Back to Journals » Journal of Inflammation Research » Volume 19
Biological Therapies for Urate Lowering and Inflammation Control in Gout Management
Authors Zhou Y ![]() , Zhang H, Liu N, Yan H, Meng F, Peng J
, Zhang H, Liu N, Yan H, Meng F, Peng J
Received 31 December 2025
Accepted for publication 9 March 2026
Published 16 March 2026 Volume 2026:19 592891
DOI https://doi.org/10.2147/JIR.S592891
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ujjwol Risal
Ye Zhou,1,* Hengyan Zhang,2,* Nian Liu,1,3,* Heguo Yan,1 Fanyu Meng,1 Jiangyun Peng1,3
1First Clinical Medical College, Yunnan University of Chinese Medicine, Kunming, People’s Republic of China; 2Department of Rheumatology, Zhaotong Hospital of Traditional Chinese Medicine, Zhaotong, People’s Republic of China; 3Department of Rheumatology, Yunnan Provincial Hospital of Traditional Chinese Medicine, Kunming, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jiangyun Peng, Department of Rheumatology, Yunnan Provincial Hospital of Traditional Chinese Medicine, Kunming, People’s Republic of China, Email [email protected]
Abstract: Gout remains the most prevalent inflammatory arthritis worldwide, driven by hyperuricemia and monosodium urate crystal-induced inflammation. Conventional therapies manage most patients effectively, but refractory disease and contraindications in special populations create unmet clinical needs. This gap is particularly evident among patients with advanced chronic kidney disease or organ transplants. Biologic therapies targeting key pathophysiologic mechanisms have emerged as specialized options for these difficult-to-treat cases. Pegloticase achieves sustained urate reduction in refractory gout. Concomitant immunomodulatory therapy using methotrexate or mycophenolate substantially improves response rates by mitigating antidrug antibody formation. Anti-inflammatory biologics targeting the interleukin-1 (IL-1) pathway underwent a prolonged clinical translation process. Anakinra accumulated extensive off-label evidence over two decades without formal approval, while rilonacept faced regulatory rejection in 2012 despite demonstrating efficacy. Canakinumab received Food and Drug Administration (FDA) approval in August 2023 after its initial rejection in 2011, becoming the first biologic formally indicated for gout in the United States. NLR family pyrin domain-containing protein 3 (NLRP3) inflammasome inhibitors represent a recent area of investigation. OLT1177 has advanced through clinical development after incorporating safety lessons from the hepatotoxicity experience associated with MCC950. Diverse pipeline compounds further reflect active research in this therapeutic class. Biologics function as rescue options for patients with inadequate responses to conventional treatment rather than as first-line alternatives. Remaining challenges include immunogenicity management, treatment costs that limit accessibility, and incomplete long-term safety characterization. Future progress depends on refining patient selection through predictive biomarkers and ensuring appropriate access for patients most likely to benefit from these advances.
Keywords: gout, biological products, urate oxidase, interleukin-1, NLR family, pyrin domain-containing 3 protein, hyperuricemia
Introduction
Gout represents the most prevalent inflammatory arthritis worldwide, affecting millions of individuals with increasing incidence linked to aging populations, metabolic syndrome prevalence, and dietary patterns.1 The disease follows a chronic progressive course characterized by recurrent acute inflammatory flares and gradual accumulation of monosodium urate crystal deposits in joints and soft tissues. Persistent hyperuricemia provides the biochemical foundation, with sustained serum urate elevation above saturation thresholds enabling crystal formation and deposition.2 These crystals trigger intense inflammatory responses mediated primarily through the NLR family pyrin domain-containing protein 3 (NLRP3) inflammasome and interleukin-1 (IL-1) pathways, producing the acute joint pain, swelling, and disability characteristic of gout attacks.3 The dual pathophysiology involving both sustained hyperuricemia and crystal-induced inflammation establishes distinct therapeutic targets. Effective long-term management requires addressing hyperuricemia to prevent further crystal deposition and promote existing deposit dissolution. Acute management necessitates suppressing inflammatory responses to crystals already present in tissues.4 This mechanistic understanding informs two complementary therapeutic strategies that have guided drug development for decades.
Conventional pharmacotherapy successfully manages most patients with gout. It comprises oral urate-lowering medications including allopurinol and febuxostat combined with anti-inflammatory agents such as nonsteroidal anti-inflammatory drugs (NSAIDs), colchicine, and corticosteroids.5 However, a subset of patients experiences inadequate disease control despite optimized conventional therapy. Refractory gout describes situations where patients fail to achieve target serum urate levels below 6 mg/dL or continue experiencing recurrent flares.6 Progressive tophaceous disease may also develop despite appropriate treatment attempts. Multiple factors contribute to refractory disease. These include severe baseline hyperuricemia requiring urate reduction beyond conventional drug capacity, medication intolerances limiting dose optimization, and patient adherence challenges with chronic oral regimens.7 Beyond refractoriness, specific clinical scenarios present therapeutic dilemmas where conventional options face substantial limitations. Advanced chronic kidney disease restricts allopurinol dosing and contraindicates NSAIDs while reducing colchicine clearance. Solid organ transplant recipients require careful navigation of drug interactions with immunosuppressive regimens.8 Patients with multiple comorbidities frequently accumulate contraindications to standard agents. These difficult-to-treat populations represent substantial unmet medical needs where alternative therapeutic approaches could provide meaningful clinical benefit.
Biologic therapies have emerged as specialized treatment options for gout patients inadequately served by conventional approaches.9 These agents target key pathophysiologic mechanisms through recombinant protein technologies to achieve highly selective interventions at well-defined molecular targets. Uricase enzymes catalyze uric acid oxidation to more soluble metabolites. This enables profound urate reduction unattainable through xanthine oxidase inhibition alone.10 Anti-inflammatory biologics intercept the IL-1 signaling cascade responsible for acute gout flare pathophysiology. They function either by blocking cytokine-receptor interactions or by preventing upstream inflammasome activation.11 This narrative review examines biologic therapies developed for gout management across these two mechanistic categories. Relevant literature was identified through searches of PubMed and MEDLINE, with priority given to controlled clinical trials, real-world evidence, and translational studies bearing on regulatory development and clinical application. We discuss clinical translation experiences across three major therapeutic classes. These include uricase variants such as pegloticase and emerging alternatives, IL-1 pathway inhibitors encompassing receptor antagonists and neutralizing antibodies, and NLRP3 inflammasome inhibitors representing more recent investigational approaches. For each therapeutic class, we discuss development histories including regulatory challenges encountered during gout indication approval processes. We also examine current clinical evidence from controlled trials and real-world experience, and review evolving strategies to optimize efficacy and safety profiles. The objective is to provide clinicians and researchers with a practical understanding of how these biologics function within contemporary gout management paradigms.
Biological Therapies for Urate Lowering
Development and Clinical Translation of Recombinant Uricases
The exploration of uricase replacement therapy originated from the recognition that humans lost the uricase gene through evolution.12 This enzyme deficiency distinguishes humans from most other mammals that can metabolize uric acid into the more soluble allantoin.13 Rasburicase emerged as the first recombinant uricase approved for tumor lysis syndrome. While it demonstrated rapid urate reduction in acute settings, its short half-life of approximately 18 hours and high immunogenicity limited its chronic application in gout management.14 Clinical experience showed that rasburicase required continuous or frequent infusions to maintain therapeutic effects, making it impractical for long-term treatment of patients with refractory gout.15 These limitations catalyzed the development of modified uricase preparations suitable for chronic disease management.
Pegloticase represented a significant advancement through polyethylene glycol (PEG) conjugation technology. The FDA approved pegloticase in September 2010 as the first biologic therapy specifically indicated for chronic refractory gout in adults. PEGylation extended the half-life to 10 to 12 days, enabling a practical biweekly infusion schedule.16 Pivotal Phase III trials demonstrated that 42% of patients receiving pegloticase 8 mg every two weeks achieved sustained serum urate levels below 6 mg/dL during month 6. Beyond urate lowering, pegloticase treatment resulted in significant tophus resolution and improvements in physical function and quality-of-life measures. However, clinical application soon revealed a critical challenge. Approximately 40% to 50% of patients developed antidrug antibodies against the PEG moiety or the uricase protein itself.17,18 These antibodies led to loss of therapeutic efficacy and increased risk of infusion reactions. Current prescribing guidelines recommend discontinuing pegloticase when patients show loss of urate-lowering response, as continued treatment in the presence of high antibody titers substantially increases infusion reaction risk.
Recognizing immunogenicity as the central barrier to successful uricase therapy, investigators explored immunomodulatory strategies.19 SEL-212 combines pegadricase with ImmTOR, a proprietary rapamycin-containing nanoparticle designed to induce antigen-specific immune tolerance. Phase 2 dose-finding studies established that SEL-037 (pegadricase) at 0.2 mg/kg combined with SEL-110 (ImmTOR) at 0.15 mg/kg achieved optimal results.20 When pegadricase was administered alone, rapid urate reductions were not sustained beyond 30 days in most participants due to antibody formation. In contrast, 66% of evaluable participants achieved sustained urate control at week 20 when receiving five monthly doses of the combination therapy. Another study showed that ImmTOR inhibited the development of uricase-specific antibodies in a dose-dependent manner, thereby enabling sustained enzyme activity and sustained reduction in serum uric acid levels.21 The Comparative Randomized Assessment of Pegloticase vs SEL-212 (COMPARE) trial directly compared SEL-212 with pegloticase in a head-to-head Phase 2 study. While the primary endpoint showed numerically higher response rates with SEL-212 compared to pegloticase during months 3 and 6 combined, this difference did not reach statistical significance.22 However, percentage reductions in serum urate levels were statistically greater with SEL-212. Importantly, SEL-212 required only monthly infusions compared to biweekly pegloticase administration, potentially reducing treatment burden and cumulative glucocorticoid exposure for flare prophylaxis.
Beyond SEL-212, multiple alternative approaches to addressing uricase immunogenicity have entered development. For example, co-administration of high molecular weight free PEG with pegloticase demonstrated 4-fold to 12-fold reductions in anti-PEG antibodies in murine models.23 This strategy employs competitive inhibition, where free PEG blocks PEGylated protein binding to PEG-specific B cell receptors. Another innovative approach involves engineered red blood cell-based delivery. Loading uricase into red blood cells provided immunological masking, leading to maintained urate-lowering performance after repeated infusions through reduced antibody-mediated macrophage clearance.24 Recent studies also highlight novel uricase constructs. PAT101, featuring recombinant human albumin conjugated to Aspergillus flavus uricase through click chemistry, exhibited more than double the half-life of pegloticase in transgenic mice.25 After 4 weeks of repeated administration in rats, rasburicase retained only 24% of uricase activity while PAT101 maintained 86%. Similarly, another study showed that PEGylated porcine-human recombinant uricase (PEG-PHRU) demonstrated effective urate degradation and mitigation of hyperuricemia-induced renal damage in animal models.26
The developmental trajectory from rasburicase through pegloticase to current investigational agents illustrates iterative problem-solving in biologic drug development (Table 1). Each generation addressed specific limitations of its predecessor while revealing new challenges. Pegloticase overcame the short half-life of rasburicase but encountered immunogenicity barriers. Current strategies targeting immune tolerance through various mechanisms represent responses to this obstacle. Two Phase 3 trials of SEL-212 are ongoing, with results expected to clarify whether immunomodulatory co-administration can meaningfully improve sustained response rates compared to pegloticase monotherapy. The diversity of approaches under investigation reflects both the clinical importance of solving this problem and the complexity of achieving sustained immune tolerance to therapeutic foreign proteins.
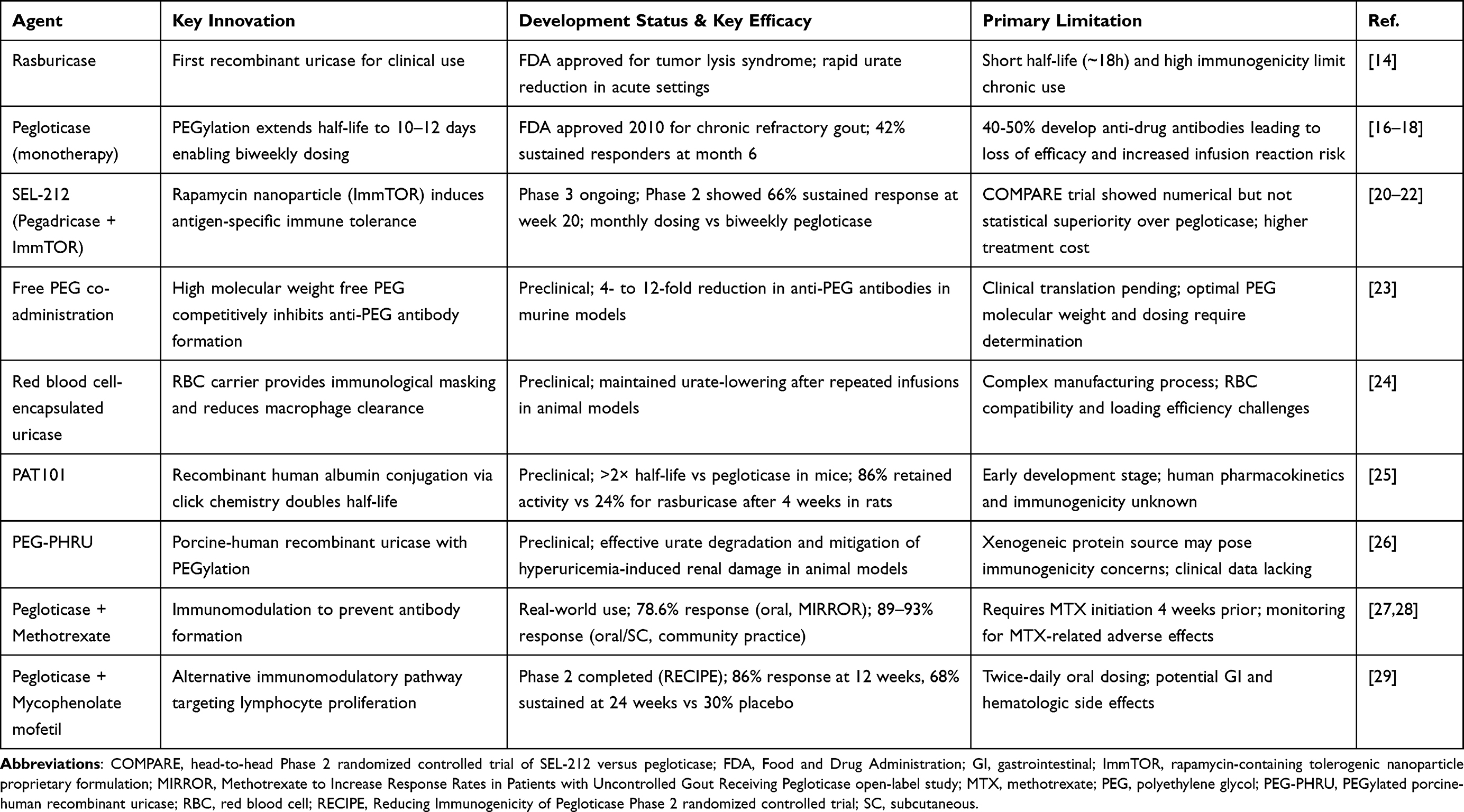 |
Table 1 Development and Clinical Characteristics of Uricase-Based Therapies for Refractory Gout |
Clinical Considerations and Management Strategies
The recognition that antidrug antibodies represent the primary cause of pegloticase treatment failure prompted investigation of immunomodulatory co-therapy strategies.30 This approach builds on established principles from other biologic therapies where concomitant immunosuppression reduces antibody formation. Effective immunomodulation must achieve sustained tolerance without causing unacceptable toxicity or infection risk. This requirement is particularly relevant given the chronic nature of gout and the frequent presence of multiple comorbidities in affected patients.31 Unlike broad immunosuppression that affects the entire immune system, the goal involves preventing specific antibody responses to the therapeutic enzyme while preserving general immune surveillance.
Methotrexate has emerged as the most extensively studied immunomodulatory agent for pegloticase co-therapy.32 The Methotrexate to Increase Response Rates in Patients with Uncontrolled Gout Receiving Pegloticase (MIRROR) study evaluated weekly oral methotrexate at 15 mg combined with folic acid supplementation in 14 patients with uncontrolled gout.27 This open-label trial demonstrated that 78.6% of patients met the responder definition of serum urate below 6 mg/dL for at least 80% of the time during month 6. This response rate substantially exceeded the historical 42% observed with pegloticase monotherapy. All patients tolerated methotrexate without unexpected safety concerns.33 The methotrexate regimen began 4 weeks prior to the first pegloticase infusion and continued throughout treatment, allowing time for immunomodulatory effects to develop before enzyme exposure. Recent real-world experience from two community rheumatology practices provided additional evidence supporting this strategy.28 Among 34 patients receiving various immunomodulators with pegloticase, subcutaneous methotrexate at 15.4 mg weekly achieved a 93% response rate while oral methotrexate at 15.3 mg weekly achieved 89%. These findings suggest that both administration routes can effectively prevent antibody formation, though subcutaneous delivery may offer more consistent bioavailability.
Other immunomodulatory agents have also demonstrated efficacy in preventing pegloticase immunogenicity. The Reducing Immunogenicity of Pegloticase (RECIPE) trial evaluated mycophenolate mofetil at 1000 mg twice daily in a randomized, double-blind, placebo-controlled design.29 Results showed that 86% of patients in the mycophenolate arm achieved serum urate levels at or below 6 mg/dL at 12 weeks compared to 40% in the placebo arm. By week 24, sustained response rates were 68% versus 30% respectively. Notably, the placebo arm experienced more infusion reactions at 30% compared to none in the mycophenolate group. Another study reported successful long-term pegloticase treatment over 98 weeks when combined with low-dose azathioprine at 50 mg daily.17 This case demonstrated significant tophus reduction of 77% with maintained low serum urate levels and absence of infusion reactions. Two transient increases in serum urate correlated with azathioprine non-compliance and resolved upon medication reinstitution. The community practice experience mentioned previously included 3 patients receiving mycophenolate mofetil at 1000 mg daily, all of whom responded successfully, and 2 patients on azathioprine at 100 mg daily with a 50% response rate.28 These data suggest that multiple immunomodulatory pathways can effectively prevent antibody formation, though optimal agent selection may depend on individual patient factors including comorbidities and contraindications.
Patient selection and baseline characteristics significantly influence pegloticase treatment outcomes. Analysis comparing patients with and without clinically apparent tophi revealed important differences.34 Patients with tophaceous gout were significantly older, had longer disease duration, greater numbers of tender and swollen joints, and lower estimated glomerular filtration rate. Despite more severe baseline disease, both groups experienced significant clinical benefit with pegloticase treatment. However, patients with tophi showed improvements in additional parameters including swollen joints and functional scores that were not significant in non-tophaceous patients. Another analysis examined the 2-year period before pegloticase initiation in 408 patients.35 During this time, the percentage of patients with tophi increased from 15.4% to 61.5%, those with at least one flare increased from 49% to 84%, and mean flare frequency rose from 1.0 to 2.1 per patient. Healthcare resource utilization also increased substantially across all categories except emergency department visits. These findings underscore the progressive nature of uncontrolled gout and suggest that earlier identification and intervention may help prevent disease escalation.
Real-world data provide important insights into pegloticase utilization patterns and post-treatment management. A retrospective analysis of nearly 3 million gout patients identified only 483 pegloticase initiators, highlighting limited real-world use.36 The median number of infusions was four and median treatment duration was 3 months. During follow-up, anaphylaxis occurred in 0.6% of patients while heart failure hospitalizations affected 6.4%. These rates differ somewhat from clinical trial data, potentially reflecting differences in patient populations and clinical settings. Regarding long-term benefits, one analysis demonstrated that responders to biweekly pegloticase who maintained persistently low serum urate levels experienced significant reductions in both systolic and diastolic blood pressure, independent of changes in renal function.37 After pegloticase discontinuation, management becomes challenging. Analysis of 375 patients who discontinued treatment showed that 86% subsequently received oral urate-lowering therapy, but only 51% of them achieved target serum urate below 6 mg/dL.38 The median time to starting or restarting therapy was 92 days. For patients who responded to initial pegloticase treatment, retreatment after a gap in therapy appears feasible. A case series described four patients successfully retreated with pegloticase following treatment gaps ranging from 12 to 156 weeks.39 Three of four patients achieved serum urate below 1.0 mg/dL during retreatment without infusion reactions or significant adverse events. These observations suggest that prior response predicts future response, though careful monitoring remains essential given the immunogenic potential of re-exposure after a treatment gap.
Biological Therapies for Inflammation Control
IL-1 Pathway Inhibitors
Interleukin-1β (IL-1β) plays a central role in the inflammatory cascade triggered by monosodium urate crystals, making IL-1 pathway inhibition a mechanistically rational therapeutic strategy.40 Although IL-1β serves as the principal mediator of acute crystal-induced inflammation, interleukin-1α (IL-1α) contributes to the propagation and persistence of the inflammatory response. This contribution is particularly evident in the context of chronic tophaceous disease. This distinction has practical consequences for therapeutic design. Agents that block both IL-1α and IL-1β exert broader suppression of the IL-1 signaling axis. In contrast, selective IL-1β neutralization preserves the IL-1α-dependent components of innate immune surveillance, a consideration of potential relevance in patients with elevated infection risk. Two distinct approaches have emerged to interrupt IL-1 signaling. Receptor antagonists and trap proteins block IL-1 binding to cell surface receptors in a nonselective manner, affecting both IL-1α and IL-1β. Monoclonal antibodies, by contrast, provide selective IL-1β neutralization.41 These mechanistic differences are accompanied by substantial variation in pharmacokinetic profiles. Half-lives range from hours to weeks across available agents, translating into dosing schedules that span daily administration for acute management to quarterly injections suited to long-term prophylaxis. These therapeutic modalities underwent a prolonged clinical translation from initial approvals in other indications to formal regulatory recognition in gout. This process involved the accumulation of extensive off-label clinical experience and multiple regulatory setbacks. Ultimately, these efforts yielded approvals that validated targeted IL-1 inhibition as a viable approach for patients with inadequate responses to conventional treatments.
IL-1 Receptor Antagonists and Trap Proteins
Among the broad-spectrum IL-1 inhibitors, anakinra entered clinical use earliest and has accumulated the most extensive evidence in gout.42 The agent functions as a recombinant IL-1 receptor antagonist that competes with both IL-1α and IL-1β for receptor binding, directly interrupting the inflammatory response to monosodium urate crystals. Following FDA approval in November 2001 for rheumatoid arthritis (RA), anakinra found off-label use in acute gout despite never obtaining formal approval for this indication.43 The short half-life necessitates daily subcutaneous administration, which offers flexibility for acute management where rapid treatment initiation and discontinuation provide clinical advantages. Multiple controlled trials established anakinra efficacy across diverse patient populations. A randomized non-inferiority study comparing anakinra to conventional treatments including colchicine, naproxen, or prednisone in 88 patients with crystal-proven gout demonstrated comparable symptom reduction in both groups.44 A Phase II trial evaluated anakinra at two doses against triamcinolone injection, enrolling 165 patients and covering 301 flares.45 Although statistical superiority was not achieved for the primary pain reduction endpoint, anakinra showed comparable efficacy and favorable performance on most secondary outcomes.
Real-world evidence demonstrates anakinra utility particularly in patients with contraindications to conventional therapies. A retrospective analysis of 91 hospitalized cases, including 77 gout episodes and 11 pseudogout episodes, found that half of patients had comorbidities limiting standard treatment options.46 Anakinra achieved therapeutic success in 92% of gout flares and 79% of pseudogout flares with good tolerability. This population represents precisely the clinical scenarios where alternative treatments become essential. Another study specifically examined 31 patients with stage 4 to 5 chronic kidney disease or those who had undergone kidney transplantation.47 Anakinra proved efficacious in all cases without significant renal function deterioration. Ten patients required prolonged treatment due to symptom recurrence upon discontinuation, while only one serious infection occurred three months after therapy initiation. These findings address a significant clinical need, as advanced renal impairment frequently accompanies severe gout while contraindicating most conventional anti-inflammatory agents.
A systematic review synthesized anakinra experience from 38 studies encompassing 551 patients and 648 analyzed flares.48 The cohort reflected typical severe gout characteristics, with mean age 57.9 years and 82.9% male representation. Polyarticular presentation occurred in 47.5% of cases while tophaceous disease affected 66.9%, and more than half of patients carried multiple comorbidities. Effectiveness reached 94% for acute flares and 91% for long-term treatment. The analysis described successful use in complex scenarios including 65 patients with active infection, 41 solid organ transplant recipients, and 14 patients receiving hemodialysis. Adverse effects occurred in 6.7% of acute treatments, though rates increased with long-term administration. The ability to administer anakinra during active infection represents a particular advantage. Hospitalized patients frequently develop gout flares concurrent with infections, creating therapeutic dilemmas when conventional agents carry infection risks or face contraindications. The Anakinra for Synovial fluid analysis in Gout and Related Diseases trial (ASGARD) currently investigates anakinra efficacy and safety specifically in patients with chronic kidney disease, a population with substantial unmet therapeutic needs.49
Rilonacept occupies a distinct clinical position within the same mechanistic class. Where anakinra’s short half-life and daily dosing suit acute on-demand management, rilonacept’s extended pharmacokinetic profile was designed for sustained prophylactic use. This design is particularly relevant during the period of urate-lowering therapy initiation when flare risk is transiently elevated. Rilonacept employs a trap protein mechanism, binding both IL-1α and IL-1β before receptor engagement through a dimeric fusion protein architecture combining IL-1 receptor components with immunoglobulin domains.50 This design enables weekly dosing compared to anakinra’s daily regimen. The agent received FDA approval in February 2008 for cryopyrin-associated periodic syndromes (CAPS).51 A Phase III trial randomized 241 patients to placebo or rilonacept at 80 mg or 160 mg weekly for 16 weeks while initiating allopurinol.52 Both rilonacept doses significantly reduced mean gout flares from 1.06 per patient with placebo to 0.29 and 0.21 respectively.53 Proportions experiencing multiple flares decreased from 46.8% to 18.8% and 16.3%. Adverse events remained generally balanced except for increased injection site reactions with rilonacept.
Despite these positive efficacy results, an FDA Advisory Committee voted 11 to 0 against approval for gout in May 2012. The committee determined that evidence did not demonstrate benefits outweighing risks. Specific concerns included indication breadth, insufficient data from truly treatment-refractory patients, and limited long-term safety information. This outcome illustrated an important distinction between statistical efficacy and the evidentiary standards required for regulatory approval in a biologic indication. These standards were particularly stringent when the target population and benefit-risk boundaries had not been defined with sufficient precision. The rilonacept experience directly informed subsequent IL-1 inhibitor development strategies, reinforcing the importance of restrictive patient selection criteria and comprehensive long-term safety characterization as prerequisites for regulatory success. Rilonacept remains available only for periodic fever syndromes. Anakinra therefore remains the primary IL-1 receptor antagonist option for gout, despite its own lack of formal approval for this indication.
IL-1β-Specific Monoclonal Antibodies
The regulatory lessons drawn from rilonacept’s rejection shaped the development strategy for the next generation of IL-1 pathway inhibitors. Stricter patient selection criteria and a more precisely defined benefit-risk population guided subsequent applications. Canakinumab represents a selective approach targeting IL-1β through fully human monoclonal antibody technology.54 This selectivity carries mechanistic implications beyond the distinction noted in the preceding section. By sparing IL-1α-mediated signaling, canakinumab preserves a broader component of innate immune function compared to receptor-level antagonists, a property that may be relevant in patients requiring long-term treatment. Its extended half-life and quarterly subcutaneous dosing position it specifically for sustained prophylactic use rather than acute on-demand management. This profile makes it most suitable for patients with a history of frequent flares who require durable inflammatory suppression.
Following FDA approval in June 2009 for CAPS, canakinumab entered investigation for gout management.55 The regulatory path proved prolonged and challenging. An FDA Advisory Committee rejected the initial gout application in 2011, citing insufficient evidence that benefits outweighed risks despite demonstrated flare reduction efficacy in clinical trials. European authorities assessed the evidence differently, granting approval in August 2013 specifically for patients with contraindications or inadequate responses to conventional treatments in whom urate-lowering therapy had not achieved target levels.56 The divergent outcomes reflected differing interpretations of benefit-risk boundaries and patient population definitions across regulatory agencies, echoing the evidentiary tensions encountered with rilonacept. After continued evidence generation and regulatory dialogue, canakinumab obtained FDA approval for symptomatic gout treatment in August 2023.57 The approved dosing of 150 mg subcutaneously every 12 weeks offers substantial convenience compared to the more frequent administration schedules required by other IL-1 inhibitors.
Important mechanistic insights emerged from cardiovascular research examining IL-1β inhibition in patients with prior myocardial infarction and elevated inflammatory markers. The Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) trial provided two complementary analyses characterizing canakinumab’s influence on gout-related outcomes. A causal mediation analysis investigated relationships between canakinumab treatment, serum urate changes, and incident gout development.58 This analysis demonstrated that canakinumab significantly reduced incident gout risk independent of changes in serum urate levels. Statistical modeling revealed that urate reduction mediated only a small proportion of the observed effect on gout prevention. A separate exploratory analysis examined 10,059 patients over a median 3.7 years of follow-up.52 Among participants receiving placebo, incident gout attack rates increased progressively with higher baseline urate concentrations, ranging from 0.49 per 100 person-years in those with urate below 404.5 μmol/L to 2.94 per 100 person-years in those with urate at or above 535.4 μmol/L. Quarterly canakinumab administration significantly reduced gout attack risk across all baseline urate strata without altering serum uric acid levels over time. Taken together, these findings support a conceptually important point. Suppressing IL-1β-mediated inflammation can prevent acute gout manifestations even when hyperuricemia and crystal deposition persist. This observation provides a mechanistic rationale for selective IL-1β inhibition as a standalone strategy in patients for whom urate-lowering targets remain difficult to achieve. The CANTOS data also demonstrated acceptable safety in patients with substantial cardiovascular comorbidity, extending the evidence base for canakinumab beyond dedicated gout trial populations.
NLRP3 Inflammasome Inhibitors
NLRP3 inflammasome inhibitors represent a more recent area of investigation compared to the decades-long development of IL-1 pathway blockers.59 Their mechanistic appeal lies in targeting a step upstream of IL-1β maturation. Where IL-1 receptor antagonists and monoclonal antibodies intercept cytokine signaling after IL-1β has already been processed and released, NLRP3 inhibition acts earlier in the inflammatory cascade by preventing inflammasome assembly and the subsequent cleavage of pro-IL-1β into its active form. This positioning raises the possibility of broader suppression across multiple downstream inflammatory mediators beyond IL-1β alone. Whether this theoretical advantage translates into clinically meaningful benefits over direct IL-1 blockade remains to be established. The field has not yet produced agents with sufficient clinical evidence to allow direct comparison. The field has experienced both notable setbacks and promising advances as researchers have refined their understanding of optimal inhibitor characteristics and safety profiles.
Lead Compounds: From Setbacks to Progress
MCC950 was among the earliest small molecule NLRP3 inhibitors to enter clinical development, advancing to Phase II trials for RA around 2015 to 2017 on the basis of preclinical efficacy across multiple inflammatory disease models.60 However, development terminated when high-dose administration at approximately 1200 mg daily produced hepatotoxicity signals including transaminase elevations in trial participants.61 This clinical failure represented a significant setback for the field but also provided valuable lessons regarding dose selection, hepatic safety monitoring, and therapeutic window considerations.
Rather than abandoning the NLRP3 target, the research community intensified mechanistic investigations to understand MCC950’s actions and guide next-generation compound development. Subsequent biochemical and structural studies, conducted primarily in cell-free and cell-based systems, established three important findings. Proteomic analysis confirmed NLRP3 as the direct binding target of MCC950.62 Structural studies revealed that MCC950 functions by closing the active conformation of NLRP3, preventing the protein from adopting the oligomerized state necessary for inflammasome assembly.63 More detailed mechanistic work identified that MCC950 specifically targets the Walker B motif within the NLRP3 NACHT (nucleoside triphosphate-binding domain present in NAIP, CIITA, HET-E, and TP1 proteins).64 Together, these findings established proof of concept for direct NLRP3 inhibition while highlighting that sufficient selectivity and an adequate safety margin are prerequisites for clinical translation. The knowledge gained from MCC950 characterization continues to inform structure-based design efforts for improved inhibitors.
The development of OLT1177, also known as dapansutrile, reflected a deliberate response to the MCC950 experience. Dose selection and safety monitoring protocols were designed from the outset to address the hepatotoxicity concerns that had terminated the earlier program. Preclinical evaluation in a monosodium urate crystal-induced peritonitis mouse model demonstrated that OLT1177 effectively reduced neutrophil infiltration and inflammatory cytokine production.65 The compound showed dose-dependent anti-inflammatory effects with favorable pharmacokinetic properties, and toxicology assessments at therapeutically relevant doses did not reveal hepatotoxicity signals. Critically, whereas MCC950 had been advanced to doses exceeding 1000 mg daily before hepatotoxic effects became apparent, the OLT1177 program incorporated hepatic safety endpoints at each dose escalation step. Conservative upper dose boundaries were established prior to first-in-human studies. This design reflects a direct application of the therapeutic window lessons derived from the MCC950 experience.
Clinical evaluation then proceeded through structured dose-finding studies. Phase I testing established tolerability up to 1000 mg daily for 8 days without hepatotoxicity signals, contrasting favorably with the MCC950 experience.66 A subsequent Phase 2a trial in acute gout enrolled patients with crystal-proven disease and evaluated multiple dose levels ranging from 300 mg to 2000 mg daily. All tested doses demonstrated efficacy in reducing pain and inflammatory markers compared to baseline, and the compound maintained acceptable tolerability across this broad dose range with no evidence of significant liver enzyme elevations. These results suggested a wider therapeutic window relative to MCC950. The trial design incorporated frequent hepatic monitoring specifically informed by prior hepatotoxicity concerns. OLT1177’s oral bioavailability and convenient dosing schedule offer practical advantages for outpatient gout management. Based on Phase 2a results, a Phase 2/3 trial is currently underway to further define optimal dosing and establish efficacy in larger patient populations. The progression from MCC950’s clinical termination through mechanistic characterization to OLT1177’s advancing clinical programme illustrates how a well-defined safety failure can be converted into actionable design principles for subsequent candidates, provided that the underlying target mechanism retains sufficient biological validity.
Pipeline Compounds and Mechanistic Innovations
Building on the design principles established through the MCC950 and OLT1177 experience, multiple research groups have pursued parallel strategies to develop NLRP3 inhibitors with improved selectivity, potency, and safety profiles. These efforts fall broadly into two categories. One involves compounds that act directly on the NLRP3 protein itself, while the other encompasses compounds that intervene at alternative or upstream points in the inflammasome activation pathway.
Among direct NLRP3 inhibitors, structural optimization has been a primary strategy. One study synthesized and evaluated 22 derivatives to establish structure-activity relationships guiding compound refinement.67 This work identified specific structural features associated with improved potency and reduced off-target effects. Separately, scaffold hopping strategies generated compounds with novel chemical frameworks while retaining NLRP3 inhibitory activity, aiming to escape potential liability features present in earlier scaffolds.68 Focused optimization efforts have yielded specific compounds showing promise in preclinical evaluation. A series of N-sulfonylurea derivatives produced compound 4b, which demonstrated potent NLRP3 inhibition with favorable pharmacokinetic properties in rodent inflammatory models.69 More recently, computational approaches have accelerated inhibitor discovery, with machine learning algorithms trained on existing NLRP3 inhibitor data predicting novel compound CSC-6 with nanomolar potency.70 Experimental validation confirmed the computational predictions, demonstrating that artificial intelligence methods can complement traditional medicinal chemistry in identifying optimized lead compounds. All of these candidates remain at the preclinical stage, and their pharmacokinetic and safety profiles in humans have not yet been characterized.
A second category of pipeline compounds targets alternative intervention points in the inflammasome activation pathway. The shared rationale is that disrupting upstream or parallel nodes may achieve comparable anti-inflammatory effects while avoiding some of the on-target liabilities associated with direct NLRP3 binding. The compound SLC3037 specifically disrupts NIMA-related kinase 7 (NEK7)-NLRP3 binding, preventing inflammasome oligomerization without directly engaging the NLRP3 protein itself.71 This approach may preserve some regulatory mechanisms that direct inhibitors would suppress. In preclinical models, SLC3037 demonstrated anti-inflammatory activity in monosodium urate crystal-induced gout models, though human pharmacology data are not yet available. Another innovation involves simultaneously targeting multiple nodes in the inflammatory cascade. The compound CBED exhibits dual inhibitory activity against both NLRP3 inflammasome assembly and gasdermin D pore formation in preclinical studies.72 Since gasdermin D mediates pyroptotic cell death downstream of inflammasome activation, dual inhibition provides a degree of redundancy that may limit compensatory pathway activation. Whether this translates into superior efficacy over single-target inhibition remains to be tested clinically. Addressing an upstream trigger rather than the inflammasome itself, the compound Z1456467176 functions as an allosteric modulator of purinergic receptor P2X7 (P2X7R), which serves as an upstream sensor triggering NLRP3 activation in response to extracellular adenosine triphosphate.73 By dampening P2X7R signaling, this compound indirectly reduces NLRP3 inflammasome activation, offering the potential advantage of targeting an established druggable receptor class. Natural products have also attracted attention as potential sources of NLRP3 inhibitor scaffolds. Cucurbitacin B, derived from traditional medicinal plants, demonstrated NLRP3 inhibitory activity in cellular and animal models relevant to gout pathophysiology in preclinical screening assays. It may serve as a starting point for semisynthetic derivatives with improved drug-like properties.74
The breadth of mechanistic approaches represented in this pipeline reflects the degree to which the field has moved beyond first-generation direct NLRP3 binding strategies. Direct inhibitors offer well-characterized target engagement but must navigate the selectivity and hepatic safety constraints illustrated by the MCC950 experience. Indirect and multi-target approaches introduce additional mechanistic flexibility, though at the cost of greater complexity in predicting clinical pharmacology. A limitation common to all compounds discussed in this section is their exclusive reliance on preclinical evidence. Whether the mechanistic diversity observed in cell and animal studies will translate into differentiated clinical profiles remains an open question. Table 2 summarizes the key characteristics of both IL-1 pathway inhibitors and NLRP3 inflammasome-targeted agents discussed in this review, providing a comparative reference across regulatory status, mechanism, and available evidence.
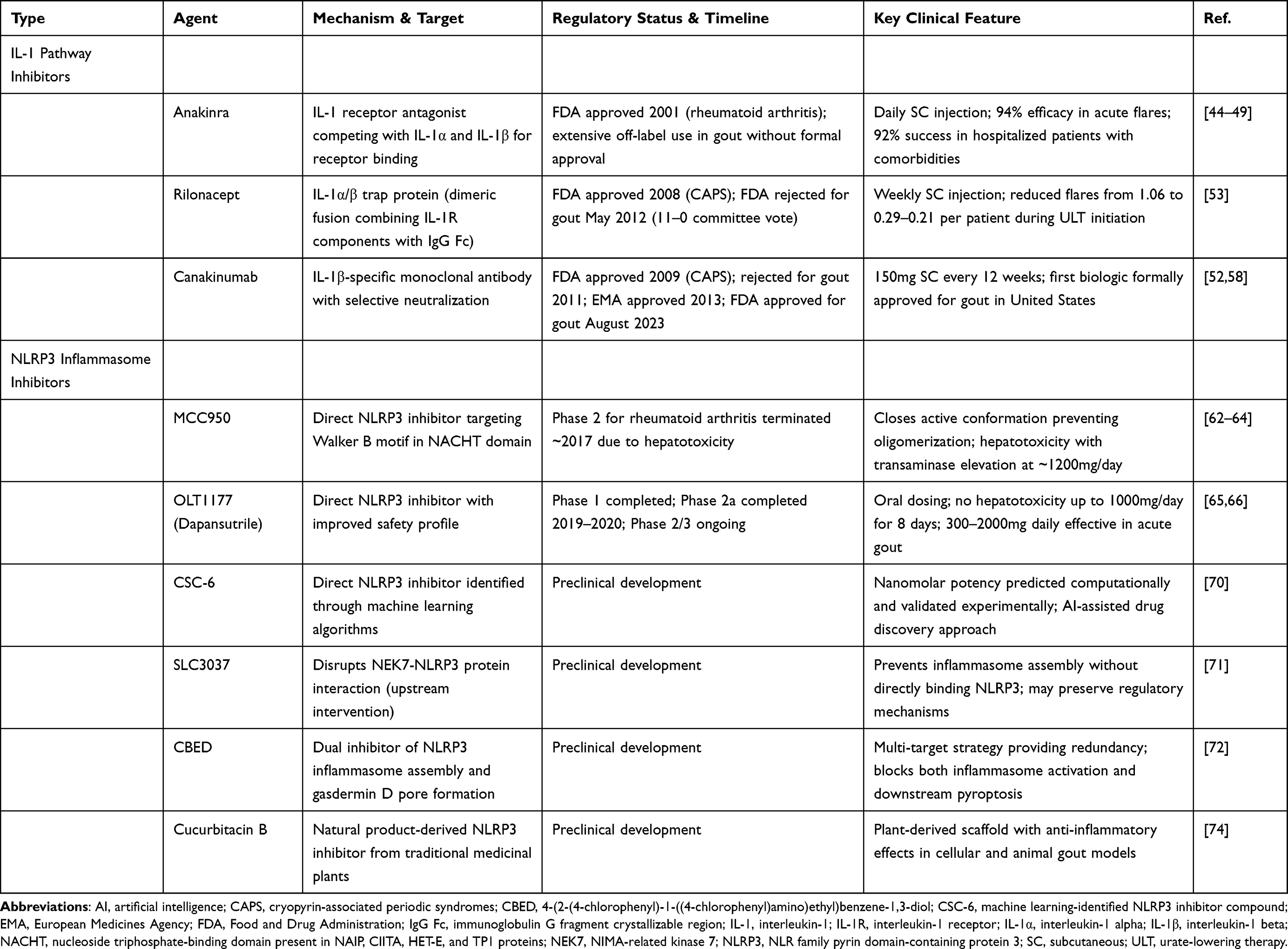 |
Table 2 Anti-Inflammatory Biologic and Small Molecule Therapies Targeting IL-1 and NLRP3 Pathways in Gout |
Clinical Decision-Making and Practical Considerations
Treatment Selection Framework
Biologic therapies for gout function as specialized options for specific patient populations rather than replacements for conventional treatments. Oral urate-lowering medications and standard anti-inflammatory agents remain first-line therapy for most patients. Biologics enter consideration when conventional treatments fail to achieve therapeutic targets, produce intolerable adverse effects, or face contraindications. Refractory gout with persistently elevated urate despite optimized oral therapy represents the primary indication for uricase biologics. Frequent acute flares uncontrolled by standard anti-inflammatory agents justify IL-1 pathway inhibitors or NLRP3 inhibitors. Advanced chronic kidney disease exemplifies clinical scenarios where multiple conventional options face simultaneous limitations, rendering biologics valuable alternatives. This treatment paradigm positions biologics as rescue therapies. The designation reflects both their specialized mechanisms and practical considerations including cost and administration burden.
Within biologic classes, agent selection requires matching the therapeutic mechanism to the individual patient’s clinical profile and treatment objectives. Patients needing sustained urate reduction benefit from pegloticase or related uricase therapies. Those requiring flare suppression despite adequate urate control benefit from anti-inflammatory biologics. Among IL-1 pathway inhibitors, the practical distinctions in administration frequency carry direct implications for patient suitability. The daily subcutaneous dosing required by anakinra suits acute on-demand management in hospital or clinic settings. By contrast, the quarterly schedule of canakinumab is better suited to patients requiring sustained prophylaxis in an outpatient context. These practical differences should inform selection alongside the mechanistic considerations discussed in the preceding section. Patient comorbidities including infection history, immunosuppression status, and organ function guide safety assessments. Economic constraints and insurance coverage patterns create practical limitations, typically requiring documented conventional treatment inadequacy before biologic approval. Shared decision-making incorporating patient preferences regarding administration routes and visit frequency ensures selected treatments align with individual circumstances.
Monitoring and Safety Management
Uricase therapy monitoring centers on serial serum uric acid measurements at each infusion visit. Successful treatment typically achieves levels below 6 mg/dL, often reaching below 2 mg/dL. Persistent urate elevation above 6 mg/dL or two consecutive elevated measurements suggest antidrug antibody development and impending treatment failure. Where immunomodulatory co-therapy is employed, additional laboratory surveillance including blood counts and hepatic function tests should be conducted at intervals appropriate to the chosen agent. Anti-inflammatory biologic monitoring emphasizes flare frequency and severity through patient symptom documentation. Clinical efficacy is reflected in reduced joint symptoms and improved function, though standardized response criteria remain lacking. Monitoring frequency is generally higher during treatment initiation and can be extended once stable responses are established.
The principal safety considerations differ by administration route and mechanism. Infusion reactions represent the most immediate concern with intravenous biologics, requiring protocols for recognition and management. These protocols should address presentations ranging from mild reactions to anaphylaxis. Premedication and post-infusion observation periods serve to mitigate these risks. Subcutaneous agents produce injection site reactions that are generally manageable through technique education. Across both routes, immunosuppression increases infection susceptibility, necessitating baseline screening for latent infections and clear patient education about prompt symptom reporting. Vaccination status review ensures appropriate immunization before therapy when feasible. Special populations including those with advanced kidney disease or prior organ transplantation warrant individualized monitoring reflecting elevated baseline risks. Systematic documentation of clinical responses and adverse events supports ongoing benefit-risk assessment and enables timely treatment modification when safety signals emerge.
Conclusion
Biologic therapies now occupy defined roles in gout management for patients who cannot achieve adequate disease control through conventional treatments. Uricase-based approaches have advanced beyond the immunogenicity limitations of early recombinant enzymes, with concomitant immunomodulatory therapy improving sustained response rates to pegloticase by reducing antidrug antibody formation. IL-1 pathway inhibitors completed a prolonged regulatory trajectory, culminating in canakinumab FDA approval in August 2023 as the first biologic formally indicated for gout in the United States. Anakinra retains practical utility in patients with contraindications to conventional anti-inflammatory agents. NLRP3 inflammasome inhibitors remain at an earlier stage of clinical development. The field has incorporated lessons from the MCC950 hepatotoxicity experience into more cautious dose selection and safety monitoring for subsequent candidates. Across all three classes, biologics function as specialized rescue options for patients with refractory or contraindicated conventional therapy rather than as first-line alternatives. Shared challenges including immunogenicity management, treatment costs, and limited long-term safety data continue to constrain broader clinical adoption.
Several limitations of this review warrant acknowledgment. As a narrative rather than a systematic review, literature selection was guided by clinical and translational relevance rather than a predefined protocol, and some degree of selection bias cannot be excluded. The sections addressing NLRP3 inflammasome inhibitors draw predominantly on preclinical data, as most pipeline compounds have not yet entered or completed clinical trials. Conclusions regarding their therapeutic potential therefore remain preliminary pending human validation. The published literature on long-term safety for several agents discussed here also remains limited. This limitation constrains the strength of conclusions that can be drawn regarding extended treatment risk profiles.
Future progress will depend on advances in several converging areas. Predictive biomarkers capable of identifying patients most likely to respond to specific biologic classes would improve treatment efficiency and reduce unnecessary exposure to costly or immunosuppressive regimens. For uricase therapies, ongoing Phase 3 evaluation of SEL-212 will clarify whether immunomodulatory co-administration can reliably extend sustained response rates beyond those achievable with pegloticase monotherapy. For IL-1 pathway inhibitors, real-world safety data accumulating under post-approval canakinumab experience will be important in defining the long-term benefit-risk profile across diverse patient populations. These populations include those with cardiovascular comorbidities and chronic kidney disease. In NLRP3 inhibitor development, successful clinical translation of oral candidates such as OLT1177 would introduce a mechanistically distinct treatment option. Such an option would avoid the administration burden and immunogenicity concerns associated with injectable biologics. Addressing the economic and access barriers that currently limit biologic use to a narrow subset of eligible patients will ultimately determine whether the advances discussed in this review reach those who stand to benefit from them.
Abbreviations
ASGARD, Anakinra for Synovial fluid analysis in Gout and Related Diseases; CANTOS, Canakinumab Anti-inflammatory Thrombosis Outcomes Study; CAPS, cryopyrin-associated periodic syndromes; COMPARE, Comparative Randomized Assessment of Pegloticase vs SEL-212; FDA, Food and Drug Administration; IL-1, interleukin-1; IL-1α, interleukin-1 alpha; IL-1β, interleukin-1 beta; ImmTOR, rapamycin-containing tolerogenic nanoparticle proprietary formulation; MIRROR, Methotrexate to Increase Response Rates in Patients with Uncontrolled Gout Receiving Pegloticase; NEK7, NIMA-related kinase 7; NLRP3, NLR family pyrin domain-containing protein 3; NSAIDs, nonsteroidal anti-inflammatory drugs; PEG, polyethylene glycol; PEG-PHRU, PEGylated porcine-human recombinant uricase; RA, rheumatoid arthritis; RECIPE, Reducing Immunogenicity of Pegloticase.
Data Sharing Statement
No new data were created or analyzed in this study.
Author Contributions
Ye Zhou: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Supervision. Hengyan Zhang: Conceptualization, Validation, Writing – review & editing. Nian Liu: Conceptualization, Writing – original draft, Writing – review & editing, Visualization. Heguo Yan: Investigation, Writing – original draft, Writing – review & editing, Data curation. Fanyu Meng: Conceptualization, Supervision, Writing – review & editing. Jiangyun Peng: Conceptualization, Resources, Writing – review & editing, Supervision, Project administration, Funding acquisition. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Yunnan Clinical Research Center for Rheumatism in Traditional Chinese Medicine (202405AJ310004), the Yunnan Science and Technology Talent and Platform Program (202105AG070012), the Yunnan University of Chinese Medicine–Hospital Joint Fund Projects (XYLH2024042 and XYLH2024030), the Science Research Foundation of Education Department of Yunnan Province (2025Y0638 and 2024Y380), the Expert Workstation of Zhangxuan in Yunnan Province (202305AF150175), the Funding of Yunnan Applied Basic Research Projects-Union Foundation (202401AZ070001-052) and the High level Key Discipline of TCM Construction Project, Rheumatology, of National Administration of Traditional Chinese Medicine (zyyzdxk-2023189).
Disclosure
The authors declare no competing interests in this work.
References
1. Mainak B, Satinath M. Gout. N Engl J Med. 2023;388(13). doi:10.1056/NEJMc2216467
2. Tabi-Amponsah AD, Stewart S, Stamp LK, Taylor WJ, Terkeltaub R, Dalbeth N. The gout, hyperuricemia and crystal-associated disease network (G-CAN) definition of clinical remission in gout. Ann Rheumatic Dis. 2025;S0003496725010271. doi:10.1016/j.ard.2025.05.017
3. So AK, Martinon F. Inflammation in gout: mechanisms and therapeutic targets. Nat Rev Rheumatol. 2017;13(11):639–16. doi:10.1038/nrrheum.2017.155
4. Neilson J, Bonnon A, Dickson A, Roddy E. Gout: diagnosis and management—summary of NICE guidance. BMJ. 2022;o1754. doi:10.1136/bmj.o1754
5. Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective – a review. J Adv Res. 2017;8(5):495–511. doi:10.1016/j.jare.2017.04.008
6. Jatuworapruk K, Louthrenoo W. Emerging therapeutic options for refractory gout. Nat Rev Rheumatol. 2024;20(2):73–74. doi:10.1038/s41584-023-01066-5
7. Gérard B, Leask M, Merriman TR, et al. Hyperuricaemia and gout in the Pacific. Nat Rev Rheumatol. 2025;21(4):197–210. doi:10.1038/s41584-025-01228-7
8. Terkeltaub R. Emerging urate-lowering drugs and pharmacologic treatment strategies for gout: a narrative review. Drugs. 2023;83(16):1501–1521. doi:10.1007/s40265-023-01944-y
9. Stamp LK, Gaffo A. What future do biological therapies have in the treatment of gout? Expert Opin Biol Ther. 2023;23(12):1151–1154. doi:10.1080/14712598.2023.2273936
10. Schlesinger N, Kaufmann D. Updates in uricase therapy for gout. Curr Opin Rheumatol. 2025;37(6):422–429. doi:10.1097/BOR.0000000000001122
11. Arnold DD, Yalamanoglu A, Boyman O. Systematic review of safety and efficacy of IL-1-targeted biologics in treating immune-mediated disorders. Front Immunol. 2022;13:888392. doi:10.3389/fimmu.2022.888392
12. Liu Y, Jarman JB, Low YS, et al. A widely distributed gene cluster compensates for uricase loss in hominids. Cell. 2023;186(16):3400–3413.e20. doi:10.1016/j.cell.2023.06.010
13. Roman YM. The role of uric acid in human health: insights from the uricase gene. JPM. 2023;13(9):1409. doi:10.3390/jpm13091409
14. Richette P, Bardin T. Successful treatment with rasburicase of a tophaceous gout in a patient allergic to allopurinol. Nat Rev Rheumatol. 2006;2(6):338–342. doi:10.1038/ncprheum0214
15. Xu H, Feldman GM, Max EE. High-dose IV administration of rasburicase suppresses anti-rasburicase antibodies, depletes rasburicase-specific lymphocytes, and upregulates treg cells. AAPS J. 2020;22(4):80. doi:10.1208/s12248-020-00461-0
16. Lyseng-Williamson KA. Pegloticase: in treatment-refractory chronic gout. Drugs. 2011;71(16):2179–2192. doi:10.2165/11202830-000000000-00000
17. Berhanu AA, Krasnokutsky S, Keenan RT, Pillinger MH. Pegloticase failure and a possible solution: immunosuppression to prevent intolerance and inefficacy in patients with gout. Semin Arthritis Rheumatism. 2017;46(6):754–758. doi:10.1016/j.semarthrit.2016.09.007
18. Schlesinger N, Padnick-Silver L, LaMoreaux B. Enhancing the response rate to recombinant uricases in patients with gout. BioDrugs. 2022;36(2):95–103. doi:10.1007/s40259-022-00517-x
19. Xiong H, Zhou Y, Zhou Q, et al. Nanocapsule assemblies as effective enzyme delivery systems against hyperuricemia. Nanomed Nanotechnol Biol Med. 2016;12(6):1557–1566. doi:10.1016/j.nano.2016.02.010
20. Kivitz A, DeHaan W, Azeem R, et al. Phase 2 dose-finding study in patients with gout using SEL-212, a novel PEGylated uricase (SEL-037) combined with tolerogenic nanoparticles (SEL-110). Rheumatol Ther. 2023;10(4):825–847. doi:10.1007/s40744-023-00546-0
21. Sands E, Kivitz A, DeHaan W, Leung SS, Johnston L, Kishimoto TK. Tolerogenic nanoparticles mitigate the formation of anti-drug antibodies against pegylated uricase in patients with hyperuricemia. Nat Commun. 2022;13(1):272. doi:10.1038/s41467-021-27945-7
22. Baraf HSB, Khanna PP, Kivitz AJ, et al. The COMPARE head-to-head, randomized controlled trial of SEL-212 (pegadricase plus rapamycin-containing nanoparticle, ImmTORTM) versus pegloticase for refractory gout. Rheumatology. 2024;63(4):1058–1067. doi:10.1093/rheumatology/kead333
23. Li Z, Shen L, Ma A, et al. Pegloticase co-administered with high MW polyethylene glycol effectively reduces PEG-immunogenicity and restores prolonged circulation in mouse. Acta Biomater. 2023;170:250–259. doi:10.1016/j.actbio.2023.08.052
24. Ban Z, Sun M, Ji H, et al. Immunogenicity-masking delivery of uricase against hyperuricemia and gout. J Control Release. 2024;372:862–873. doi:10.1016/j.jconrel.2024.06.042
25. Cho J, Yang B, Lee JH, et al. In vivo study of newly developed albumin-conjugated urate oxidase for gout treatment. Arthritis Res Ther. 2023;25(1):247. doi:10.1186/s13075-023-03231-3
26. Wang X, Lu H, Rong J, et al. PEGylated porcine–human recombinant uricase: a novel fusion protein with improved efficacy and safety for the treatment of hyperuricemia and renal complications. Open Life Sci. 2024;19(1):20220799. doi:10.1515/biol-2022-0799
27. Botson JK, Tesser JRP, Bennett R, et al. Pegloticase in combination with methotrexate in patients with uncontrolled gout: a multicenter, open-label study (MIRROR). J Rheumatol. 2021;48(5):767–774. doi:10.3899/jrheum.200460
28. Broadwell A, Albert JA, Padnick-Silver L, LaMoreaux B. Community practice experiences with a variety of immunomodulatory agents co-administered with pegloticase for the treatment of uncontrolled gout. Rheumatol Ther. 2022;9(6):1549–1558. doi:10.1007/s40744-022-00492-3
29. Khanna P, Khanna D, Cutter G, et al. Reducing immunogenicity of pegloticase (Recipe) with concomitant use of mycophenolate mofetil in patients with refractory gout: a phase ii randomized controlled trial. Ann Rheumatic Dis. 2021;80:279–280. doi:10.1136/annrheumdis-2021-eular.3111
30. Lipsky PE, Calabrese LH, Kavanaugh A, et al. Pegloticase immunogenicity: the relationship between efficacy and antibody development in patients treated for refractory chronic gout. Arthritis Res Ther. 2014;16(2):R60. doi:10.1186/ar4497
31. Holladay EE, Mudano AS, Xie F, et al. Real‐world effectiveness of pegloticase associated with use of concomitant immunomodulatory therapy. Arthritis Care Res. 2024;76(10):1361–1370. doi:10.1002/acr.25361
32. Botson J, Peloso PM, Obermeyer K, Lamoreaux B, Weinblatt ME, Peterson J. Pegloticase response improvement by co-treatment with methotrexate: results from the mirror open-label clinical trial in patients with uncontrolled gout. Ann Rheumatic Dis. 2020;79:446. doi:10.1136/annrheumdis-2020-eular.3932
33. Botson JK, Saag K, Peterson J, et al. A randomized, placebo‐controlled study of methotrexate to increase response rates in patients with uncontrolled gout receiving pegloticase: primary efficacy and safety findings. Arthritis Rheumatol. 2023;75(2):293–304. doi:10.1002/art.42335
34. Lawrence Edwards N, Singh JA, Troum O, Yeo AE, Lipsky PE. Characterization of patients with chronic refractory gout who do and do not have clinically apparent tophi and their response to pegloticase. Rheumatology. 2019;58(8):1422–1431. doi:10.1093/rheumatology/kez017
35. Morlock RJ, Dalal D, Divino V, et al. Characteristics and management of uncontrolled gout prior to pegloticase therapy: a 2-year claims analysis. Rheumatol Ther. 2025;12(1):37–51. doi:10.1007/s40744-024-00723-9
36. Chen SK, Liu J, Kim SC. Real-world patterns of pegloticase use for treatment of gout: descriptive multidatabase cohort study. BMJ Open. 2020;10(12):e041167. doi:10.1136/bmjopen-2020-041167
37. Johnson RJ, Choi HK, Yeo AE, Lipsky PE. Pegloticase treatment significantly decreases blood pressure in patients with chronic gout. Hypertension. 2019;74(1):95–101. doi:10.1161/HYPERTENSIONAHA.119.12727
38. Holladay EE, Mudano AS, Xie F, et al. Urate-lowering therapy, serum urate, inflammatory biomarkers, and renal function in patients with gout following pegloticase discontinuation. Arthritis Res Ther. 2024;26(1):86. doi:10.1186/s13075-024-03318-5
39. Morton AH, Hosey T, LaMoreaux B. Retreatment with pegloticase after a gap in therapy in patients with gout: a report of four cases. Rheumatol Ther. 2018;5(2):583–594. doi:10.1007/s40744-018-0111-9
40. Powers NE, Swartzwelter B, Marchetti C, et al. PASylation of IL-1 receptor antagonist (IL-1Ra) retains IL-1 blockade and extends its duration in mouse urate crystal-induced peritonitis. J Biol Chem. 2020;295(3):868–882. doi:10.1016/S0021-9258(17)49941-8
41. Schlesinger N, Pillinger MH, Simon LS, Lipsky PE. Interleukin-1β inhibitors for the management of acute gout flares: a systematic literature review. Arthritis Res Ther. 2023;25(1):128. doi:10.1186/s13075-023-03098-4
42. Doaré E, Robin F, Racapé H, et al. Features and outcomes of microcrystalline arthritis treated by biologics: a retrospective study. Rheumatol Ther. 2021;8(3):1241–1253. doi:10.1007/s40744-021-00335-7
43. Ottaviani S, Molto A, Ea HK, et al. AB0642 Efficacy of anakinra in gouty arthritis in real life population: a report of 36 cases. Ann Rheumatic Dis. 2013;72:A985. doi:10.1136/annrheumdis-2013-eular.2964
44. Janssen CA, Oude Voshaar MAH, Vonkeman HE, et al. Anakinra for the treatment of acute gout flares: a randomized, double-blind, placebo-controlled, active-comparator, non-inferiority trial. Rheumatology. 2019;58(8):1344–1352. doi:10.1093/rheumatology/key402
45. Saag KG, Khanna PP, Keenan RT, et al. A randomized, phase ii study evaluating the efficacy and safety of anakinra in the treatment of gout flares. Arthritis Rheumatol. 2021;73(8):1533–1542. doi:10.1002/art.41699
46. Desmarais J, Chu CQ. Utility of anakinra in acute crystalline diseases: a retrospective study comparing a university hospital with a veterans affairs medical center. J Rheumatol. 2019;46(7):748–750. doi:10.3899/jrheum.180393
47. Loustau C, Rosine N, Forien M, et al. Effectiveness and safety of anakinra in gout patients with stage 4–5 chronic kidney disease or kidney transplantation: a multicentre, retrospective study. Joint Bone Spine. 2018;85(6):755–760. doi:10.1016/j.jbspin.2018.03.015
48. Jeria-Navarro S, Gomez-Gomez A, Park HS, et al. Effectiveness and safety of anakinra in gouty arthritis: a case series and review of the literature. Front Med. 2023;9:1089993. doi:10.3389/fmed.2022.1089993
49. Balasubramaniam G, Parker T, Turner D, et al. Feasibility randomised multicentre, double-blind, double-dummy controlled trial of anakinra, an interleukin-1 receptor antagonist versus intramuscular methylprednisolone for acute gout attacks in patients with chronic kidney disease (ASGARD): protocol study. BMJ Open. 2017;7(9):e017121. doi:10.1136/bmjopen-2017-017121
50. Wang TKM, Klein AL. Rilonacept (Interleukin-1 Inhibition) for the treatment of pericarditis. Curr Cardiol Rep. 2022;24(1):23–30. doi:10.1007/s11886-021-01621-0
51. Church LD, McDermott MF. Rilonacept in cryopyrin-associated periodic syndromes: the beginning of longer-acting interleukin-1 antagonism. Nat Rev Rheumatol. 2009;5(1):14–15. doi:10.1038/ncprheum0959
52. Schlesinger N. Relationship of interleukin-1β blockade with incident gout and serum uric acid levels. Ann Intern Med. 2019;170(10):737–738. doi:10.7326/L19-0123
53. Schumacher HR, Evans RR, Saag KG, et al. Rilonacept (interleukin‐1 trap) for prevention of gout flares during initiation of uric acid–lowering therapy: results from a phase III randomized, double‐blind, placebo‐controlled, confirmatory efficacy study. Arthritis Care Res. 2012;64(10):1462–1470. doi:10.1002/acr.21690
54. Landi L, Ravaglia C, Russo E, et al. Blockage of interleukin-1β with canakinumab in patients with Covid-19. Sci Rep. 2020;10(1):21775. doi:10.1038/s41598-020-78492-y
55. Dhimolea E. Canakinumab. mAbs. 2010;2(1):3–13. doi:10.4161/mabs.2.1.10328
56. Schlesinger N. Canakinumab in gout. Expert Opin Biol Ther. 2012;12(9):1265–1275. doi:10.1517/14712598.2012.705825
57. Zhang W, Chen Y, Yao Z, Ouyang M, Sun M, Zou S. Post-marketing pharmacovigilance of canakinumab from the FDA adverse event reporting system (FAERS). Pharmaceuticals. 2025;18(1):114. doi:10.3390/ph18010114
58. Yoshida K, Glynn RJ, Choi HK, et al. Canakinumab’s effect against subsequent gout flares and high‐sensitivity C‐reactive protein levels: a causal mediation analysis. Arthritis Care Res. 2023;75(4):817–824. doi:10.1002/acr.24832
59. Tian Y, He X, Li R, Wu Y, Ren Q, Hou Y. Recent advances in the treatment of gout with NLRP3 inflammasome inhibitors. Bioorg Med Chem. 2024;112:117874. doi:10.1016/j.bmc.2024.117874
60. Li H, Guan Y, Liang B, et al. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome. Eur J Pharmacol. 2022;928:175091. doi:10.1016/j.ejphar.2022.175091
61. Zheng Y, Zhang X, Wang Z, et al. MCC950 as a promising candidate for blocking NLRP3 inflammasome activation: a review of preclinical research and future directions. Arch Pharm. 2024;357(11):e2400459. doi:10.1002/ardp.202400459
62. Zhao H, Kumar P, Sobreira TJP, et al. Integrated proteomics characterization of NLRP3 inflammasome inhibitor MCC950 in monocytic cell line confirms direct MCC950 engagement with endogenous NLRP3. ACS Chem Biol. 2024;19(4):962–972. doi:10.1021/acschembio.3c00777
63. Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol. 2019;15(6):560–564. doi:10.1038/s41589-019-0278-6
64. Coll RC, Hill JR, Day CJ, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15(6):556–559. doi:10.1038/s41589-019-0277-7
65. Marchetti C, Swartzwelter B, Koenders MI, et al. NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res Ther. 2018;20(1):169. doi:10.1186/s13075-018-1664-2
66. Klück V, Jansen TLTA, Janssen M, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020;2(5):e270–e280. doi:10.1016/S2665-9913(20)30065-5
67. Wang W, Pang J, Ha EH, et al. Development of novel NLRP3-XOD dual inhibitors for the treatment of gout. Bioorg Med Chem Lett. 2020;30(4):126944. doi:10.1016/j.bmcl.2019.126944
68. Shi C, Lyu W, Yu J, et al. Scaffold hopping-driven optimization for the identification of NLRP3 inhibitors as potential gout therapeutics. Eur J Med Chem. 2024;279:116881. doi:10.1016/j.ejmech.2024.116881
69. Narros-Fernández P, Chioua M, Petcu SA, et al. Synthesis and pharmacological evaluation of New N -Sulfonylureas as NLRP3 inflammasome inhibitors: identification of a hit compound to treat gout. J Med Chem. 2022;65(8):6250–6260. doi:10.1021/acs.jmedchem.2c00149
70. Shi C, Zhang X, Chi X, et al. Discovery of NLRP3 inhibitors using machine learning: identification of a hit compound to treat NLRP3 activation-driven diseases. Eur J Med Chem. 2023;260:115784. doi:10.1016/j.ejmech.2023.115784
71. Park K, Shin I, Kim Y, et al. A novel NLRP3 inhibitor as a therapeutic agent against monosodium urate-induced gout. Front Immunol. 2024;14:1307739. doi:10.3389/fimmu.2023.1307739
72. Zhou M, Li S, Song L, Hu Q, Liu W. 4-(2-(4-chlorophenyl)-1-((4-chlorophenyl)amino)ethyl)benzene-1, 3-diol is a potential agent for gout therapy as a dual inhibitor of XOD and NLRP3. Phytomedicine. 2018;42:9–17. doi:10.1016/j.phymed.2018.03.007
73. Li X, Liu Y, Luo C, Tao J. Z1456467176 alleviates gouty arthritis by allosterically modulating P2X7R to inhibit NLRP3 inflammasome activation. Front Pharmacol. 2022;13:979939. doi:10.3389/fphar.2022.979939
74. Xue Y, Li R, Fang P, et al. NLRP3 inflammasome inhibitor cucurbitacin B suppresses gout arthritis in mice. J Mol Endocrinol. 2021;67(2):27–40. doi:10.1530/JME-20-0305
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.