Back to Archived Journals » Current Biomarker Findings » Volume 5
Biomarker discovery in systemic sclerosis: state of the art
Authors Bonella F, Patuzzo G, Lunardi C
Received 14 January 2015
Accepted for publication 8 April 2015
Published 8 July 2015 Volume 2015:5 Pages 47—68
DOI https://doi.org/10.2147/CBF.S60446
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hung Khong
Francesco Bonella,1 Giuseppe Patuzzo,2 Claudio Lunardi2
1Interstitial and Rare Lung Disease Unit, Ruhrlandklinik University Hospital, University of Duisburg-Essen, Essen, Germany; 2Department of Medicine, University of Verona, Verona, Italy
Abstract: Systemic sclerosis (SSc) is an autoimmune disease characterized by immune dysfunction and by abnormalities of the microvasculature with vascular obliteration, eventually leading to fibrosis of the skin, gastrointestinal tract, lungs, heart, and kidney. The etiology and pathogenesis of SSc remain unclear, despite recent significant progress in the field. Immune activation and microangiopathy are followed by widespread organ fibrosis, leading to organ failure and increased mortality. The production of inflammatory cytokines and growth factors after tissue injury, as well as the presence of circulating autoantibodies, provide a source of biomarkers with potential diagnostic and prognostic applications in the clinical routine. Two principal approaches exist to discover and characterize biomarkers. The proof-of-concept approach verifies the ability of known proteins, generally involved in the pathogenesis, to correlate with disease phenotype and outcome. A proteomic approach does not need prior knowledge of the proteins or of their function, but it requires high-performance and time-consuming techniques. In this review, we highlight the most recent findings in biomarkers used to characterize SSc organ involvement, to stratify the patients, and to assess the response to treatment.
Keywords: systemic sclerosis, biomarkers, proteomics, gene expression profiling
Introduction
Systemic sclerosis (SSc) is a rare and complex autoimmune disease that involves the connective tissue of skin and internal organs, with remarkable heterogeneity in the clinical features and course of the disease, resulting in high morbidity and mortality.1 The disease is characterized by severe and diffuse endothelial cell damage and by an overproduction and accumulation of collagen and other extracellular matrix (ECM) proteins, resulting in the thickening of the skin and fibrosis of the affected organs. The pathogenetic mechanisms involve three interactive components represented by vascular dysfunction and injury, immune system activation, and increased secretion of collagen by fibroblasts.1 The prevalence of SSc is between 50 and 300 cases per one million inhabitants, and an incidence ranging from 2.3 to 22.8 cases per million inhabitants per year in the general population.2
The complexity of the underlying pathogenetic mechanisms suggests that disease prediction may depend on a combinatorial analysis of many mediators rather than single molecules.3 As a sign of the times, the new classification criteria for SSc by the American College of Rheumatology/European League Against Rheumatism incorporate autoantibodies (Abs), specifically anti-Scl70, anti-RNA polymerase (RNA-pol) III, and anticentromere (ACA), as supportive tools for the classification of SSc.4 This is a milestone in the evolution of thinking regarding the role of serology in the clinical management of SSc. Multicohort studies have reported variable antibody frequency and distribution based on race, but the clinical associations remain very similar.5–10 It is worthwhile to point out that in the last 2 years, two new Abs have been identified through mass spectrometry (MS) and included in the serology panel of SSc.11,12
In a near future perspective, the application of new technologies like gene expression profiling and proteomic wide analysis will offer the opportunity to improve discrimination of SSc phenotypes and personalize treatment. Particularly promising are the proteomic studies, because proteome results from the interaction between genetic background and environmental factors, and it may be considered the signature of a disease, avoiding the limitations of RNA transcriptional and post-transcriptional changes.13
Several attempts have been made in the last years to identify biomarkers able to predict disease outcome and response to treatment, but the majority of these studies originate from single centers and need further validation in large cohorts.
Pathogenesis of SSc
The majority of biomarkers investigated in SSc are not only epiphenomena, but molecules involved in the pathogenesis of the disease. Therefore, an introduction to the pathogenetic mechanisms underlying SSc is necessary.
As for other autoimmune diseases, genetic and environmental factors play a pivotal role in the pathogenesis of the disease. Among environmental factors, infectious agents may be involved in the pathogenesis of the disease with different mechanisms. The molecular mimicry mechanism (ie, the similarity between autoantigens and proteins of viral origin) has been the most widely studied.14 It is worth mentioning that there is a certain structural similarity between retroviral proteins and topoisomerase I, the antigen recognized by anti-Sc1-70 Abs. Moreover, human cytomegalovirus may contribute to the onset of SSc through its ability to infect endothelial cells and to induce cell activation and apoptosis. Patients affected by SSc present with antibodies directed against the human cytomegalovirus-derived protein UL94; such antibodies are able to induce apoptosis of human endothelial cells and to stimulate fibroblast proliferation – two typical aspects of SSc.14–17
Endothelial cell damage is considered the primary event in the pathogenesis of SSc, leading to the increased expression of adhesion molecules; recruitment and activation of leucocytes; thickening of the basal lamina; proliferation of the pericytes, fibroblasts, and smooth muscle cells; platelet adhesion and activation; and formation of the perivascular infiltrate.18,19 Perivascular infiltrate is initially characterized by myeloid dendritic cells, by CD4+ T-lymphocytes, and by mastocytes, and subsequently by polymorphonucleates, B-lymphocytes, and T-lymphocytes.18,20–23 Growth factors, oxidative stress, and cytokines stimulate transdifferentiation of the fibroblasts into myofibroblasts within the vessel wall, with consequent thickening of the vessel wall itself and further reduction of the regional blood flow by narrowing the vessel lumen. This is exacerbated by activation of platelets and of the coagulation system, with the formation of microthrombi. Chronic perivascular inflammation, in advanced sclerodermic lesions, is replaced by fibrosis, which contributes to the permanent alteration of the vessel architecture, with thickening of the vessel wall, and progressive narrowing of the lumen. The composition of the ECM varies according to the stage of the disease: proteoglycans and elastic fibers such as fibrillin are typical of the first phases of the fibrotic process; while type I collagen is found in the advanced phases.
Irrespective of the progressive loss of capillaries and of the high plasma levels of vascular endothelial growth factor (VEGF) in response to tissue hypoxia, a deficient angiogenesis is observed.19 Many consequences of endothelial damage are related to an imbalance in vasoactive factors, with hyperproduction of endothelin (ET)-1 and hypoproduction of nitric oxide (NO) and prostacyclin. An imbalance of these mediators has an effect not only on vascular tone, but also on inflammation, platelet activation, myofibroblastic induction, and fibrosis.24,25
Tools and techniques used in biomarker discovery
In Table 1, the principal tools and techniques used in biomarker discovery are shown. For the sake of brevity, we will describe only some of the tools in detail.
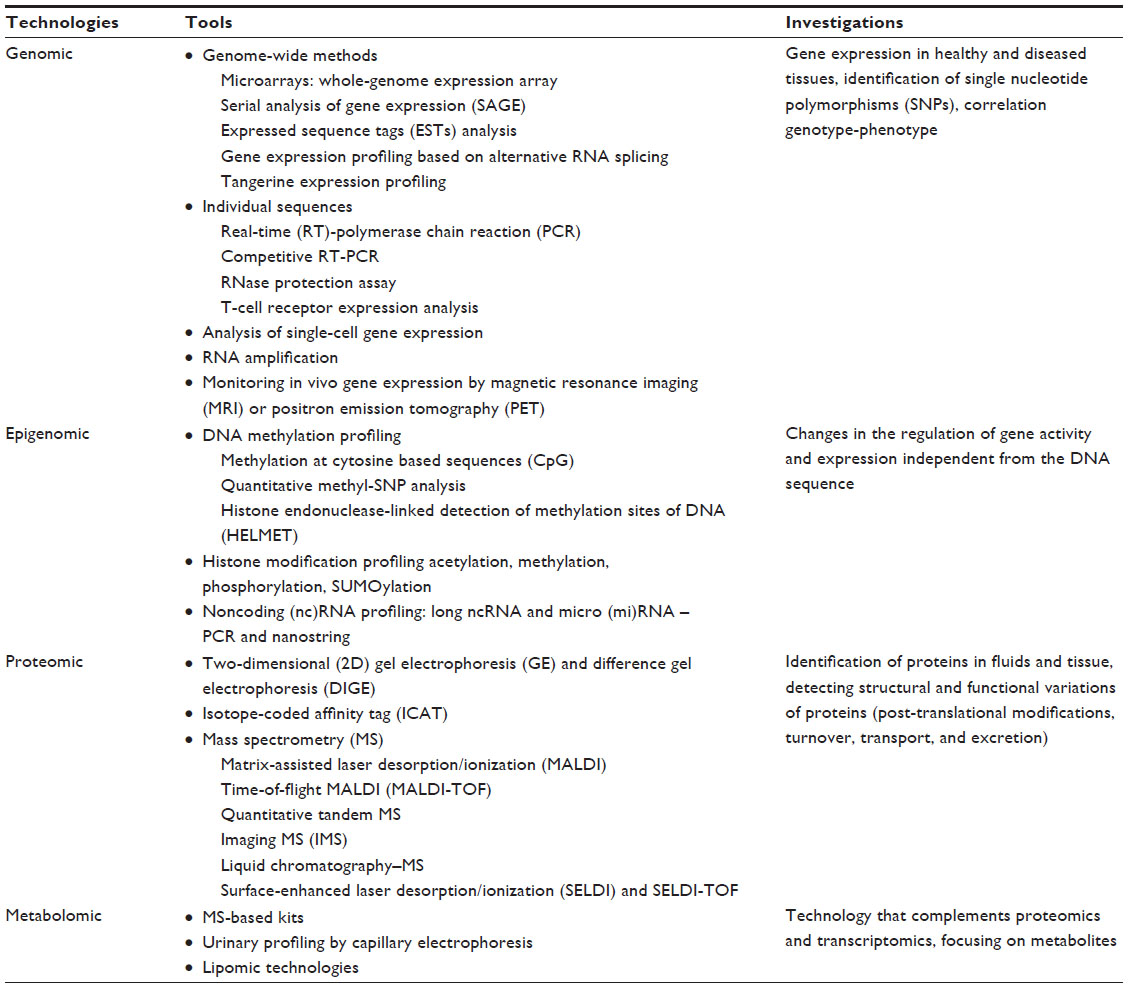 | Table 1 Principal tools and techniques used in biomarker discovery |
Gene expression and candidate gene studies
Gene expression profiling has become an increasingly applied tool, not only to reveal pathogenetic aspects of disease, but also to better characterize SSc subsets.26 In one of the first studies in this sense, Milano et al27 analyzed the genome-wide patterns of gene expression with DNA microarrays in skin biopsies from distinct scleroderma subsets (17 patients with diffuse SSc [dSSc], seven patients with limited cutaneous SSc [lSSc], three patients with morphea, and six healthy controls). Analysis by hierarchical clustering demonstrates nearly identical patterns of gene expression in 17 out of 22 of the forearm and back skin pairs of SSc patients. Distinct patterns of gene expression separate patients with dSSc from those with lSSc without overlapping with healthy controls. Moreover, a 177-gene signature was associated with the severity of skin disease in dSSc. In a further study,28 the same group reported a transforming growth factor (TGF)-β-responsive gene signature isolated from primary dermal fibroblasts, which was associated with increased disease severity in dSSc patients. Comparative studies with classical serum biomarkers are needed to confirm the value of gene signatures in SSc.
With regard to SSc-interstitial lung disease (ILD), the gene expression profiles of SSc fibrotic lung seem to differ significantly from idiopathic pulmonary fibrosis (IPF). Seibold et al29 found a strong correlation of polymorphism in the mucin 5B (MUC5B) promoter region with pulmonary fibrosis, and Putman et al30 found a correlation of the minor allele with fibrotic abnormalities in the general population. By contrast, no association was found for MUC5B with SSc-ILD in three different cohorts,31–33 suggesting different pathophysiology.
In the skin biopsies of 59 SSc patients from the Genetics versus Environment in Scleroderma Outcome Study (GENISOS),34 several transcripts including genes involved in extravasation and adhesion of inflammatory cells (the most representative, ELP, CCL2, and MMP3) correlated with the severity of ILD. In the GENISOS cohort, genome-wide association study (GWAS) was performed in samples from more than 5,000 individuals with SSc. A new susceptibility locus for SSc was demonstrated at CD247 and MHC, and IRF5 and STAT4 were confirmed as genetic risk factors for SSc.35 In a second study from the GENISOS cohort and two additional validation cohorts,36 single nucleotide polymorphisms (SNPs) from the three susceptibility loci obtained through GWAS were investigated. A SNP in the IRF5 promoter region (rs4728142), associated with lower IRF5 transcript levels, was predictive of longer survival and milder ILD in patients with SSc. Moreover, the IRF5 rs4728142 minor allele correlated with higher forced vital capacity (FVC)% predicted at enrolment (P=0.019).36
It is known that ethnicity may significantly impact disease traits. A recent GWAS study in a Han Chinese cohort of 387 SSc patients did not confirm the association between the rs2056626 of CD247 and SSc or any SSc subtypes.37 Moreover, another study demonstrated an association of gene polymorphisms in RHOB and FAM167A-BLK, which are proteins involved in intracellular protein trafficking, with susceptibility to SSc in a Chinese Han population.38 Interestingly, the loci of the SNPs in the RHOB region were reported to be different between Asian and Caucasian populations.39–41 The results of all these genetic association studies suggest, once again, the complex nature of genetics of SSc.
New data confirm that the gene expression profiles of SSc-ILD and SSc-pulmonary hypertension (PH) lung tissue are different. Hsu et al42 characterized the molecular fingerprint of lung tissue of 33 SSc patients who underwent lung transplantation. Fibrotic lungs were enriched not only for genes for collagen types I and III, IGFBPs, tissue inhibitors of metalloproteases 1 (TIMPs-1), and interferon gamma receptor (IFNGR), which are classically involved in fibrosis, but also for insulin growth factor (IGF). Conversely, SSc-PH lungs showed specific genes including chemokines (CCL-2, CXCL10) and HLA (-DR and -A) genes.42
Gene expression profiling of peripheral blood mononuclear cells (PBMCs) is a powerful and less invasive tool for the identification of biomarkers involved in disease processes.43 Comparing the transcript profiles of PBMCs from 30 patients with idiopathic PH, 19 SSc without PH, and 42 with SSc-PH, Cheadle et al43 found that a specific signature for erythrocyte maturation was enriched in patients with PH, suggesting a reaction to the increasing hypoxemia in this condition. A similar study on PBMC gene expression in 36 SSc patients reported the increased expression of nine genes (ICAM1, IFNGR1, IL1B, IL13Ra1, JAK2, AIF1, CCR1, ALAS2, and TIMP2) in lSSc-PH patients.44 Markers of vascular injury (VCAM-1, VEGF, and von Willebrand factor [vWF]) were also found to be elevated in the serum of SSc-PH patients.44
Proteomic approach
Multiplex protein technology has the potential to identify biomarkers for the differentiation, classification, and improved understanding of the pathogenesis of complex diseases. In comparison to the proof-of-concept approach, protein technology is time consuming and more expensive. This is probably one of the reasons why proteome-wide studies in SSc are limited.13,45,46
Proteome-wide analysis through protein chip array (generally, surface-enhanced laser desorption/ionization [SELDI-TOF]) followed by MS for the characterization of the peaks allows for the screening of fluids, tissues, and cells for a large number of putative transcripts at the same time. In the study cited earlier on plasma S100A8/A9 as a biomarker for SSc, the authors efficaciously used SELDI-TOF-MS to identify proteins differentially expressed in SSc patients and healthy controls.13 Similarly, the discovery and validation of serum CXCL4 as a biomarker for SSc was reached through a complex proteome-wide analysis.46
microRNA expression profiling
Epigenetic mechanisms, including alterations in DNA methylation, histone code modifications, and changes in microRNA (miRNA) expression levels, can be defined as the “missing bridge” in understanding the pathogenesis of SSc.47–50 miRNAs are highly stable small noncoding RNAs, which function in RNA silencing and post-transcriptional regulation of gene expression.51 Since they can be detected in biologic fluids and tissue, and given that they are resistant to variations in sample handling and temperature, miRNAs represent potential biomarkers. Recent reports have described the differential expression of numerous regulatory miRNAs in SSc mainly in skin fibroblasts,47 several of which are important for TGF-β pathways and for downstream signaling cascades (Table 2).47,52
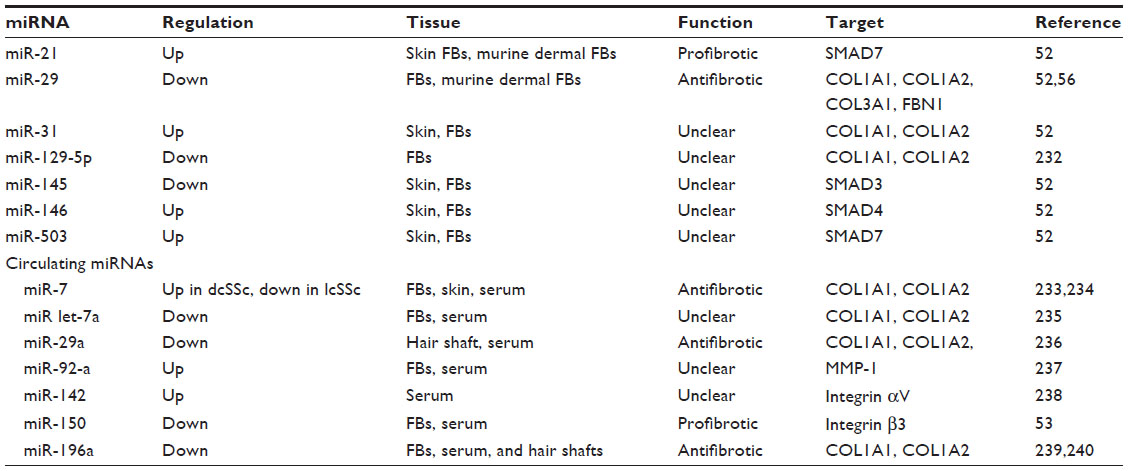 | Table 2 miRNAs investigated in SSc, their potential function, and molecular targets |
Among the investigated miRNAs, some of them are likely to correlate with disease subsets and severity. For example, SSc patients with lower serum miR-150 levels have more severe clinical manifestations, like digital ulcers (DUs) and PH, but not ILD.53 Similarly, patients with lower serum miR-196a levels show a significantly higher modified Rodnan Skin Score (mRSS) and a higher prevalence of pitting scars than patients with higher miR-196a levels.54
MiR-29 has been extensively studied, and its antifibrotic effects in several major fibrotic disorders, including SSc, are well known.55 The downregulation of miR-29 is correlated with the upregulation of COL1A1 messenger (m)RNA.52 Maurer et al56 identified miR-29 as a key regulator of collagen expression in SSc. Interestingly, SSc patients presenting with lower miR-29a levels had significantly higher right ventricular systolic pressure, as estimated by Doppler echocardiography, than those with normal miR-29a levels, suggesting a role in the pathogenesis of PH.57 The identification of specific miRNAs as markers of disease subsets and severity could lead, in the future, to the development of disease- and stage-specific targeted therapies.
Biomarkers for diagnosis, prognosis, and treatment response
Clinical manifestations and diagnosis of SSc
Raynaud’s phenomenon is usually the first manifestation of SSc and it may precede the development of other clinical features by months or years, depending on the subset of the disease (ie, the dSSc or lSSc form, respectively). From a clinical point of view, Raynaud’s phenomenon occurs when the delicate balance between vasodilatation and vasoconstriction is disturbed, in favor of vasoconstriction.58 Raynaud’s phenomenon is an exaggerated physiological response of the extremities to exposure to cold or emotional stress. Its classical presentation is characterized by the fingers turning white (ischemia), then blue (cyanosis) and, finally, red (reperfusion). Antinuclear antibodies are usually present at the time of Raynaud’s phenomenon onset. Indeed, the finding of a positive antinuclear antibody in a patient with Raynaud’s phenomenon suggests the need for further screening in order to identify SSc at an early stage.
Chronic ischemia leads to a reduction of the finger pad substance with consequent loss of small areas of tissue. DUs and digital gangrene are caused by a severe degree of ischemia. Ulcers that spontaneously occurs on the fingertips are due exclusively to ischemia, whereas those over the extensor surface of the proximal interphalangeal, metacarpophalangeal, ulnar styloid, and elbow joints are due to a combination of poor perfusion in areas of stretched skin or in areas of repeated minor trauma.59
The fibrosing process results in thickened skin; gastrointestinal dysmotility; heart involvement of the pericardium, myocardium, and cardiac conduction system; renal failure; and lung involvement with ILD and pulmonary arterial hypertension (PAH).60 Moreover, arthralgia and peripheral neuropathy due to nerve entrapment are frequently present.
The thickening of the skin is the hallmark feature of SSc. At the cutaneous level, fibrosis begins at the deep derma and extends to involve the entire derma and the more superficial layers. The earliest findings may be puffiness, swelling, and decreased flexibility to the joint and tendons.61 Skin thickening is frequently accompanied by hyperpigmentation, giving a salt-and-pepper appearance. As SSc advances to the fibrotic stage, the skin becomes more thickened until atrophy occurs.
The obliterative small-vessel vasculopathy is also responsible for PAH and scleroderma renal crisis (SRC). Usually, in SSc patients, PAH develops late in the course of the disease. It is more frequent in patients with lSSc than in patients with dSSc. Typically, the development of PAH is related to the presence of ACA antibodies and anti-Th/To antibody, which are associated with lSSc, and with anti-U3RNP antibodies.62–64 PAH may be initially silent. Early symptoms are nonspecific (a sense of generalized weakness on exertion) with dyspnea appearing later. Echocardiography plays a pivotal role in the follow-up of SSc patients in order to identify patients with possible PAH, and right-heart catheterization is the gold standard to confirm the diagnosis.
SRC was the most common cause of death in SSc patients prior to the introduction of angiotensin-converting enzyme inhibitors. It is characterized by an abrupt rise in blood pressure over days to weeks, as well as by rapidly progressive renal failure, if untreated. SRC occurs in the diffuse form of the disease. Risk factors include the use of corticosteroids and cyclosporine A, and the presence of anti-RNA-pol antibodies.65 Clinical signs and symptoms are those of severe hypertension and can include headaches, stroke, and heart failure. The creatinine and plasma levels of renin are elevated; urinalysis shows proteinuria and microscopic hematuria.
ILD is a frequent complication of SSc.66 In SSc patients, ILD most commonly occurs within 10 years after diagnosis and it may be seen in patients with both the limited and diffuse subsets of the disease. The presence of antitopoisomerase I antibodies (ATA) and anti-U11/U12 antibody is associated with an increased risk of ILD.63 Early lung disease is frequently asymptomatic; however, dry cough and dyspnea in a patient with SSc should raise the possibility of ILD, and a work up for ILD should be performed.
With regard to the diagnostic criteria, the new combined American College of Rheumatology (ACR)/European League Against Rheumatism clinical classification criteria,4 have been designed to improve the deficiencies of the earlier 1980 ACR clinical classification criteria using the advances in diagnostic techniques for Abs and nailfold videocapillaroscopy (NVC). Indeed, the new criteria also incorporate Abs – specifically, anti-Scl70 (or ATA), RNAP, and ACA – which provide support for the classification and subtypization of SSc.4
Autoantibody subsets and organ involvement in SSc
In Table 3, the Abs occurring with higher frequencies (>5%) in SSc patients and their association with organ involvement are reported. For frequently occurring antibodies, such as ACA and ATA, reliable detection systems based on enzyme-linked immunosorbent assay (ELISA) or other binding tests have been developed.6 For other antibodies like RNAPs, there are no standardized testing procedures, and the choice of the detection method is a critical point, impacting the sensitivity/specificity and reliability of the results.6
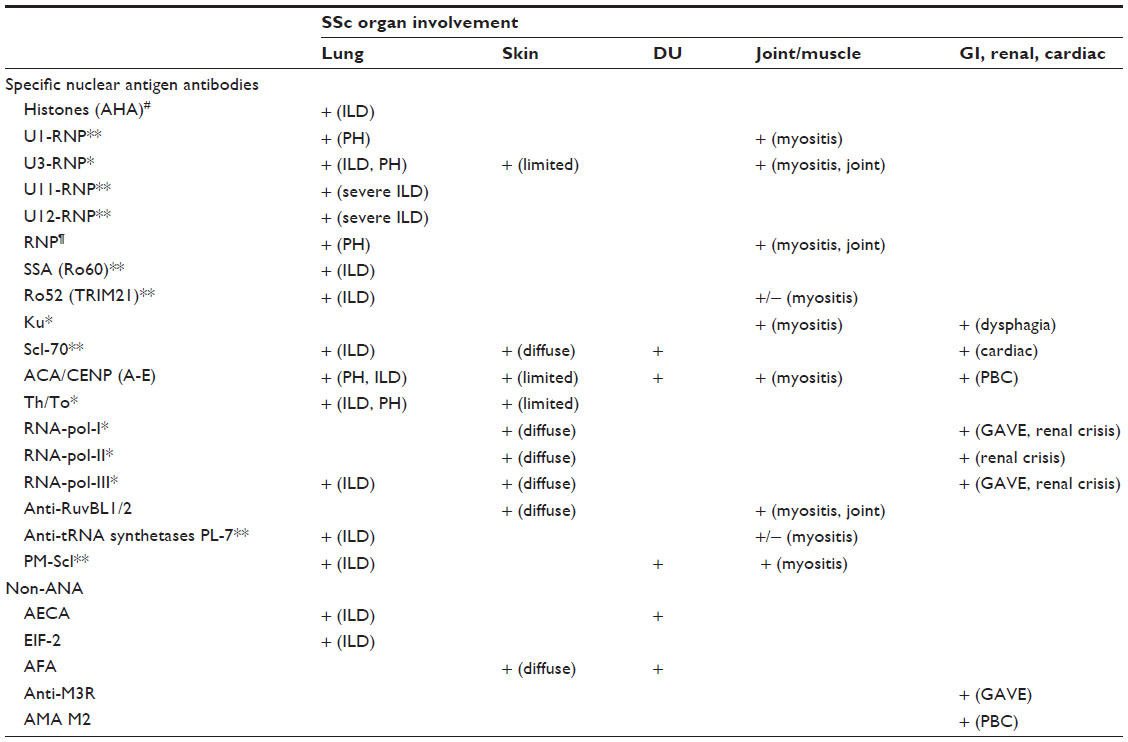 | Table 3 Specific nuclear antigen and nonantinuclear autoantibodies and organ involvement in SSc |
ATA have been consistently found to be associated with dSSc, pulmonary fibrosis, and severe renal crisis, while ACA are present in up to 90% of patients with lSSc and are linked to PH and, rarely, to ILD.6,67,68 ACA-positive patients show a better prognosis than ATA-positive patients.69
Serum Abs to small nuclear ribonucleoproteins (RNPs) have been found in patients with SSc and other connective tissue diseases (CTDs).63 Among the anti-RNP Abs, anti-U1 and anti-U3 (also called antifibrillarin) are the most frequent in SSc patients, while anti-U5 and anti-U4/U6 are rare.6,63 Lung fibrosis has been reported in 79% of the anti-U11/U12 RNP AB-positive SSc patients, while PH has been described in anti-U3-positive patients;70 moreover, anti-U11/U12 RNP-positive patients showed a higher mortality from pulmonary fibrosis than antibody-negative patients.63
Anti-RNA-pol I, II, and III Abs are associated with severe clinical manifestations of SSc, especially anti-RNA-pol III.71 Although a reduced incidence of lung fibrosis has been found in anti-RNA-pol III-positive patients,72 these Abs are strongly associated with the development of SRC and the progression of skin disease.71,73 Additionally, anti-RNA-pol Abs have been found to be associated with a twofold hazard ratio (HR) for the development of cancer within 36 months of the clinical onset of SSc.74
Antiendothelial cell Abs (AECA) are reported in 22%–86% of patients with SSc.75 These Abs are involved in the pathogenesis of autoimmune diseases with vascular involvement, but some AECA may bind to fibroblasts in vivo, triggering fibroblast activation.76 AECA positivity has also been noted in patients with other CTDs (rheumatoid arthritis [RA], systemic lupus erythematosus [SLE], and Wegener granulomatosis).75 Their presence has been associated with a higher frequency of pulmonary fibrosis and impairment of lung function tests, particularly the diffusing capacity of the lung for carbon monoxide (DLCO).77
Recently, a new set of Abs, called anti-RuvBL1/2, two highly conserved eukaryotic proteins implicated in transcription, DNA repair, and small nucleolar RNP assembly, has been associated with SSc in overlap with myositis; they occur with a frequency of 2% in Asian and North American populations.11
Biomarkers correlating with presence and extension of skin fibrosis
The pathogenetic mechanisms underlying dermal fibrosis include inflammation, fibroblast recruitment, differentiation, and the deposition of ECM, and they are paradigmatic for other organs involved in SSc.78–80
The gold standard for assessing SSc skin fibrosis is the mRSS, which is based on the determination of skin thickness at 17 distinct areas of the body (grading from 0 to 3 per area).81 The mRSS is widely used during evaluation in clinical routine, and as a surrogate endpoint in clinical trials.82 Despite the good intraobserver variability of the mRSS, its interobserver reproducibility is low.83 Therefore, biomarker investigations for skin and lung fibrosis in SSc are arising and include a broad spectrum of applications, ranging from the measurement of mediators in the peripheral blood and imaging (NVC), to gene expression profiling and proteomics.68 Several analytes have been identified as potential serum and plasma SSc biomarkers, mostly derived from matrix remodeling and fibrosis molecular pathways. Serum osteopontin (OPN), matrix metalloproteinase (MMP), and Krebs von den Lungen 6 (KL-6) have been found to correlate directly with the extent of skin fibrosis; moreover, adiponectin concentrations are lower in SSc patients with the diffuse cutaneous form (dcSSc). None of these analytes are currently used during clinical routine.68
OPN is a matrix protein with proinflammatory and profibrotic properties, binding to cell surface integrins and CD44; it modulates signaling in a wide variety of cell types, like fibroblasts and myofibroblasts.84 Wu et al85 demonstrated high plasma OPN levels in SSc patients, both with limited and diffuse disease, as compared to healthy controls. Recently, an OPN SNP, previously reported to be associated with SLE,86 has been found to be more frequent in SSc patients than in healthy controls, suggesting a role of OPN in SSc susceptibility.87
MMPs belong to the family of proteases, and they are enzymes capable of degrading all kinds of ECM proteins and processing a number of bioactive molecules. Serum levels of MMP-9 and MMP-12 have been found to be elevated in SSc patients with the dcSSc form, and they also correlate with mRSS, ILD, and the presence of DUs.88,89 In particular, MMP-12 has been found to be strongly expressed in keratinocytes, dermal endothelial cells, myofibroblasts, and inflammatory cells in the skin of patients with SSc, and it correlates with peripheral vascular damage.89
KL-6 is a glycoprotein belonging to MUC class I.90 KL-6 is mainly produced by regenerating pneumocytes type II and, in Japan, it is routinely used to assess the severity of lung fibrosis and other conditions. KL-6 is not only an epiphenomenon; it shows profibrotic and antiapoptotic effects on lung fibroblasts.91 The precise epitope structure recognized by the anti-KL-6 monoclonal (m)Ab is still unclear, but candidate carbohydrate epitopes for KL-6 mAb have been identified in O-linked glycans of MUC1 containing 6′sulfo-Gal/GalNAc.92 In a small cohort, Bonella et al93 found that KL-6, but not surfactant proteins (SP)-D, strongly correlated to skin involvement (mRSS) and a disease activity index. This finding led the authors to speculate that circulating levels of KL-6, similar to other pleonastic molecules, may directly contribute to chronic, persistent systemic fibrosis in a TGF-β-additive way in SSc.93
Adiponectin is a downstream target of PPAR-gamma, a nuclear receptor involved in obesity and metabolism. Serum adiponectin concentrations are measured in diabetic patients to assess response to treatment with thioglitazones.94 Recent studies found an inverse correlation between the extent of skin disease and serum adiponectin levels in SSc patients.95,96 Adiponectin was decreased in dcSSc patients when compared to healthy controls and other rheumatic diseases.96
Biomarkers associated with DUs
DUs in SSc are the main clinical manifestation of peripheral vascular disease and a severe complication requiring early medical intervention.97–99 High levels of serum placental growth factor, low counts of circulating endothelial progenitor cells (EPCs),100 and high levels of interleukin (IL)-6101 have been found to be independent predictors of the occurrence of new DUs. Elevated serum IL-6 in early dcSSc has also been shown to identify a subgroup of patients with more severe skin disease, ILD, and reduced survival.102
The Capillaroscopy Skin Ulcer Index was proposed in 2009 by Sebastiani et al,103 and it was recently validated in 229 SSc patients as a tool to identify patients with a high risk for developing DUs at 3 months from the initial evaluation, independently from the video capillaroscopy devices.104,105 A simpler score based on mean capillary loss at each finger has been also developed to predict digital trophic lesions in SSc patients with a follow-up of 6–12 months.106
The number of studies investigating possible correlations between NVC severity patterns and SSc disease activity are increasing. In a recent investigation of two independent cohorts, NVC patterns were found to predict novel future severe organ involvement with a stronger odds ratio according to the more severe NVC patterns (early, active, and late).107
Biomarkers of ILD
ILD occurs more often in SSc than in any other CTD and it is radiologically evident in up to two-thirds of patients with SSc. The most prevalent histologic pattern identified in SSc is fibrotic Nonspecific interstitial pneumonia followed by the usual interstitial pneumonia lung injury pattern.66
The Scleroderma Lung Study (SLS) and GENISOS collected the most significant cohorts aimed to identify biomarkers for predicting SSc-ILD and its outcomes.108,109 A number of Abs (Table 3) and serum proteins (Table 4) correlating with the presence or severity of ILD in the context of SSc have been investigated. Even if the majority of lung-derived biomarkers are shared between idiopathic ILD and SSc-ILD, some differences exist, reflecting the peculiar pathogenetic mechanisms of the two conditions.110,111
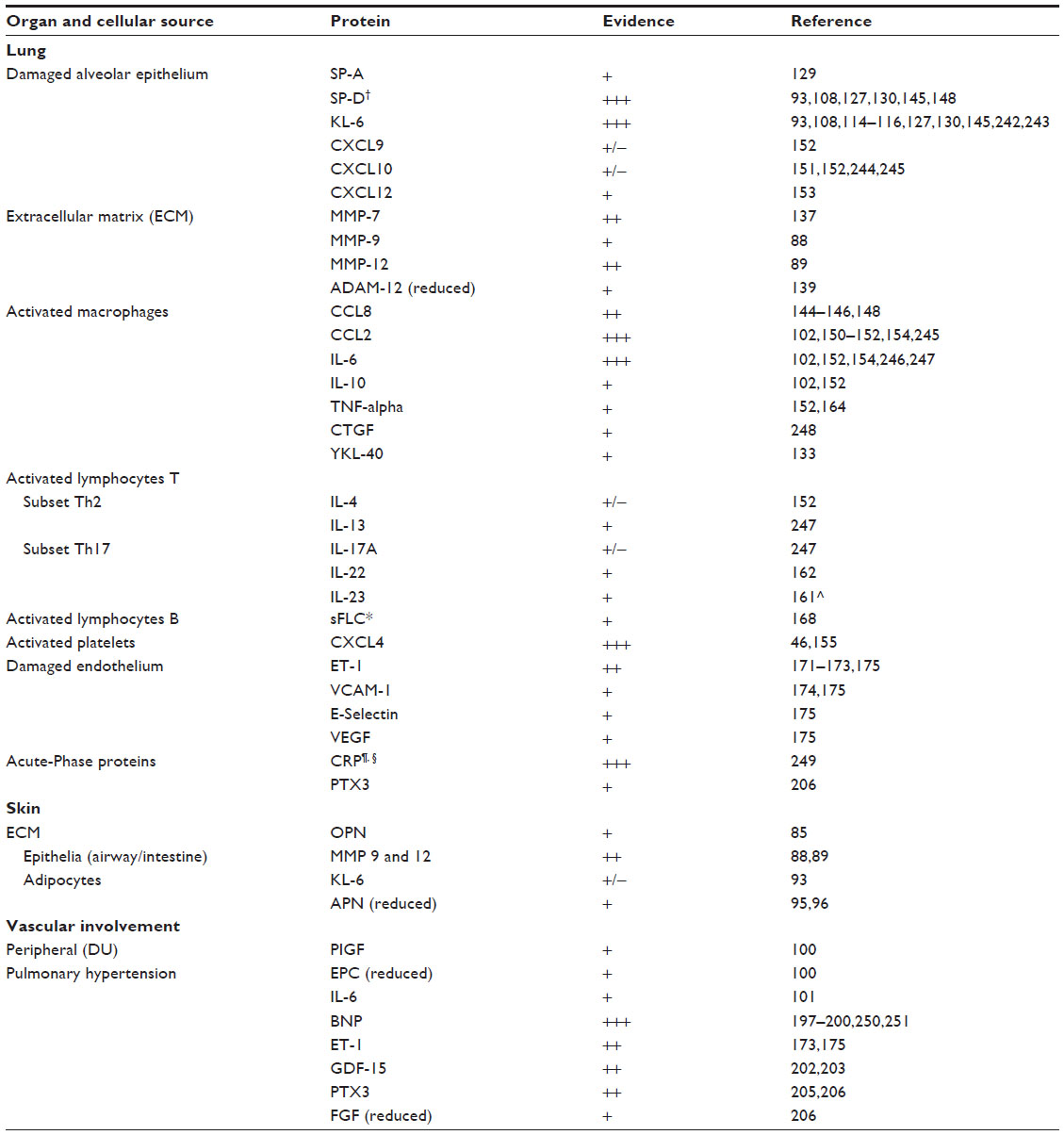 | Table 4 Circulating biomarkers associated with organ involvement in SSc and their cellular source |
Abs associated with the presence of lung fibrosis
While there is enough evidence that ATA positivity is associated with a greater risk of lung fibrosis, it remains unclear whether it is associated with more progressive ILD and further investigations are needed.
Serum Abs to small nuclear RNPs have been found not only in SSc patients, but also in other CTDs.63 Some Abs recognize individual RNPs, while others are directed against a complex of RNPs. Among them, anti-U1 and anti-U3 are the most frequent in SSc patients, while anti-U5 and anti-U4/U6 are rare.63 Lung fibrosis has been reported in 79% of the anti-U11/U12 RNP Ab-positive SSc patients; moreover, anti-U11/U12 RNP positivity seems to be linked to higher mortality in SSc-ILD patients.63 Despite more severe skin involvement, patients with anti-RNA-pol antibody postivity have significantly lower frequencies of telangiectasias, inflammatory myopathy, restrictive lung disease, and serious cardiac abnormalities than those with ATA positivity.72
AECA are a heterogeneous class of antibodies whose role in the pathogenesis of autoimmune diseases with vascular involvement has been extensively studied.75 Some AECA may directly activate fibroblasts and cause fibrosis.76 AECA positivity, reported in 22%–86% of patients with SSc, has also been seen in other CTDs.75 AECA have been associated with a higher frequency of ILD and lower levels of DLCO.77
Human leukocyte antigens haplotypes
Human leukocyte antigens (HLA), PTPN22, CTGF, TGF-β, and PDGF are established molecules involved in the pathophysiology of SSc112,113 but the number of studies investigating these molecules as biomarkers for SSc-ILD is limited.113 Odani et al113 reported that the prevalence of HLA-DRB5 gene carriers was increased in Japanese SSc patients with ILD relative to those without ILD or to healthy controls in both cohorts. The HLA-DRB5*01:05 allele was significantly more frequent in SSc patients with ILD than in SSc patients without ILD or in healthy controls.113 These associations were also confirmed in a validation cohort. The authors concluded that the HLA-DRB5*01:05 allele could be linked to ILD susceptibility in SSc patients.113
Lung epithelium derived proteins
Although the studies on lung epithelial derived proteins provided the largest evidence for these markers as predictors of SSc-ILD, there are no specific biomarkers to distinguish idiopathic ILD from SSc-ILD. Most of these proteins, especially KL-6, SP-D, and CCL-18, correlate with the extent of lung fibrosis and can predict short-term decline of lung function.
KL-6 (MUC1) (the general characteristics of KL-6 have been presented earlier) is probably the most established biomarker for the assessment of ILD, at least in the Japanese population.90 Serum KL-6 is elevated not only in idiopathic interstitial pneumonia, but also in ILD of known origin, including hypersensitivity pneumonitis, drug-induced pneumonitis, sarcoidosis, and CTD-ILD.90 KL-6 has been investigated in several studies in SSc-ILD patients,93,108,114–119 and its serum levels seem to correlate inversely with DLCO.93,108 The presence of the MUC-1 568 adenosine to guanine polymorphism (rs4072037) has been found to influence serum KL-6 levels in Caucasian and Japanese ethnicities.120,121 In the most recent study on KL-6 in SSc, Kumánovics et al119 assessed serum KL-6 levels over time from 173 SSc patients. The negative correlation of serum KL-6 with lung function parameters, independently of the time of investigation, as well with the probability of death were confirmed.119 However, no significant differences were seen in the deterioration and improvement rates between groups with normal and elevated KL-6 levels at baseline.119 The authors concluded that even if KL-6 seems to be useful as a marker of lung fibrosis in SSc, its role as disease activity marker for SSc remains questionable.119
Cube cell 16-kDa (CC16) is a low-molecular-weight protein produced by bronchiolar Cube cells122 and released in consequence to lung injury.122–124 Increased CC16 levels have also been found in serum and bronchoalveolar lavage fluid (BALF) of patients with sarcoidosis.122,125,126 Recently, Hasegawa et al127 compared the value of CC16, KL-6, and SP-D serum levels for the diagnosis and monitoring of pulmonary fibrosis SSc, SLE, and healthy controls. A CC16 level of 46.0 ng/mL or higher was diagnostic of ILD (sensitivity [Se]: 52%; specificity [Sp]: 89%), slightly inferior to KL-6 (Se: 85%; Sp: 85%), but comparable with that of SP-D (Se: 71%; Sp: 77%).127 Serum levels of CC16 correlated inversely with pulmonary function tests (FVC and DLCO) and disease activity in patients with SSc-ILD.127
SP-D and SP-A are secreted by type II alveolar epithelial cells and airway Cube cells; they contribute to stabilize the alveolar surface tension and play a pivotal role in the homeostasis of the host–defense system.128 Serum levels of SP-A and SP-D are significantly higher in SSc patients with ILD than in those without.93,129,130 Serum SP-D serum levels are negatively correlated with FVC and DLCO in SSc-ILD patients.129,130 In one study of 42 SSc patients, SP-D levels were more sensitive than SP-A in detecting ILD, as defined by high-resolution computed tomography (HRCT), but they are less specific (Se is 77% and Sp is 83% for SP-D versus 33% and 100% for SP-A).129 In 66 SSc patients from the SLS cohort, SP-D levels >90 ng/mL demonstrated good Se (89%) and Sp (80%) in the assessment of “alveolitis”, as defined by bronchoalveolar lavage (BAL) neutrophilia and/or HRCT ground glass density.108 A positive correlation between SP-D serum levels and ILD worsening has been described in a small prospective study of 35 patients with SSc-ILD followed up to 10 years.130 Moreover, Yanaba et al116 showed retrospectively that longitudinal SP-D levels were useful to assess the response to immunosuppressive therapy, decreasing by clinical improvement.
Chitinase-like protein YKL-40 is produced by activated macrophages and it acts as a growth factor for connective tissue cells. It is elevated in the sera of patients with diseases characterized by inflammation, tissue remodeling, or fibrosis.131 Serum and BALF YKL-40 levels are predictors of survival in IPF.132 YKL-40 is increased in the sera of SSc patients and correlates with airway obstruction, reduced DLCO, and poor prognosis.133 The presence of single nucleotide polymorphisms (SNPs) in the gene encoding YKL-40 (CHIl3-329 and CHII3-321) is associated with the interindividual variability of the levels of this marker in serum.132
Calcium-binding proteins S100A8 and S100A9 (also named calgranulin A, myeloid-related protein [MRP]8, and calgranulin B, MRP14) are low-molecular-weight members of the S100 family of calcium-binding proteins involved in the initiation and amplification of inflammation.134 They are located in the cytoplasm and nucleus of a wider range of cells. Xu et al134 found that plasma concentrations of S100A8 and S100A9 were significantly higher in dcSSc patients than in normal controls and lcSSc patients, but above all in those with dcSSc with lung and kidney involvement. Recently, van Bon et al13 confirmed in a double-cohort study that S100A8/A9 levels were elevated in the plasma of SSc patients, although more so in lcSSc than in dcSSc. This discrepancy between the findings of Xu et al134 and van Bon et al13 could be ascribed to ethnicity or to differences in the assay used. Since S100A8/A9 are considered to be ligands of the Toll-like receptor (TLR)-4, and since they are able to induce fibroblast proliferation and pulmonary fibrosis, van Bon et al13 conclude that the correlation between lung fibrosis and S100A8/A9 levels may not only be an epiphenomenon.
Biomarkers derived from ECM
Characteristics of MMPs and their inhibitors (TIMP) have been described. Among SSc patients, higher serum levels of MMP-7, an established prognostic biomarker for IPF,135 and MMP-9 (this one was also elevated in BALF) were found in patients with ILD when compared to those without, and they were associated with lower DLCO. The association of these MMPs with ILD progression was not evaluated in these studies.88,136,137
Circulating levels of MMP-12 have been reported to correlate with FVC (r=−0.82), and the expression of MMP-12 is significantly increased in SSc-ILD lungs when compared to normal controls.89 TIMP-1 levels were found to be associated with the presence of ILD in 62 SSc patients and to correlate inversely with DLCO.138
A disintegrin and metalloproteinase (ADAM)12 is one of the MMP-type ADAMs and exerts extracellular proteinase and cell-binding functions. ADAM12 is expressed in two alternative forms, which are a membrane-anchored form (ADAM12-L) and a short secreted form (ADAM12-S).139 Taniguchi et al139 measured serum ADAM12-S levels in 61 SSc patients and found a negative correlation with HRCT fibrosis score and a positive one with the ground glass score, indicating interstitial inflammation. ADAM12-S levels showed a positive correlation with FVC% predicted, but not with DLCO. On the basis of these results, the authors concluded that serum ADAM12-S seems to be more linked to inflammation than fibrosis.139
In a recent study of Oka et al,140 MMP-1, MMP-8, MMP-9, and TIMP-3 were upregulated in the sera of 25 CTD patients (RA, SSc, and polymyositis/dermatomyositis) with acute-onset ILD. In contrast, MMP-3 and TIMP-2 were downregulated. The ratio of MMP-3 to MMP-1 serum levels, reduced in patients who died, has been proposed by the authors as a predictor of poor outcomes in these patients.140 These results need validation in a larger cohort of CTD-ILD patients.
Lysyl oxidase-like (LOXL) enzymes are proteins necessary for stability of the ECM, cross-linking type I collagen fibrils. LOXL-2 is highly expressed in the fibrotic lungs of IPF patients and is implicated in maintaining the fibrotic microenvironment.141,142 A Phase II clinical trial with simtuzumab (GS-6624), a mAb targeting soluble LOXL-2, is currently ongoing in IPF (NLM identifier: NCT01769196). Serum levels of LOXL-2 have also been found to be significantly elevated in SSc, particularly in patients with dcSSc disease.143 Interestingly, serum LOXL-2 concentration correlated with the mRSS in patients without lung fibrosis.143 Whether targeting anti-LOXL-2 could be a potential therapeutic strategy for SSc-ILD should be addressed by a specific clinical trial.
Chemokines and ligands
Among the studied chemokines, CCL18 and CXCL4 are those with the largest evidence as biomarkers for detecting lung fibrosis and for assessing lung disease severity. While CCL18 shows some predictive value for ILD progression, but is not specific for SSc, CXCL4 strongly correlates with the presence of fibrosis in SSc patients.
CCL18, previously known as pulmonary and activation-regulated chemokine, is constitutively expressed at high levels in the lungs, mainly by alveolar macrophages, and is chemotactic for a variety of mononuclear cells.144 CCL18 is profibrotic and has been found increased in the serum and BALF of IPF and SSc-ILD patients.144 Three longitudinal studies in a total of 116 patients with SSc-ILD investigated the correlation of serum CCL18 with ILD activity, as determined by HRCT, lung function, and BAL analysis, with promising results.144–146 Tiev et al146 analyzed serum CCL18 levels in 83 SSc patients with ILD over a 4-year follow-up period. At a cutoff of 187 ng/mL, serum CCL18 was independently predictive of ILD worsening (HR: 5.4; P=0001).146 These results were similar to those by Prasse et al147 in IPF patients. However, Elhaj et al148 compared the predictive significance of CCL18 and SP-D for the disease outcomes of SSc-ILD in the GENISOS cohort, in 266 patients.109,148 For 83 patients, it was possible to collect a complete follow-up for biomarkers over 4 years. In contrast to the previous studies,144,146 Elhaj et al148 did not show a significant predictive significance of baseline serum CCL18 for long-term changes in FVC. Moreover, the correlation between baseline serum CCL18 and concomitant or 1-year follow-up FVC or DLCO was not confirmed. A subgroup analysis based on disease type (limited/diffuse) or ATA positivity did not yield significant results for short- or long-term predictive values of SP-D or CCL18.148
CCL2, CXCL10, and CXCL12 were found to be upregulated in the serum of SSc patients and to correlate with the presence of ILD.149–153 CCL2 concentrations were also found to be elevated in BALF and they were associated with the presence of ILD, correlating with lung function parameters and computed tomography scores.154
CXCL4 is an angiogenic and profibrotic factor that inhibits the expression of the antifibrotic cytokine interferon-γ and upregulates profibrotic cytokines IL-4 and IL-13.155 A large multicohort study of 779 SSc patients (462 with lcSSc and 317 with dcSSc) demonstrated that CXCL4 is the predominant protein secreted by plasmacytoid dendritic cells in SSc, both in circulation and in the skin.46 CXCL4 levels correlated with skin and lung fibrosis and with PAH. Moreover, only CXCL4 among the studied biomarkers (CCL2, CXCL10, CCL5, vWF, and CCL18) predicted the risk and progression of SSc.
ILs and their ligands
ILs are cytokines with pleiotropic function, and they cannot be considered specific of organ involvement. Among ILs investigated in SSc patients with lung fibrosis, IL-6 seems to be the most promising for application in clinical routine to assess disease severity and to predict outcomes.
Elevated serum and BALF levels of IL-6, a cytokine linked to the Th2 lymphocyte subset, have been described in several lung diseases.156–158 Recently, the prognostic value of serum IL-6 was assessed in a large cohort of 212 patients with SSc-ILD.102 In the exploratory analysis, only serum IL-6 among IL-8, IL-10, CCL2, CXCL10, VEGF, fibroblast growth factor (FGF)-2, and CX3CL1 was an independent predictor of DLCO decline in both IPF and SSc-ILD. At a cutoff of 7.7 pg/mL, serum IL-6 was predictive of a decline in FVC (HR: 2.58) and in DLCO (HR: 3.2) within the 1st year, and predictive of death within the first 30 months (HR: 2.69).102
Wuttge et al159 found that serum levels of another pleiotropic cytokine, IL-15, a survival and growth factor for T- and B-lymphocytes, were strongly correlated with impaired lung function and ILD in SSc.
Levels of IL-17A and IL-23, connected to the Th17 lymphocyte subset, are elevated in SSc and associated with the presence of ILD.160,161 Circulating IL-22- and IL-17-producing T-cells are increased in patients with SSc-ILD when compared to those without.162 These findings support the hypothesis that Th22 may also be involved in pathological processes leading to SSc; the increased frequency of Th22 cells appears to be of interest as a biomarker in SSc.162
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL), a member of the TNF family of cytokines linked with apoptosis, is involved in the pathophysiology of some autoimmune diseases and cancer.163 Mean serum TRAIL levels were found to be significantly higher in SSc patients than in RA patients and healthy controls (P<0.001). Moreover, serum TRAIL levels correlated with the HRCT score.164
Biomarkers from activated B-cells
It is increasingly thought that humoral immunity and B-cells play an important role in the pathophysiology of SSc, producing specific Abs and profibrotic cytokines.165,166 SSc patients have abnormal blood B-cell compartments with an expansion of naïve B-cells and activated memory B-cells.167 In a recent study by Lanteri et al,168 levels of free light chains of immunoglobulins (sFLC) were assessed in the serum from 134 SSc patients and 401 healthy controls. In comparison to other markers of B-cell activation (immunoglobulin [Ig]G, IgA, beta2-microglobulin, and B-cell activating factor), sFLC serum levels correlated with mRSS and were independently associated with the presence of ILD and its severity.168 Since sFLC can bind antigens or activate some cells, such as mast cells, involved in the pathogenesis of lung fibrosis,169 these findings suggest that sFLC could have a direct role in the pathogenesis of SSc-ILD.168
Biomarkers derived from endothelial cells
Endothelial cell injury is involved in the pathogenesis of fibrosis.170 ETs are potent peptides regulating both cell proliferation and turnover of the ECM. Increased ET-1 levels have been found in BALF and in lung tissue from patients with SSc.171,172 Increased serum levels of soluble vascular cell adhesion molecule-1, soluble E-selectin, VEGF, and ET-1 have been described in patients with SSc and lung fibrosis.173–175 AECA have been discussed earlier.
Circulating cells as biomarkers
Circulating endothelial cells (CECs) and EPCs have been studied in different conditions characterized by vasculopathy. High levels of CECs and EPCs have been reported in patients with SSc and seem to correlate with disease activity, with DUs, and with PAH. Moreover EPCs are higher in the early stages of the disease, suggesting an attempt to repair endothelial cell damage and to favor revascularization.176,177
Fibrocytes are circulating precursors for fibroblasts. The fibrocytes, and their CD14-positive mononuclear precursors, are recruited from the blood to organs through multiple chemokine signaling pathways (particularly, the CCL2/CCR2 and CXCL12/CXCR4 pathways).178,179 Fibrocytes have been found increased in the blood of patients with IPF, ranging from 2.7% of the nucleated cells in stable IPF to 14.5% in the acute exacerbation of IPF (1% fibrocytes are present in healthy subjects).180 Recently, Borie et al181 detected fibrocytes in the alveolar space in 55% of SSc patients through BAL. Since fibrocyte detection was not associated with the capacity to grow fibroblasts from BALF in cell culture, the physiological role and their interaction with circulating fibrocytes remain unclear.181
BAL cell profiles
BAL differential cell counts in SSc are of questionable diagnostic and prognostic value.182 Nevertheless, a study on 73 SSc patients showed an association with BAL cellularity and the ILD pattern.183 A positive correlation between BAL neutrophils, the extent of disease on HRCT, and poor survival has also been described in SSc-ILD.184–186 In the study by Goh et al,187 BAL cellular profiles in 141 patients with SSc-ILD did not show a prognostic value for disease progression, once disease severity had been quantified by pulmonary function tests and HRCT. BAL neutrophilia, independent of disease severity, was associated with early mortality (within 2 years), but not with long-term prognosis.
In the SLS cohort, BAL cell counts were performed in 201 patients as part of a randomized placebo-controlled trial of cyclophosphamide versus placebo.188 Abnormal cellularity was present in 101 patients, and a subgroup was identified with more severe lung function, more extensive ground glass opacity, and more extensive fibrosis in the right middle lobe. The cell differential was not an independent predictor of disease progression or response to cyclophosphamide at 1 year.189
Nailfold capillary patterns and ILD
NVC is a noninvasive diagnostic tool that permits the detection of the main local microvascular alterations in SSc, characterized by enlargement of the capillary loops, a loss of capillaries, disruption of the orderly appearance of the capillary bed, and distortion of the capillaries.190 Two studies found a positive correlation between the vascular deletion score in NVC and skin thickness, the presence of anti-Scl-70 antibodies, signs of peripheral ischemia, esophageal dysfunction, and pulmonary disease.191 Recently, Corrado et al192 highlighted the differences in the nailfold pattern between SSc-ILD and IPF. They pointed out that capillaroscopic changes in patients with lung fibrosis could indicate the first signs of a systemic disease involving the lungs, thus requiring particular attention in detecting the appearance of immunological/clinical signs of CTD.192
Biomarkers of PH associated with SSc
Pulmonary (arterial) hypertension in SSc (SSc-PH) can occur as a primary lung manifestation in association with ILD and/or as a consequence of left-sided heart disease. Patients with combined SSc-ILD and PH have a worse prognosis in comparison to those with ILD alone, with a 3-year survival of 47%.193 A severe discrepancy in the reduction in DLCO compared to lung volumes and the development of hypoxia can be early signs of PH.194,195 Since echocardiography has low Sp for the diagnosis of PH,196 and given that right-sided cardiac catheterization is invasive and cannot be used routinely for monitoring purposes, several serum biomarkers have been investigated. Among the studied biomarkers, brain natriuretic peptide (BNP) is widely applied in the clinical routine for detecting right ventricular dysfunction and indirectly monitoring PH.
Natriuretic peptides include BNP and N-terminal pro-BNP (NT-pro-BNP), and they are released in response to ventricular stretch by cardiomyocytes. Natriuretic peptides have been established as markers of right ventricular dysfunction in PH.197,198 BNP was also found to be associated with poor prognosis in patients with chronic lung diseases.199 In a study of 90 ILD patients − including 18 CTD-ILD − Corte et al200 found that serum BNP correlated with pulmonary pressures estimated by echocardiography. Furthermore, a serum BNP ≥20 pmol/L was associated with a 14-fold increased mortality compared to patients with BNP <4 pmol/L.200 However, since natriuretic peptide levels are released in response to right ventricle strain, markers of earlier pulmonary vasculopathy are still needed.
ET-1 has been proposed as a biomarker for both vascular involvement and lung fibrosis development in patients with SSc.173,175 However, Camargo et al showed that serum levels of ET-1 and vWF (a carrier protein for coagulation factor VIII), which can predict the development of pulmonary hypertension in SSc patients, were not correlated with SSc disease stage.201
Growth differentiation factor (GDF)-15 is a secreted member of the TGF-β cytokine superfamily and its levels are elevated in the serum of patients with cardiovascular diseases. Meadows et al202 demonstrated that patients with SSc-PH had significantly higher mean circulating levels of GDF-15 in plasma compared with patients with SSc without PH (422±369.5 pg/mL versus 108±193 pg/mL, respectively; P=0.004). GDF-15 levels correlated positively with estimated right ventricular systolic pressure on echocardiogram and plasma levels of the amino terminal propeptide form of BNP.202 GDF-15 levels >125 pg/mL were associated with reduced survival. In a further study of 119 SSc patients, increased GDF-15 serum levels correlated with disease activity and the extent of lung fibrosis, including lung function, at follow-up.203
Pentraxin 3 (PTX3) is a multifunctional pattern recognition protein that can suppress FGF-2, has nonredundant roles in antimicrobial innate immunity, and is a component of the ECM, contributing to fibrosis.204 In a recent study from a Japanese cohort of 171 SSc patients, Shirai et al205 found PTX3 elevated in the serum of SSc patients who had DUs or PH, while the FGF-2 concentration was reduced. Multivariate analysis selected PTX3 as an independent parameter for the presence of DUs and PAH.205 Since, in a previous study, a correlation between elevated PTX3 and pulmonary fibrosis, but not PH,206 was seen, the study by Shirai et al205 is the first to address serum PTX3 as a biomarker for PH in SSc.
Biomarkers for liver involvement
Primary biliary cirrhosis (PBC) is the most common liver disease usually associated with scleroderma.207 It is a chronic liver disease characterized by cholestasis and immune-mediated nonsuppurative cholangitis, mainly affecting interlobular and septal bile ducts.208 PBC usually presents with pruritus followed by progressive jaundice. Antimitochondrial antibodies (AMA) are diagnostic.208 The reported prevalence of clinically evident PBC among patients with SSc is 2%–2.5%.209,210 The prevalence of AMA positivity among SSc patients is estimated to be 5%,210 and ACA are present in 25% of patients with PBC. However, a high prevalence of AMA-negative PBC in patients with SSc has also been described.211 Dual isotype ELISA, which detects multiple mitochondrial and nuclear Abs specific for PBC, has been developed to enhance the detection of PBC in patients that were found to be AMA negative by immunofluorescence.212 Prince et al213 demonstrated that AMA can precede clinical symptoms of PBC with a median time of 5 years; therefore, patients require long-term monitoring of liver function tests and abdominal ultrasound.213
Multiple biomarker assessment
SSc is a biologically complex disease; its multiple pathogenetic patterns suggest that the best predictive tool should come from the combination of many mediators.3 The multibiomarker disease activity tool for RA is based on 12 biomarkers and was found to be consistently associated with clinical disease activity at baseline and over time, and to correlate with damage progression, as measured by radiography.214,215
Several attempts have been made to compare or combine different biomarkers at the same time for assessing disease activity and organ involvement in SSc, but mostly with a limited number of molecules.93,118,129
Beirne et al216 investigated a panel of 30 circulating biomarkers in order to differentiate between the presence or absence of lung fibrosis in 20 SSc patients, as compared with healthy controls and sarcoidosis patients. Interestingly, only a 17-analyte biomarker model correctly classified 90% of study individuals to their respective disease group, and another 14-biomarker panel correctly identified the presence of lung fibrosis in SSc patients.216 Although promising, these results need validation in a large cohort.
Biomarkers for prognosis and treatment response
The management of a patient with SSc depends greatly on disease subset, duration, and organ involvement. Full clinical assessment, including serological profile, capillaroscopy, gastroenteric assessment, chest X-ray and high-resolution tomography, pulmonary function testing, echocardiography, and electrocardiography are mandatory in every new SSc patient.
The therapeutic goals in SSc include minimization of damage from early inflammation and autoimmunity, restoration of vascular homeostasis, promotion of repair of structural connective tissue, and resolution of scarring. Treatment depends on the active problems, and immunosuppressive agents are used when there is active progressive skin or organ disease. A pivotal role is played by the local and general treatment of DUs and skin ulcers, which greatly limit the quality of life of the patients.217 Critical digital ischemia or recurrent DUs are now treated with phosphodiesterase type 5 inhibitors, prostacyclin analogs, and ET receptor antagonists. The currently used immunosuppressive drugs for SSc act on two important aspects of the pathogenesis of the disease – inflammation and fibrosis – although, nonspecific drugs able to block or revert fibrosis have been identified so far.218 Cyclophosphamide is the only immunosuppressive agent that has been shown to be of benefit for SSc-associated lung fibrosis and skin disease. Retrospective cohort analyses and prospective open-label studies have suggested that mycophenolate mofetil may improve skin disease and lung fibrosis. In end-stage disease, lung transplantation represents a viable therapeutic option to consider for patients who have limited extrapulmonary manifestations, and for those with severe PAH when ET-1 receptor antagonists (bosentan, macitentan, ambrisentan), phosphodiesterase-5 inhibitors (sildenafil and tadalafil), the soluble guanylate cyclase stimulator, riociguat, and prostanoids (epoprostenol, treprostinil, and iloprost) have failed.219
Among biologics, rituximab – an anti-CD20 mAb – has emerged as a potential treatment for SSc, improving lung function and skin scores in pilot and prospective open-label studies.
Autologous hematopoietic stem cell transplantation was investigated in a small series of patients with dcSSc who obtained a sustained improvement of skin fibrosis and stabilization of major organ function. Its utility still needs to be proven in larger clinical trials.
Although numerous exploratory studies have been published on serum and genetic markers to predict disease outcomes,68 markers to assess the response to treatment, or to drive clinical decisions, have been poorly investigated.
In a recent study of 173 SSc patients from a single center, Kumánovics et al120 investigated the predictive value of KL-6 serum levels for the outcome of ILD, including the response to treatment with cyclophosphamide. Serum levels of KL-6 significantly decreased in patients receiving cyclophosphamide treatment in spite of the fact that the spirometry results (FVC and DLCO) did not show a significant change.120
In a recent small prospective study, plasma ET-1 and NO were assessed at baseline and after 24 weeks of treatment with bosentan in ten SSc patients with PH.220 Plasma ET-1 levels were significantly decreased after 24 weeks of bosentan therapy (P<0.0001), and ET-1 levels of SSc patients with PH decreased to a level comparable to that in patients without PH. Although a decrement in plasma ET-1 was induced by bosentan, plasma ET-1 levels remained high when compared with those of healthy controls.220 These findings reflect the necessity to interpret biomarkers, not on the basis of absolute cutoffs, but considering the percentage changes from baseline in single patients.
Since repeating blood sampling over time can be problematic in advanced SSc patients, noninvasive biomarkers, like exhaled compounds, are alluring. NO, a key cellular mediator, is both a powerful endogenous vasodilator and a highly reactive biomarker of inflammation. NO is produced in high quantities during alveolar inflammation.221 It has been shown that exhaled NO is related to the presence and severity of SSc-ILD.222,223 A recent study by Tiev et al224 assessed the efficacy of immunosuppressive treatment in 19 nonsmoking SSc-ILD patients, by measuring changes of exhaled NO over time after six cycles of cyclophosphamide. Patients with high levels of exhaled NO (>8.5 ppb), were the most responsive to cyclophosphamide therapy, reflecting that alveolar inflammation identifies SSc patients with a greater chance of benefitting from cyclophosphamide treatment.
Gene expression profiling has been also introduced as a tool to assess response to treatment in SSc. Chung et al225 compared gene expression patterns in two SSc patients before and after 1 month of therapy with imatinib mesylate, a tyrosine kinase inhibitor. A set of 1,050 genes modified by imatinib in both patients was demonstrated, involving multiple functional pathways (eg, cell proliferation, matrix and vascular remodeling, and growth factor signaling). Another recent, prospective study by Hinchcliff et al226 analyzed the molecular profile of skin biopsies in SSc patients treated with mycophenolate mofetil (MMF). Of the seven patients treated with MMF, four were improvers and three nonimprovers. A specific, baseline 321-gene expression signature was identified in the skin and was found to correlate with mRSS improvement during MMF treatment, but only in responders.226 DNA microarray seems to be promising in identifying potential responders to treatment, but it still needs further investigation in a larger cohort.
True perspectives: personalized medicine in SSc
The clinical management of patients with SSc remains a challenge and it currently involves known and newly proposed therapeutic approaches. Indeed, deeper knowledge of the cellular and molecular regulators of vasculopathy, immune response, and fibrosis has led to the identification of novel targets that possibly interrupt the vascular damage and the fibrotic process in SSc.227–229
We suggest that the development, validation and application of some of the biomarkers we have described, singly or more likely in combination, will have a deep impact on SSc, and will play a pivotal role in early diagnosis, classification, and management, as well as in the design of clinical trials with novel therapeutic agents.
It is worthwhile to mention that a novel treatment directed against integrins has been shown to control fibrosis in an animal model of SSc.230 However, combination therapy may be necessary to control the complex biological network active in SSc, leading to a control of disease progression. Finally, the knowledge of the genetic background and of mechanisms that play a major role in each patient may lead to an individually tailored treatment.
Disclosure
The authors report no conflicts of interest in this work.
References
Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360(19):1989–2003. | |
Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum. 2008;37(4):223–235. | |
Bonella F, Caramaschi P. The ambitious goal of validating prognostic biomarkers for systemic sclerosis-related interstitial lung disease. J Rheumatol. 2013;40(7):1034–1036. | |
van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–2747. | |
Domsic RT. Scleroderma: the role of serum autoantibodies in defining specific clinical phenotypes and organ system involvement. Curr Opin Rheumatol. 2014;26(6):646–652. | |
Mierau R, Moinzadeh P, Riemekasten G, et al. Frequency of disease-associated and other nuclear autoantibodies in patients of the German Network for Systemic Scleroderma: correlation with characteristic clinical features. Arthritis Res Ther. 2011;13(5):R172. | |
Sujau I, Ng CT, Sthaneshwar P, et al. Clinical and autoantibody profile in systemic sclerosis: baseline characteristics from a West Malaysian cohort. Int J Rheum Dis. 2015;18(4):459–465. | |
Hashimoto A, Endo H, Kondo H, et al. Clinical features of 405 Japanese patients with systemic sclerosis. Mod Rheumatol. 2012;22:272–279. | |
Hasegawa M, Hatta Y, Matsushita T, et al. Clinical and laboratory features dependent on age at onset in Japanese systemic sclerosis. Mod Rheumatol. 2013;23:913–919. | |
Vanthuyne M, Smith V, De Langhe E, et al. The Belgian Systemic Sclerosis Cohort: correlations between disease severity scores, cutaneous subsets, and autoantibody profile. J Rheumatol. 2012;39:2127–2133. | |
Kaji K, Fertig N, Medsger TA Jr, et al. Autoantibodies to RuvBL1 and RuvBL2: a novel systemic sclerosis-related antibody associated with diffuse cutaneous and skeletal muscle involvement. Arthritis Care Res (Hoboken). 2014;66(14):575–584. | |
Betteridge Z, Woodhead F, Bunn C, et al. Anti-EIF2B: a novel interstitial lung disease associated autoantibody in patients with systemic sclerosis [abstract]. Arthritis Rheum. 2012;64 Suppl 10:834. | |
van Bon L, Cossu M, Loof A, et al. Proteomic analysis of plasma identifies the Toll-like receptor agonists S100A8/A9 as a novel possible marker for systemic sclerosis phenotype. Ann Rheum Dis. 2014;73(8):1585–1589. | |
Dolcino M, Puccetti A, Barbieri A, et al. Infections and autoimmunity: role of human cytomegalovirus in autoimmune endothelial cell damage. Lupus. 2015;24(4-5):419–432. | |
Lunardi C, Bason C, Navone R, et al. Systemic sclerosis immunoglobulin G autoantibodies bind the human cytomegalovirus late protein UL94 and induce apoptosis in human endothelial cells. Nat Med. 2000;6(10):1183–1186. | |
Namboodiri AM, Rocca KM, Pandey JP. IgG antibodies to human cytomegalovirus late protein UL94 in patients with systemic sclerosis. Autoimmunity. 2004;37(3):241–244. | |
Lunardi C, Dolcino M, Peterlana D, et al. Antibodies against human cytomegalovirus in the pathogenesis of systemic sclerosis: a gene array approach. PLoS Med. 2006;3(1):e2. | |
Prescott RJ, Freemont AJ, Jones CJ, Hoyland J, Fielding P. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol. 1992;166(3):255–263. | |
Kuwana M, Okazaki Y, Yasuoka H, Kawakami Y, Ikeda Y. Defective vasculogenesis in systemic sclerosis. Lancet. 2004;364(9434):603–610. | |
Ishikawa O, Ishikawa H. Macrophage infiltration in the skin of patients with systemic sclerosis. J Rheumatol. 1992;19(8):1202–1206. | |
Kräling BM, Maul GG, Jimenez SA. Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology. 1995;63(1):48–56. | |
Whitfield ML, Finlay DR, Murray JI, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A. 2003;100(21):12319–12324. | |
Roumm AD, Whiteside TL, Medsger TA Jr, Rodnan GP. Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification, subtyping, and clinical correlations. Arthritis Rheum. 1984;27(6):645–653. | |
Liakouli V, Cipriani P, Marrelli A, Alvaro S, Ruscitti P, Giacomelli R. Angiogenic cytokines and growth factors in systemic sclerosis. Autoimmun Rev. 2011;10(10):590–594. | |
Pattanaik D, Brown M, Postlethwaite AE. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105–125. | |
Sargent JL, Whitfield ML. Capturing the heterogeneity in systemic sclerosis with genome-wide expression profiling. Expert Rev Clin Immunol. 2011;7(4):463–473. | |
Milano A, Pendergrass SA, Sargent JL, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3(7):e2696. | |
Sargent JL, Milano A, Bhattacharyya S, et al. A TGFbeta-responsive gene signature is associated with a subset of diffuse scleroderma with increased disease severity. J Invest Dermatol. 2010;130(3):694–705. | |
Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364(16):1503–1512. | |
Putman RK, Rosas IO, Hunninghake GM. Genetics and early detection in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014; 189(7):770–778. | |
Peljto AL, Steele MP, Fingerlin TE, et al. The pulmonary fibrosis-associated MUC5B promoter polymorphism does not influence the development of interstitial pneumonia in systemic sclerosis. Chest. 2012;142(6):1584–1588. | |
Borie R, Crestani B, Dieude P, et al. The MUC5B variant is associated with idiopathic pulmonary fibrosis but not with systemic sclerosis interstitial lung disease in the European Caucasian population. PLoS One. 2013;8(8):e70621. | |
Stock CJ, Sato H, Fonseca C, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68(5):436–441. | |
Assassi S, Wu M, Tan FK, et al. Skin gene expression correlates of severity of interstitial lung disease in systemic sclerosis. Arthritis Rheum. 2013;65(11):2917–2927. | |
Radstake TR, Gorlova O, Rueda B, et al; Spanish Scleroderma Group. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet. 2010;42(5):426–429. | |
Sharif R, Mayes MD, Tan FK, et al. IRF5 polymorphism predicts prognosis in patients with systemic sclerosis. Ann Rheum Dis. 2012;71(7):1197–1202. | |
Wang J, Assassi S, Guo G, et al. Clinical and serological features of systemic sclerosis in a Chinese cohort. Clin Rheumatol. 2013;32(5):617–621. | |
Shu C, Du W, Mao X, et al. Possible single-nucleotide polymorphism loci associated with systemic sclerosis susceptibility: a genetic association study in a Chinese Han population. PLoS One. 2014;9(12):e113197. | |
Gourh P, Agarwal SK, Martin E, et al. Association of the C8orf13-BLK region with systemic sclerosis in North-American and European populations. J Autoimmun. 2010;34(2):155–162. | |
Allanore Y, Saad M, Dieudé P, et al. Genome-wide scan identifies TNIP1, PSORS1C1, and RHOB as novel risk loci for systemic sclerosis. PLoS Genet. 2011;7(7):e1002091. | |
Bossini-Castillo L, Martin JE, Broen J, et al; Spanish Scleroderma Group. Confirmation of TNIP1 but not RHOB and PSORS1C1 as systemic sclerosis risk factors in a large independent replication study. Ann Rheum Dis. 2013;72(4):602–607. | |
Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63(3):783–794. | |
Cheadle C, Berger AE, Mathai SC, et al. Erythroid-specific transcriptional changes in PBMCs from pulmonary hypertension patients. PLoS One. 2012;7(4):e34951. | |
Pendergrass SA, Hayes E, Farina G, et al. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS One. 2010;5(8):e12106. | |
Aden N, Shiwen X, Aden D, et al. Proteomic analysis of scleroderma lesional skin reveals activated wound healing phenotype of epidermal cell layer. Rheumatology (Oxford). 2008;47(12):1754–1760. | |
van Bon L, Affandi AJ, Broen J, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. 2014; 370(5):433–443. | |
Babalola O, Mamalis A, Lev-Tov H, Jagdeo J. The role of microRNAs in skin fibrosis. Arch Dermatol Res. 2013;305(9):763–776. | |
Altorok N, Almeshal N, Wang Y, Kahaleh B. Epigenetics, the holy grail in the pathogenesis of systemic sclerosis. Rheumatology (Oxford). Epub April 16, 2014. | |
Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol. 2014;26(6):615–620. | |
Broen JC, Radstake TR, Rossato M. The role of genetics and epigenetics in the pathogenesis of systemic sclerosis. Nat Rev Rheumatol. 2014; 10(11):671–681. | |
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. | |
Zhu H, Li Y, Qu S, et al. MicroRNA expression abnormalities in limited cutaneous scleroderma and diffuse cutaneous scleroderma. J Clin Immunol. 2012;32(3):514–522. | |
Honda N, Jinnin M, Kira-Etoh T, et al. miR-150 down-regulation contributes to the constitutive type I collagen overexpression in scleroderma dermal fibroblasts via the induction of integrin β3. Am J Pathol. 2013;182(1):206–216. | |
Makino T, Jinnin M, Etoh M, et al. Down-regulation of microRNA-196a in the sera and involved skin of localized scleroderma patients. Eur J Dermatol. 2014;24(4):470–476. | |
Zhu S, Pan W, Qian Y. MicroRNA in immunity and autoimmunity. J Mol Med (Berl). 2013;91(9):1039–1050. | |
Maurer B, Stanczyk J, Jüngel A, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010;62(6):1733–1743. | |
Kawashita Y, Jinnin M, Makino T, et al. Circulating miR-29a levels in patients with scleroderma spectrum disorder. J Dermatol Sci. 2011;61(1):67–69. | |
Herrick AL. The pathogenesis, diagnosis and treatment of Raynaud phenomenon. Nat Rev Rheumatol. 2012;8(8):469–479. | |
Galluccio F, Matucci-Cerinic M. Registry evaluation of digital ulcers in systemic sclerosis. Int J Rheumatol. 2010;2010:363679. | |
Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis-a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014;10(7):390–402. | |
Eisenberg ME, Nguyen BY, Karnath BM. Clinical Features of Systemic Sclerosis. Available from: http://www.turner-white.com/memberfile.php?PubCode=hp_jan08_sclerosis.pdf. Accessed January 14, 2015. | |
Tormey VJ, Bunn CC, Denton CP, Black CM. Anti-fibrillarin antibodies in systemic sclerosis. Rheumatology (Oxford). 2001;40(10):1157–1162. | |
Fertig N, Domsic RT, Rodriguez-Reyna T, et al. Anti-U11/U12 RNP antibodies in systemic sclerosis: a new serologic marker associated with pulmonary fibrosis. Arthritis Rheum. 2009;61(7):958–965. | |
Mitri GM, Lucas M, Fertig N, Steen VD, Medsger TA. A comparison between anti-Th/To- and anticentromere antibody-positive systemic sclerosis patients with limited cutaneous involvement. Arthritis Rheum. 2003;48(1):203–209. | |
Shanmugam VK, Steen VD. Renal disease in scleroderma: an update on evaluation, risk stratification, pathogenesis and management. Current Opinion in Rheumatology. 2012;24(6):669–676. | |
Olson AL, Brown KK, Fischer A. Connective tissue disease-associated lung disease. Immunol Allergy Clin North Am. 2012;32(4):513–536. | |
Grassegger A, Pohla-Gubo G, Frauscher M, Hintner H. Autoantibodies in systemic sclerosis (scleroderma): clues for clinical evaluation, prognosis and pathogenesis. Wien Med Wochenschr. 2008;158(1–2):19–28. | |
Castelino FV, Varga J. Current status of systemic sclerosis biomarkers: applications for diagnosis, management and drug development. Expert Rev Clin Immunol. 2013;9(11):1077–1090. | |
Hesselstrand R, Scheja A, Shen GQ, Wiik A, Akesson A. The association of antinuclear antibodies with organ involvement and survival in systemic sclerosis. Rheumatology (Oxford). 2003;42(4):534–540. | |
Aggarwal R, Lucas M, Fertig N, Oddis CV, Medsger TA. Anti-U3 RNP autoantibodies in systemic sclerosis. Arthritis Rheum. 2009;60(4):1112–1118. | |
Cavazzana I, Ceribelli A, Angela C, et al. Anti-RNA polymerase III antibodies: a marker of systemic sclerosis with rapid onset and skin thickening progression. Autoimmun Rev. 2009;8(7):580–584. | |
Okano Y, Steen VD, Medsger TA. Autoantibody reactive with RNA polymerase III in systemic sclerosis. Ann Intern Med. 1993;119(10):1005–1013. | |
Nguyen B, Assassi S, Arnett FC, Mayes MD. Association of RNA polymerase III antibodies with scleroderma renal crisis. J Rheumatol. 2010;37(5):1068; author reply 1069. | |
Moinzadeh P, Fonseca C, Hellmich M, et al. Association of anti-RNA polymerase III autoantibodies and cancer in scleroderma. Arthritis Res Ther. 2014;16(1):R53. | |
Mihai C, Tervaert JW. Anti-endothelial cell antibodies in systemic sclerosis. Ann Rheum Dis. 2010;69(2):319–324. | |
Hill MB, Phipps JL, Cartwright RJ, Milford Ward A, Greaves M, Hughes P. Antibodies to membranes of endothelial cells and fibroblasts in scleroderma. Clin Exp Immunol. 1996;106(3):491–497. | |
Ihn H, Sato S, Fujimoto M, et al. Characterization of autoantibodies to endothelial cells in systemic sclerosis (SSc): association with pulmonary fibrosis. Clin Exp Immunol. 2000;119(1):203–209. | |
Abraham DJ, Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol. 2005;26(11):587–595. | |
Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117(3):557–567. | |
Varga J, Whitfield ML. Transforming growth factor-beta in systemic sclerosis (scleroderma). Front Biosci (Schol Ed). 2009;1:226–235. | |
Clements P, Lachenbruch P, Siebold J, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol. 1995;22(7):1281–1285. | |
Clements PJ, Hurwitz EL, Wong WK, et al. Skin thickness score as a predictor and correlate of outcome in systemic sclerosis: high-dose versus low-dose penicillamine trial. Arthritis Rheum. 2000;43(11):2445–2454. | |
Clements PJ, Lachenbruch PA, Seibold JR, et al. Skin thickness score in systemic sclerosis: an assessment of interobserver variability in 3 independent studies. J Rheumatol. 1993;20(11):1892–1896. | |
Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R. Osteopontin expression is required for myofibroblast differentiation. Circ Res. 2008;102(3):319–327. | |
Wu M, Schneider DJ, Mayes MD, et al. Osteopontin in systemic sclerosis and its role in dermal fibrosis. J Invest Dermatol. 2012;132(6):1605–1614. | |
Wong CK, Lit LC, Tam LS, Li EK, Lam CW. Elevation of plasma osteopontin concentration is correlated with disease activity in patients with systemic lupus erythematosus. Rheumatology (Oxofrd). 2005;44(5):602–606. | |
Barizzone N, Marchini M, Cappiello F, et al. Association of osteopontin regulatory polymorphisms with systemic sclerosis. Hum Immunol. 2011;72(10):930–934. | |
Kim WU, Min SY, Cho ML, et al. Elevated matrix metalloproteinase-9 in patients with systemic sclerosis. Arthritis Res Ther. 2005;7(1):R71–R79. | |
Manetti M, Guiducci S, Romano E, et al. Increased serum levels and tissue expression of matrix metalloproteinase-12 in patients with systemic sclerosis: correlation with severity of skin and pulmonary fibrosis and vascular damage. Ann Rheum Dis. 2012;71(6):1064–1072. | |
Ishikawa N, Hattori N, Yokoyama A, Kohno N. Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir Investig. 2012;50(1):3–13. | |
Ohshimo S, Yokoyama A, Hattori N, Ishikawa N, Hirasawa Y, Kohno N. KL-6, a human MUC1 mucin, promotes proliferation and survival of lung fibroblasts. Biochem Biophys Res Commun. 2005;338(4):1845–1852. | |
Seko A, Ohkura T, Ideo H, Yamashita K. Novel O-linked glycans containing 6′-sulfo-Gal/GalNAc of MUC1 secreted from human breast cancer YMB-S cells: possible carbohydrate epitopes of KL-6(MUC1) monoclonal antibody. Glycobiology. 2012;22(2):181–195. | |
Bonella F, Volpe A, Caramaschi P, et al. Surfactant protein D and KL-6 serum levels in systemic sclerosis: correlation with lung and systemic involvement. Sarcoidosis Vasc Diffuse Lung Dis. 2011;28(1):27–33. | |
Shetty S, Kusminski CM, Scherer PE. Adiponectin in health and disease: evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol Sci. 2009;30(5):234–239. | |
Lakota K, Wei J, Carns M, et al. Levels of adiponectin, a marker for PPAR-gamma activity, correlate with skin fibrosis in systemic sclerosis: potential utility as biomarker? Arthritis Res Ther. 2012; 14(3):R102. | |
Arakawa H, Jinnin M, Muchemwa FC, et al. Adiponectin expression is decreased in the involved skin and sera of diffuse cutaneous scleroderma patients. Exp Dermatol. 2011;20(9):764–766. | |
Kowal-Bielecka O, Landewé R, Avouac J, et al; EUSTAR Co-Authors. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR). Ann Rheum Dis. 2009;68(5):620–628. | |
Matucci-Cerinic M, Allanore Y, Czirják L, et al. The challenge of early systemic sclerosis for the EULAR Scleroderma Trial and Research group (EUSTAR) community. It is time to cut the Gordian knot and develop a prevention or rescue strategy. Ann Rheum Dis. 2009;68(9):1377–1380. | |
Dobrota R, Mihai C, Distler O. Personalized medicine in systemic sclerosis: facts and promises. Curr Rheumatol Rep. 2014;16(6):425. | |
Avouac J, Meune C, Ruiz B, et al. Angiogenic biomarkers predict the occurrence of digital ulcers in systemic sclerosis. Ann Rheum Dis. 2012;71(3):394–399. | |
Khan K, Xu S, Nihtyanova S, et al. Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Ann Rheum Dis. 2012;71(7):1235–1242. | |
De Lauretis A, Sestini P, Pantelidis P, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J Rheumatol. 2013;40(4):435–446. | |
Sebastiani M, Manfredi A, Colaci M, et al. Capillaroscopic skin ulcer risk index: a new prognostic tool for digital skin ulcer development in systemic sclerosis patients. Arthritis Rheum. 2009;61(5):688–694. | |
Sebastiani M, Manfredi A, Vukatana G, et al. Predictive role of capillaroscopic skin ulcer risk index in systemic sclerosis: a multicentre validation study. Ann Rheum Dis. 2012;71(1):67–70. | |
Sebastiani M, Manfredi A, Lo Monaco A, et al. Capillaroscopic Skin Ulcers Risk Index (CSURI) calculated with different videocapillaroscopy devices: how its predictive values change. Clin Exp Rheumatol. 2013;31(2 Suppl 76):115–117. | |
Smith V, De Keyser F, Pizzorni C, et al. Nailfold capillaroscopy for day-to-day clinical use: construction of a simple scoring modality as a clinical prognostic index for digital trophic lesions. Ann Rheum Dis. 2011;70(1):180–183. | |
Smith V, Riccieri V, Pizzorni C, et al. Nailfold capillaroscopy for prediction of novel future severe organ involvement in systemic sclerosis. J Rheumatol. 2013;40(12):2023–2028. | |
Hant FN, Ludwicka-Bradley A, Wang HJ, et al; Scleroderma Lung Study Research Group. Surfactant protein D and KL-6 as serum biomarkers of interstitial lung disease in patients with scleroderma. J Rheumatol. 2009;36(4):773–780. | |
Assassi S, Sharif R, Lasky RE, et al; GENISOS Study Group. Predictors of interstitial lung disease in early systemic sclerosis: a prospective longitudinal study of the GENISOS cohort. Arthritis Res Ther. 2010;12(5):R166. | |
Baraut J, Michel L, Verrecchia F, Farge D. Relationship between cytokine profiles and clinical outcomes in patients with systemic sclerosis. Autoimmun Rev. 2010;10(2):65–73. | |
Murray LA, Rubinowitz A, Herzog EL. Interstitial lung disease: is interstitial lung disease the same as scleroderma lung disease? Curr Opin Rheumatol. 2012;24(6):656–662. | |
Delgado-Vega A, Sánchez E, Löfgren S, Castillejo-López C, Alarcón-Riquelme ME. Recent findings on genetics of systemic autoimmune diseases. Curr Opin Immunol. 2010;22(6):698–705. | |
Odani T, Yasuda S, Ota Y, et al. Up-regulated expression of HLA-DRB5 transcripts and high frequency of the HLA-DRB5*01:05 allele in scleroderma patients with interstitial lung disease. Rheumatology (Oxford). 2012;51(10):1765–1774. | |
Yamane K, Ihn H, Kubo M, et al. Serum levels of KL-6 as a useful marker for evaluating pulmonary fibrosis in patients with systemic sclerosis. J Rheumatol. 2000;27(4):930–934. | |
Yanaba K, Hasegawa M, Hamaguchi Y, Fujimoto M, Takehara K, Sato S. Longitudinal analysis of serum KL-6 levels in patients with systemic sclerosis: association with the activity of pulmonary fibrosis. Clin Exp Rheumatol. 2003;21(4):429–436. | |
Yanaba K, Hasegawa M, Takehara K, Sato S. Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis. J Rheumatol. 2004;31(6):1112–1120. | |
Doishita S, Inokuma S, Asashima H, et al. Serum KL-6 level as an indicator of active or inactive interstitial pneumonitis associated with connective tissue diseases. Intern Med. 2011;50(23):2889–2892. | |
Kumánovics G, Minier T, Radics J, Pálinkás L, Berki T, Czirják L. Comprehensive investigation of novel serum markers of pulmonary fibrosis associated with systemic sclerosis and dermato/polymyositis. Clin Exp Rheumatol. 2008;26(3):414–420. | |
Kumánovics G, Görbe E, Minier T, Simon D, Berki T, Czirják L. Follow-up of serum KL-6 lung fibrosis biomarker levels in 173 patients with systemic sclerosis. Clin Exp Rheumatol. 2014;32(6 Suppl 86):S-138–S-144. | |
Janssen R, Kruit A, Grutters JC, Ruven HJ, Gerritsen WB, van den Bosch JM. The mucin-1 568 adenosine to guanine polymorphism influences serum Krebs von den Lungen-6 levels. Am J Respir Cell Mol Biol. 2006;34(4):496–499. | |
Horimasu Y, Hattori N, Ishikawa N, et al. Different MUC1 gene polymorphisms in German and Japanese ethnicities affect serum KL-6 levels. Respir Med. 2012;106(12):1756–1764. | |
Janssen R, Sato H, Grutters JC, et al. Study of Clara cell 16, KL-6, and surfactant protein-D in serum as disease markers in pulmonary sarcoidosis. Chest. 2003;124(6):2119–2125. | |
Bernard A, Hermans C, Van Houte G. Transient increase of serum Clara cell protein (CC16) after exposure to smoke. Occup Environ Med. 1997;54(1):63–65. | |
Petrek M, Hermans C, Kolek V, Fialová J, Bernard A. Clara cell protein (CC16) in serum and bronchoalveolar lavage fluid of subjects exposed to asbestos. Biomarkers. 2002;7(1):58–67. | |
Hermans C, Petrek M, Kolek V, et al. Serum Clara cell protein (CC16), a marker of the integrity of the air-blood barrier in sarcoidosis. Eur Respir J. 2001;18(3):507–514. | |
Kucejko W, Chyczewska E, Naumnik W, Ossolinska M. Concentration of surfactant protein D, Clara cell protein CC-16 and IL-10 in bronchoalveolar lavage (BAL) in patients with sarcoidosis, hypersensivity pneumonitis and idiopathic pulmonary fibrosis. Folia Histochem Cytobiol. 2009;47(2):225–230. | |
Hasegawa M, Fujimoto M, Hamaguchi Y, et al. Use of serum clara cell 16-kDa (CC16) levels as a potential indicator of active pulmonary fibrosis in systemic sclerosis. J Rheumatol. 2011;38(5):877–884. | |
Pastva AM, Wright JR, Williams KL. Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc Am Thorac Soc. 2007;4(3):252–257. | |
Takahashi H, Kuroki Y, Tanaka H, et al. Serum levels of surfactant proteins A and D are useful biomarkers for interstitial lung disease in patients with progressive systemic sclerosis. Am J Respir Crit Care Med. 2000;162(1):258–263. | |
Asano Y, Ihn H, Yamane K, et al. Clinical significance of surfactant protein D as a serum marker for evaluating pulmonary fibrosis in patients with systemic sclerosis. Arthritis Rheum. 2001;44(6):1363–1369. | |
Johansen JS. Studies on serum YKL-40 as a biomarker in diseases with inflammation, tissue remodelling, fibroses and cancer. Dan Med Bull. 2006;53(2):172–209. | |
Korthagen NM, van Moorsel CH, Barlo NP, et al. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir Med. 2011;105(1):106–113. | |
Nordenbaek C, Johansen JS, Halberg P, et al. High serum levels of YKL-40 in patients with systemic sclerosis are associated with pulmonary involvement. Scand J Rheumatol. 2005;34(4):293–297. | |
Xu X, Wu WY, Tu WZ, et al. Increased expression of S100A8 and S100A9 in patients with diffuse cutaneous systemic sclerosis. A correlation with organ involvement and immunological abnormalities. Clin Rheumatol. 2013;32(10):1501–1510. | |
Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest. 2013;143(5):1422–1429. | |
Andersen GN, Nilsson K, Pourazar J, et al. Bronchoalveolar matrix metalloproteinase 9 relates to restrictive lung function impairment in systemic sclerosis. Respir Med. 2007;101(10):2199–2206. | |
Moinzadeh P, Krieg T, Hellmich M, et al. Elevated MMP-7 levels in patients with systemic sclerosis: correlation with pulmonary involvement. Exp Dermatol. 2011;20(9):770–773. | |
Kikuchi K, Kubo M, Sato S, Fujimoto M, Tamaki K. Serum tissue inhibitor of metalloproteinases in patients with systemic sclerosis. J Am Acad Dermatol. 1995;33(6):973–978. | |
Taniguchi T, Asano Y, Akamata K, et al. Serum levels of ADAM12-S: possible association with the initiation and progression of fibrosis and interstitial lung disease in patients with systemic sclerosis. J Eur Acad Dermatol Venereol. 2013;27(6):747–753. | |
Oka S, Furukawa H, Shimada K, et al. Serum biomarker analysis of collagen disease patients with acute-onset diffuse interstitial lung disease. BMC Immunol. 2013;14:9. | |
Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16(9):1009–1017. | |
Chien JW, Richards TJ, Gibson KF, et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur Respir J. 2014;43(5):1430–1438. | |
Rimar D, Rosner I, Nov Y, et al. Brief report: lysyl oxidase is a potential biomarker of fibrosis in systemic sclerosis. Arthritis Rheumatol. 2014;66(3):726–730. | |
Prasse A, Pechkovsky DV, Toews GB, et al. CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum. 2007;56(5):1685–1693. | |
Kodera M, Hasegawa M, Komura K, Yanaba K, Takehara K, Sato S. Serum pulmonary and activation-regulated chemokine/CCL18 levels in patients with systemic sclerosis: a sensitive indicator of active pulmonary fibrosis. Arthritis Rheum. 2005;52(9):2889–2896. | |
Tiev KP, Hua-Huy T, Kettaneh A, et al. Serum CC chemokine ligand-18 predicts lung disease worsening in systemic sclerosis. Eur Respir J. 2011;38(6):1355–1360. | |
Prasse A, Probst C, Bargagli E, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(8):717–723. | |
Elhaj M, Charles J, Pedroza C, et al. Can serum surfactant protein D or CC-chemokine ligand 18 predict outcome of interstitial lung disease in patients with early systemic sclerosis? J Rheumatol. 2013;40(7):1114–1120. | |
Hasegawa M, Sato S, Takehara K. Augmented production of chemokines (monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1alpha (MIP-1alpha) and MIP-1beta) in patients with systemic sclerosis: MCP-1 and MIP-1alpha may be involved in the development of pulmonary fibrosis. Clin Exp Immunol. 1999;117(1):159–165. | |
Carulli MT, Handler C, Coghlan JG, Black CM, Denton CP. Can CCL2 serum levels be used in risk stratification or to monitor treatment response in systemic sclerosis? Ann Rheum Dis. 2008;67(1):105–109. | |
Antonelli A, Ferri C, Fallahi P, et al. CXCL10 (alpha) and CCL2 (beta) chemokines in systemic sclerosis – a longitudinal study. Rheumatology (Oxford). 2008;47(1):45–49. | |
Hasegawa M, Fujimoto M, Matsushita T, Hamaguchi Y, Takehara K, Sato S. Serum chemokine and cytokine levels as indicators of disease activity in patients with systemic sclerosis. Clin Rheumatol. 2011;30(2):231–237. | |
Campioni D, Lo Monaco A, Lanza F, et al. CXCR4 pos circulating progenitor cells coexpressing monocytic and endothelial markers correlating with fibrotic clinical features are present in the peripheral blood of patients affected by systemic sclerosis. Haematologica. 2008; 93(8):1233–1237. | |
Schmidt K, Martinez-Gamboa L, Meier S, et al. Bronchoalveoloar lavage fluid cytokines and chemokines as markers and predictors for the outcome of interstitial lung disease in systemic sclerosis patients. Arthritis Res Ther. 2009;11(4):R111. | |
Romagnani P, Maggi L, Mazzinghi B, et al. CXCR3-mediated opposite effects of CXCL10 and CXCL4 on TH1 or TH2 cytokine production. J Allergy Clin Immunol. 2005;116(6):1372–1379. | |
Simeonova PP, Toriumi W, Kommineni C, et al. Molecular regulation of IL-6 activation by asbestos in lung epithelial cells: role of reactive oxygen species. J Immunol. 1997;159(8):3921–3928. | |
Tripathi SS, Mishra V, Shukla M, et al. IL-6 receptor-mediated lung Th2 cytokine networking in silica-induced pulmonary fibrosis. Arch Toxicol. 2010;84(12):947–955. | |
Chang CH, Hsiao CF, Yeh YM, et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int J Cancer. 2013;132(9):1977–1985. | |
Wuttge DM, Wildt M, Geborek P, Wollheim FA, Scheja A, Akesson A. Serum IL-15 in patients with early systemic sclerosis: a potential novel marker of lung disease. Arthritis Res Ther. 2007;9(5):R85. | |
Kurasawa K, Hirose K, Sano H, et al. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. 2000;43(11):2455–2463. | |
Komura K, Fujimoto M, Hasegawa M, et al. Increased serum interleukin 23 in patients with systemic sclerosis. J Rheumatol. 2008;35(1):120–125. | |
Truchetet ME, Brembilla NC, Montanari E, Allanore Y, Chizzolini C. Increased frequency of circulating Th22 in addition to Th17 and Th2 lymphocytes in systemic sclerosis: association with interstitial lung disease. Arthritis Res Ther. 2011;13(5):R166. | |
Martinez-Lostao L, Marzo I, Anel A, Naval J. Targeting the Apo2L/TRAIL system for the therapy of autoimmune diseases and cancer. Biochem Pharmacol. 2012;83(11):1475–1483. | |
Azab NA, Rady HM, Marzouk SA. Elevated serum TRAIL levels in scleroderma patients and its possible association with pulmonary involvement. Clin Rheumatol. 2012;31(9):1359–1364. | |
Hasegawa M. B lymphocytes: shedding new light on the pathogenesis of systemic sclerosis. J Dermatol. 2010;37(1):3–10. | |
Elhai M, Avouac J, Kahan A, Allanore Y. Systemic sclerosis at the crossroad of polyautoimmunity. Autoimmun Rev. 2013;12(11):1052–1057. | |
Sato S, Fujimoto M, Hasegawa M, Takehara K. Altered blood B lymphocyte homeostasis in systemic sclerosis: expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum. 2004;50(6):1918–1927. | |
Lanteri A, Sobanski V, Langlois C, et al. Serum free light chains of immunoglobulins as biomarkers for systemic sclerosis characteristics, activity and severity. Autoimmun Rev. 2014;13(9):974–980. | |
Veerappan A, O’Connor NJ, Brazin J, et al. Mast cells: a pivotal role in pulmonary fibrosis. DNA Cell Biol. 2013;32(4):206–218. | |
Günther A, Korfei M, Mahavadi P, von der Beck D, Ruppert C, Markart P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev. 2012;21(124):152–160. | |
Cambrey AD, Harrison NK, Dawes KE, et al. Increased levels of endothelin-1 in bronchoalveolar lavage fluid from patients with systemic sclerosis contribute to fibroblast mitogenic activity in vitro. Am J Respir Cell Mol Biol. 1994;11(4):439–445. | |
Abraham DJ, Vancheeswaran R, Dashwood MR, et al. Increased levels of endothelin-1 and differential endothelin type A and B receptor expression in scleroderma-associated fibrotic lung disease. Am J Pathol. 1997;151(3):831–841. | |
Vancheeswaran R, Magoulas T, Efrat G, et al. Circulating endothelin-1 levels in systemic sclerosis subsets – a marker of fibrosis or vascular dysfunction? J Rheumatol. 1994;21(10):1838–1844. | |
Ihn H, Sato S, Fujimoto M, Takehara K, Tamaki K. Increased serum levels of soluble vascular cell adhesion molecule-1 and E-selectin in patients with systemic sclerosis. Br J Rheumatol. 1998;37(11):1188–1192. | |
Kuryliszyn-Moskal A, Klimiuk PA, Sierakowski S. Soluble adhesion molecules (sVCAM-1, sE-selectin), vascular endothelial growth factor (VEGF) and endothelin-1 in patients with systemic sclerosis: relationship to organ systemic involvement. Clin Rheumatol. 2005;24(2):111–116. | |
Del Papa N, Colombo G, Fracchiolla N, et al. Circulating endothelial cells as a marker of ongoing vascular disease in systemic sclerosis. Arthritis Rheum. 2004;50(4):1296–1304. | |
Tinazzi E, Dolcino M, Puccetti A, et al. Gene expression profiling in circulating endothelial cells from systemic sclerosis patients shows an altered control of apoptosis and angiogenesis that is modified by iloprost infusion. Arthritis Res Ther. 2010;12(4):R131. | |
Bellini A, Mattoli S. The role of the fibrocyte, a bone marrow-derived mesenchymal progenitor, in reactive and reparative fibroses. Lab Invest. 2007;87(9):858–870. | |
Mattoli S, Bellini A, Schmidt M. The role of a human hematopoietic mesenchymal progenitor in wound healing and fibrotic diseases and implications for therapy. Curr Stem Cell Res Ther. 2009;4(4):266–280. | |
Moeller A, Gilpin SE, Ask K, et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009;179(7):588–594. | |
Borie R, Quesnel C, Phin S, et al. Detection of alveolar fibrocytes in idiopathic pulmonary fibrosis and systemic sclerosis. PLoS One. 2013;8(1):e53736. | |
Costabel U, Guzman J, Bonella F, Oshimo S. Bronchoalveolar lavage in other interstitial lung diseases. Semin Respir Crit Care Med. 2007; 28(5):514–524. | |
Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165(12):1581–1586. | |
Wells AU, Hansell DM, Rubens MB, et al. Fibrosing alveolitis in systemic sclerosis. Bronchoalveolar lavage findings in relation to computed tomographic appearance. Am J Respir Crit Care Med. 1994; 150(2):462–468. | |
Wells AU, Hansell DM, Haslam PL, et al. Bronchoalveolar lavage cellularity: lone cryptogenic fibrosing alveolitis compared with the fibrosing alveolitis of systemic sclerosis. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1474–1482. | |
Behr J, Vogelmeier C, Beinert T, et al. Bronchoalveolar lavage for evaluation and management of scleroderma disease of the lung. Am J Respir Crit Care Med. 1996;154(2 Pt 1):400–406. | |
Goh NS, Veeraraghavan S, Desai SR, et al. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosis-associated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. 2007;56(6):2005–2012. | |
Tashkin DP, Elashoff R, Clements PJ, et al; Scleroderma Lung Study Research Group. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007;176(10):1026–1034. | |
Strange C, Bolster MB, Roth MD, et al; Scleroderma Lung Study Research Group. Bronchoalveolar lavage and response to cyclophosphamide in scleroderma interstitial lung disease. Am J Respir Crit Care Med. 2008;177(1):91–98. | |
Sato LT, Kayser C, Andrade LE. Nailfold capillaroscopy abnormalities correlate with cutaneous and visceral involvement in systemic sclerosis patients. Acta Reumatol Port. 2009;34(2A):219–227. | |
Bredemeier M, Xavier RM, Capobianco KG, et al. Nailfold capillary microscopy can suggest pulmonary disease activity in systemic sclerosis. J Rheumatol. 2004;31(2):286–294. | |
Corrado A, Carpagnano GE, Gaudio A, Foschino-Barbaro MP, Cantatore FP. Nailfold capillaroscopic findings in systemic sclerosis related lung fibrosis and in idiopathic lung fibrosis. Joint Bone Spine. 2010;77(6):570–574. | |
Launay D, Humbert M, Berezne A, et al. Clinical characteristics and survival in systemic sclerosis-related pulmonary hypertension associated with interstitial lung disease. Chest. 2011;140(4):1016–1024. | |
Schreiber BE, Valerio CJ, Keir GJ, et al. Improving the detection of pulmonary hypertension in systemic sclerosis using pulmonary function tests. Arthritis Rheum. 2011;63(11):3531–3539. | |
Corte TJ, Wort SJ, MacDonald PS, et al. Pulmonary function vascular index predicts prognosis in idiopathic interstitial pneumonia. Respirology. 2012;17(4):674–680. | |
Arcasoy SM, Christie JD, Ferrari VA, et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med. 2003;167(5):735–740. | |
Nagaya N, Nishikimi T, Okano Y, et al. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardiol. 1998;31(1):202–208. | |
Gan CT, McCann GP, Marcus JT, et al. NT-proBNP reflects right ventricular structure and function in pulmonary hypertension. Eur Respir J. 2006;28(6):1190–1194. | |
Leuchte HH, Baumgartner RA, Nounou ME, et al. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. Am J Respir Crit Care Med. 2006;173(7):744–750. | |
Corte TJ, Wort SJ, Gatzoulis MA, et al. Elevated brain natriuretic peptide predicts mortality in interstitial lung disease. Eur Respir J. 2010;36(4):819–825. | |
Camargo CZ, Sekiyama JY, Arismendi MI, Kayser C. Microvascular abnormalities in patients with early systemic sclerosis: less severe morphological changes than in patients with definite disease. Scand J Rheumatol. 2015;44(1):48–55. | |
Meadows CA, Risbano MG, Zhang L, et al. Increased expression of growth differentiation factor-15 in systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2011;139(5):994–1002. | |
Lambrecht S, Smith V, De Wilde K, et al. Growth differentiation factor 15, a marker of lung involvement in systemic sclerosis, is involved in fibrosis development but is not indispensable for fibrosis development. Arthritis Rheumatol. 2014;66(2):418–427. | |
Cieslik P, Hrycek A. Long pentraxin 3 (PTX3) in the light of its structure, mechanism of action and clinical implications. Autoimmunity. 2012;45(2):119–128. | |
Shirai Y, Okazaki Y, Inoue Y, et al. Elevated levels of pentraxin 3 in systemic sclerosis: associations with vascular manifestations and defective vasculogenesis. Arthritis Rheumatol. 2015;67(2):498–507. | |
Iwata Y, Yoshizaki A, Ogawa F, et al. Increased serum pentraxin 3 in patients with systemic sclerosis. J Rheumatol. 2009;36(5):976–983. | |
Savarino E, Furnari M, de Bortoli N, et al. Gastrointestinal involvement in systemic sclerosis. Presse Med. 2014;43(10 Pt 2):e279–291. | |
Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353(12):1261–1273. | |
Rigamonti C, Shand LM, Feudjo M, et al. Clinical features and prognosis of primary biliary cirrhosis associated with systemic sclerosis. Gut. 2006;55(3):388–394. | |
Assassi S, Fritzler MJ, Arnett FC, et al. Primary biliary cirrhosis (PBC), PBC autoantibodies, and hepatic parameter abnormalities in a large population of systemic sclerosis patients. J Rheumatol. 2009;36(10):2250–2256. | |
Sakauchi F, Mori M, Zeniya M, Toda G. Antimitochondrial antibody negative primary biliary cirrhosis in Japan: utilization of clinical data when patients applied to receive public financial aid. J Epidemiol. 2006;16(1):30–34. | |
Liu H, Norman GL, Shums Z, et al. PBC screen: an IgG/IgA dual isotype ELISA detecting multiple mitochondrial and nuclear autoantibodies specific for primary biliary cirrhosis. J Autoimmun. 2010;35(4):436–442. | |
Prince MI, Chetwynd A, Craig WL, Metcalf JV, James OF. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53(6):865–870. | |
Centola M, Cavet G, Shen Y, et al. Development of a multi-biomarker disease activity test for rheumatoid arthritis. PLoS One. 2013;8(4):e60635. | |
Hirata S, Dirven L, Shen Y, et al. A multi-biomarker score measures rheumatoid arthritis disease activity in the BeSt study. Rheumatology (Oxford). 2013;52(7):1202–1207. | |
Beirne P, Pantelidis P, Charles P, et al. Multiplex immune serum biomarker profiling in sarcoidosis and systemic sclerosis. Eur Respir J. 2009;34(6):1376–1382. | |
Mihai C, Landewé R, van der Heijde D, et al. Digital ulcers predict a worse disease course in patients with systemic sclerosis. Ann Rheum Dis. Epub 2015 Feb 16. | |
Dimitroulas T, Daoussis D, Garyfallos A, Sfikakis PP, Kitas GD. Molecular and cellular pathways as treatment targets for biologic therapies in systemic sclerosis. Curr Med Chem. 2015;22(16):1943–1955. | |
Highland KB. Recent advances in scleroderma-associated pulmonary hypertension. Curr Opin Rheumatol. 2014;26(6):637–645. | |
Kawashiri SY, Ueki Y, Terada K, Yamasaki S, Aoyagi K, Kawakami A. Improvement of plasma endothelin-1 and nitric oxide in patients with systemic sclerosis by bosentan therapy. Rheumatol Int. 2014;34(2):221–225. | |
Ricciardolo FL, Sterk PJ, Gaston B, Folkerts G. Nitric oxide in health and disease of the respiratory system. Physiol Rev. 2004;84(3):731–765. | |
Girgis RE, Gugnani MK, Abrams J, Mayes MD. Partitioning of alveolar and conducting airway nitric oxide in scleroderma lung disease. Am J Respir Crit Care Med. 2002;165(12):1587–1591. | |
Tiev KP, Cabane J, Aubourg F, et al. Severity of scleroderma lung disease is related to alveolar concentration of nitric oxide. Eur Respir J. 2007;30(1):26–30. | |
Tiev KP, Rivière S, Hua-Huy T, Cabane J, Dinh-Xuan AT. Exhaled NO predicts cyclophosphamide response in scleroderma-related lung disease. Nitric Oxide. 2014;40:17–21. | |
Chung L, Fiorentino DF, Benbarak MJ, et al. Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheum. 2009;60(2):584–591. | |
Hinchcliff M, Huang CC, Wood TA, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol. 2013;133(8):1979–1989. | |
Topal AA, Dhurat RS. Scleroderma therapy: clinical overview of current trends and future perspective. Rheumatol Int. 2013;33(1):1–18. | |
Denton CP, Ong VH. Targeted therapies for systemic sclerosis. Nat Rev Rheumatol. 2013;9(8):451–464. | |
Castelino FV, Varga J. Emerging cellular and molecular targets in fibrosis: implications for scleroderma pathogenesis and targeted therapy. Curr Opin Rheumatol. 2014;26(6):607–614. | |
Gerber EE, Gallo EM, Fontana SC, et al. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature. 2013;503(7474):126–130. | |
Jain KK. Technology for Discovery of Biomarkers. In: The Handbook of Biomarkers. Vol 1. Totowa, NJ: Humana Press; 2010:23–72. | |
Nakashima T, Jinnin M, Yamane K, et al. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J Immunol. 2012;188(8):3573–3583. | |
Kajihara I, Jinnin M, Yamane K, et al. Increased accumulation of extracellular thrombospondin-2 due to low degradation activity stimulates type I collagen expression in scleroderma fibroblasts. Am J Pathol. 2012;180(2):703–714. | |
Etoh M, Jinnin M, Makino K, et al. microRNA-7 down-regulation mediates excessive collagen expression in localized scleroderma. Arch Dermatol Res. 2013;305(1):9–15. | |
Makino K, Jinnin M, Hirano A, et al. The downregulation of microRNA let-7a contributes to the excessive expression of type I collagen in systemic and localized scleroderma. J Immunol. 2013;190(8):3905–3915. | |
Takemoto R, Jinnin M, Wang Z, et al. Hair miR-29a levels are decreased in patients with scleroderma. Exp Dermatol. 2013;22(12):832–833. | |
Sing T, Jinnin M, Yamane K, et al. microRNA-92a expression in the sera and dermal fibroblasts increases in patients with scleroderma. Rheumatology (Oxford). 2012;51(9):1550–1556. | |
Makino K, Jinnin M, Kajihara I, et al. Circulating miR-142-3p levels in patients with systemic sclerosis. Clin Exp Dermatol. 2012;37(1):34–39. | |
Honda N, Jinnin M, Kajihara I, et al. TGF-β-mediated downregulation of microRNA-196a contributes to the constitutive upregulated type I collagen expression in scleroderma dermal fibroblasts. J Immunol. 2012;188(7):3323–3331. | |
Wang Z, Jinnin M, Kudo H, et al. Detection of hair-microRNAs as the novel potent biomarker: evaluation of the usefulness for the diagnosis of scleroderma. J Dermatol Sci. 2013;72(2):134–141. | |
Bonella F, Costabel U. Biomarkers in connective tissue disease-associated interstitial lung disease. Semin Respir Crit Care Med. 2014;35(2):181–200. | |
Nakajima H, Harigai M, Hara M, et al. KL-6 as a novel serum marker for interstitial pneumonia associated with collagen diseases. J Rheumatol. 2000;27(5):1164–1170. | |
Sato S, Nagaoka T, Hasegawa M, Nishijima C, Takehara K. Elevated serum KL-6 levels in patients with systemic sclerosis: association with the severity of pulmonary fibrosis. Dermatology. 2000;200(3):196–201. | |
Buttmann M, Merzyn C, Hofstetter HH, Rieckmann P. TRAIL, CXCL10 and CCL2 plasma levels during long-term Interferon-beta treatment of patients with multiple sclerosis correlate with flu-like adverse effects but do not predict therapeutic response. J Neuroimmunol. 2007;190(1–2):170–176. | |
Antonelli A, Fallahi P, Ferrari SM, et al. Systemic sclerosis fibroblasts show specific alterations of interferon-γ and tumor necrosis factor-α-induced modulation of interleukin 6 and chemokine ligand 2. J Rheumatol. 2012;39(5):979–985. | |
Takeda N, Ihn H, Teramoto S. Markedly increased levels of IL-6 and CA125 in pleural fluid of an elderly person with overlap syndrome of systemic sclerosis and systemic lupus erythematosus. Age Ageing. 2001;30(2):171. | |
Gourh P, Arnett FC, Assassi S, et al. Plasma cytokine profiles in systemic sclerosis: associations with autoantibody subsets and clinical manifestations. Arthritis Res Ther. 2009;11(5):R147. | |
Sato S, Nagaoka T, Hasegawa M, et al. Serum levels of connective tissue growth factor are elevated in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. J Rheumatol. 2000;27(1):149–154. | |
Muangchan C, Harding S, Khimdas S, Bonner A, Baron M, Pope J; Canadian Scleroderma Research group. Association of C-reactive protein with high disease activity in systemic sclerosis: results from the Canadian Scleroderma Research Group. Arthritis Care Res (Hoboken). 2012;64(9):1405–1414. | |
Leuchte HH, Neurohr C, Baumgartner R, et al. Brain natriuretic peptide and exercise capacity in lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2004;170(4):360–365. | |
Leuchte HH, El Nounou M, Tuerpe JC, et al. N-terminal pro-brain natriuretic peptide and renal insufficiency as predictors of mortality in pulmonary hypertension. Chest. 2007;131(2):402–409. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.