Back to Journals » Drug Design, Development and Therapy » Volume 12
Biological activity, quantitative structure–activity relationship analysis, and molecular docking of xanthone derivatives as anticancer drugs
Authors Miladiyah I ![]() , Jumina J, Haryana SM, Mustofa M
, Jumina J, Haryana SM, Mustofa M
Received 25 August 2017
Accepted for publication 17 November 2017
Published 15 January 2018 Volume 2018:12 Pages 149—158
DOI https://doi.org/10.2147/DDDT.S149973
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Isnatin Miladiyah,1,2 Jumina Jumina,3 Sofia Mubarika Haryana,4 Mustofa Mustofa5
1Pharmacology Department, Faculty of Medicine, Islamic University of Indonesia, 2Doctorate Program of Medical Science and Health, Faculty of Medicine, 3Chemistry Department, Faculty of Mathematics and Natural Sciences, 4Histology and Cell Biology Department, Faculty of Medicine, 5Pharmacology and Therapeutic Department, Faculty of Medicine, Gadjah Mada University, Yogyakarta, Indonesia
Background: Xanthone derivatives have a wide range of pharmacological activities, such as those involving antibacterial, antiviral, antimalarial, anthelmintic, anti-inflammatory, antiprotozoal, and anticancer properties. Among these, we investigated the anticancer properties of xanthone. This research aimed to analyze the biological activity of ten novel xanthone derivatives, to investigate the most contributing-descriptors for their cytotoxic activities, and to examine the possible mechanism of actions of xanthone compound through molecular docking.
Materials and methods: The cytotoxic tests were carried out on WiDR and Vero cell lines, by a 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay method. The structural features required for xanthone’s anticancer activity were conducted by using the semi-empirical Austin Model-1 method, and continued with quantitative structure-activity relationship (QSAR) analysis using BuildQSAR program. The study of the possible mechanism of actions of the selected xanthone compound was done through molecular docking with PLANTS.
Results: The three novel xanthone derivatives (compounds 5, 7, and 8) exhibited cytotoxic activity with compound 5 showed the highest degree of cytotoxicity at concentration 9.23 µg/mL (37.8 µM). The following best equation model was obtained from the BuildQSAR calculation: log 1/IC50 = -8.124 qC1 -35.088 qC2 -6.008 qC3 + 1.831 u + 0.540 logP -9.115 (n = 10, r = 0.976, s = 0.144, F = 15.920, Q2 = 0.651, SPRESS = 0.390). This equation model generated 15 proposed new xanthone compounds with better-predicted anticancer activities. A molecular docking study of compound 5 showed that xanthone formed binding interactions with some receptors involved in cancer pathology, including telomerase, tumor-promoting inflammation (COX-2), and cyclin-dependent kinase-2 (CDK2) inhibitor.
Conclusion: The results suggested that compound 5 showed the best cytotoxic activity among the xanthone derivatives tested. QSAR analysis showed that the descriptors contributed to xanthone’s cytotoxic activity were the net atomic charge at qC1, qC2, and qC3 positions, also dipole moment and logP. Compound 5 was suspected to be cytotoxic by its inhibition of telomerase, COX-2, and CDK2 receptors.
Keywords: xanthones, anticancer, semiempirical AM1, BuildQSAR, molecular docking
Introduction
Xanthone (9H-xanthene-9-one) is a heterocyclic compound with dibenzo-γ-pyrone as the main structure. The basic active compound consists of a tricyclic planar bone with one pyran ring fused with two phenyl rings on both sides.1 Natural xanthone compounds are reported to have various pharmacological activities, including antibacterial,2 antivirus,3 anthelmintics,4 antiprotozoal,5 hepatoprotective,6 anti-inflammatory,7 antimalarial,8 antitrypanosomiasis,9 and anticancer activities. Over the past decade, the anticancer activities of xanthone derivatives, mainly from natural origin, have been widely studied.
Cancer is the major cause of death in developed countries and the second leading cause in developing countries. The International Agency for Research on Cancer reported 12.7 million new cancer cases with 7.6 million deaths in 2008 alone,10 and this number increased in 2012.11 The low success rate of chemotherapeutic agents has led to increased interest in the development of new anticancer compounds. One of the discovery strategies for new anticancer drugs is to explore natural compounds, including xanthones, that possess anticancer activities. Some naturally occurring xanthones have been reported to have anticancer activities.12–14 However, natural compounds have the disadvantage of low yields obtained from the extraction processes. Therefore, it would be beneficial to develop a synthetic compound with higher yield to ensure the availability and continuity of production of the compound.
Along with the advancements in knowledge and technology, drug discovery and development also involve computational processes,15 including quantitative structure–activity relationship (QSAR) analysis and molecular docking. A QSAR model generates new compounds with better predicted biological activity,16 which can then be developed as drug candidates. Molecular docking is based on a structure-based approach and computational method using mathematical algorithms (the scoring function) to evaluate the binding tightness between the docked compound and target protein (receptor). Both QSAR analysis and molecular docking can be used individually or in combination.17 Recently, QSAR analysis and molecular docking are among the important strategies to design new molecules and identify hit molecules in medicinal chemistry.
A previous study identified some polyhydroxyxanthone derivatives (1,6-dihydroxy-, 1,3,7-trihydroxy-, and 1,3,6,8-tetrahydroxyxanthones) as potent in vitro anticancer agents. The cytotoxic activity did not increase linearly with an increasing number of hydroxyl groups, which suggested that the position of the substituted group influenced the activity.18 In addition, ten novel hydroxyl- and halogen-substituted xanthone derivatives identified by a QSAR study were synthesized.19 Some of these compounds have been published earlier and described as having anticancer18 and antimalarial effects.20 These compounds have not been evaluated for efficacy against colorectal cancer.
The aim of this study was to evaluate these novel xanthones and determine the most prominent descriptor for cytotoxic activity to aid the development of more active anticancer agents. We also investigated the most probable mechanisms of action of xanthone against cancer on the basis of common principles of cancer through in silico molecular docking.
Materials and methods
Tested compounds and cancer cell culture
The synthesized xanthone compounds were the property of Yuanita (Laboratory of Organic Chemistry of the Faculty of Mathematics and Natural Sciences, Gadjah Mada University) and are listed in Table 1. All tested compounds were dissolved in 1% dimethyl sulfoxide and diluted with media to the desired treatment concentrations. Colorectal cancer WiDR cells and normal Vero cells were obtained from the Laboratory of Parasitology, Faculty of Medicine, Gadjah Mada University. Cells were cultured in RPMI 1640 (for WiDR) and M199 (for Vero) (Sigma-Aldrich Co., St Louis, MO, USA) supplemented with 0.1 mg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) and maintained in an incubator with 5% CO2 at 37°C. Only confluent cells were used for the experiment. This study received ethics approval from the Medical and Health Research Committee Faculty of Medicine, Gadjah Mada University, and Dr Sardjito General Hospital, number KE/FK/399/EC/2016.
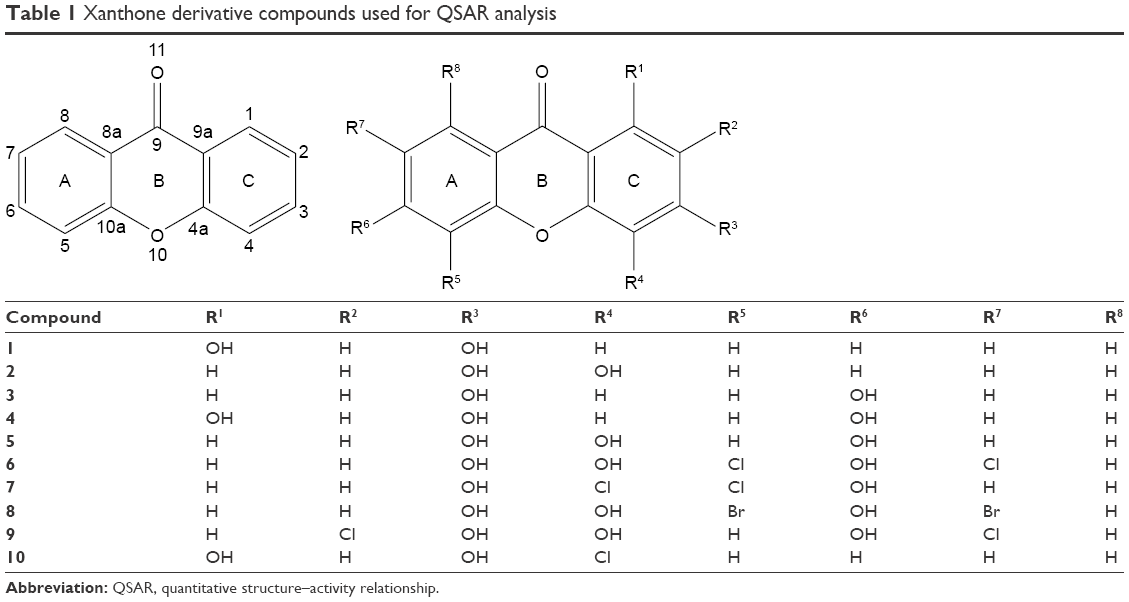 | Table 1 Xanthone derivative compounds used for QSAR analysis |
Data set
The data set for the QSAR analysis was obtained from the cytotoxic activity test results. The in vitro cytotoxic activities of xanthones were expressed as the inhibitory concentration 50% (IC50) values that were converted to log 1/IC50 and used as dependent variables in the QSAR analysis.
Instrumentation
A personal computer with an Intel® Pentium® 2117U @ 1.80 GHz CPU, 4.00 GB RAM, 500 GB hard disk drive, and a Windows 8® operating system was used. All quantum mechanical calculations of xanthones were executed by using Hyperchem 8®, and QSAR analysis was performed by using the BuildQSAR program.21 Preparation of the receptor protein and docking validation method was performed by using YASARA (http://www.yasara.org), and preparation of the ligand was performed by using the Chemdraw Ultra 12.0.2 and Marvinsketch programs. The molecular docking process was performed by using the PLANTS/Protein–Ligand Ant System.22 Meanwhile, the Pymol program (www.pymol.org) was used to see the hydrogen bonds in amino acid residues between the ligand and receptors.23
Procedures
Cytotoxic activity
The in vitro cytotoxic activities were evaluated by using the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) method as described earlier24 with modifications. Cells were seeded at 1×104 cells/well in 96-well plates and incubated for 24 h. They were then treated with 100 μL of medium (as untreated control) or 100 μL of varying concentrations of individual compounds (500, 250, 125, 62.5, 31.25, 15.625, 7.813, and 3.906 μg/mL for the WiDR cell line, and 1,000, 500, 250, 125, 62.5, 31.25, 15.625, and 7.813 μg/mL for the Vero cell line), 100 μL of various concentrations of doxorubicin as a positive control (100, 50, 25, 12.5, 6.25, and 3.125 μg/mL), and 100 μL of medium without cells as a medium control. The plates were incubated at 5% CO2 and 37°C for 24 h. After treatment, the medium was removed, MTT solution (10 μL per 100 μL medium) was added to all wells, and plates were incubated at 5% CO2 37°C for 4 h. To the cells was then added 100 μL of 10% sodium dodecyl sulfate in HCl 0.01 N; the cells were then incubated overnight to ensure that all formazan crystals were dissolved. The plates were read on a microplate enzyme-linked immunosorbent assay reader at 595 nm. The percentage of viable cells was determined according to the following formula:
|
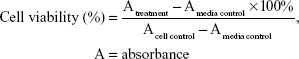
Cytotoxic activity was represented as the inhibitory concentration 50% (IC50) of each compound that was determined by probit analysis of the sample concentration versus cell viability. The selectivity index (SI) was calculated from the ratio of IC50 in Vero versus that in WiDR cells. SI values >3 were considered as having high selectivity.25
QSAR analysis
The structure of xanthone derivatives was sketched on Chemdraw Ultra 12.0.2 software. These structures were converted to their 3D forms by using Hyperchem® version 8.0 software, and a model was built to obtain the most stereochemically stable structure of each compound. The Austin Model 1 (AM1) semiempirical method was applied for geometric optimization of each compound. The obtained descriptors were arranged in a data set that was used in the BuildQSAR program to generate the most appropriate QSAR equation model.21
Docking molecular preparation
To explore the possibility of an action mechanism of xanthone derivatives, in silico molecular docking was applied to the most active compound. Molecular docking was performed against a number of cancer pathologies (the hallmarks of cancer) as described earlier (Table 2).26 Three-dimensional (3D) structures of enzyme receptors investigated were downloaded from the Protein Data Bank (PDB) database (www.rcsb.org) with each corresponding to PDB identities (PDB IDs). The 3D structures of those enzymes were then prepared by using YASARA in the standard setting. Only the A-chain of the protein was extracted from each PDB file, and hydrogen atoms were added in the preparation process. The results were saved in the .mol2 format and were the virtual targets for docking simulation. The downloaded native ligands of each enzyme were prepared by using Marvinsketch to configure them into two-dimensional (2D) formats. The pKa values were 7.4, and ligand conformations were performed. The 10 conformers from each ligand were then saved in the .mol2 format for the next docking process.
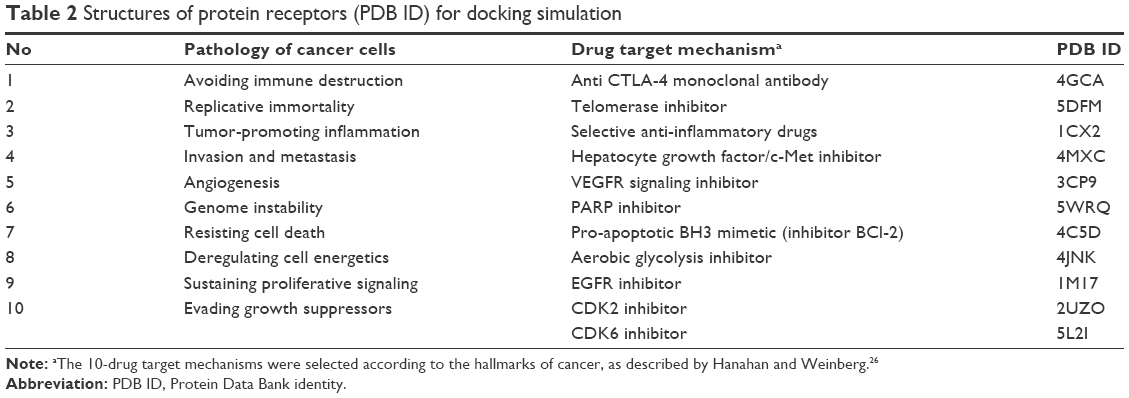 | Table 2 Structures of protein receptors (PDB ID) for docking simulation |
The most active xanthone compound was used as a test ligand. The 2D xanthone structure was constructed by using Marvinsketch 5.2.5.1.27 This ligand preparation was performed in a similar manner as used for the native ligands, and the 10 conformers generated were saved in a .mol2 format for the docking process.
Docking molecular process
In silico molecular docking of xanthone against a number of enzymes was performed by using the PLANTS/Protein–Ligand Ant System.22 The YASARA program was used to assess docking validation as measured by the root median square deviation (RMSD) parameter. An RMSD of <2 Å indicates that a docking program is valid and suitable for being reproduced.28 Smaller RMSD values indicate that the coordinate difference between two ligands is smaller and the overlap of the crystal structure and ligand copy is more similar.29 In the PLANTS program, the binding site definition file was set to 5 Å from the coordinate location where native ligand was bound to the enzyme. The binding pose with the best free binding energy (the lowest score) was considered as the best predictive binding position. Amino acid residues that formed hydrogen bonds with the redocking compound from Pymol visualization were compared with those that interacted with the crystal molecule. The minor difference between the free binding energy of the native ligand and selected xanthone as well as the similarity of the amino acid residues of the hydrogen bonds suggested the possible mechanism underlying the anticancer activity of xanthone.
Results and discussion
Cytotoxic effects of xanthone derivatives on WiDR cancer cells and their selectivity
Ten novel xanthones (Table 1) were evaluated for their cytotoxicity against WiDR (colorectal cancer) and Vero (normal) cell lines by using the MTT method. Doxorubicin was used as a positive control group because it had a nucleus structure similar to that of xanthones.18 The aglycon structure of doxorubicin comprises a tetracyclic ring with quinine–hydroquinone, methoxy, and carbonyl substituents. Over the last 30 years, doxorubicin has become one of the most potent chemotherapeutic agents against many kinds of cancer.30
Some of the xanthone compounds tested in this study had potent antimalarial activity.20 Previous studies have shown that compounds with antimalarial activity, including artemisinin,31 chalcone,32 sesquiterpene lactones,33 and phenanthroline,34 had potential as anticancer agents. Those existing antimalarial agents had a wide range of known anticancer properties that could be distinct from their antimalarial properties. Most of the naturally derived antimalarial agents were found to have anticancer properties, and nearly half of them entered the clinical phase of drug development. The potential anticancer activity in antimalarial agents might be contributed by the multifaceted nature of the cellular response to some antimalarials, such as artemisinin.35
The IC50 values of the WiDR and Vero cell lines treated with the xanthones are summarized in Table 3. Compound 5 exhibited the best cytotoxic activity against the WiDR cell line with an IC50 value of 9.23 μg/mL, which was categorized as moderate cytotoxicity.36 All other xanthones with IC50 values >50 μg/mL showed insignificant cytotoxic activity. The SI of xanthones was calculated from the ratio of the IC50 of Vero cells versus that of WiDR cells. In the tumor selectivity analysis, we found that compound 5 exhibited high tumor selectivity with an SI of 66.40. Compounds with SI values >3 were considered to have high selectivity.25 The good tumor selectivity demonstrated by compound 5 suggested that it might have the potential to be further investigated and developed as an antitumor agent.
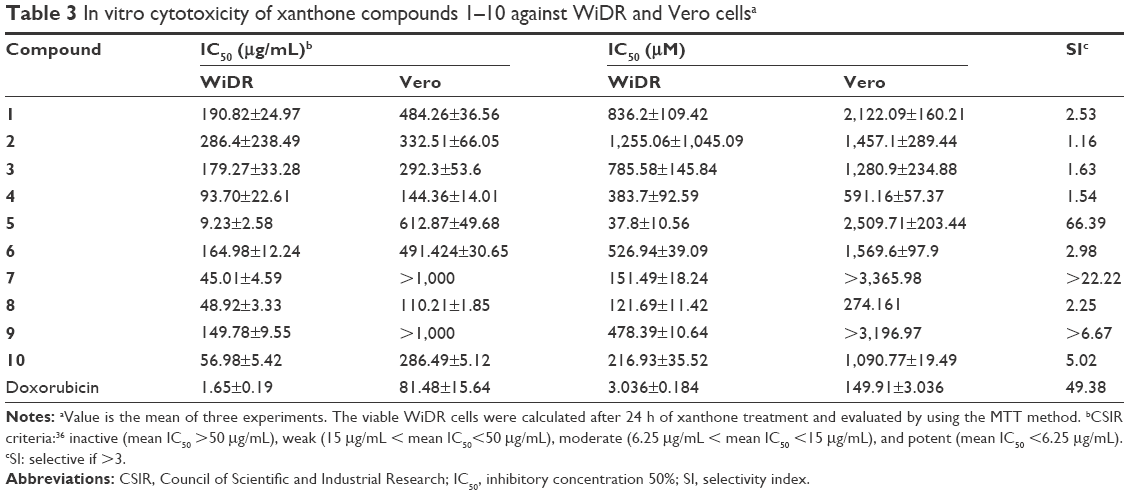 | Table 3 In vitro cytotoxicity of xanthone compounds 1–10 against WiDR and Vero cellsa |
This study revealed that only compound 5 (3,4,6-trihydroxyxanthone) showed a statistically significant moderate cytotoxicity effect weaker than that of doxorubicin as the control drug (p<0.05). A compound with moderate cytotoxic activity could be selected for further laboratory investigation.36 The high SI of compound 5 made it also promising for selection as a model compound for other in vitro and in silico screenings, including analysis of the possible mechanisms of action by docking simulation (to be discussed later).
A previous study has shown that the same compound 5 also exhibited good antimalarial activity, although its IC50 was slightly higher than that of the standard drug chloroquine.20 As previously reported, 93% of the naturally derived antimalarial compounds had potential for development as anticancer agents.35 Therefore, this result is in line with efforts to discover new anticancer agents among the established antimalarial compounds.
QSAR analysis process
Since colorectal cancer is one of the most frequent and deadly cancers, it is important to develop more potent agents against this cancer. Further structural optimization of some antitumor compounds needs QSAR analysis based on their cytotoxic effects. The following QSAR analysis was mainly based on the cytotoxic activity (in log 1/IC50) of xanthones in WiDR cell lines and some descriptors of the xanthones’ structures to predict their activity.
Xanthone structures were geometrically optimized by using the AM1 semiempirical method. The AM1 method was selected because it is a simple geometrical optimization that requires no complex mathematical calculation, unlike the ab initio method.37 Structural optimization using the AM1 method is rapid, and electronic structures are generated easily. This method is able to predict large molecules and multivalent compounds38 with good accuracy.
To determine the influence of a side chain on the cytotoxic activity of compounds, BuildQSAR was performed on a series of xanthones. BuildQSAR is a free QSAR program that enables researchers to build and analyze quantitative models through regression analysis. Multiple regressions performed by using BuildQSAR correlate the physicochemical descriptors and the cytotoxic activity of xanthone derivatives. This program automatically detects collinearity between descriptors, and only descriptors with non-collinearity are included in the regression equation. The use of BuildQSAR requires no statistical analysis because the analysis had already been included in the program.21
Six QSAR models each with statistical properties obtained from BuildQSAR are listed in Table 4. All of the models showed good correlation between biological activity and descriptors (R>0.75). Model 4 of the genetic algorithm method was similar to model 1 of the systematic research. Because of the closeness of R values among all models, determination of the best model could not be established only by comparing the R value. Generally, the best derived QSAR models are selected on the basis of a large correlation coefficient (R), Fcalc/Ftable, and Q2 and a small “s” and SPRESS.39 The ranks of the selected QSAR model performed by using the Microsoft Excel program are summarized in Table 5.
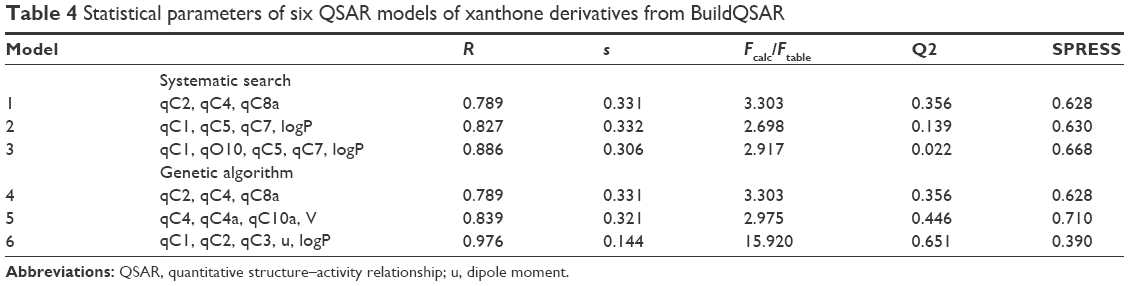 | Table 4 Statistical parameters of six QSAR models of xanthone derivatives from BuildQSAR |
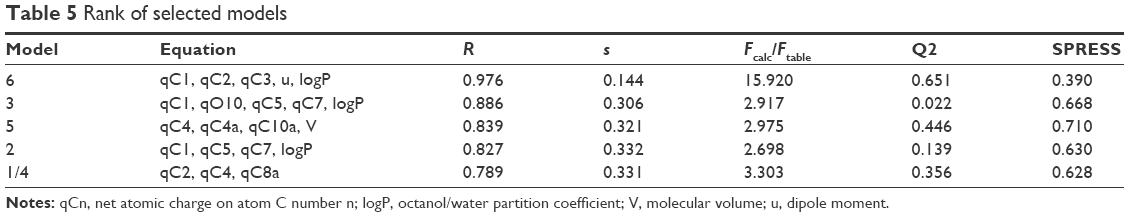 | Table 5 Rank of selected models |
qCn represents the net atomic charge on atom C number n. This is the most important descriptor in a QSAR equation because such electrostatic interactions are driven by electrical charges in the molecule. Thus, the electron charges are important in many physicochemical properties of compounds and widely used as chemical reactivity indices. u represents the dipole moment and serves as the most often used descriptor to describe the polarity of a molecule. This descriptor has a role in the molecule–receptor interaction. V represents the molecular volume that has a role in the binding free energies. LogP represents the octanol/water partition coefficient. This descriptor shows the affinity of a molecule to partition into the non-polar fraction (eg, lipids) instead of into the polar fraction (water).40 Model 6 was selected; the complete equation is given below:
|
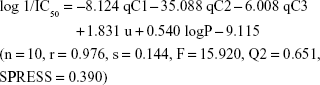
The equation of model 6 is statistically the best; thus, all of the following discussions are based on model 6. In this model, n represents the number of compounds contributed to build the model. The R value is the correlation coefficient; the closer R is to 1, the better the goodness of fit of the equation.39 The Fcalc/Ftable value represents the ratio between the variance of the calculated and tabulated values and, therefore, indicates that the regression relationships were statistically significant and did not occur by chance. Q2 serves as a criterion of robustness and predictive ability of the regression equation. The high Q2 value (>0.5) suggests the model’s ability to give an accurate prediction. The low s (the standard error of estimates) and SPRESS values suggest that the model is statistically significant for activity prediction.39
The r value of 0.976 from model 6 demonstrated that there was a strong correlation between the independent variables (descriptors) and cytotoxic activities of the xanthones. We confirmed that 97.6% of the altered cytotoxic activity of the studied xanthones was caused by the modification of qC1, qC2, qC3, moment dipole, and logP. These findings were relevant to a previous study that revealed that structural modification of the aromatic ring C, especially at the C1 and C2 positions, predicted improvement of the cytotoxic activity of the xanthone compounds. Therefore, structural modification is suggested on ring A or C.41
Model 6 has been used to predict the activity of the compounds to enable comparison with experimental results (observed activity). Observed versus predicted log 1/IC50 values based on the selected model 6 are presented in Table 6, and its scatter plot is presented in Figure 1. The correlation result showed that model 6 could predict the cytotoxic activity of 10 xanthone compounds very well, with a slope and correlation coefficient (R2) of 0.976 and 0.951, respectively.
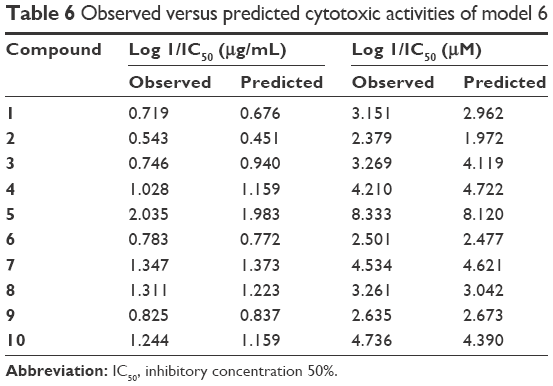 | Table 6 Observed versus predicted cytotoxic activities of model 6 |
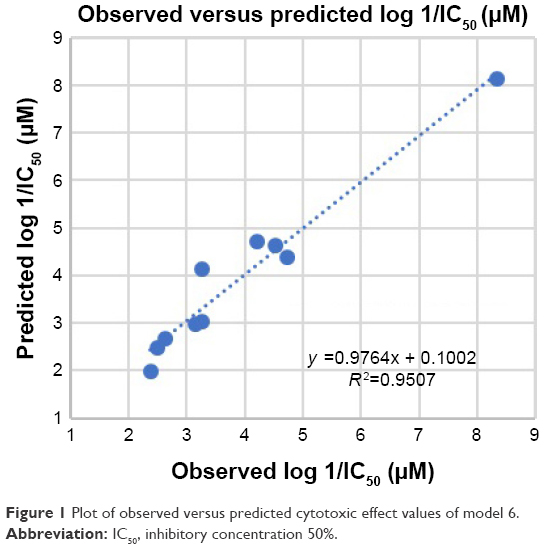 | Figure 1 Plot of observed versus predicted cytotoxic effect values of model 6. |
Design of novel xanthone derivatives
The basic structure of xanthone (Table 1) indicates that phenyl ring B cannot be substituted. Alteration of the net charge of adjacent atoms (qO11, qC9, qC9a, qC4a, qO10, qC10a, and qC8) will only be obtained through electronic density induction of charge modification of atoms in phenyl rings A and C. On the basis of the structure of compound 5 (as it had the best cytotoxic activity), this alteration could be achieved by modifications at qC5, qC7, and qC8 (ring A) and at qC1 and qC2 (ring C). Therefore, these two phenyl rings must be considered in designing a new xanthone with better cytotoxic activity.41
The best selected QSAR model is used to predict the cytotoxic activities of new synthetic xanthone compounds. The better cytotoxic activities of xanthones as IC50 values are given by the more positive value of log 1/IC50. Modification of new xanthones on the basis of the selected model 6 was performed by using the structure of compound 5 (3,4,6-trihydroxyxanthone) as the model because of the highest value of the cytotoxic activity. The more negative net atomic charge of qC1, qC2, and qC3, along with the more positive value of the dipole moment and logP, was recommended to increase the more positive value of log 1/IC50. Efforts such as substitution of electron-donating groups, such as R, OH, OR, NH2, NR2, NHCOR, OCOR, or CHCR2 groups, at the C1 and C2 positions (C3 position remained unchanged as the previous structure of compound 5) could be made. Structural modifications of compound 5 generated some formulas of new xanthones with better predicted cytotoxic activities, as listed in Table 7.
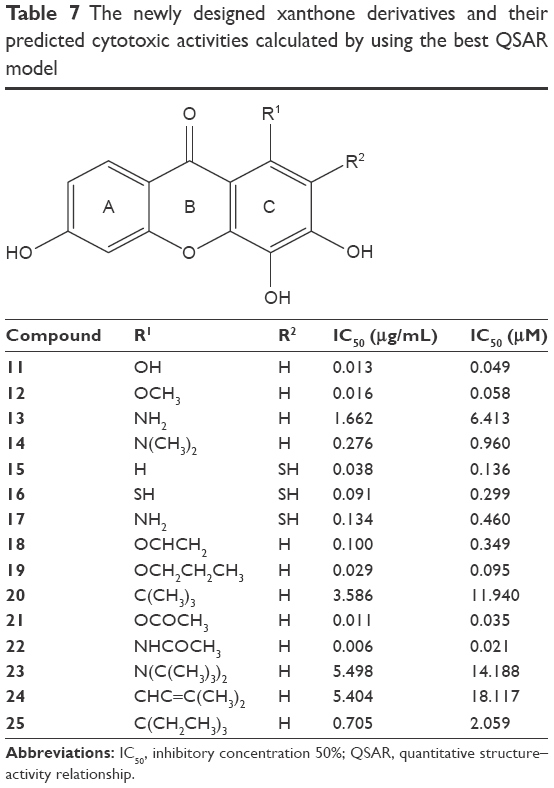 | Table 7 The newly designed xanthone derivatives and their predicted cytotoxic activities calculated by using the best QSAR model |
These electron-donating substitution groups give a negative atomic charge of qC1 and qC2 and encourage electron resonance toward the C1 and C2 positions as well as increase the electron density of these atoms. The increased electron density in qC1 and qC2 will give a higher value of log 1/IC50, which means that the IC50 is predicted as better than that of the model compound (compound 5). Molecular design of xanthone derivatives using equation of model 6 generated more potent compounds than the model structure (compound 5). All of these modifications were predicted to increase the inhibitory activity of new xanthone models. It is possible to synthesize the proposed novel compounds; however, the synthesis of the new xanthones that had the best anticancer activity in this study has not been reported.
Molecular docking of possible mechanisms
Molecular docking simulation was performed to explore the possible mechanism of xanthone compounds as cytotoxic agents. The critical points are highlighted in the pathology of cancer known as the hallmarks of cancer.26 The structures of the protein receptors downloaded from PDB for applying docking simulation are presented in Table 2. The results of docking process with hydrogen bonds where compound 5 interacted with the receptors are presented in Table 8.
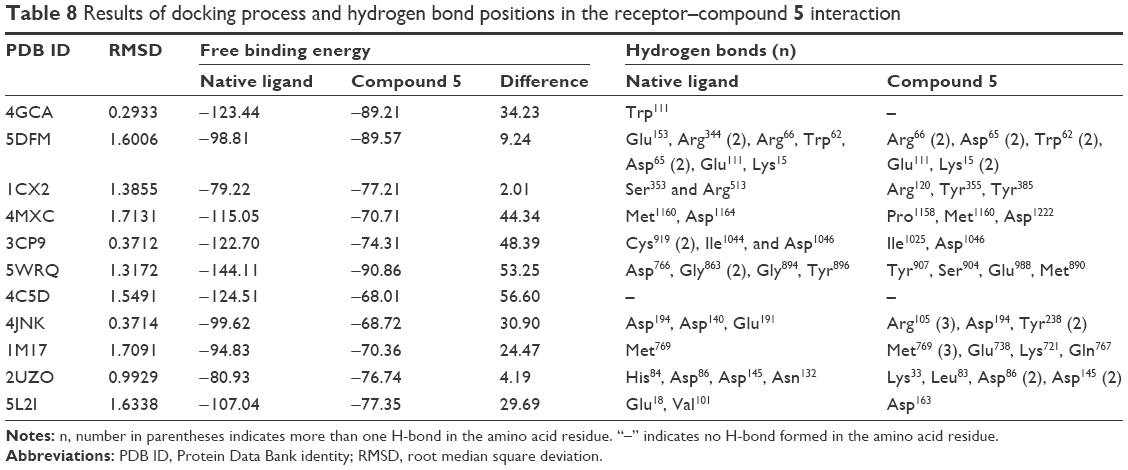 | Table 8 Results of docking process and hydrogen bond positions in the receptor–compound 5 interaction |
All RMSD values of the 10 receptors met the validity criteria for docking validation (<2 Å)28 and the docking process could be continued. Among the 10 hallmarks of cancer, better docking scores (free binding energy) relative to those of native ligands were achieved by PDB ID 5DFM (telomerase inhibition), 1CX2 (tumor promoting inflammation inhibition), and 2UZO (CDK2 inhibition). A lower free binding energy and a larger number of hydrogen bonds are associated with a more stable interaction between a compound and its receptor.42 The hydrogen bonds between the ligand and enzyme can predict the strength and catalytic activity of the complex.43
The binding site of compound 5 formed interactions with Arg120, Tyr355, and Tyr385 of the COX-2 enzyme. Arg120 and Tyr355 are the important parts of amino acid residues of the active site of the COX-2 enzyme and have a major role as the gateway entry of the ligand to the active site of the enzyme and initiation of the inhibitory effect.44 The hydrogen bonds in these residues are associated with better COX-2 inhibitory activity. Compound 5 was located in an area bounded by several amino acid residues, such as Trp385, Trp387, Phe518, Val523, His90, Leu352, Leu531, Ala527, Val349, Gly526, and Ser530 (image not displayed), and these positions were similar to the inhibitor of COX-2, SC558.45 This protein–ligand interaction determined the inhibition activities of COX-2 enzymes although the inhibition was not selective.46 The similarities between amino acids involved in a protein–ligand interaction of the native ligand and compound 5 showed that this compound was able to occupy the active site of the COX-2 enzyme and would have a role in the inhibitory activity against the COX-2 enzyme.
The native ligand of the CDK2 structure formed four hydrogen bonds with the protein receptor, as listed in Table 8 and Figure 2A. The binding site of compound 5 formed six hydrogen bonds at the amino acid residues of Lys33, Leu83, Arg86 (2 bonds), and Asp145 (2 bonds) of the CDK2 structure, as shown in Table 8 and Figure 2B. It is believed that Arg86 is an important residue in the CDK2 inhibition mechanism.47 These results are also supported by the finding that Lys33, Arg88, and Asp145 are involved in polar interactions and have a significant site role in CDK2 inhibition and that Leu83 is part of the hinge interaction site.48 Thus, hydrogen bonds of the given amino acid residues had an important role in the catalytic activity of CDK2 inhibition. These similarities indicate the hypothetical mechanisms of CDK2 inhibition of the xanthone compound, but more thorough evaluation is required. In vitro and in vivo experimental laboratory, investigations should be conducted to prove this hypothetical mechanism of action.
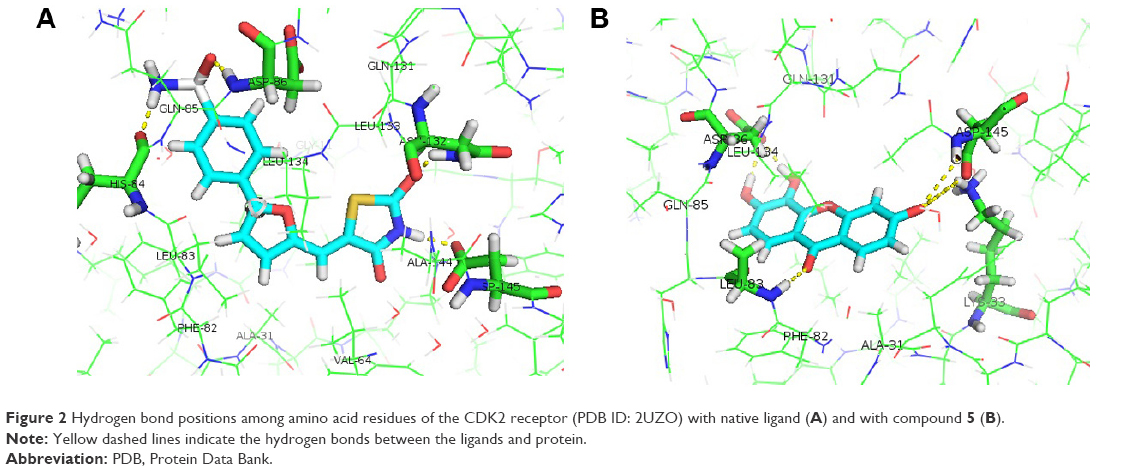 | Figure 2 Hydrogen bond positions among amino acid residues of the CDK2 receptor (PDB ID: 2UZO) with native ligand (A) and with compound 5 (B). |
Conclusion
In this study, a series of xanthone derivatives were studied as cytotoxic agents against the WiDR cell line (colorectal cancer). The inhibitory concentration of xanthones ranged between 9.23 and 286.4 μg/mL. The best compound was compound 5 with an SI of 66.40. The QSAR study revealed that the descriptors of the net atomic charges at qC1, qC2, and qC3; dipole moment; and logP were hypothetically the most responsible for the compounds’ cytotoxic activities. The best QSAR equation was defined as: log 1/IC50 = −8.124 qC1 − 35.088 qC2 − 6.008 qC3 + 1.831 u + 0.540 logP − 9.115 (n=10, r=0.976, s=0.144, F=15.920, Q2 =0.651, SPRESS =0.390). This QSAR model generated proposed novel xanthone compounds with better predicted cytotoxic activities within the range of 0.013–5.498 μg/mL. Analysis of the possible action mechanisms by performing a docking molecular study suggested that xanthone interacted with the receptors of some inhibition signaling processes involved in cancer pathology, including telomerase, tumor-promoting inflammation (COX-2), and cyclin-dependent kinase inhibitor.
Acknowledgments
This work was financially supported by the Ministry of Research, Technology, and Higher Education through Beasiswa Pendidikan Pascasarjana Dalam Negeri (BPPDN) for the PhD scholarship program and by the PhD Dissertation Grant (number 004/DirDPPM/70/DPPM/DisertasiDoktor-KEMENRISTEK DIKTI/IV/2017). We would like to thank Ms Rumbiwati for her laboratory assistance, Ms Emmy Yuanita for the xanthone materials support, and Mr Hari Purnomo (Organic Pharmacy Department, Faculty of Pharmacy, Universitas Gadjah Mada) for his help in the BuildQSAR analysis. The authors would like to thank Enago (www.enago.com) for the English language review.
Disclosure
The authors report no conflicts of interest in this work.
References
Yang ZM, Huang J, Qin JK, et al. Design, synthesis and biological evaluation of novel 1-hydroxyl-3-aminoalkoxy xanthone derivatives as potent anticancer agents. Eur J Med Chem. 2014;85:487–497. | ||
Yasunaka K, Abe F, Nagayama A, et al. Antibacterial activity of crude extracts from Mexican medicinal plants and purified coumarins and xanthones. J Ethnopharmacol. 2005;97(2):293–299. | ||
Zhang H, Tao L, Fu W, et al. Prenylated benzoylphloroglucinols and xanthones from the leaves of Garcinia oblongifolia with antienteroviral activity. J Nat Prod. 2014;77(4):1037–1046. | ||
Keiser J, Vargas M, Winter R. Anthelminthic properties of mangostin and mangostin diacetate. Parasitol Int. 2012;61(2):369–371. | ||
Al-Massarani SM, El Gamal AA, Al-Musayeib NM, et al. Phytochemical, antimicrobial and antiprotozoal evaluation of Garcinia mangostana pericarp and α-mangostin, its major xanthone derivative. Molecules. 2013;18(9):10599–10608. | ||
Das J, Ghosh J, Roy A, Sil PC. Mangiferin exerts hepatoprotective activity against D-galactosamine induced acute toxicity and oxidative/nitrosative stress via Nrf2-NFκB pathways. Toxicol Appl Pharmacol. 2012;260(1):35–47. | ||
Chen LG, Yang LL, Wang CC. Anti-inflammatory activity of mangostins from Garcinia mangostana. Food Chem Toxicol. 2008;46(2):688–693. | ||
Hay AE, Hélesbeux JJ, Duval O, Labaïed M, Grellier P, Richomme P. Antimalarial xanthones from Calophyllum caledonicum and Garcinia vieillardii. Life Sci. 2004;75(25):3077–3085. | ||
Caleare AO, Lazarin-bidóia D, Garcia DA, et al. Parasitology international isolated from Kielmeyera coriacea. Parasitol Int. 2013;62:405–411. | ||
Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
International Agency for Research on Cancer [IARC]. Latest World Cancer Statistics Global Cancer Burden Rises to 14. 1 Million New Cases in 2012: Marked Increase in Breast Cancers Must Be Addressed. IARC; 2013. Available from: http://www.iarc.fr/en/media-centre/pr/2013/pdfs/pr223_E.pdf. Accessed August 25, 2017. | ||
Cheng JH, Huang A-M, Hour TC, Yang SC, Pu YS, Lin CN. Antioxidant xanthone derivatives induce cell cycle arrest and apoptosis and enhance cell death induced by cisplatin in NTUB1 cells associated with ROS. Eur J Med Chem. 2011;46(4):1222–1231. | ||
Chan KM, Hamzah R, Rahaman AA, et al. The pyranoxanthone inophyllin A induces oxidative stress mediated-apoptosis in Jurkat T lymphoblastic leukemia cells. Food Chem Toxicol. 2012;50(8):2916–2922. | ||
Kuete V, Sandjo LP, Nantchouang JL, Fouotsa H, Wiench B, Efferth T. Cytotoxicity and modes of action of three naturally occurring xanthones (8-hydroxycudraxanthone G, morusignin I and cudraxanthone I) against sensitive and multidrug-resistant cancer cell lines. Phytomedicine. 2014;21(3):315–322. | ||
Andrade CH, Pasqualoto KFM, Ferreira EI, Hopfinger AJ. 4D-QSAR: perspectives in drug design. Molecules. 2010;15(5):3281–3294. | ||
Parekh B. QSAR Modeling for Drug Discovery and Development: Applications and Methodology Bhagavati Parekh Category Methods for regression problems Methods 1. Multiple Linear Regression 2. Partial Least Squares 3. Feedforward backpropagation neural network 4. Int J Sci Res. 2015;4:329–331. | ||
Lavecchia A, Giovanni CD. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. 2013;20(23):2839–2860. | ||
Su Q, Liu Y, Cai Y, Sun Y, Wang B, Xian L. Anti-tumour effects of xanthone derivatives and the possible mechanisms of action. Invest New Drugs. 2011;29(6):1230–1240. | ||
Yuanita E, Pranowo HD, Jumina J, Mustofa M. Design of hydroxy xanthones derivatives as anticancer using quantitative structure-activity relationship. Asian J Pharm Clin Res. 2016;9(2):3–8. | ||
Syahri J, Yuanita E, Nurohmah B, et al. Xanthone as antimalarial: QSAR analysis, synthesis, molecular docking and in-vitro antimalarial evaluation. Orient J Chem. 2017;33(1):29–40. | ||
De Oliveira D, Gaudio A. BuildQSAR: a new computer program for QSAR analysis. Quant Struct Act Relat. 2001;19(6):599–601. | ||
Korb O, Stutzle T, Exner TE. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inf Model. 2009;49(1):84–96. | ||
DeLano WL. The case for open-source software in drug discovery. Drug Discov Today. 2005;10(3):213–217. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Prayong P, Barusrux S, Weerapreeyakul N. Cytotoxic activity screening of some indigenous Thai plants. Fitoterapia. 2008;79(7–8):598–601. | ||
Hanahan D, Weinberg RA. Review hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Hafeez A, Saify ZS, Naz A, Yasmin F, Akhtar N. Molecular docking study on the interaction of riboflavin (vitamin B 2) and cyanocobalamin (vitamin B 12) coenzymes. J Comput Med. 2013;2013:5. | ||
Huang S, Zou X. Efficient molecular docking of NMR structures: application to HIV-1 protease. Protein Sci. 2007;16(1):43–51. | ||
Kartasasmita RE, Herowati R, Harmastuti N, Gusdinar T. Quercetin derivatives docking based on study of flavonoid interaction to cyclooxygenase-2. Indo J Chem. 2009;9(2):297–302. | ||
Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action. J Pharm Pharmacol. 2013;65(2):157–170. | ||
Buragohain P, Surineni N, Barua NC, et al. Synthesis of a novel series of fluoroarene derivatives of artemisinin as potent antifungal and anticancer agent. Bioorg Med Chem Lett. 2015;25(16):3338–3341. | ||
Pingaew R, Saekee A, Mandi P, et al. Synthesis, biological evaluation and molecular docking of novel chalcone-coumarin hybrids as anticancer and antimalarial agents. Eur J Med Chem. 2014;85:65–76. | ||
Ghantous A, Gali-Muhtasib H, Vuorela H, Saliba NA, Darwiche N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov Today. 2010;15(15–16):668–678. | ||
Sholikhah EN, Mustofa M, Miladiyah I, et al. Cytotoxic activity of N-alkyl and N-benzyl 1,10-phenanthroline derivatives in human cancer cell lines. Berkala Ilmu Kedokteran. 2007;39:1–6. | ||
Duffy R, Wade C, Chang R. Discovery of anticancer drugs from antimalarial natural products: a MEDLINE literature review. Drug Discov Today. 2012;17(17–18):942–953. | ||
Fouche G, Cragg GM, Pillay P, Kolesnikova N, Maharaj VJ, Senabe J. In vitro anticancer screening of South African plants. J Ethnopharmacol. 2008;119(3):455–461. | ||
Stewart JJP. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J Mol Model. 2013;19(1):1–32. | ||
Gunasekaran S, Anita B, Seshadri S. Vibrational spectroscopy study of diethyl carbamazine using semi-empirical calculations. Indian J Pure Appl Phys. 2010;48:183–191. | ||
Moorthy NSHN, Ramos MJ, Fernandes PA. Topological, hydrophobicity, and other descriptors on α-glucosidase inhibition: a QSAR study on xanthone derivatives. J Enzyme Inhib Med Chem. 2011;26:755–766. | ||
Karelson M, Lobanov V. Quantum-chemical descriptors in QSAR/QSPR studies. Chem Rev. 1996;96:1027–1043. | ||
Miladiyah I, Tahir I, Jumina J, Mubarika S, Mustofa M. Quantitative structure-activity relationship analysis of xanthone derivates as cytotoxic agents in liver cancer cell line HepG2. J Mol. 2016;11(1):143–157. | ||
Musfiroh I, Muhtadi A, Kartasasmita RE, Tjahjono DH. In silico study of asiatic acid interaction with inducible nitric oxide synthase (INOS) and cyclooxygenase-2 (COX-2). Int J Pharm Sci. 2013;5:204–207. | ||
Meenambiga SS, Rajagopal K, Durga R. In silico docking studies on the components of inonotus sp, a medicinal mushroom against cyclooxygenase-2 enzyme. Asian J Pharm Clin Res. 2015;8(3):142–145. | ||
Kumar V, Gupta GK, Kaur K, Singh R. 4-Fluorophenylhydrazones as potential COX-2 inhibitors: a novel, efficient, one pot solid phase synthesis, docking study and pharmacological evaluation. Med Chem Res. 2013;22(12):5890–5900. | ||
Kurumbail W, Stevens AM, Gierse JK, et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384(6610):400–402. | ||
Miladiyah I, Jumina J, Haryana SM, Mustofa M. In silico molecular docking of xanthone derivatives as cyclooxygenase-2 inhibitor agents. Int J Pharm Sci. 2017;9(3):98–104. | ||
Tang H, Chen CY. Drug design of cyclin-dependent kinase 2 inhibitor for melanoma from traditional chinese medicine. Biomed Res Int. 2014;2014:17. | ||
McGrath CF, Pattabiraman N, Kellogg GE, et al. Homology model of the CDK1/cyclin B complex. J Biomol Struct Dyn. 2005;22(5):493–502. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.