggests that B16F10 cells with internalized rQDs-GSH increase migration at lower NAC concentrations, likely as the consequence of increased intracellular ROS levels (Figure 2D) that are known to promote cancer cell migration.34 Therefore, NAC pretreatment permitted ameliorating undesirable effects observed following QDs-GSH internalization related to intracellular ROS levels, and allowed obtaining QDs-GSH-labeled B16F10 cells that migrated like the unlabeled cells.
 | Figure 3 Migration and invasion of B16F10 cells labeled with rQDs-GSH in the presence of NAC. |
Likewise, rQDs-GSH-labeled B16F10 cells were still able to invade in a matrigel invasion assay (Figure 3C), maintaining the labeling associated with QDs-GSHr (Figure 3C and D), but did so much less effectively. rQDs-GSH presence significantly decreased invasion for both B16F10QDs-GSH-4NAC and B16F10QDsQDs-GSH-10NAC, to 41% and 6%, respectively (Figure 3E). These results suggest that greater amounts of internalized rQDs-GSH (Figure 2F) correlate with a reduction in cell invasiveness. Thus, because augmenting intracellular ROS levels increases cell metastasis,34 our results indicate that internalization of rQDs-GSH in B16F10 cells decreases invasiveness in a ROS-independent fashion. A possible explanation might be that Cd2+ in rQDS-GSH reduces the activity of matrix metalloproteinases (MMPs), which are zinc-containing endopeptidases capable of degrading extracellular matrix (ECM) proteins.35 MMPs are exocytosed by metastatic cells and mediate tumor microenvironment changes during cancer progression, favoring metastatic cell invasion.36 B16F10 exposure to Cd+2 has been shown to reduce cell invasiveness due to upregulation of tissue inhibitor of metalloproteinase-1 (TIMP-1) expression, which is Cd+2-dependent.37 In addition, our results show that the methodology employed here permits obtaining rQDs-GSH-labeled B16F10 cells that migrate similarly to unlabeled cells, but display dramatically reduced invasiveness. These results suggest that the invasiveness of rQDs-GSH-labeled cells is reduced by a cadmium-dependent mechanism, but further experimentation beyond the scope of this study is required to confirm this possibility.
Therefore, rQDs-GSH-labeled cells can be useful for in vivo studies of metastasis steps where migration is relevant (ie, distribution of metastatic cells throughout the body and transendothelial migration). With this in mind, we envisioned that B16F10QDS-GSH-10NAC cells might be useful to monitor early steps in metastasis. To this end, viability, proliferation, and QD-clearance were evaluated for 5 days after labeling using B16F10QDS-GSH-10NAC cells.
Proliferation of B16F10QDS-GSH-10NAC and persistence of labeling
Intracellular NP content may be reduced by different mechanisms, such as cell death, cell proliferation, NP diffusion, lysosomal degradation, and/or transcytosis and exocytosis.38 For example, D-penicillamine-coated QDs (DPA-QDs) were not degraded in the lysosomal compartment during the first 24 hours post-internalization,39 but rather were rapidly exocytosed (half-life of 21 minutes). Therefore, it was important here to determine rQD-GSH persistence in B16F10 cells prior to the subsequent evaluation in in vivo metastasis assays. B16F10QDs-GSH-10NAC cells were cultured in RPMI supplemented with 10% BFS for 5 days and the total cell number (quantification of proliferation), cell viability, percentage of viable labeled cells, and MFI were determined after cell labeling (Figure 4). B16F10QDs-GSH-10NAC viability was not substantially reduced during a 5-day period post-labeling (Figure 4A), although a slight decline in viability was observed after the third day (to 79%), which then remained stable up to day 5. A significant reduction in MFI was observed from the second day onwards, which resulted in a reduction of the fluorescence intensity by 43% after 5 days (Figure 4C). The data obtained suggest that B16F10QDs-GSH-10NAC cells remain viable with a stable percentage of viable labeled cells for at least 3 days (Figure 4B), and that the MFI does not vary significantly until the second-day post-labeling. Thus, the labeling methodology described here employing biomimetic QDs-GSH allows for more prolonged cell labeling than other QDs-based methodologies described in the literature (on the order of minutes).39 Interestingly, B16F10QDs-GSH-10NAC cells appear unable to proliferate, since no increase in the total number of cells was observed during the 5 days of the assay (Figure 4D). For rQDs-GSH-labeled B16F10 cells, in the absence of NAC treatments (lower case labeling in Figure 2), the total number of cells was found to increase 24 hours post-labeling in a manner similar to unlabeled cells (Figure S3D), whereby viability, percentage of labeled cells, and MFI were maintained (Figure S3A–C). These results are in agreement with previous publications indicating that NP internalization may affect cellular proliferation40 and that Cd2+ inhibits cell proliferation in a dose-dependent manner.41,42 Thus, the methodology described here has the potential to be used as a cell-labeling strategy to study early steps in vivo involved in accumulation/distribution of metastatic cells in distant organs.
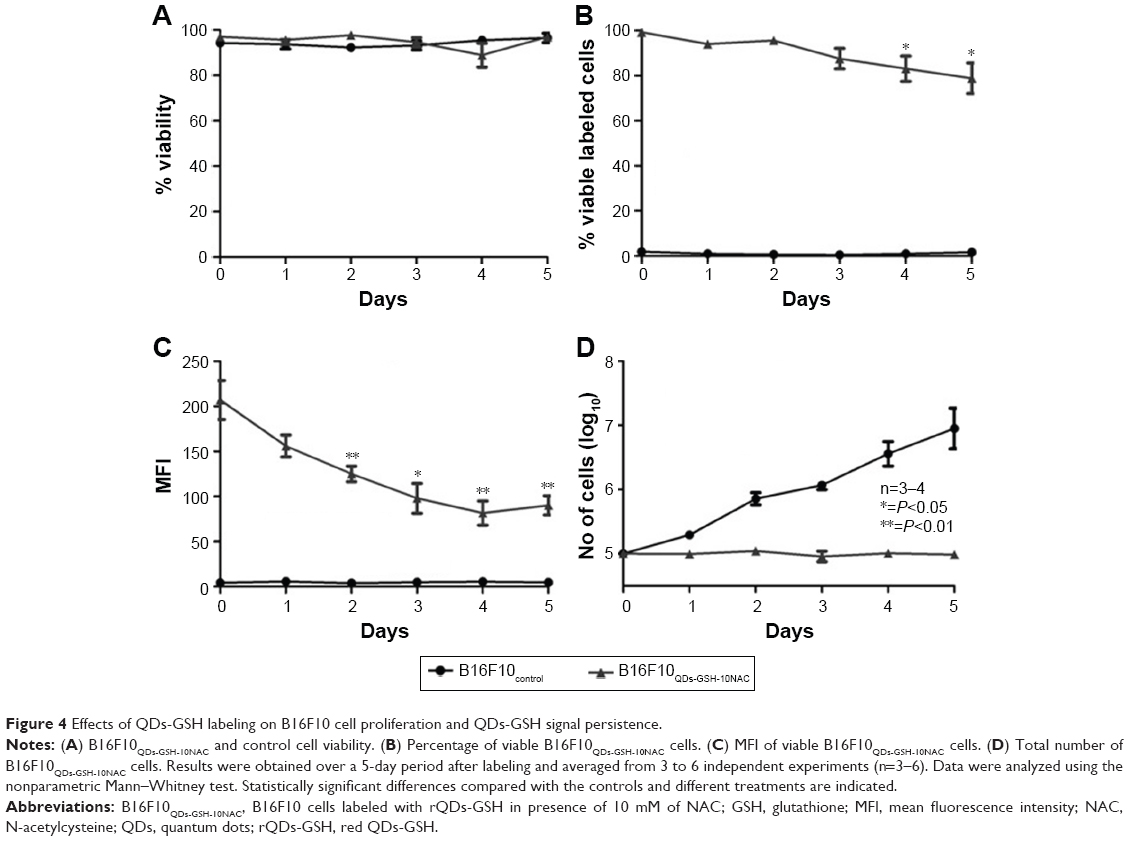 | Figure 4 Effects of QDs-GSH labeling on B16F10 cell proliferation and QDs-GSH signal persistence. |
In vivo metastasis assays
For the in vivo studies, we employed a syngeneic metastasis model in C57BL/6 mice with an intact immune system. B16F10QDs-GSH-10NAC cells were injected into the tail vein of mice and 21 days post-injection mouse survival and tumor mass in the lungs were determined (Figure 5A). Administration of B16F10QDs-GSH-10NAC cells did not alter mouse survival (Figure 5B). After necropsy, metastasis remained undetectable in over 90% of the B16F10QDs-GSH-10NAC treated mice; lung tumor mass in the only mouse with metastasis was equivalent to only 1.4% of total lung mass. To the contrary, metastatic lung nodules were detected in all control mice injected with the same number of B16F10 cells (Figure 5C) and accounted for 14.5% of lung mass, similar to what we have described in our previous publications.25,28 These results show that incorporating QDs into B16F10 cells, as is the case for B16F10QDs-GSH-10NAC, significantly reduces the ability of these cells to form metastatic nodules in vivo 21 days post-injection. This is in agreement with the in vitro data showing that labeled cells neither invade nor proliferate (Figures 3E and 4D). On the other hand, the use of B16F10 cells labeled with CdSe/ZnS QDs capped with dihydrolipoic acid (DHLA) was reported using lipofectamine-aided transfection for internalization of QDs-DHLA. However, in those experiments, the authors observed that B16F10 cells formed metastatic nodules 40 days post-injection.12 These differences may be attributable to variations in the amount of QDs-GSH present and Cd2+ release inside B16F10QDs-GSH-10NAC cells. Thus, using our methodology, QDs-GSH-labeled cells were obtained that migrated similarly to unlabeled cells, but were unable to invade and proliferate.
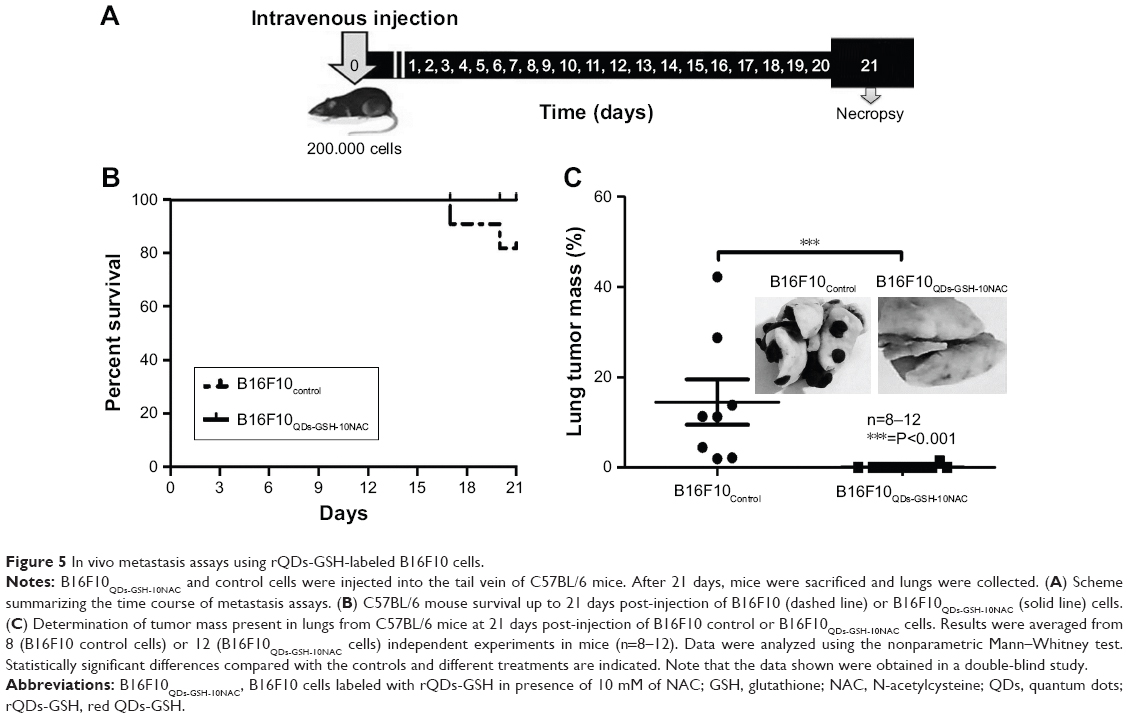 | Figure 5 In vivo metastasis assays using rQDs-GSH-labeled B16F10 cells. |
Over the last 30 years, the repertoire of compounds containing transition metals with anti-tumor properties has expanded to include compounds containing Zn, Co, Ni, Sn, Ag, and Cd.43–49 Among these elements, cadmium has been ascribed elevated anti-tumor effects in comparison with other transition metals.43 In 2016, Sreekanth et al reported that cadmium oxide (CdO) NPs inhibited the proliferation of normal and tumor cell lines, whereby the tumor cells were more severely affected by CdO NPs.44 Our study shows that QDs-GSH have potent anti-tumor effects, as evidenced by the dramatic inhibition of cell proliferation and invasion following internalization of QDs-GSH. Moreover, the ability of such QD-GSH-labeled cells to form metastatic nodules in C57BL/6 mice was ablated within the time frame analyzed. Data from the literature favor the notion that these anti-metastatic effects are related to the presence of cadmium in the QDs-GSH core. Our results highlight the need to further investigate the anti-tumor effects of QDs-GSH, bearing in mind that selective tumor accumulation should permit using these QDs-GSH both as diagnostic agents and in anti-tumor therapy (theranostics).
Here, however, we focused on evaluating the utility of B16F10QDs-GSH-10NAC in metastatic cells tracking at early stages post-injection, since they were able to migrate, but did not invade or proliferate.
In vivo distribution of B16F10QDs-GSH-10NAC cells
Previously, studies have shown that QDs can be followed by fluorescence microscopy in vitro and also to track interactions between different cell populations in vivo.12,50 For instance, this approach has permitted visualizing the clogging of blood vessels produced by B16-BL6 melanoma cells during metastasis.45 However, it was still not possible to track the accumulation or distribution of metastatic cells by in vivo fluorescence imaging.
To assess the potential utility of B16F10QDs-GSH-10NAC cells in recording early steps of metastasis by in vivo fluorescence imaging, cells were injected (Figure 6A and B) into the tail vein of C57BL/6 mice and whole animal images were obtained using the IVIS. After injection of the cells into mice, the fluorescence signal was not detectable (Figure S4), likely because a number of biological molecules, including hemoglobin, absorb light between 400 and 600 nm, thereby interfering with the B16F10QDs-GSH-10NAC signal. Thus, animals were sacrificed and after necropsy at the indicated time points, organs were placed in Fekete’s solution to eliminate blood traces and then fixed as described.46 Examples of lungs from C57BL/6 mice obtained after 6 hours inoculation with B16F10QDs-GSH-10NAC cells are shown (Figure 6C). Fluorescence due to B16F10QDs-GSH-10NAC presence in lungs was clearly distinguishable from the background fluorescence of controls (Figure 6D), suggesting the presence of B16F10QDs-GSH-10NAC in the lungs of C57BL/6 mice.
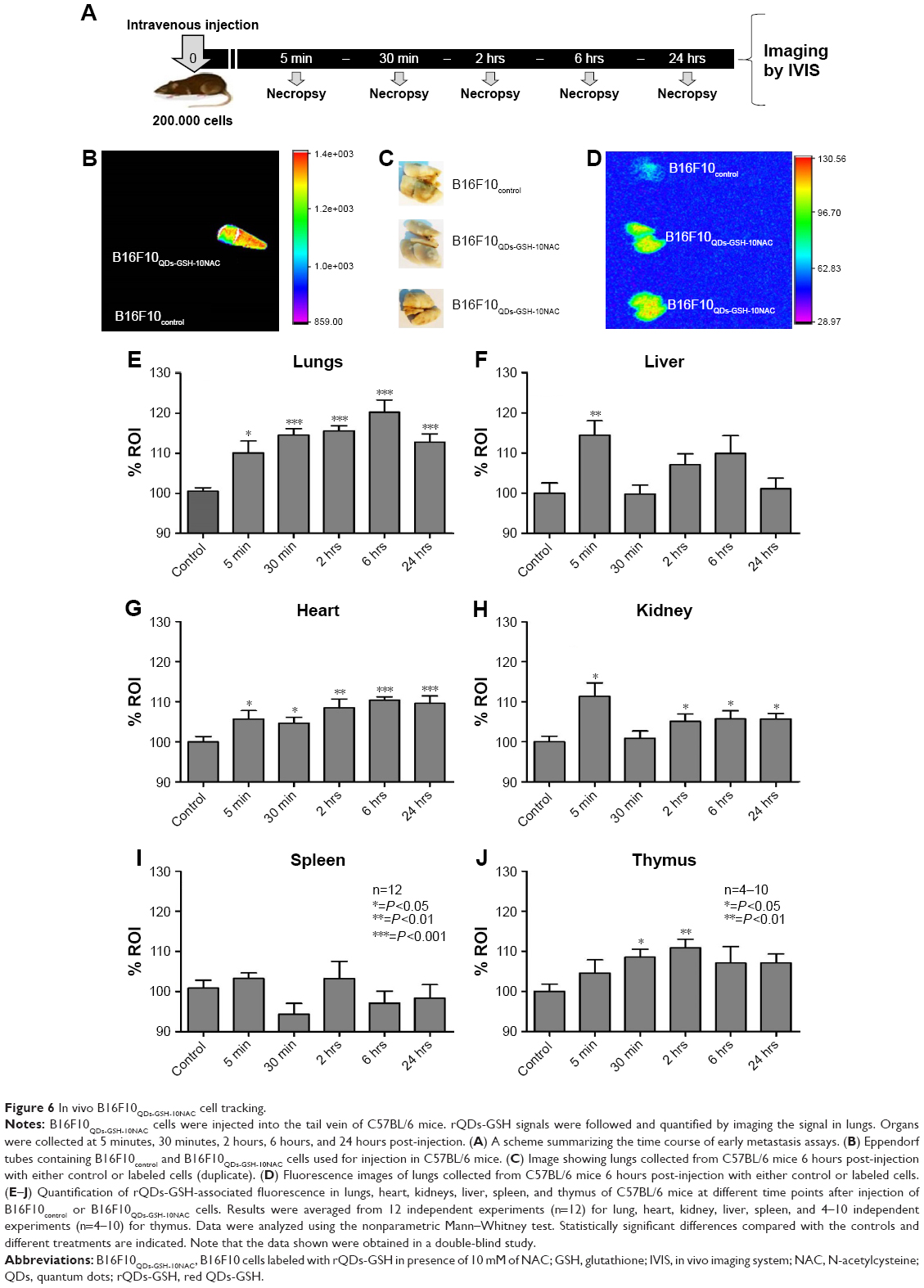 | Figure 6 In vivo B16F10QDs-GSH-10NAC cell tracking. |
Using this approach, we determined the kinetics of B16F10 accumulation, particularly in the lungs of C57BL/6 mice. To this end, 2×105 B16F10QDs-GSH-10NAC cells were injected into the tail vein of mice and then the following organs were analyzed: 1) lungs (metastatic cell target); 2) spleen, liver, and kidneys (principal QD storage organs post-intravenous injection) and; 3) heart and thymus (organs where no extensive accumulation of QDs has been previously described).47,48 Mice were sacrificed and organs were harvested at 5 minutes, 30 minutes, 2 hours, 6 hours, and 24 hours post-injection, and the fluorescence was detected by direct imaging of the organs (Figure 6E–J). The greatest difference was observed in lungs harvested 6 hours after B16F10QDs-GSH-10NAC injection (Figure 6E). Although a significant increase in fluorescence was detectable as soon as 5 minutes post-injection, the intensity increased until reaching a maximum 6 hours post-injection. B16F10 cells preferentially metastasize to the lungs of C57BL/6 mice, possibly due to their preference for laminin and/or fibronectin, two ECM proteins present surrounding small vessels of the lung, in the basal membrane and alveolar epithelium.28 These data correlate with previous studies, using QDs and fluorescence microscopy, indicating that 5 minutes after administration of B16-BL6 cells into the blood stream, these cells accumulated in small lung vessels.45
Several studies have shown that the majority of QDs present in the blood stream of mice rapidly accumulate in reticuloendothelial system organs (liver and spleen) and are excluded from heart and thymus.51–54 Nevertheless, no significant fluorescence increase in the spleen was detected when B16F10QDs-GSH-10NAC cells were injected (Figure 6I), while fluorescence accumulation in the thymus was detected between 30 minutes and 2 hours post-injection (Figure 6J). These results suggest that rQDs-GSH remain confined within B16F10 cells and are in agreement with a previous report by Voura et al, showing that QDs were not released from QDs-DHLA-labeled B16F10 cells.12
The thymus is a lymphatic, endocrine, and immune organ where T cells mature and are released to the circulation.53 The cytotoxic T-cells, as well as natural killer T-cells, specifically participate in the early clearance of tumor cells from the blood stream.54,55 Given that the blood–thymus barrier prevents direct contact between the thymus and components of the blood stream,56,57 the observed pronounced accumulation of fluorescence in the thymus suggests that B16F10QDs-GSH-10NAC are being rapidly recognized by T cells.
Our results indicated that B16F10QDs-GSH-10NAC cells were rapidly distributed (5 minutes) to the heart, liver, kidneys, and lungs of C57BL/6 mice and likely accumulated there by an embolic arrest process.24 However, despite the initial generic distribution, fluorescence intensity decreases in the other organs and a selective enrichment of B16F10QDs-GSH-10NAC cells in lungs is observed within the first few hours post-injection. This suggests that B16F10QDs-GSH-10NAC cells maintain the “homing” reported previously,24,25,45 and thus have the potential to be used as a tool for assessing early accumulation/distribution of metastatic cells.
To confirm the presence of B16F10QDs-GSH-10NAC cells in the lungs of C57BL/6 mice, lung sections obtained 6 hours post-injection of cells into the tail vein of C57BL/6 mice were analyzed by immunohistochemistry (Figure 7). Fluorescence due to B16F10QDs-GSH-10NAC presence was observed in histological slices from mice and was clearly distinguishable from the background fluorescence of controls (Figure 7A), confirming the presence of B16F10QDs-GSH-10NAC cells in the lungs of C57BL/6 mice. During colonization of distant organs, the extravasation process is crucial.58 Given that B16F10 cells specifically target the lung in metastasis assays, it was important to determine whether B16F10QDs-GSH-10NAC cells were capable of transmigrating through the vascular endothelium and colonizing the lungs in vivo. With this in mind, B16F10QDs-GSH-10NAC and Calcein AM-labeled B16F10 (B16F10Calcein) cells were compared by histological analysis of lung sections 6 and 24 hours post-injection of cells into the tail vein of C57BL/6 mice. Specifically, we determined the fluorescence associated with B16F10QDs-GSH-10NAC and B16F10Calcein cells in the endothelial vasculature, alveolae, and bronchi (Figure S5A and B). B16F10QDs-GSH-10NAC and B16F10Calcein cells were mostly found in the perialveolar area, after 6 and 24 hours (Figure 7B and C). In addition, the number of cells present in those areas increased after 24 hours (Figure 7C), suggesting that the B16F10QDs-GSH-10NAC cells transmigrated to the same extent as B16F10Calcein cells. These results are consistent with previously published studies indicating that QDs-DHLA-labeled B16F10 cells transmigrate in a manner similar to non-labeled B16F10 cells. However, the mechanism by which transmigration of QDs-labeled cells occurs had not been elucidated yet.
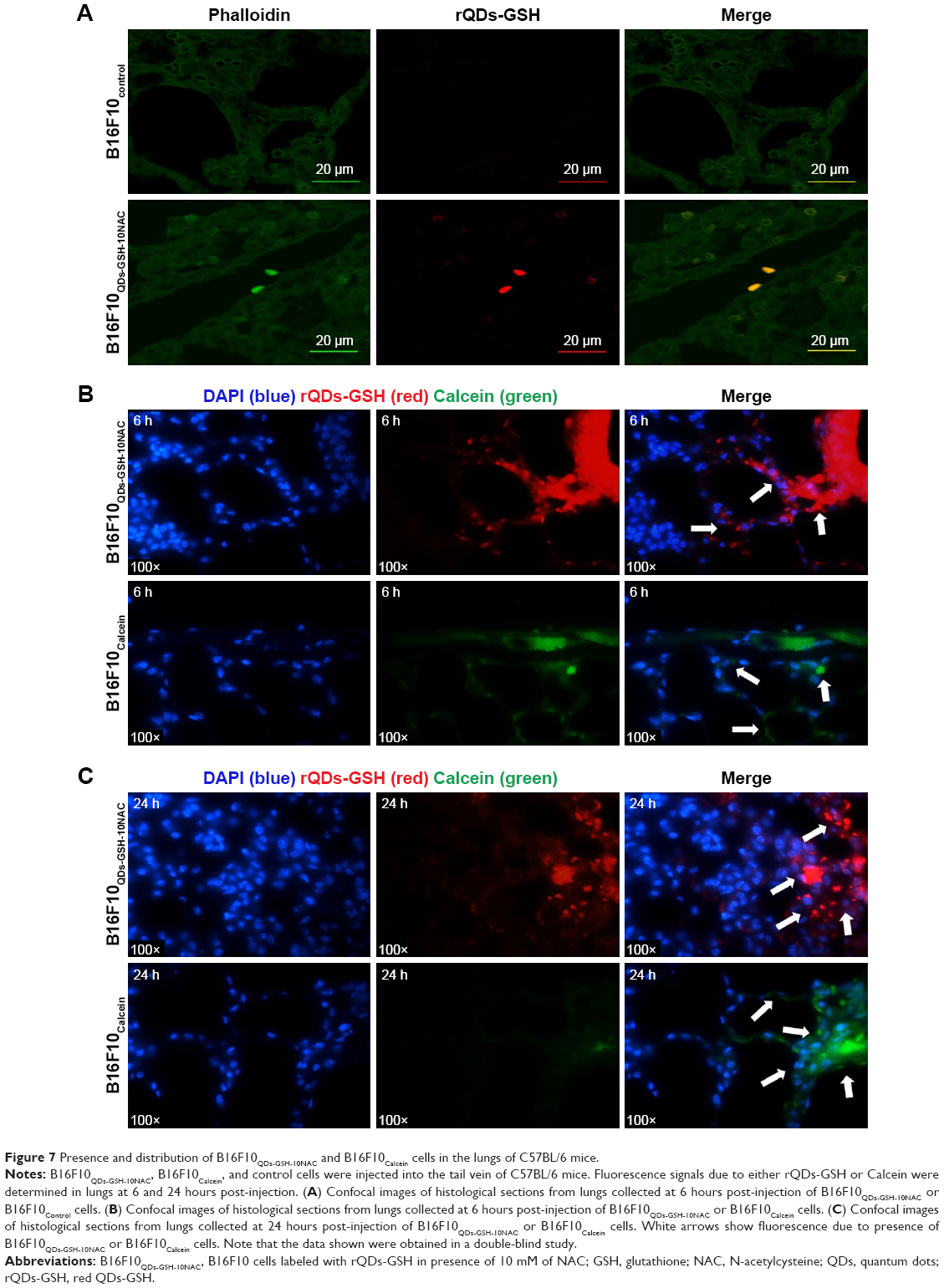 | Figure 7 Presence and distribution of B16F10QDs-GSH-10NAC and B16F10Calcein cells in the lungs of C57BL/6 mice. |
In this context, it is worth noting that transmigration can occur by at least three different processes or combinations of thereof: 1) mesenchymal cell invasion (protease dependent), 2) collective cell invasion (protease dependent), and 3) amoeboid cell invasion (protease independent).58 Melanoma cells can invade using all three mechanisms and/or may switch from protease-dependent to amoeboid cell invasion.58,59 Amoeboid invasion may explain why B16F10QDs-GSH-10NAC cells, that are non-invasive in a protease-dependent invasion assay (Figure 3C and E), display the same in vivo transmigration ability as B16F10Calcein control cells (Figure 7B and C).
As shown in Figure S6, B16F10QDs-GSH-10NAC cells (Figure S6A, Quadrant LR) and B16F10Calcein cells (Figure S6B, Quadrant UL) exhibit similar MFI. Therefore, a similar intensity would be expected when these cells are present in organs. A decrease in fluorescence intensity was observed between 6 and 24 hours for rQDs-GSH and Calcein-labeled cells (Figure S6C). This result could explain the decrease in fluorescence observed in lungs after 24 hours (Figure 6E) and indicates that B16F10QDs-GSH-10NAC cells are still present in the lungs at this time but with decreased fluorescence intensity. Also, these results indicate that 6 hours post-injection represents the optimal time point for the tracking of organ-specific B16F10 cell accumulation and permits avoiding the possible loss of signal due to decreases in B16F10 cell fluorescence during the tracking process.
Of note, the fluorescence intensity due to rQDs-GSH was significantly higher than that associated with Calcein after 24 hours (Figure S6C), suggesting that the rQDs-GSH labeling is more stable over time than fluorescence labeling using Calcein.
Finally, these results demonstrate that our methodology of QDs-GSH-labeling cells allows obtaining nearly 100% viable labeled cells with increased fluorescence signal. In addition, these cells can migrate, transmigrate, and maintain their “homing,” similar to the unlabeled B16F10 cells. Thus, B16F10QDs-GSH-10NAC cells can be used to track the early distribution/accumulation of B16F10 cells in C57BL/6 organs by in vivo fluorescence imaging, without the need for fluorescence microscopy and, in doing so, monitor early steps involved in the colonization of distant organs.
Proof of concept
According to Sahai 2007, “the development of imaging studies can be important for preclinical evaluation of drugs that target steps in the metastatic process.”4 Our results showed that B16F10QDs-GSH-10NAC cells can be used to track in vivo migration and the accumulation/distribution of metastatic cells in C57BL/6 mice independently of the invasion process. We believe that this model has the potential to be used as a tool to determine more specifically how expression of different proteins in these cells and/or the application of drugs affect migration, accumulation, and/or distribution of B16F10QDs-GSH-10NAC cells in C57BL/6 mice.
CAV-1 is an integral membrane protein, which has been shown to increase the migration, invasion, and metastasis of B16F10 cells.25,60 With this in mind, we performed a proof-of-concept experiment, using B16F10 cells stably transfected with either the empty pLacIOP vector [B16F10(Mock) cells] or the vector containing the CAV1-encoding insert [B16F10(CAV-1) cells] that were then labeled with rQDs-GSH as described in Figure 8A (B16F10QDs-GSH-10NAC). For the sake of simplicity, such labeled cells are referred to here as B16F10(Mock) and B16F10(CAV1) cells. Following labeling, we found that overall viability (Figure 8B), as well as the viability of labeled cells (Figure 8C), as determined by flow cytometry, were similar for B16F10(Mock) and B16F10(CAV1) cells. Moreover, the MFI was also similar (Figure 8D) for both types of cells as determined by flow cytometry. These results indicate that overexpression of CAV-1 in B16F10 cells neither altered the labeling with rQDs-GSH nor the viability of labeled B16F10 cells. B16F10 cells do not express PTRF/Cavin-1,28,61 and also lack the presence of caveolae-like structures at the cell surface. Moreover, immunofluorescence studies have shown that the large majority of the CAV1 protein in B16F10(CAV1) cells is intracellular, predominantly in a perinuclear location,28 consistent with the notion that the presence of CAV1 in B16F10(CAV1) cells does not modulate QDs-GSH internalization.
 | Figure 8 Proof-of-concept experiment to evaluate the effect of CAV1 expression in labeled B16F10 cells following injection into C57BL/6 mice. |
Subsequently, 2×105 viable and rQDs-GSH-labeled B16F10(Mock) or B16F10(CAV-1) cells were injected into the tail vein of C57BL/6 mice, and 6 hours post-injection lungs from those recipient mice were collected for ex vivo fluorescence tracking using IVIS. As shown in Figure 8E, fluorescence intensity observed in lungs was greater for mice injected with B16F10(CAV-1) cells than those that received B16F10(Mock) cells. This result suggests that enhanced expression of CAV-1 in B16F10 melanoma cells promotes experimental lung metastasis described previously by our group by enhancing the accumulation of B16F10 cells in the lungs of recipient mice. In doing so, these experiments validate the potential of using rQDs-GSH-labeled cells to monitor early steps in metastasis.
Conclusion
In the present study, we describe for the first time the effects of biomimetic QDs-GSH incorporation on the viability and metastatic behavior of B16F10 cells. Also, we define a novel methodology that permits ameliorating detrimental consequences of QDs-GSH uptake. QDs-GSH’ incorporation augments ROS and cell migration in B16F10 cells; however, these effects can be corrected by NAC treatment. In addition, NAC treatment allows increasing significantly the viability and fluorescence intensity of rQDs-GSH-labeled cells. However, proliferation and invasiveness of B16F10 cells, as well as the formation of metastatic lung nodules, were all attenuated by QDs-GSH internalization, probably as a consequence of cadmium present in the QDs-GSH core. Moreover, we show that QDs-GSH-labeled B16F10 cells can be used to track the in vivo migration and early distribution/accumulation of these cells in C57BL/6 organs. Our study reveals that these cells preferentially accumulated in lungs of C57BL/6 mice as early as 5 minutes post-injection. Thus, although QDs-GSH internalization blocks the ability of B16F10 cells to form nodules in the lung, QDs-GSH-labeled B16F10 cells can be successfully tracked during the early steps involved in the colonization of distant organs.
Finally, our study provides a cell-labeling methodology that should be useful for characterizing the early steps of tissue-specific accumulation and distribution of B16F10 cells in C57BL/6 mice, as well as help to evaluate the effects of drugs and/or different biological factors (cytokines, signaling proteins, etc.) on these processes and, in doing so, improve our understanding of these events. With this in mind, we propose that QDs-GSH can be expected to have considerable potential for future use in cancer theranostics.
Acknowledgments
The authors appreciate the support of Cellular Communication Laboratory, Faculty of Medicine, Universidad de Chile; The BioNanotechnology and Microbiology Laboratory, Faculty of Biological Sciences, Universidad Andres Bello; The Department of Pharmacological and Toxicological Chemistry, Faculty of Chemical and Pharmaceutical Sciences, Universidad de Chile and Fundación Ciencia y Vida, Santiago, Chile. The authors also appreciate the personal support of the Díaz-García family (VMDG) and Erika Elcira Donoso Lopez (JMPD). The study was funded by CONICYT-FONDAP 15130011 (AQ), FONDECYT 1130250, 1170925 (AQ), Anillo ACT 1111 (AQ, JMPD), FONDECYT 1151255 (JMPD), INACH RT-25–16 (JMPD), AFOSR FA9550-15-1-0140 (JMPD), and Basal de FCV PFB-16 (LLG). A post-doctoral CONICYT award 3140463 (SG) and a CONICYT PhD fellowship 21120816 (VMDG) are also acknowledged.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. | ||
Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–292. | ||
Sahai E. Illuminating the metastatic process. Nat Rev Cancer. 2007;7(10):737–749. | ||
Chinen AB, Guan CM, Ferrer JR, Barnaby SN, Merkel TJ, Mirkin CA. Nanoparticle Probes for the Detection of Cancer Biomarkers, Cells, and Tissues by Fluorescence. Chem Rev. 2015;115(19):10530–10574. | ||
Fang M, Peng CW, Pang DW, Li Y. Quantum dots for cancer research: current status, remaining issues, and future perspectives. Cancer Biol Med. 2012;9(3):151–163. | ||
Chen C, Peng J, Sun SR, Peng CW, Li Y, Pang DW. Tapping the potential of quantum dots for personalized oncology: current status and future perspectives. Nanomedicine. 2012;7(3):411–428. | ||
Pérez-Donoso JM, Monrás JP, Bravo D, et al. Biomimetic, mild chemical synthesis of CdTe-GSH quantum dots with improved biocompatibility. PLoS One. 2012;7(1):e30741. | ||
Díaz V, Ramírez-Maureira M, Monrás JP, et al. Spectroscopic properties and biocompatibility studies of CdTe Quantum Dots capped with biological thiols. Sci Adv Mater. 2012;4(5):609–616. | ||
Zheng Y, Gao S, Ying JY. Synthesis and Cell-Imaging Applications of Glutathione-Capped CdTe Quantum Dots. Adv Mater. 2007;19(3):376–380. | ||
Rosenblum LT, Kosaka N, Mitsunaga M, Choyke PL, Kobayashi H. In vivo molecular imaging using nanomaterials: general in vivo characteristics of nano-sized reagents and applications for cancer diagnosis. Mol Membr Biol. 2010;27(7):274–285. | ||
Voura EB, Jaiswal JK, Mattoussi H, Simon SM. Tracking metastatic tumor cell extravasation with quantum dot nanocrystals and fluorescence emission-scanning microscopy. Nat Med. 2004;10(9):993–998. | ||
Derfus AM, Chan WCW, Bhatia SN. Probing the Cytotoxicity Of Semiconductor Quantum Dots. Nano Lett. 2004;4(1):11–18. | ||
Hardman R. A toxicologic review of quantum dots: toxicity depends on physicochemical and environmental factors. Environ Health Perspect. 2006;114(2):165–172. | ||
Rzigalinski BA, Strobl JS. Cadmium-containing nanoparticles: perspectives on pharmacology and toxicology of quantum dots. Toxicol Appl Pharmacol. 2009;238(3):280–288. | ||
Ochi T, Otsuka F, Takahashi K, Ohsawa M. Glutathione and metallothioneins as cellular defense against cadmium toxicity in cultured Chinese hamster cells. Chem Biol Interact. 1988;65(1):1–14. | ||
Anderson ME. Glutathione: an overview of biosynthesis and modulation. Chem Biol Interact. 1998;111–112:1–14. | ||
Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12(10):1161–1208. | ||
Peng L, He M, Chen B, Qiao Y, Hu B. Metallomics Study of CdSe/ZnS Quantum Dots in HepG2 Cells. ACS Nano. 2015;9(10):10324–10334. | ||
Loo G. Redox-sensitive mechanisms of phytochemical-mediated inhibition of cancer cell proliferation (review). J Nutr Biochem. 2003;14(2):64–73. | ||
Xu G, Zeng S, Zhang B, Swihart MT, Yong KT, Prasad PN. New Generation Cadmium-Free Quantum Dots for Biophotonics and Nanomedicine. Chem Rev. 2016;116(19):12234–12327. | ||
Pérez-Donoso JM, Monrás JP. Osorio-Roman IO, Vásquez CC; Universidad de Santiago de Chile. Synthesis of Highly Fluorescent GSH-CdTe Nanoparticles (Quantum Dots). United States patent US20130284979. 2017 Aug 15. | ||
Gautier JL, Monrás JP, Osorio-Román IO, et al. Surface characterization of GSH-CdTe quantum dots. Mater Chem Phys. 2013;140(1):113–118. | ||
Fidler IJ. Biological behavior of malignant melanoma cells correlated to their survival in vivo. Cancer Res. 1975;35(1):218–224. | ||
Lobos-González L, Aguilar L, Diaz J, et al. E-cadherin determines Caveolin-1 tumor suppression or metastasis enhancing function in melanoma cells. Pigment Cell Melanoma Res. 2013;26(4):555–570. | ||
Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82(1):70–77. | ||
Riener CK, Kada G, Gruber HJ. Quick measurement of protein sulfhydryls with Ellman’s reagent and with 4,4′-dithiodipyridine. Anal Bioanal Chem. 2002;373(4–5):266–276. | ||
Ortiz R, Díaz J, Díaz N, et al. Extracellular matrix-specific Caveolin-1 phosphorylation on tyrosine 14 is linked to augmented melanoma metastasis but not tumorigenesis. Oncotarget. 2016;7(26):40571–40593. | ||
Yildirimer L, Thanh NT, Loizidou M, Seifalian AM. Toxicology and clinical potential of nanoparticles. Nano Today. 2011;6(6):585–607. | ||
Lovrić J, Bazzi HS, Cuie Y, Fortin GR, Winnik FM, Maysinger D. Differences in subcellular distribution and toxicity of green and red emitting CdTe quantum dots. J Mol Med. 2005;83(5):377–385. | ||
Schaeublin NM, Braydich-Stolle LK, Schrand AM, et al. Surface charge of gold nanoparticles mediates mechanism of toxicity. Nanoscale. 2011;3(2):410–420. | ||
Panté N, Kann M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol Biol Cell. 2002;13(2):425–434. | ||
Catalan-Figueroa J, Palma-Florez S, Alvarez G, Fritz HF, Jara MO, Morales JO. Nanomedicine and nanotoxicology: the pros and cons for neurodegeneration and brain cancer. Nanomedicine (Lond). 2016;11(2):171–187. | ||
Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25(4):695–705. | ||
Verma RP, Hansch C. Matrix metalloproteinases MMPs: chemical-biological functions and (Q)SARs. Bioorg Med Chem. 2007;15(6):2223–2268. | ||
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. | ||
Khokha R, Zimmer MJ, Graham CH, Lala PK, Waterhouse P. Suppression of invasion by inducible expression of tissue inhibitor of metalloproteinase-1 (TIMP-1) in B16-F10 melanoma cells. J Natl Cancer Inst. 1992;84(13):1017–1022. | ||
Fröhlich E. Cellular elimination of nanoparticles. Environ Toxicol Pharmacol. 2016;46:90–94. | ||
Jiang X, Röcker C, Hafner M, Brandholt S, Dörlich RM, Nienhaus GU. Endo- and exocytosis of zwitterionic quantum dot nanoparticles by live HeLa cells. ACS Nano. 2010;4(11):6787–6797. | ||
Mahmoudi M, Azadmanesh K, Shokrgozar MA, Journeay WS, Laurent S. Effect of nanoparticles on the cell life cycle. Chem Rev. 2011;111(5):3407–3432. | ||
Yang PM, Chiu SJ, Lin KA, Lin LY. Effect of cadmium on cell cycle progression in Chinese hamster ovary cells. Chem Biol Interact. 2004;149(2–3):125–136. | ||
Bertin G, Averbeck D. Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review). Biochimie. 2006;88(11):1549–1559. | ||
Kareem A, Zafar H, Sherwani A, Mohammad O, Khan TA. Synthesis, characterization and in vitro anticancer activity of 18-membered octaazamacrocyclic complexes of Co(II), Ni(II), Cd(II) and Sn(II). J Mol Struct. 2014;1075:17–25. | ||
Sreekanth TV, Pandurangan M, Dillip GR, Kim DH, Lee YR. Toxicity and efficacy of CdO nanostructures on the MDCK and Caki-2 cells. J Photochem Photobiol B. 2016;164:174–181. | ||
Saitoh Y, Terada N, Ohno N, et al. Imaging of thrombosis and microcirculation in mouse lungs of initial melanoma metastasis with in vivo cryotechnique. Microvasc Res. 2014;91:73–83. | ||
Wexler H. Accurate identification of experimental pulmonary metastases. J Natl Cancer Inst. 1966;36(4):641–645. | ||
Choi HS, Liu W, Misra P, et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25(10):1165–1170. | ||
Ye L, Yong KT, Liu L, et al. A pilot study in non-human primates shows no adverse response to intravenous injection of quantum dots. Nat Nanotechnol. 2012;7(7):453–458. | ||
Schipper ML, Cheng Z, Lee SW, et al. MicroPET-based biodistribution of quantum dots in living mice. J Nucl Med. 2007;48(9):1511–1518. | ||
Smith AM, Duan H, Mohs AM, Nie S. Bioconjugated quantum dots for in vivo molecular and cellular imaging. Adv Drug Deliv Rev. 2008;60(11):1226–1240. | ||
Schipper ML, Iyer G, Koh AL, et al. Particle size, surface coating, and PEGylation influence the biodistribution of quantum dots in living mice. Small. 2009;5(1):126–134. | ||
Haque MM, Im H-Y, Seo J-E, Hasan M, Woo K, Kwon O-S. Effects of folic acid and polyethylene glycol coated quantum dots on toxicity and tissue uptake to precision-cut spleen slices of rats. J Pharm Investig. 2013;43(5):375–383. | ||
Miller JF. The discovery of thymus function and of thymus-derived lymphocytes. Immunol Rev. 2002;185(1):7–14. | ||
Andersen MH, Pedersen LO, Capeller B, Bröcker EB, Becker JC, thor Straten P. Spontaneous cytotoxic T-cell responses against survivin-derived MHC class I-restricted T-cell epitopes in situ as well as ex vivo in cancer patients. Cancer Res. 2001;61(16):5964–5968. | ||
Whiteside TL, Herberman RB. The role of natural killer cells in immune surveillance of cancer. Curr Opin Immunol. 1995;7(5):704–710. | ||
Kendall MD. Functional anatomy of the thymic microenvironment. J Anat. 1991;177(1):1–29. | ||
Nishino M, Ashiku SK, Kocher ON, Thurer RL, Boiselle PM, Hatabu H. The thymus: a comprehensive review. Radiographics. 2006;26(2):335–348. | ||
van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728(1–2):23–34. | ||
Hegerfeldt Y, Tusch M, Bröcker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002;62(7):2125–2130. | ||
Diaz-Valdivia N, Bravo D, Huerta H, et al. Enhanced caveolin-1 expression increases migration, anchorage-independent growth and invasion of endometrial adenocarcinoma cells. BMC Cancer. 2015;15(1):463. | ||
Hill MM, Daud NH, Aung CS, et al. Co-regulation of cell polarization and migration by caveolar proteins PTRF/Cavin-1 and caveolin-1. PLoS One. 2012;7(8):e43041. |
Supplementary materials
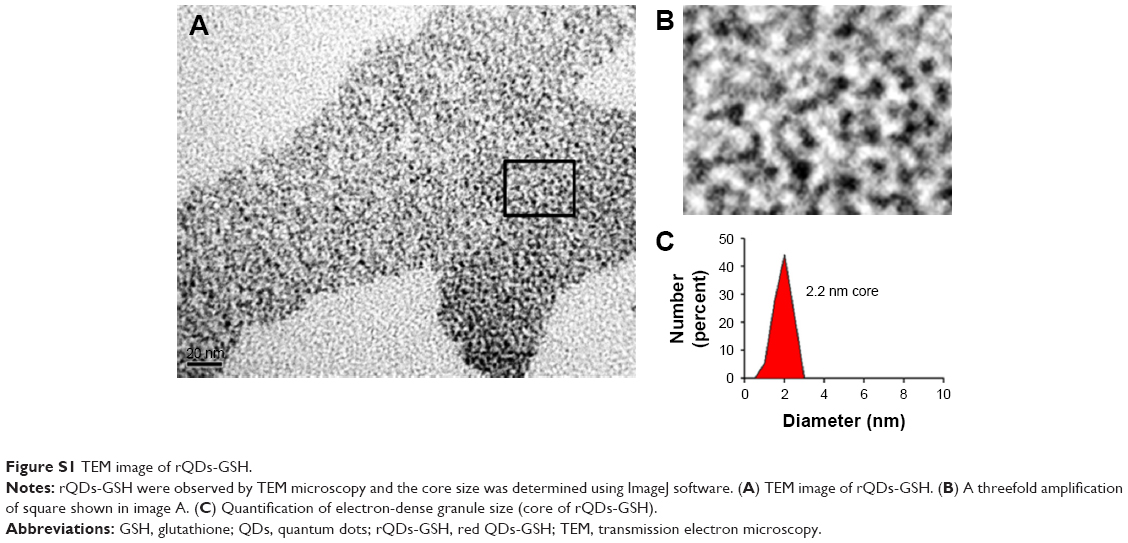 | Figure S1 TEM image of rQDs-GSH. |
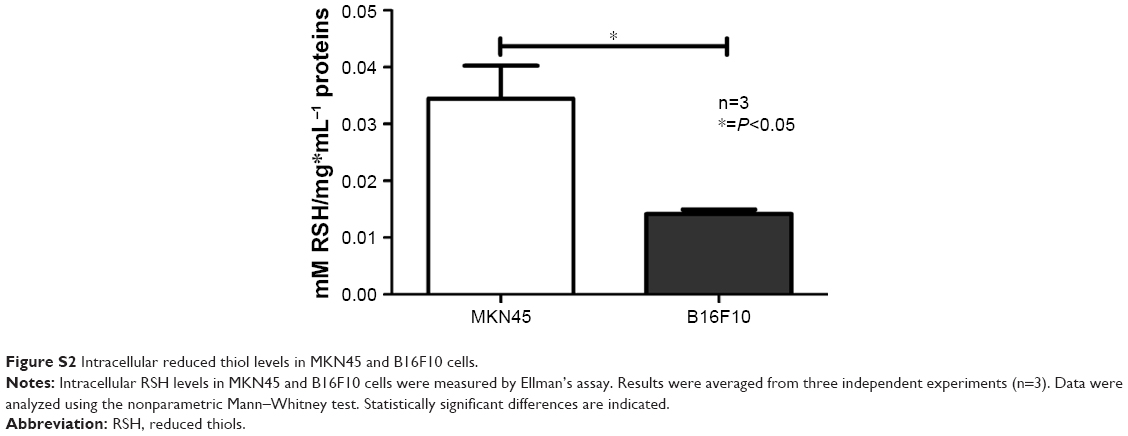 | Figure S2 Intracellular reduced thiol levels in MKN45 and B16F10 cells. |
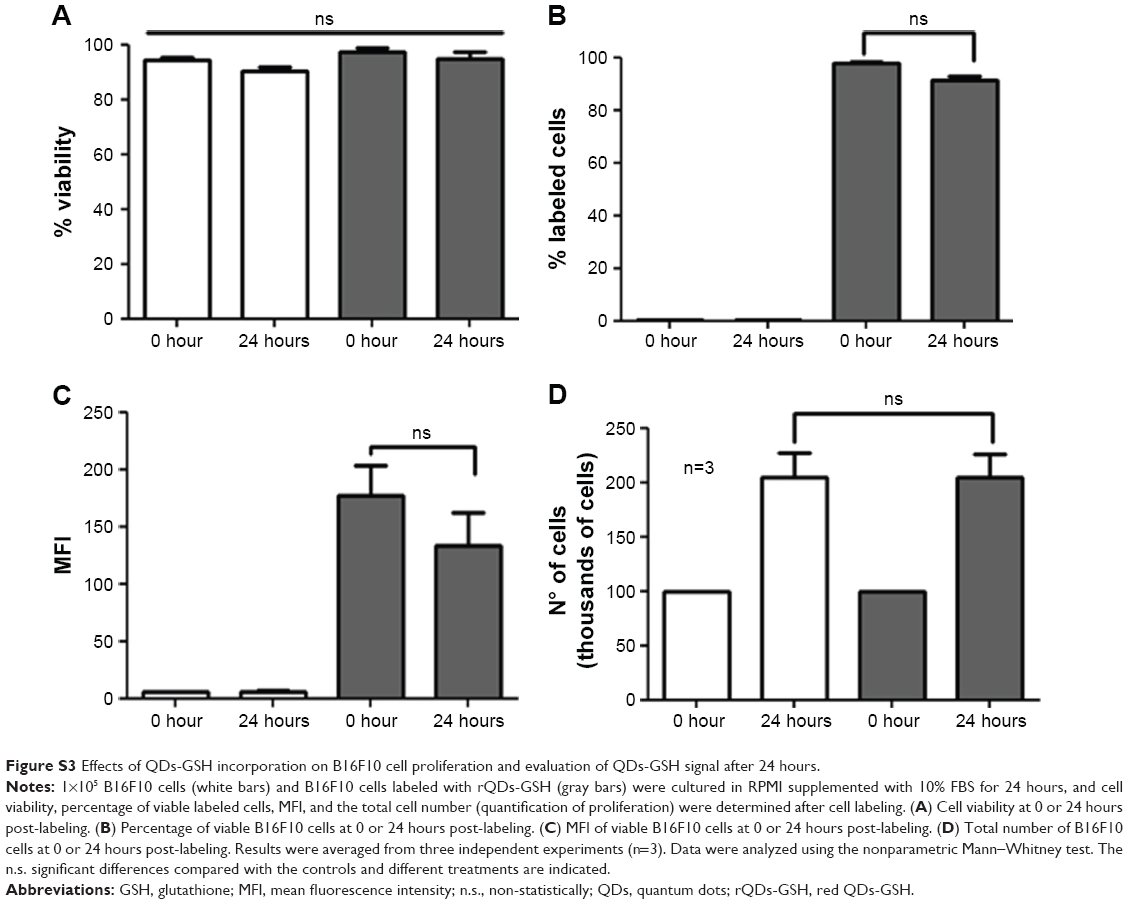 | Figure S3 Effects of QDs-GSH incorporation on B16F10 cell proliferation and evaluation of QDs-GSH signal after 24 hours. |
 | Figure S4 In vivo imaging of C57BL/6 mice treated with B16F10QDs-GSH-10NAC and B16F10 control cells. |
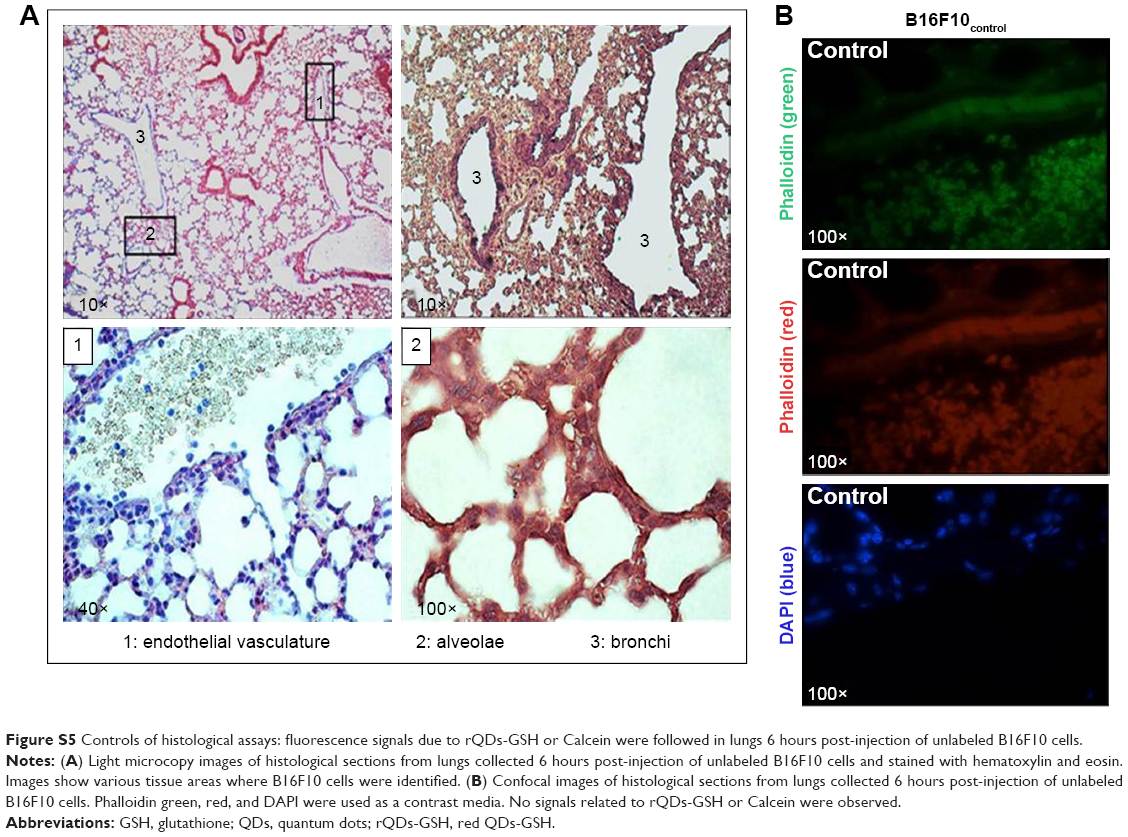 | Figure S5 Controls of histological assays: fluorescence signals due to rQDs-GSH or Calcein were followed in lungs 6 hours post-injection of unlabeled B16F10 cells. |
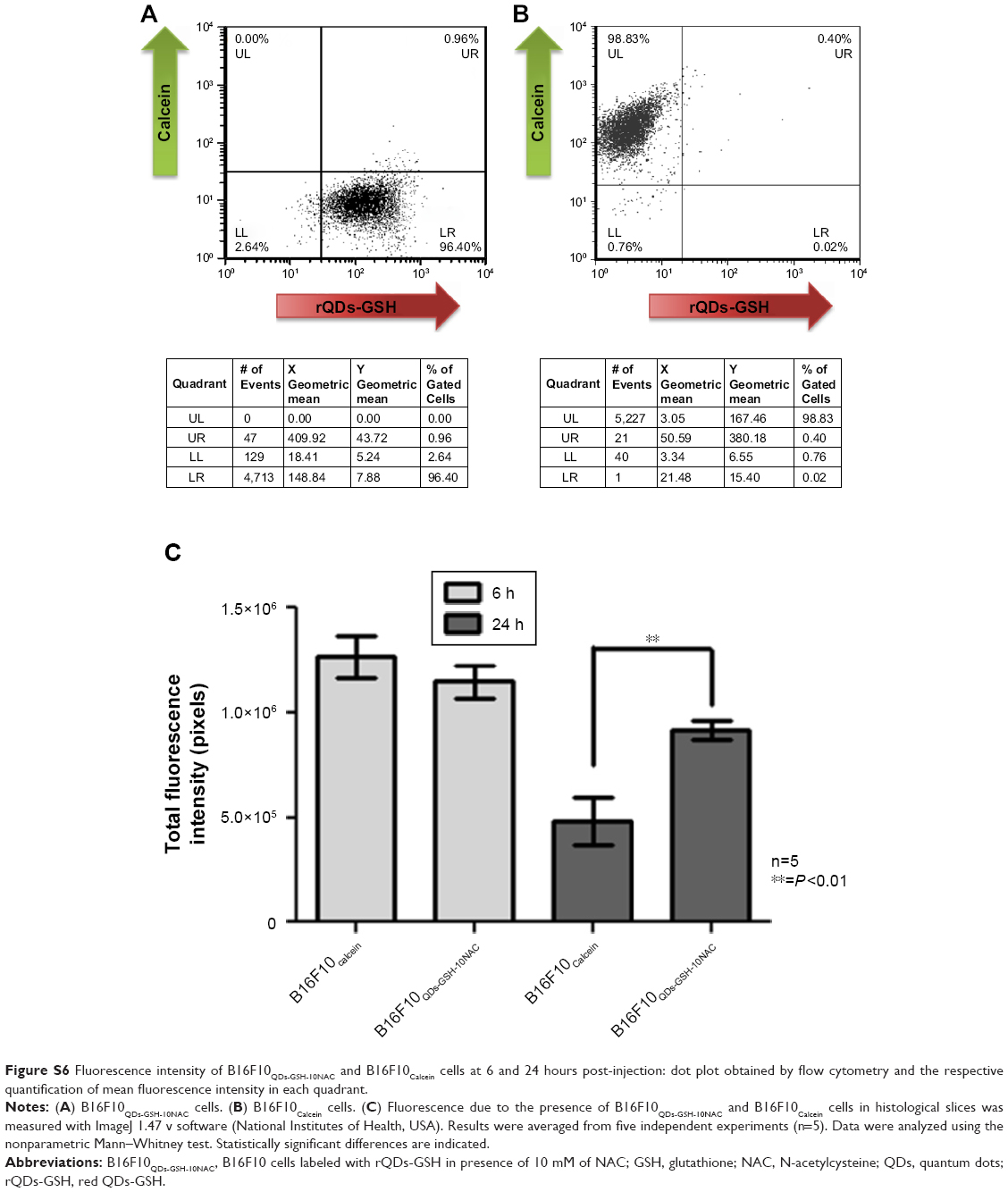 | Figure S6 Fluorescence intensity of B16F10QDs-GSH-10NAC and B16F10Calcein cells at 6 and 24 hours post-injection: dot plot obtained by flow cytometry and the respective quantification of mean fluorescence intensity in each quadrant. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.