Back to Journals » OncoTargets and Therapy » Volume 9
Biomarkers predicting resistance to epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer with wild-type KRAS
Authors Liu J, Hu J, Cheng L, Ren W, Mi Y, Liu B, Xie L, Qian X
Received 20 April 2015
Accepted for publication 31 August 2015
Published 27 January 2016 Volume 2016:9 Pages 557—565
DOI https://doi.org/10.2147/OTT.S86966
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Daniele Santini
Jiang Liu,* Jing Hu,* Lei Cheng, Wei Ren, Mi Yang, Baorui Liu, Li Xie, Xiaoping Qian
The Comprehensive Cancer Center of Drum-Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, Nanjing, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Abstract: EGFR pathway is an important therapeutic target in human tumors, including metastatic colorectal cancer (mCRC). The advent of EGFR-targeted monoclonal antibodies panitumumab and cetuximab has generated promise for the treatment of mCRC and has largely improved patients’ progression-free survival (PFS) and overall survival (OS). However, treatment with anti-EGFR monoclonal antibodies is only effective in a subset of mCRC patients with wild-type KRAS. This indicates that there are other factors affecting the efficacy of anti-EGFR monoclonal antibodies. Existing studies have demonstrated that among colorectal cancer patients with wild-type KRAS, harboring mutations of BRAF, PIK3CA, NRAS, or PTEN-null may demonstrate resistance to anti-EGFR-targeted therapy, and biomarkers detection can provide better-personalized treatment for mCRC patients. How to identify and reverse the secondary resistance to anti-EGFR monoclonal antibody therapy is also another great challenge to improve the anti-EGFR efficacy in wild-type KRAS mCRC patients. Finally, both of the molecular mechanisms of response and acquired resistance would be important for the directions of future research. This review focuses on how to further improve the predictive value of anti-EGFR therapies and how to also try and avoid futile treatment for wild-type KRAS colorectal cancer patients.
Keywords: colorectal cancer, EGFR, BRAF, RAS, cetuximab, panitumumab
Introduction
EGFR is a transmembrane tyrosine kinase receptor belonging to the human epidermal growth factor receptor (HEGFR) family to which ten different ligands can selectively bind.1,2 When ligands bind to the EGFR molecules, the receptor structure is changed, causing receptor autophosphorylation through receptor tyrosine kinase activity.2 The latter triggers a battery of intracellular signaling pathways, including RAS/RAF/MEK/MAPK and the PI3K/AKT pathways, which leads to tumor cell proliferation, inhibition of apoptosis, activation of invasion and metastasis, and stimulation of tumor-induced neovascularization.1,2 Therefore, EGFR is an important therapeutic target in human cancers including metastatic colorectal cancer (mCRC).
Colorectal cancer is the second most common epithelial tumor and in 2012 was also the second leading cause of death, due to cancer, in Europe.3 Over the past decade, systemic chemotherapy has made tremendous progress in the treatment of mCRC patients, and the median overall survival (OS) has increased from less than 9 months without treatment, to more than 20 months with treatment.4 The emergence of the EGFR-targeted monoclonal antibodies panitumumab and cetuximab, is a milestone in the history of the treatment of mCRC and indicates future directions for personalized treatment. Panitumumab and cetuximab have brought promise for the treatment of mCRC and have largely improved progression-free survival (PFS) or OS, as well as quality of life, but the treatment with anti-EGFR monoclonal antibodies is effective only in a subset of mCRC patients.5–9 Up-to-date, KRAS mutational status has been extensively studied to predict the clinical outcome of anti-EGFR-targeted therapy in mCRC patients.5–8 Cetuximab or panitumumab monotherapy,10–12 as well as combination therapy with chemotherapy,13–20 have been evaluated in several studies. Cetuximab in combination with standard chemotherapy13,15,16 in mCRC patients carrying wild-type KRAS has proven to improve patients’ OS, PFS, and objective response rate significantly. Similarly, the PFS and objective response rate for mCRC patients with wild-type KRAS have been remarkably improved, when panitumumab is applied in combination with chemotherapy.18,19,21
However, not all mCRC patients carrying wild-type KRAS respond to anti-EGFR therapy. Thus, batteries of other potential predictive markers have also been investigated to guide this therapy.9,22–29 Two retrospective studies9,30 have shown that among wild-type KRAS patients receiving cetuximab or panitumumab, BRAF mutations were significantly and independently associated with patient survival. PIK3CA mutations and the loss of PTEN expression have been reported as predictive markers underlying the response to cetuximab or panitumumab in wild-type KRAS mCRC patients in a number of other studies.24,31–33 In recent years, the relationship between NRAS mutations and the efficacy of anti-EGFR antibodies therapy has also been evaluated.29,34,35 Moreover, the acquired resistance to anti-EGFR antibodies therapy is another urgent problem to improve the efficacy and life quality in mCRC patients. Its underlying mechanism may also relate to BRAF, PIK3CA, NRAS, and PTEN status. In this paper, we reviewed these studies and tried to figure out the most useful biomarkers to help the use of anti-EGFR monoclonal antibodies.
BRAF mutations
BRAF is a downstream effector of the RAS signaling pathway. It has been reported that BRAF mutations are related to the resistance of cetuximab or panitumumbab in approximately 10% of the cases of colorectal cancer.30,36 The most common BRAF mutation in tumors was the BRAF V600E mutation that was mutually exclusive with KRAS mutations.36,37 Therefore, the combination of KRAS with BRAF status, can identify further optimized populations that may benefit from anti-EGFR antibodies therapy. Two retrospective studies have reported, whereby BRAF mutations impaired the response to cetuximab or panitumumab in mCRC patients.29,36 Di Nicolantonio et al discovered that HT-29 and COLO-205 (both BRAF V600E mutation and wild-type for KRAS) were highly resistant to cetuximab or panitumumab therapy, and the BRAF inhibitor sorafenib could restore sensitivity to anti-EGFR therapy.36 This implies that combining EGFR and BRAF inhibitors may be more effective for wild-type KRAS/BRAF-mutation populations. Furthermore, Di Nicolantonio et al have also shown that in eleven patients with wild-type KRAS/BRAF-mutation receiving cetuximab or panitumumab treatment, PFS and OS were significantly shorter than both wild-type populations,36 which was inconsistent with two other retrospective studies.9,30 However, Karapetis et al retrospectively analyzed the role of activating mutations of the EGFR signaling pathway in predicting the efficacy of cetuximab-based treatment using the sample obtained from the NCIC CTG/AGITG CO.17 study, and did not discover the predictive significance of BRAF V600E mutation25 in the year of 2013. Furthermore, the 2013 ASCO (American Society of Clinical Oncology) Annual Meeting reported that in a retrospective analysis of the role of RAS and RAF mutations in the Phase III PRIME study, BRAF V600E mutation found no predictive value.35 It was also surprising that OS was prolonged in a small number of wild-type KRAS/BRAF-mutant (n=11) patients who were receiving cetuximab and FOLFOX-4, compared with patients treated with only FOLFOX-4 in the OPUS study.16 This phenomenon is likely related to the heterogeneity of cancer and the small-sample-size. Owing to the small number of BRAF mutation cases and lack of perspective studies, it is difficult to conclude the predictive value of anti-EGFR therapies in colorectal cancer at the current station.
In recent years, three large randomized clinical studies, including the CRYSTAL (cetuximab combined with FOLFIRI as first-line therapy for mCRC) study,13 the COIN (cetuximab combined with oxaliplatin-based first-line chemotherapy for treatment of advanced colorectal cancer) trial,38 and the NORDIC-VII (cetuximab with Nordic FLOX versus FLOX alone in the first-line therapy for mCRC) study,39 have consistently demonstrated that the BRAF V600E mutation predicts poor prognosis, which is supported by the data from the pooled analysis of the CRYSTAL and OPUS randomized clinical trials.40 In the CRYSTAL study, although the addition of cetuximab to FOLFIRI did not show any significant difference in wild-type KRAS/BRAF-mutant patterns, it improved PFS and OS slightly. Thus, cetuximab treatment may not be completely forbidden in terms of BRAF mutations, this is because there is still a survival benefit for such populations. In a word, BRAF mutations are robustly and negatively prognostic factors in mCRC populations according to the studies mentioned earlier. However, it is controversial with respect to its predictive value in anti-EGFR treatment.
NRAS mutations
NRAS, a member of RAS family, is often mutated in human tumors. An experimental research study showed that activating mutations in NRAS robustly stimulates tumorigenesis by suppressing apoptosis in the condition of inflammation.41 Thus, NRAS mutations might be predictors of treatment effect as well as a treatment target. Since KRAS and NRAS mutations were completely exclusive,29,34 NRAS mutational status might be a valuable predictor to wild-type KRAS patients receiving anti-EGFR therapy. Similar to KRAS, the common mutational sites of NRAS were codons 12, 13, and 61, and the mutational frequency was approximately 3% in wild-type KRAS populations.42 The data from two small-sample-size studies showed that wild-type KRAS patients carrying NRAS mutations, had lower response rates for anti-EGFR therapy compared with those with dual wild-type genes.43,44 A poor prognostic effect was observed in patients with NRAS mutations in the COIN trial.38 Moreover, in a retrospective randomized Phase III study evaluating response to panitumumab, treatment with panitumumab resulted in improved PFS in patients with wild-type KRAS/NRAS rather than those with wild-type KRAS/mutational NRAS.45 Although the mutational frequency of NRAS is very low, there is a strong trend toward a negative response to anti-EGFR antibodies therapy in populations for wild-type KRAS/mutational NRAS.
PIK3CA mutations and PTEN-null
In addition to the RAS/RAF/MEK/MAPK signaling pathway, the PI3K/AKT/mTOR pathway also plays an important role in the development of malignant tumors. The most common mutation sites of phosphoinositide 3-kinase (PI3K) are located on exons 9 and 20.24,46,47 It was observed that cell lines carrying wild-type PIK3CA/PTEN expression were more sensitive to cetuximab than cell lines with PIK3CA mutations or loss of PTEN expression.31 This implied the potential correlation between cell response to anti-EGFR antibody therapy and the two genes level, in vitro. However, clinical results were not consistent. Before this study, in a small-sample-size clinical study, the association between PIK3CA mutations and response to cetuximab was not identified for unselected patients5 that was in agreement with another report.46 Subsequently, Sartore-Bianchi et al indicated PIK3CA mutations were independently associated with a poorer clinical outcome of anti-EGFR antibodies therapy in mCRC patients, and this effect was enlarged in the wild-type KRAS subgroup.24 No conclusions were made regarding the predictive value of mutations at PI3K in the PICCOLO trial comparing panitumumab plus irinotecan, versus only irinotecan for patients with wild-type KRAS.20 Simultaneously, a retrospective analysis also did not find any association in terms of PIK3CA mutations and the efficacy of cetuximab.25 It was difficult to draw conclusions from these conflicting findings due to small-sample-size and very few mutational PI3K cases. Thus, to further explore the relationship between PIK3CA mutations and efficacy of anti-EGFR antibodies therapy in colorectal cancer patients, exons 9 and 20 of PI3K should be analyzed respectively. A large cohort study containing 1,022 mCRC patients treated with cetuximab revealed a poorer clinical outcome for patients with exon 20 mutations rather than exon 9 mutations in wild-type KRAS populations (Table 1).29 It was likely that similar findings were observed in other tumors found in, for example, breast cancer patients.48,49 In 2011, a meta-analysis containing 13 studies had suggested that compared with exon 9 mutations, as well as all other mutations, the predictive power of exon 20 mutations were more significant with respect to objective response rate, PFS, and OS in the wild-type KRAS subgroup.50 In the time following, another meta-analysis revealed that PIK3CA mutations as a whole, might be negative predictive markers for PFS and OS in wild-type KRAS mCRC patients treated with anti-EGFR therapy (odds ratio [OR]=0.42; PFS: hazard ratio [HR]=1.54; OS: HR=1.4).51 Because the negative effect of PI3K exon 9 mutations was not observed in this meta-analysis, the whole effect largely depended on exon 20 mutations. Hence, PI3K exon 20 mutations are of more predictive value for anti-EGFR therapy.
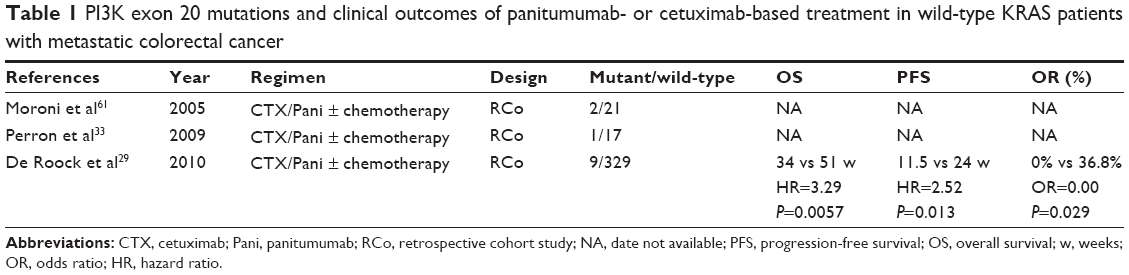 | Table 1 PI3K exon 20 mutations and clinical outcomes of panitumumab- or cetuximab-based treatment in wild-type KRAS patients with metastatic colorectal cancer |
The loss of the PTEN is widespread in human tumors. The absence of PTEN function could constitutively activate downstream effectors of the PI3K signaling pathway in cancer cells.52 This suggested dysfunction of PTEN might be related to the reduced efficacy of antitumor therapies and the shorter survival rate of patients. The PTEN loss detected by immunohistochemistry decreased OS instead of PFS in wild-type KRAS patients treated with cetuximab.9 On the contrary, a cohort study has reported wild-type KRAS patients with PTEN-positive on metastatic, rather than primary tumors, had longer PFS.26 This interesting finding implied that biological characteristics of primary tumors and metastases was to some extent different, which is contradictive to the result found in another study.53 Two other retrospective studies have revealed that PTEN expression was related to longer time to progression (TTP) in patients carrying wild-type KRAS.54,55 PTEN expression loss was found to cause poor PFS and OS in wild-type KRAS colorectal cancer patients undergoing anti-EGFR antibodies therapy (Table 2),56 which was in accordance with the results found in a meta-analysis study.57 Nevertheless, there was no statistical significance with respect to the association between PTEN-null and the clinical outcome of cetuximab in the NCIC CTG/AGITG CO.17 trial.25 An important problem that influenced the results of the clinical trials was that interpretation of immunohistochemistry results may be determined by multiple factors, such as the academic level of analysts and/or different scoring systems. Another question that remained uncertain was whether PTEN protein expression of primary tumors and metastases was different. If these problems are resolved, the combination of PTEN expression and KRAS mutational status might be a better predictor of the efficacy of anti-EGFR antibodies therapy.
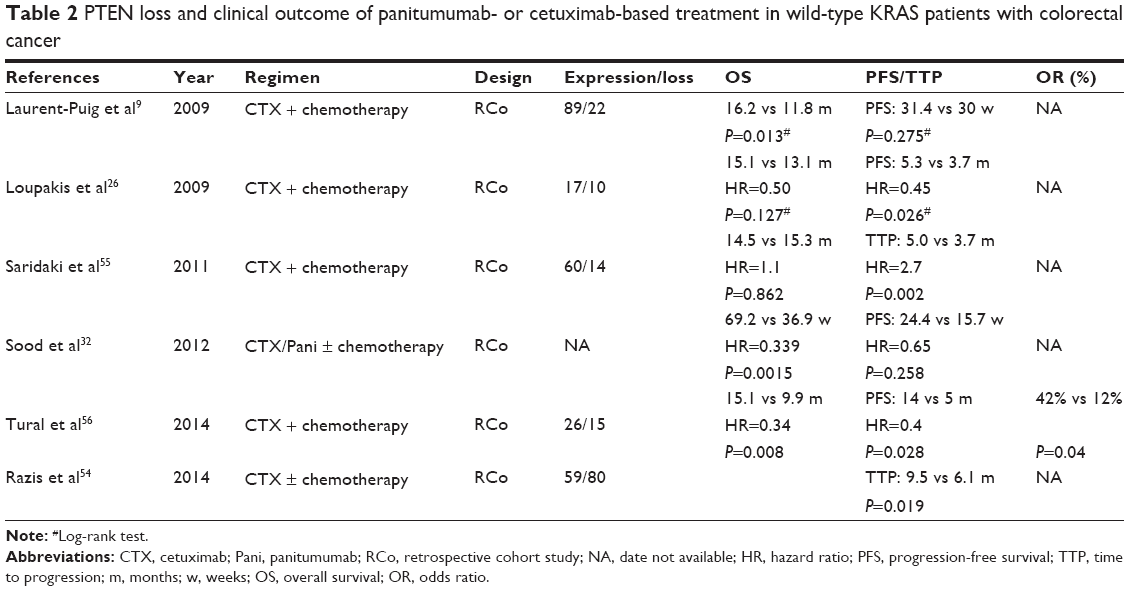 | Table 2 PTEN loss and clinical outcome of panitumumab- or cetuximab-based treatment in wild-type KRAS patients with colorectal cancer |
In addition, several researchers found that PI3K mutations and PTEN expression were joint predictors of the clinical outcome of anti-EGFR therapy.31–33 The wild-type KRAS patients with PTEN-null/PI3K mutations had a significantly shorter OS, and there was very strong trend toward the decrease in PFS.32 However, the data were limited for the combination of PI3K mutations and PTEN loss as predictive markers.
EGFR and its ligands
Activation mutations of the kinase domain in EGFR, such as mutations of exons 18, 19, and 21, have been reported to be associated with the response to tyrosine kinase inhibitors (TKIs) (gefitinib or erlotinib) in lung cancer,58,59 but such mutations were rare in colorectal cancer.60 In contrast, the relationship between EGFR gene copy number as well as expression, and anti-EGFR antibodies therapy in colorectal cancer, has been largely concerned with. Unselected patients’ tumors which had an increased EGFR gene copy number, detected by fluorescence in situ hybridization, were more likely to respond to cetuximab,61–64 which was inconsistent with the results of two meta-analysis published in 2013.65,66 In addition, for wild-type KRAS mCRC patients treated with cetuximab, EGFR amplification had also been reported to be associated with better clinical outcomes,54 and EGFR-negative cases, confirmed by fluorescence in situ hybridization, might be resistant to cetuximab.67 Interestingly, increased EGFR gene copy number could not always be translated into the corresponding protein,61,62 so the relationship between increased EGFR gene copy number and response has remained unclear.68 In most studies, the sample-size was too small and EGFR different scoring systems were used. Finally, heterogeneity (ie, baseline characteristics of patients, receiving cetuximab or panitumumab monotherapy, or combination with chemotherapy) existed among these studies. In short, based on the above mentioned factors, EGFR gene copy number is difficult to apply in clinical practice regardless of KRAS status.
Over-expression of EGFR ligands, such as epiregulin and amphiregulin, may also stimulate tumor growth and promote tumor cell survival through an autocrine loop.23 For unselected patients treated with cetuximab, higher expressions of mRNA for epiregulin and amphiregulin were related to higher disease control rate (P<0.0001), and longer PFS was statistically significantly (epiregulin: P=0.0002, median PFS=103.5 vs 57 days; amphiregulin: P<0.0001, median PFS=115.5 vs 57 days) when compared with groups of lower expression levels for these ligands.23 In 2009, a prospective study concluded that epiregulin and amphiregulin mRNA over-expression in primary tumors were significantly associated with better outcomes in wild-type KRAS mCRC patients treated with cetuximab and irinotecan,69 which was supported by another recently published study.70 Interestingly, objective responses were also observed in some patients with low EGFR ligand expression,69 which suggested other signaling pathways might be involved. Moreover, in 2014, a retrospective study from a Phase III clinical trial of cetuximab and best supportive care versus best supportive care alone, underscored PFS and OS benefit in the presence of high expressed epiregulin in wild-type KRAS patients.71 Although these studies have produced gratifying results, their role as predictive biomarkers in wild-type KRAS patients still requires more reliable evidence and standardized detection methods.
MET
MET as a proto-oncogene, is activated by its ligand, hepatocyte growth factor. MET amplification led to resistance to EGFR TKIs through activation of the ERBB3/PI3K/AKT signaling pathway in EGFR mutant lung cancer cells.72,73 Cetuximab resistance resulting from activated MET signal was evaluated in not only lung cancer cells but also colorectal cancer cells.74–76 Inhibiting or interfering MET expression could restore sensitivity to cetuximab in cetuximab-resistant cells.74,77 These preclinical results provide strong evidence for the simultaneous targeting of EGFR and MET signaling pathway clinically. Moreover, in two clinical studies, among subpopulations for wild-type KRAS, MET over-expression was significantly related to poor clinical outcome.78,79 In consideration of the limited data, whether MET amplification is reliable biomarker for anti-EGFR therapy in wild-type KRAS colorectal cancer patients remains uncertain.
Acquired drug resistance to anti-EGFR therapy
Tumors sensitive to anti-EGFR therapy would eventually develop resistance to this therapy. Thus, identifying the molecular bases and mechanisms of acquired resistance to this therapy is critical to guide-personalized therapy and reverse drug resistance. Currently, the viewpoints about this molecular bases and mechanisms have not reached a consensus. KRAS mutations were considered to be driving factors of acquired resistance to cetuximab in colorectal cancers, which implied early combination with mitogen-activated protein kinase (MAPK) inhibitors might be a rational therapy strategy to reverse drug resistance.80 Nevertheless, several preclinical studies showed that secondary resistance to anti-EGFR antibodies was closely associated with abnormal changes of HER family members, such as dysregulation of EGFR internalization/degradation, nuclear translocation of the EGFR, and activation of HER2 or HER3.81–83 The Src gene and MET gene as proto-oncogenes, have recently been given widespread attention. Wheeler et al reported that Src family kinases played a role in the acquired resistance to cetuximab in non-small-cell lung cancer cell lines.84 MET amplification might mediate secondary resistance to anti-EGFR antibodies therapy in patient-derived colorectal cancer xenografts, and this result could be eliminated by MET inhibitors.75,85 When considered together, the understanding of acquired resistance to anti-EGFR antibodies therapy is poor and it is difficult to draw a clear conclusion from these studies. For example, most data regarding acquired resistance come from preclinical, as opposed to clinical studies. Furthermore, each result only partly explains this phenomenon and the data were limited.
Other potential biomarkers
Tyrosine kinase domain of each member of the HER family is highly conserved, and the structure and function is of high homology, which underlies the molecular basis of both the interaction of the receptors and cross-activation. HER2 amplification was prospectively identified as a predictive marker of resistance to anti-EGFR antibodies therapy, and in response to combination therapy, against HER2 and EGFR from mCRC xenografts.86 Subsequently, this phenomenon also existed among mCRC patients in a small-sample clinical study.87 In addition, wild-type KRAS colorectal cancer patients with positive HER3, detected by immunohistochemistry, had poor clinical outcomes in two retrospective studies.27,88 It would seem that HER2 and HER3 are possible biomarkers for anti-EGFR antibodies therapy, but these studies were also limited due to a small samplesize.
Integrins can affect cell growth and repair via their receptors.89,90 The integrin pathway is activated in the absence of HER3 activation, implying that it may have a role in HER3 negative tumors. Upon univariate or multivariate analysis, integrin β4 rs8669 genotyping might be superior in selecting colorectal cancer patients who are more likely to benefit from anti-EGFR therapies in the HER3-negative/wild-type KRAS subgroup.91
Other downstream effectors of EGFR have also been recently evaluated. For example, phosphorylated protein kinase B and MAPK expression in metastatic tumors, were related to poorer clinical outcomes in 72 mCRC patients receiving irinotecan-cetuximab.92 Mitogen-activated protein kinase phosphatases (MKPs) can inhibit MAPK activity via crosstalk between distinct MAPK pathways, or between MAPK signaling and other intracellular signaling modules.93 MKP1 is the best-characterized member of MKP family. In vitro, ectopic expression of MKP1 could inhibit the anti-EGFR drug. AG1478 induced apoptosis is activated via suppressing JNK (a MAPK) activation in the lung cancer cell PC-9.94 Thus, MKP1 over-expression may be a potential biomarker of resistance to anti-EGFR agents. In a small-sample-size clinical study, including 48 mCRC patients, patients with MKP1 over-expression in the wild-type KRAS subgroup had a low response (7% vs 44%) and poor PFS (13 vs 32 weeks).95
Insulin-like growth factor 1 (IGF1) is closely related to cell growth and proliferation. The expression of IGF1 and its receptor may play a significant role in both the occurrence and growth of colorectal cancer.96 The downstream pathway of IGFR and EGFR was completely overlapped,97 which implied that tumor cell growth might be stimulated by the activation of each of IGFR and EGFR. Hence, IGF1 expression may induce resistance to anti-EGFR therapy in colorectal cancer. For example, polymorphisms of genes in the IGF1 pathway may act as potential biomarkers for cetuximab efficacy in mCRC patients for wild-type KRAS.28 However, the evidence regarding these predictive markers was insufficient and limited.
Antibody-dependent cell-mediated cytotoxicity (ADCC) is an immune mechanism in which specific antibodies are directed against a targeted antigen on tumor cells, causing their lysis via innate immune effector cells. The Fragment c (Fc) domain of the IgG1 monoclonal antibody was shown to induce ADCC. The Fc portion of cetuximab can activate ADCC through interactions with the Fc receptors (FcγR) on the effector immune cells. Bibeau et al98 discovered combined FcRIIa/FcRIIIa polymorphisms are prognostic factors for disease progression in mCRC patients treated with cetuximab and irinotecan beyond KRAS mutation status. This implies that ADCC might play an important role in cetuximab efficacy.
Conclusion and prospection
The advent of anti-EGFR monoclonal antibodies has far-reaching implications for the treatment of refractory colorectal cancer. However, KRAS mutation status has been defined as a negative predictive factor for this therapy, a considerable proportion of patients with wild-type KRAS have no response to the treatment. This indicates that there are other factors affecting the efficacy of anti-EGFR monoclonal antibodies. Thus, it is necessary to further improve predictive accuracy and avoid futile treatment. Combination with other biomarkers is a rational strategy for further selecting appropriate populations. In recent years, more evidence reveals that KRAS and NRAS, as a whole, is more predictive of the efficacy of anti-EGFR monoclonal antibodies therapy. BRAF, which is prone to be a prognostic instead of predictive factor, is not very significant in terms of identifying whether anti-EGFR monoclonal antibodies are added in colorectal treatment. There is strong evidence that combined detection of NRAS, PI3K (especially exon 20), and the PTEN gene status in wild-type KRAS colorectal cancer patients, can identify more patients unlikely to respond to anti-EGFR monoclonal antibodies. It would seem that over-expression of EGFR ligands predict superior clinical outcomes for anti-EGFR antibodies therapy in mCRC patients with wild-type KRAS. The predictive evidence of other potential biomarkers such as MET, HER2, HER3, MKP1, and IGF1 is both limited and inadequate.
There are still some problems that will need to be solved, before implementing these predictive markers in clinics. Firstly, lower variant frequency of these markers limits the clinical application and requires more reliable evidence from prospective studies with larger sample-sizes. Secondly, different detection methods and cutoff values used to detect the same markers in some of the studies may interfere with the comparability of these findings. Hence, establishing standardized methods and unifying the optimal cutoff value are rather necessary. Thirdly, immunohistochemistry as a semiquantitative method is affected by multiple factors and should therefore have standardized criteria, or be replaced by more sensitive and specific detection methods. Finally, most evidence on the acquired resistance is limited to preclinical studies.
Acknowledgments
This work was supported by the Grants 81172094 and 8110186 from the National Science Foundation of China, and Grant 2011-WS-005 from the Six Talents Peak Foundation of Jiangsu Province.
Disclosure
The authors report no conflicts of interest in this work.
References
Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol. 2009;6(9):519–527. | ||
Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–1174. | ||
Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, et al. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013;49(6):1374–1403. | ||
Wolpin BM, Mayer RJ. Systemic treatment of colorectal cancer. Gastroenterology. 2008;134(5):1296–1310. | ||
Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66(8):3992–3995. | ||
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67(6):2643–2648. | ||
Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26(3):374–379. | ||
Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. | ||
Laurent-Puig P, Cayre A, Manceau G, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27(35):5924–5930. | ||
Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351(4):337–345. | ||
Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25(13):1658–1664. | ||
Jonker DJ, O’Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357(20):2040–2048. | ||
Van Cutsem E, Köhne CH, Lang I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011–2019. | ||
Bokemeyer C, Bondarenko I, Makhson A, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27(5):663–671. | ||
Van Cutsem E, Köhne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417. | ||
Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22(7):1535–1546. | ||
Köhne CH, Hofheinz R, Mineur L, et al. First-line panitumumab plus irinotecan/5-fluorouracil/leucovorin treatment in patients with metastatic colorectal cancer. J Cancer Res Clin Oncol. 2012;138(1):65–72. | ||
Douillard JY, Siena S, Cassidy J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28(31):4697–4705. | ||
Peeters M, Price TJ, Cervantes A, et al. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28(31):4706–4713. | ||
Seymour MT, Brown SR, Middleton G, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. Lancet Oncol. 2013;14(8):749–759. | ||
Douillard JY, Oliner KS, Siena S, et al. Panitumumab–FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023–1034. | ||
Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23(9):1803–1810. | ||
Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25(22):3230–3237. | ||
Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009;69(5):1851–1857. | ||
Karapetis CS, Jonker D, Daneshmand M, et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer – results from NCIC CTG/AGITG CO.17. Clin Cancer Res. 2014;20(3):744–753. | ||
Loupakis F, Pollina L, Stasi I, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol. 2009;27(16):2622–2629. | ||
Scartozzi M, Giampieri R, Maccaroni E, et al. Analysis of HER-3, insulin growth factor-1, nuclear factor-kB and epidermal growth factor receptor gene copy number in the prediction of clinical outcome for K-RAS wild-type colorectal cancer patients receiving irinotecan-cetuximab. Ann Oncol. 2012;23(7):1706–1712. | ||
Winder T, Zhang W, Yang D, et al. Germline polymorphisms in genes involved in the IGF1 pathway predict efficacy of cetuximab in wild-type KRAS mCRC patients. Clin Cancer Res. 2010;16(22):5591–5602. | ||
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. | ||
Tol J, Dijkstra JR, Klomp M, et al. Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer. 2010;46(11):1997–2009. | ||
Jhawer M, Goel S, Wilson AJ, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res. 2008;68(6):1953–1961. | ||
Sood A, McClain D, Maitra R, et al. PTEN gene expression and mutations in the PIK3CA gene as predictors of clinical benefit to anti-epidermal growth factor receptor antibody therapy in patients with KRAS wild-type metastatic colorectal cancer. Clin Colorectal Cancer. 2012;11(2):143–150. | ||
Perrone F, Lampis A, Orsenigo M, et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol. 2009;20(1):84–90. | ||
Bando H, Yoshino T, Shinozaki E, et al. Simultaneous identification of 36 mutations in KRAS codons 61 and 146, BRAF, NRAS, and PIK3CA in a single reaction by multiplex assay kit. BMC Cancer. 2013;13:405. | ||
Oliner KS, Douillard J, Siena S, et al. Analysis of KRAS/NRAS and BRAF mutations in the phase III PRIME study of panitumumab (pmab) plus FOLFOX versus FOLFOX as first-line treatment (tx) for metastatic colorectal cancer (mCRC). J Clin Oncol. 2013;31(suppl; abstr 3511). | ||
Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–5712. | ||
Frattini M, Balestra D, Suardi S, et al. Different genetic features associated with colon and rectal carcinogenesis. Clin Cancer Res. 2004;10(12):4015–4021. | ||
Maughan TS, Adams RA, Smith CG, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377(9783):2103–2114. | ||
Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII study. J Clin Oncol. 2012;30(15):1755–1762. | ||
Bokemeyer C, Van Cutsem E, Rougier P, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48(10):1466–1475. | ||
Wang Y, Velho S, Vakiani E, et al. Mutant N-Ras protects colorectal cancer cells from stress-induced apoptosis and contributes to cancer development and progression. Cancer Discov. 2013;3(3):294–307. | ||
Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50(5):307–312. | ||
Andrè T, Blons H, Mabro M, et al. Panitumumab combined with irinotecan for patients with KRAS wild-type metastatic colorectal cancer refractory to standard chemotherapy: a GERCOR efficacy, tolerance, and translational molecular study. Ann Oncol. 2013;24(2):412–419. | ||
Di Bartolomeo M, Pietrantonio F, Perrone F, et al. Lack of KRAS, NRAS, BRAF and TP53 mutations improves outcome of elderly metastatic colorectal cancer patients treated with cetuximab, oxaliplatin and UFT. Target Oncol. 2014;9(2):155–162. | ||
Peeters M, Oliner KS, Parker A, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19(7):1902–1912. | ||
Prenen H, De Schutter J, Jacobs B, et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res. 2009;15(9):3184–3188. | ||
Velho S, Oliveira C, Ferreira A, et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur J Cancer. 2005;41(11):1649–1654. | ||
Kalinsky K, Jacks LM, Heguy A, et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009;15(16):5049–5059. | ||
Lai YL, Mau BL, Cheng WH, Chen HM, Chiu HH, Tzen CY. PIK3CA exon 20 mutation is independently associated with a poor prognosis in breast cancer patients. Ann Surg Oncol. 2008;15(4):1064–1069. | ||
Mao C, Yang ZY, Hu XF, Chen Q, Tang JL. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol. 2012;23(6):1518–1525. | ||
Huang L, Liu Z, Deng D, et al. Anti-epidermal growth factor receptor monoclonal antibody-based therapy for metastatic colorectal cancer: a meta-analysis of the effect of PIK3CA mutations in KRAS wild-type patients. Arch Med Sci. 2014;10(1):1–9. | ||
Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22(14):2954–2963. | ||
Atreya CE, Sangale Z, Xu N, et al. PTEN expression is consistent in colorectal cancer primaries and metastases and associates with patient survival. Cancer Med. 2013;2(4):496–506. | ||
Razis E, Pentheroudakis G, Rigakos G, et al. EGFR gene gain and PTEN protein expression are favorable prognostic factors in patients with KRAS wild-type metastatic colorectal cancer treated with cetuximab. J Cancer Res Clin Oncol. 2014;140(5):737–748. | ||
Saridaki Z, Tzardi M, Papadaki C, et al. Impact of KRAS, BRAF, PIK3CA mutations, PTEN, AREG, EREG expression and skin rash in ≥2 line cetuximab-based therapy of colorectal cancer patients. PloS one. 2011;6(1):e15980. | ||
Tural D, Batur S, Erdamar S, et al. Analysis of PTEN, BRAF and PI3K status for determination of benefit from cetuximab therapy in metastatic colorectal cancer patients refractory to chemotherapy with wild-type KRAS. Tumor Biol. 2014;35(2):1041–1049. | ||
Wang ZH, Gao QY, Fang JY. Loss of PTEN expression as a predictor of resistance to anti-EGFR monoclonal therapy in metastatic colorectal cancer: evidence from retrospective studies. Cancer Chemother Pharmacol. 2012;69(6):1647–1655. | ||
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. | ||
Sequist LV, Joshi VA, Jänne PA, et al. Response to treatment and survival of patients with non-small cell lung cancer undergoing somatic EGFR mutation testing. Oncologist. 2007;12(1):90–98. | ||
Barber TD, Vogelstein B, Kinzler KW, Velculescu VE. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N Engl J Med. 2004;351(27):2883. | ||
Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6(5):279–286. | ||
Cappuzzo F, Finocchiaro G, Rossi E, et al. EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol. 2008;19(4):717–723. | ||
Sartore-Bianchi A, Moroni M, Veronese S, et al. Epidermal growth factor receptor gene copy number and clinical outcome of metastatic colorectal cancer treated with panitumumab. J Clin Oncol. 2007;25(22):3238–3245. | ||
Campanella C, Mottolese M, Cianciulli A, et al. Epidermal growth factor receptor gene copy number in 101 advanced colorectal cancer patients treated with chemotherapy plus cetuximab. J Transl Med. 2010;8:36, doi10.1186/1479-5876-8-36. | ||
Jiang Z, Li C, Li F, Wang X. EGFR gene copy number as a prognostic marker in colorectal cancer patients treated with cetuximab or panitumumab: a systematic review and meta analysis. PloS one. 2013;8(2):e56205. | ||
Shen WD, Chen HL, Liu PF. EGFR gene copy number as a predictive biomarker for resistance to anti-EGFR monoclonal antibodies in metastatic colorectal cancer treatment: a meta-analysis. Chin J Cancer Res. 2014;26(1):59–71. | ||
Li YH, Wang F, Shen L, et al. EGFR fluorescence in situ hybridization pattern of chromosome 7 disomy predicts resistance to cetuximab in KRAS wild-type metastatic colorectal cancer patients. Clin Cancer Res. 2011;17(2):382–390. | ||
Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol. 2010;28(7):1254–1261. | ||
Jacobs B, De Roock W, Piessevaux H, et al. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol. 2009;27(30):5068–5074. | ||
Baker JB, Dutta D, Watson D, et al. Tumour gene expression predicts response to cetuximab in patients with KRAS wild-type metastatic colorectal cancer. Br J Cancer. 2011;104(3):488–495. | ||
Jonker DJ, Karapetis CS, Harbison C, et al. Epiregulin gene expression as a biomarker of benefit from cetuximab in the treatment of advanced colorectal cancer. Br J Cancer. 2014;110(3):648–655. | ||
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | ||
Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88. | ||
Krumbach R, Schüler J, Hofmann M, Giesemann T, Fiebig HH, Beckers T. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur J Cancer. 2011;47(8):1231–1243. | ||
Troiani T, Martinelli E, Napolitano S, et al. Increased TGF-alpha as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19(24):6751–6765. | ||
Luraghi P, Reato G, Cipriano E, et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res. 2014;74(6):1857–1869. | ||
Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011;17(3):472–482. | ||
Inno A, Di Salvatore M, Cenci T, et al. Is there a role for IGF1R and c-MET pathways in resistance to cetuximab in metastatic colorectal cancer? Clin Colorectal Cancer. 2011;10(4):325–332. | ||
Kishiki T, Ohnishi H, Masaki T, et al. Overexpression of MET is a new predictive marker for anti-EGFR therapy in metastatic colorectal cancer with wild-type KRAS. Cancer Chemother Pharmacol. 2014;73(4):749–757. | ||
Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. | ||
Wheeler DL, Huang S, Kruser TJ, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27(28):3944–3956. | ||
Yonesaka K, Zejnullahu K, Okamoto I, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3(99):99ra86, doi10.1126/scitranslmed.3002442. | ||
Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene. 2009;28(43):3801–3813. | ||
Wheeler DL, Iida M, Kruser TJ, et al. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol Ther. 2009;8(8):696–703. | ||
Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–673. | ||
Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1(6):508–523. | ||
Martin V, Landi L, Molinari F, et al. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br J Cancer. 2013;108(3):668–675. | ||
Scartozzi M, Mandolesi A, Giampieri R, et al. The role of HER-3 expression in the prediction of clinical outcome for advanced colorectal cancer patients receiving irinotecan and cetuximab. Oncologist. 2011;16(1):53–60. | ||
Scartozzi M, Loretelli C, Bearzi I, et al. Allele polymorphisms of tumor integrins correlate with peritoneal carcinosis capability of gastric cancer cells in radically resected patients. Ann Oncol. 2011;22(4):897–902. | ||
Jin H, Varner J. Integrins: roles in cancer development and as treatment targets. Br J Cancer. 2004;90(3):561–565. | ||
Scartozzi M, Giampieri R, Loretelli C, et al. Role of β4 integrin in HER-3-negative, K-RAS wild-type metastatic colorectal tumors receiving cetuximab. Future Oncol. 2013;9(8):1207–1214. | ||
Scartozzi M, Giampieri R, Maccaroni E, et al. Phosphorylated AKT and MAPK expression in primary tumours and in corresponding metastases and clinical outcome in colorectal cancer patients receiving irinotecan-cetuximab. J Transl Med. 2012;10:71, doi10.1186/1479-5876-10-71. | ||
Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J. 2013;280(2):489–504. | ||
Takeuchi K, Shin-ya T, Nishio K, Ito F. Mitogen-activated protein kinase phosphatase-1 modulated JNK activation is critical for apoptosis induced by inhibitor of epidermal growth factor receptor-tyrosine kinase. FEBS J. 2009;276(5):1255–1265. | ||
Montagut C, Iglesias M, Arumi M, et al. Mitogen-activated protein kinase phosphatase-1 (MKP-1) impairs the response to anti-epidermal growth factor receptor (EGFR) antibody cetuximab in metastatic colorectal cancer patients. Br J Cancer. 2010;102(7):1137–1144. | ||
Zhang R, Xu GL, Li Y, et al. The role of insulin-like growth factor 1 and its receptor in the formation and development of colorectal carcinoma. Journal of International Medical Research. 2013;41(4):1228–1235. | ||
Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101(19):1308–1324. | ||
Bibeau F, Lopez-Crapez E, Di Fiore F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27(7):1122–1129. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.