Back to Journals » International Journal of Nanomedicine » Volume 14
Biological Activity Of miRNA-27a Using Peptide-based Drug Delivery Systems
Authors Schachner-Nedherer AL, Werzer O ![]() , Kornmueller K, Prassl R
, Kornmueller K, Prassl R ![]() , Zimmer A
, Zimmer A ![]()
Received 12 March 2019
Accepted for publication 27 June 2019
Published 25 September 2019 Volume 2019:14 Pages 7795—7808
DOI https://doi.org/10.2147/IJN.S208446
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Anna-Laurence Schachner-Nedherer,1 Oliver Werzer,1 Karin Kornmueller,2 Ruth Prassl,2 Andreas Zimmer1
1Department of Pharmaceutical Technology and Biopharmacy, Institute of Pharmaceutical Sciences, University of Graz, Graz 8010, Austria; 2Gottfried Schatz Research Center for Cell Signaling, Metabolism and Aging, Biophysics, Medical University of Graz, Graz 8010, Austria
Correspondence: Andreas Zimmer
Institute of Pharmaceutical Sciences, Department of Pharmaceutical Technology and Biopharmacy, University of Graz, Graz 8010, Austria
Email [email protected]
Background: Endogenously expressed microRNAs (miRNAs) have attracted attention as important regulators in post-transcriptionally controlling gene expression of various physiological processes. As miRNA dysregulation is often associated with various disease patterns, such as obesity, miRNA-27a might therefore be a promising candidate for miRNA mimic replacement therapy by inhibiting adipogenic marker genes. However, application of naked nucleic acids faces some limitations concerning poor enzymatic stability, bio-membrane permeation and cellular uptake. To overcome these obstacles, the development of appropriate drug delivery systems (DDS) for miRNAs is of paramount importance.
Methods: In this work, a triple combination of atomic force microscopy (AFM), brightfield (BF) and fluorescence microscopy was used to trace the cellular adhesion of N-TER peptide-nucleic acid complexes followed by time-dependent uptake studies using confocal laser scanning microscopy (cLSM). To reveal the biological effect of miRNA-27a on adipocyte development after transfection treatment, Oil-Red-O (ORO)- staining was performed to estimate the degree of in lipid droplets accumulated ORO in mature adipocytes by using light microscopy images as well as absorbance measurements.
Results: The present findings demonstrated that amphipathic N-TER peptides represent a suitable DDS for miRNAs by promoting non-covalent complexation through electrostatic interactions between both components as well as cellular adhesion of the N-TER peptide – nucleic acid complexes followed by uptake across cell membranes and intracellular release of miRNAs. The anti-adipogenic effect of miRNA-27a in 3T3-L1 cells could be detected in mature adipocytes by reduced lipid droplet formation.
Conclusion: The present DDS assembled from amphipathic N-TER peptides and miRNAs is capable of inducing the anti-adipogenic effect of miRNA-27a by reducing lipid droplet accumulation in mature adipocytes. With respect to miRNA mimic replacement therapies, this approach might provide new therapeutic strategies to prevent or treat obesity and obesity-related disorders.
Keywords: drug delivery system, DDS, miRNA-27a, amphipathic peptides, anti-adipogenic effect, 3T3-L1 cells
Introduction
MicroRNAs (miRNAs) represent a promising class of endogenously expressed regulators controlling gene expression of various biological processes, including proliferation, cell differentiation, apoptosis and metabolism. As miRNA dysregulation is often associated with the onset and progression of various diseases, miRNA-based medicines might provide a new therapeutic approach in the treatment of genetic, metabolic and immunological disorders.1–3
With respect to miRNA processing, long double-stranded RNA molecules are undergoing consecutive cleavage events in the nucleus and the cytoplasm, which are promoted by RNA polymerases to form double-stranded mature miRNAs. After miRNA incorporation into the cytoplasmic multi-protein complex termed as RNA-induced silencing complex (RISC), these short non-coding miRNAs of approximately 22 nucleotides in length post-transcriptionally regulate gene expression to adjust protein levels. This is accomplished by separation of the miRNA duplex into the passenger strand, which is cleaved and expelled by a RISC-specific protein (Argonaute-2), and the guide strand, which is responsible for the recognition of complementary mRNA sequences remaining part of the RISC machinery. The mechanism of gene silencing (mRNA degradation or translational inhibition) is determined by the degree of complementarity with respect to Watson–Crick base pairing between the miRNA guide strand and the target mRNA.4–7 However, abnormal miRNA expression profiles diverging from physiological levels might result in the development of a variety of diseases.8–11
One strategy for miRNA-based medicines could address miRNA replacement therapy. For this purpose, short double-stranded miRNAs are extracellularly introduced into cells to mimic endogenous miRNA functions in the cytoplasm via incorporation into RISC followed by gene regulation.4,6,7 This knowledge opens up new possibilities in developing therapeutic strategies to treat or prevent diseases, in particular obesity. Generally, adipose tissue fulfils important physiological tasks as energy reservoir as well as metabolic and endocrine functions by secreting active molecules (adipocytokines). However, excess accumulation of body fat has become a serious worldwide health problem, which is often associated with obesity-related disorders, such as diabetes, dyslipidemia, hypertension, or coronary heart disease. Adipose tissue is a very heterogeneous tissue containing various cell populations such as lipid droplet storing mature adipocytes but also mesenchymal stem cells that are capable of differentiating into adipogenic, myogenic or chondrogenic cells.12,13 At this point, miRNA replacement therapy could provide a therapeutic alternative by affecting the conversion of stem cells, which are committed to the adipose lineage, from preadipocytes into mature adipocytes and thus, reducing lipid droplet formation and subsequent expansion of adipose tissue. Anti-adipogenic miRNA-27a is a specific miRNA mimic, which is a negative regulator in fat metabolism by suppressing adipogenic marker genes, such as PPARɣ (peroxisome proliferator-activated receptor ɣ). The development of obesity is often associated with reduced miRNA-27a levels and therefore, this miRNA might represent a promising candidate for miRNA mimic replacement therapy.14–17
Although nucleic acid-based therapies provide great potential to turn miRNAs into medicine, application of hydrophilic molecules, such as naked miRNAs, faces some major obstacles comprising protection against enzymatic degradation, improvement of bio-membrane permeability and intracellular release. As the biological effectiveness of miRNA delivery strongly depends on intracellular uptake and release, the development of appropriate drug delivery systems (DDS) is of paramount importance. DDS for miRNAs have to meet some requirements that include enzymatic protection against RNases, cell membrane interaction, cell uptake, intracellular cargo release of the complex as well as distribution. Different kinds of carrier systems have already been discussed in the literature that allow promising nucleic acid delivery into cells, including liposomes,18–20 lipid nanoparticles,21,22 proteins23,24 or polymers.25–27
One such approach for nucleic acid delivery might also include the application of cell penetrating peptides (CPP).28–30 Also known as protein translocation domains, CPP represent a class of small cationic peptides consisting of <30 amino acids. The cationic nature of CPP enables electrostatic interactions with anionic nucleic acids to form self-assembled particles, which represents a technically simple process to obtain a DDS for miRNAs. This phenomenon of spontaneously induced self-assembly complexation has already been reported for different peptides of the CPP family comprising cationic peptides, such as protamine,31–34 as well as amphipathic peptides, such as MPG35,36 standing for May, Pierre and Gilles (the names of the people who discovered the peptide). The primary amphipathic peptide MPG is a 27 amino acid peptide that is arranged in a sequential assembly of hydrophilic and hydrophobic domains. The hydrophilic domain, derived from the nuclear localization sequence of SV40 large T antigen (PKKKRKV), primarily contains positively charged lysine residues that promote electrostatic interactions with negatively charged nucleic acids as well as proteoglycans being present at the cell membrane surface. The hydrophobic domain, derived from HIV-1 gp41 (GALFLGFLGAAGSTMGA), promotes hydrophobic interactions with phospholipids to encourage cell membrane penetration. The linker sequence WSQ improves the flexibility between both the hydrophobic and hydrophilic domain of the peptide MPG, hereinafter referred to the trading name “N-TER peptide”. The proposed translocation mechanisms for MPG-cargo complexes suggest the formation of transient transmembrane structures. This passive membrane crossing integrates electrostatic interactions with cell-surface glycosaminoglycans followed by strong hydrophobic phospholipid–peptide interactions being responsible to form β-barrel pore-like structures.36–39 Gerbal-Chaloin et al described MPG-induced local membrane destabilization, which involves clustering of glycosaminoglycans followed by actin remodeling and consequently, increased membrane fluidity.40
With respect to drug delivery applications, the amphipathic properties of N-TER peptides make these CPP promising candidates for nucleic acid delivery by promoting non-covalent complexation with miRNAs through electrostatic interactions as well as translocation into cells through hydrophobic interactions. As described in our previous studies,41 the manufacturing of the DDS composed of N-TER peptides and nucleic acids has been successfully achieved by simply mixing aqueous solutions of both components resulting in self-assembled particles. Furthermore, physicochemical characterizations of this N-TER peptide-based DDS have been performed to provide additional information about system-specific properties regarding size, shape and charge. All of these parameters might influence the mode of action of this DDS regarding cellular adhesion and uptake and thus, the biological effectiveness of miRNA-27a.
Herein, it is demonstrated that the amphipathic N-TER peptide represents a suitable DDS for anti-adipogenic miRNA-27a. In-vitro transfection experiments of this N-TER peptide-based DDS are performed using 3T3-L1 cells, which represent a common cell culture model to investigate the degree of lipid droplet formation during adipocyte differentiation. To trace the delivery of miRNAs over time, different developmental stages during adipocyte differentiation will be presented in this paper to discuss cellular adhesion/uptake of N-TER peptide-nucleic acid complexes in preadipocytes as well as the biological effect of miRNA-27a in mature adipocytes.
Materials And Methods
Materials
N-TER peptide with the sequence GALFLGFLGAAGSTMGAWSQPKKKRKV and siRNA dilution buffer are offered as “N-TER Nanoparticle siRNA Transfection System“ from Sigma-Aldrich (Vienna, Austria).35,42 Three different nucleic acids from Dharmacon (GE Healthcare, Vienna, Austria) were used to prepare complexes with N-TER peptide, respectively:
- Double-stranded miRNA mimic mmu-miR-27a-3p (miRNA-27a) comprising the sequence UUCACAGUGGCUAAGUUCCGC (MW 13 454 g/mol) to mimic the function of endogenous miRNAs
- miRNA mimic negative control comprising the sequence UCACAACCUCCUAGAAAGAGUAGA (MW 14 074.31 g/mol) to act as a non-targeting control (NTC) in order to distinguish between specific miRNA mimic activity and background effects in miRNA mimic experiments
- miRNA mimic transfection control with Dy547 (FluoNTC) comprising the sequence CUCUUUCUAGGAGGUUGUGA 5´ Dy547 (MW 14 566.9 g/mol) to perform fluorescence experiments in-vitro
RNase-free water (VWR, Vienna, Austria) was utilized to prepare N-TER peptide-nucleic acid complexes. To perform in-vitro cell culture experiments, DMEM from Gibco (Life Technologies Corporation, Paisley, UK) was used containing 1 g/L or 4.5 g/L D-Glucose, hereinafter referred to as “low glucose DMEM” or “high glucose DMEM” respectively. With respect to fluorescence imaging experiments, low glucose DMEM without phenol red was used (Gibco). PBS (pH 7.4), HEPES buffer solution (1 M), L-glutamine (200 nM) and penicillin/streptomycin (10,000 U/mL) were also ordered from Gibco. FBS, insulin solution human, dexamethasone and isobutylmethylxanthine (IBMX) were purchased from Sigma-Aldrich. The following materials were required to perform the Oil-Red-O (ORO)-staining procedure: lipophilic dye ORO (Sigma-Aldrich), glycerol 85% (Herba Chemosan Apotheker-AG, Vienna, Austria), 2-propanol (VWR Chemicals Prolabo, France) and formaldehyde 37% (Carl Roth, Karlsruhe, Germany). The sample preparations and in-vitro transfection studies were carried out under aseptic conditions using a laminar flow box (Herasafe KS, Thermo Fisher Scientific, Vienna, Austria). RNase AWAY (Sigma-Aldrich) decontamination reagent was used for surface disinfection.
Sample Preparation
Stock solutions of three different nucleic acids (miRNA-27a, NTC or FluoNTC) were prepared in RNase-free water and stored at −80°C. Working solutions of 5 µM nucleic acids were obtained by diluting the stock solutions with RNase-free water followed by further dilution with siRNA dilution buffer to obtain 1.3 µM solutions. N-TER peptide (8 µL) was diluted with RNase-free water (42 µL). To obtain standard N-TER peptide–nucleic acid complexes, the working solutions of N-TER peptide and the respective nucleic acid were intermixed using equal volumes. These complexes were incubated at room temperatures (about 23°C) for 15 to 20 mins containing finally a nucleic acid concentration of 650 nM followed by 5 mins of sonication (Emmi-D100, EMAG Technologies, Mörfelden-Walldorf, Germany). To obtain concentrations suitable for in-vitro transfection studies, the standard N-TER peptide–nucleic acid complexes (650 nM) were diluted with serum-free low glucose DMEM. In the following, the stated in-vitro concentrations are always referred to the nucleic acids (miRNA-27a, NTC or FluoNTC).
In-vitro Transfection And Differentiation
Mouse embryonic fibroblast-derived 3T3-L1 preadipocytes were originally purchased from ATCC (Manassas, VA, USA) and kindly provided by the Graz University of Technology (Graz, Austria) from the research group of Dr. Scheideler. The cells were cultivated at 37°C under 5% CO2 water-saturated atmosphere in complete proliferation medium (PM) consisting of low glucose DMEM supplemented with 10% FBS, 1% L-glutamine, 1% HEPES and 1% penicillin/streptomycin. Transient transfection of N-TER peptide–nucleic acid complexes was performed in non-confluent 3T3-L1 preadipocytes. For this case, the cells were cultivated in complete PM with a seeding density of approximately 7×103 cells/well into 96-well plates (Greiner Bio-One GmbH, Frickenhausen, Germany) or 7×104 cells onto glass bottom dishes (WillCo Wells B.V., Amsterdam, Netherlands) 24 hrs before transfection. The glass bottom dishes were always used for fluorescence imaging due to the transmission microscopy.
In-vitro transfection was performed by diluting the standard N-TER peptide–nucleic acid complexes (650 nM) with serum-free low glucose DMEM followed by an incubation period of 4 hrs on 3T3-L1 cells. Afterwards, equal volumes of complete transfection medium were added consisting of antibiotic-free low glucose DMEM supplemented with 20% FBS, 1% L-glutamine and 1% HEPES, which finally results in nucleic acid concentrations of 5 nM, 10 nM, 25 nM, 50 nM or 80 nM, respectively. After a total incubation period of 24 hrs, the transfection medium was completely removed followed by washing with PBS to get rid of unbound sample material. To induce the in-vitro differentiation of transfected preadipocytes into mature adipocytes, freshly prepared induction medium (IM) was added consisting of high glucose DMEM supplemented with 10% FBS, 1% L-glutamine, 1% HEPES, 1% penicillin/streptomycin and some hormonal substances including insulin, dexamethasone and IBMX. Final concentrations of 10 µg/mL insulin, 1 µM dexamethasone and 500 µM IBMX were used to extracellularly trigger adipocyte differentiation, which is from now on termed as day 0 (d0). The medium was changed and replaced every second day (d2, d4) with differentiation medium consisting of high glucose DMEM supplemented with 10% FBS, 1% L-glutamine, 1% HEPES, 1% penicillin/streptomycin and insulin (5 µg/mL). ORO-staining was performed in mature adipocytes after a total differentiation period of 6 days (d6).
To investigate the effect of N-TER peptide–nucleic acid complexes on lipid droplet formation during adipocyte differentiation, concentration-dependent transfection series were performed starting from 10 nM up to 80 nM final miRNA-27a or NTC concentrations. Furthermore, the sample conditions also comprise control groups including the single complex components, such as N-TER peptide, miRNA-27a or NTC, and non-transfected cells, termed as cells only (co). The controls were treated exactly the same as described for N-TER peptide–nucleic acid complexes regarding the in-vitro transfection and differentiation procedure. Each sample condition was performed at least in triplicates on 96-well plates.
ORO-Staining Procedure
The lipophilic dye ORO is often used to assess the degree of differentiation by staining lipid droplets in mature adipocytes.16,17,43 A stock solution of ORO was prepared by dissolving 0.35 g dye in 100 mL 2-propanol stored at 4°C. The working solution was prepared by mixing 6 mL ORO stock solution with 4 mL MQ-water (Milli-Q-Gradient system from Merck-Millipore, Darmstadt, Germany) and stored at room temperatures (about 23°C) for at least 30 mins followed by filtering. The differentiated cells at d6 were washed with PBS and fixated in 10% formaldehyde (in 10X PBS) for at least 1 hr. After removing the fixative, the cells were washed with MQ-water, incubated with 60% (v/v) 2-propanol (in MQ-water) for 5 mins and completely air-dried after removing the 2-propanol. After 10 mins incubation of the filtered ORO working solution, the cells were repeatedly washed with MQ-water and covered with 50% (v/v) glycerol (in MQ-water) to avoid sample drying. To compare the staining intensities of ORO between the various sample conditions, light microscopic images were taken using a Leica microscope Type 090-135.002 (Leica Microsystems GmbH, Vienna, Austria) equipped with a Canon EOS 70D digital camera (Canon GmbH, Vienna, Austria). For semi-quantitative evaluation of in lipid droplets accumulated ORO, absorbance measurements were performed at an excitation wavelength of 500 nm in matrix scan mode using a CLARIOstar microplate reader (BMG LABTECH, Ortenberg, Germany). Glycerol 50% (v/v) was used as blank in empty wells. The results of absorbance measurements are presented as blank-corrected mean values SD.
SD.
Combinatorial Atomic Force Microscopy (AFM), Brightfield (BF) And Fluorescence Imaging
After seeding the cells onto glass bottom dishes, in-vitro transfection was performed using 5 nM (low) and 100 nM (high) final nucleic acid concentrations of N-TER peptide–FluoNTC complexes followed by formaldehyde fixation of 3T3-L1 cells 4-hr post-transfection. The samples were investigated by using a combinatorial set up including AFM, BF and fluorescence imaging. Mounting the atomic force microscope on top of a Zeiss Axio Observer (Göttingen, Germany), this instrumental set up combines the three different microscopic techniques enabling simultaneous investigations at the very same sample area. The AFM investigations were performed with a FlexAFM atomic force microscope, equipped with a C3000 controller (Nanosurf, Switzerland). Height images were recorded in tapping mode utilizing Tap300 Al-G cantilevers (Budgetsensors, Sofia, Bulgaria). All recorded AFM data were processed and examined with the software package Gwyddion.44 For fluorescence microscopy, filter settings were chosen according to the absorption/emission spectra (557 nm/570 nm) of the fluorescence marker Dy547. Combined images of all techniques were generated with the software GIMP.
Confocal Laser Scanning Microscopy (cLSM)
To trace fluorescent-labelled NTC after in-vitro transfection, cLSM was used for cellular uptake studies of N-TER peptide–nucleic acid complexes over time (4 hrs, 24 hrs, 48 hrs) at 37°C. Sample conditions were prepared according to the transfection and differentiation protocol as described before. The initial nucleic acid concentrations of 10 nM and 100 nM were diluted with complete transfection medium after an incubation period of 4 hrs. This finally resulted in concentrations of 5 nM and 50 nM with respect to incubation periods of 24 hrs and 48 hrs. The cLSM (510 Meta, Carl Zeiss GmbH) utilized the ZEN2009 software package to record fluorescence images at different focus positions. From these z-stacks, three-dimensional data is extracted. Cells incubated for 48 hrs with N-TER peptide–FluoNTC complexes were already stimulated with IM, representing d1 of differentiation. The transfected 3T3-L1 cells were washed with PBS to get rid of unbound sample material followed by formaldehyde fixation. Alexa Fluor 488 Phalloidin (Thermo Fisher Scientific) was used to stain the actin cytoskeleton and was excited at 488 nm and detected using a bandpass filter (BP 505/550nm) for the green channel. Hoechst 33342 (Thermo Fisher Scientific) was used to counterstain the nucleus and was excited at 405 nm and detected using a bandpass filter (BP 420/480nm) for the blue channel. The fluorescent-labelled NTC was detected at 543 nm excitation wavelength using a longpass filter (LP 560 nm) for the red channel.
Statistical Analysis
In mature adipocytes at d6, absorbance of in lipid droplets accumulated ORO was analyzed by one-way ANOVA to verify statistical significance. To compare mean values, F-test was performed whether unequal or equal variances had to be assumed followed by two-sample t-test. P-values <0.05 and <0.001 were considered to be statistically significant and statistically highly significant, respectively.
Results And Discussion
In-vitro Transfection And Differentiation Of 3T3-L1 Cells
Differentiation of 3T3-L1 preadipocytes into adipocytes is tightly controlled by a complex network of various transcription factors (eg, CCAAT enhancer-binding proteins and PPARɣ), which correspond to extracellular stimuli such as insulin, dexamethasone and IBMX. These substances induce the adipogenic program, in which PPARɣ is considered to be the master regulator of adipocyte differentiation.45,46 Previous studies have already demonstrated that intracellularly available miRNA-27a is a negative regulator of adipocyte differentiation by directly targeting PPARɣ and thus, resulting in decreased lipid droplet formation during adipocyte development.15,16 The aim of the present experiments has been the delivery of extracellular miRNA-27a into 3T3-L1 preadipocytes by applying the N-TER peptide–nucleic acid complexes, which represent the peptide-based DDS for miRNAs. To determine whether miRNA-27a induces an anti-adipogenic response in transfected cells, the degree of lipid droplet formation in mature adipocytes is investigated by ORO-staining. In order to discriminate between specific miRNA mimic activity and background effects, N-TER peptide complexes containing miRNA-27a or NTC are always carried out in parallel. Non-transfected cells (cells only) serve as a control to monitor the general impact of transfection treatment on adipocyte differentiation as well as cell vitality. The endpoint of these in-vitro studies is terminated in mature adipocytes at d6 of differentiation by evaluating the staining intensities of in lipid droplets accumulated ORO with both light microscopy (Figure 1) and absorbance measurements (Figure 2).
Figure 1A shows differentiated 3T3-L1 cells, which are transfected with N-TER peptide–NTC complexes (left part of the light microscopic image) and N-TER peptide–miRNA-27a complexes (right part of the light microscopic image) ranging from 10 nM up to 80 nM (nucleic acids). It can be clearly noted that NTC-transfected cells exhibit a more intensive red coloring than cells transfected with miRNA-27a. This visually noticeable difference between NTC and miRNA-27a increases with concentration and is most likely related to the anti-adipogenic effect of miRNA-27a, which is known to reduce/inhibit the lipid droplet formation during adipocyte differentiation. In contrast to the corresponding NTC-transfected cells, miRNA-27a treatment ≥25 nM leads to decreased formation of lipid droplets in mature adipocytes and thus, reduced accumulation of ORO and red coloring. The control wells of cells transfected with free NTC or miRNA-27a (Figure 1B, left image) show no difference regarding the intensity of red coloring at the tested concentrations (10 nM, 25 nM and 80 nM) in comparison to cells only (Figure 1B, right part of the right image). On the contrary, the control wells of cells transfected with free N-TER peptide (Figure 1B, left part of the right image) reveal a concentration-dependent decrease of the ORO-staining intensities in comparison to the cells only, although this effect seems to be not as prominent as compared to N-TER peptide complexed with NTC (see Figure 1A). This issue can possibly be explained by a cytotoxic effect of the amphipathic N-TER peptide partly driven by its positively charged lysine residues. The positively charged peptide domains are largely neutralized after complexation with the negatively charged nucleic acids. This neutralization phenomenon could be responsible for the aforementioned differences between free and complexed N-TER peptides by influencing the degree of cell membrane interaction.
 |
Figure 1 Exemplary light microscopic images of transfected and differentiated 3T3-L1 cells at d6 of differentiation after ORO-staining. The staining intensities of in lipid droplets accumulated ORO are presented including different sample conditions: N-TER peptide–nucleic acid complexes containing NTC or miRNA-27a (A) and the control groups comprising free NTC, miRNA-27a, N-TER peptide as well as cells only (B). Cells only serve as a control to assess the impact of the transfection treatment on adipocyte differentiation. |
To verify the observations obtained from light microscopy images (Figure 1), corresponding absorbance measurements enable semi-quantitative determination of in lipid droplets accumulated ORO (Figure 2). In Figure 2A, the semi-quantitative absorbance levels of in lipid droplets accumulated ORO are compared between N-TER peptide–NTC complexes and N-TER peptide– miRNA-27a complexes. The results at 10 nM show very similar absorbance levels for both types of N-TER peptide–nucleic acid complexes while at higher concentrations (25 nM, 50 nM and 80 nM) the differences clearly enhance. In case of 25 nM and 50 nM, the absorbance values are about 0.15 for N-TER peptide–NTC complexes and 0.1 for N-TER peptide–miRNA-27a complexes. These results indicate decreased ORO accumulation in miRNA-27a-transfected cells compared to NTC-transfected cells. However, enhancement of this miRNA-27a effect is probably not achieved with continued increase of the miRNA concentrations. This is suggested by relatively stable absorbance values (approximately 0.1) detected for the higher miRNA-27a concentrations comprising 25 nM, 50 nM as well as 80 nM. Nevertheless, the concentration-dependent response after miRNA-27a-treatment strongly supports the acceptance that N-TER peptide-based DDS are able to successfully deliver miRNA-27a into preadipocytes, which is emphasized by exerting its desired anti-adipogenic effect during adipocyte differentiation. The experiments with N-TER peptide–NTC complexes suggest a slight impact of the DDS itself on adipocyte differentiation in comparison to cells only, which seems to be concentration-dependent (see Figure 2A). By comparing cells only and N-TER peptide–NTC complexes, the ORO absorbance values are very comparable at 10 nM whereas a decrease is observed from about 0.2 (cells only) to 0.15 at higher concentrations of N-TER peptide–NTC complexes (25 nM, 50 nM and 80 nM). At this point, it is not completely clear whether the decrease is related to the peptide, which is supposed to deliver nucleic acids into cells, or the NTC, which is acting as a non-targeting control.
For clarification purposes, in-vitro transfection experiments with corresponding control groups are performed to monitor the influence of free NTC, miRNA-27a as well as N-TER peptide on adipocyte differentiation (Figure 2B). As expected for naked RNAs, both free nucleic acid controls (NTC and miRNA-27a) show no influence on lipid droplet formation during adipocyte differentiation as compared to cells only. This assumption is emphasized by nearly constant absorbance values of about 0.2 matching for all tested nucleic acid concentrations (10 nM, 25 nM, 50 nM and 80 nM). These observations are perfectly in accordance with the literature describing poor bio-membrane permeability and cellular uptake for naked RNAs, which are hydrophilic molecules that exhibit negatively charged phosphodiester backbones provoking electrostatic repulsion from the anionic cell membrane surface.1,4 Therefore, miRNAs need suitable DDS as carrier vehicles to cross cell membranes. In contrast to the free nucleic acid, free N-TER peptide causes a concentration-dependent decrease of absorbance values starting from 0.2 at lower concentrations (comparable to the other controls including free nucleic acids and cells only) down to 0.1 at higher concentrations (see Figure 2B). These observations indicate that N-TER peptide itself is possibly affecting adipocyte differentiation. The effect could be explained by a commonly known concentration-dependent, cytotoxic effect of amphipathic CPP based on the peptides` structure consisting of hydrophobic and charged amino acids, which synergistically promote translocation through cell membranes via distinct membrane perturbation mechanisms. In particular, amphipathic peptides possess the ability to promote electrostatic interactions through their hydrophilic lysine-rich domain with the negatively charged cell membrane components (eg, glycosaminoglycans or phospholipids) while their hydrophobic domain is responsible for intercalating plasma membranes. Consequently, these translocation mechanisms of amphipathic CPP result in breaking up the integrity of the cell membrane via diverse processes including pore formation or membrane thinning, which successfully enable drug delivery across cell membranes. However, if the peptide concentrations exceed a certain threshold, cytotoxic effects might occur by disrupting cell membranes through extensive leakage that can hardly be overcome by repair mechanisms of the cells.47–53 As a concentration-dependent consequence, the cell membrane damage is irreversible possibly leading to impaired adipocyte differentiation, as shown for higher concentrations of transfection complexes (>50 nM, see Figure 2B).
Furthermore, adipocyte differentiation might be also affected by the agglomeration tendency of this peptide-based DDS, which has already been demonstrated in former experiments focusing on physicochemical characterization of the DDS using dynamic light scattering technologies and AFM.41 Amphipathic peptides, such as N-TER peptide, show increased propensity for peptide–peptide interactions due to their hydrophobic amino acid sequence and thus, agglomeration.36,54,55 The possible influence of agglomeration on adipocyte differentiation will be discussed in more detail in the next section.
 |
Figure 2 Semi-quantitative absorbance measurements of in lipid droplets accumulated ORO at d6 of differentiation. The degree of lipid accumulation in triple transfected and differentiated cells is evaluated according to N-TER peptide–nucleic acid complexes (A) and controls (B) comprising free NTC, miRNA-27a, N-TER peptide as well as cells only. Cells only serve as a control to assess the impact of the transfection treatment on adipocyte differentiation. All results are presented as mean±SD (n=6). Statistical significance is marked as *(p<0.05) and ***(p<0.001). |
Both methods (light microscopy images and absorbance measurements) reveal that N-TER peptide-based DDS are capable of inducing the miRNA-27a-specific biological response, which is characterized by reduced lipid droplet formation in mature adipocytes. As demonstrated before, free miRNA-27a is unable to exert its anti-adipogenic effect on cellular level, which strongly indicates that these hydrophilic molecules need suitable carrier vehicles across cell membranes, such as N-TER peptides. The results presented up to now reflect the specific effect of complexed miRNA-27a at an advanced stage of adipocyte differentiation (mature adipocytes). The following in-vitro experiments concentrate on tracing the complexed miRNAs at an earlier stage of transfection (preadipocytes) and adipocyte differentiation (cells stimulated with IM at d1 of differentiation) to shed light on the initial stages of cellular adhesion and actual uptake of N-TER peptide–nucleic acid complexes.
Topography, Cellular Adhesion And Uptake Of N-TER Peptide–Nucleic Acid Complexes
The concomitant application of BF illumination (40×) and fluorescence imaging allows the distinction between cellular structures (cytoskeleton or nucleus) as well as the exact allocation of N-TER peptide–FluoNTC complexes on formaldehyde-fixated 3T3-L1 preadipocytes. Additionally, the AFM height images reveal the topography of the cell surface-adhered complexes at the same sample position as BF and fluorescence. Using this triple combination of AFM, BF and fluorescence microscopy, the cellular adhesion and topography of N-TER peptide–FluoNTC complexes (blue colored) on 3T3-L1 preadipocytes are identified for low (5 nM) and high (100 nM) nucleic acid concentrations, respectively (Figure 3). Starting with the high nucleic acid concentrations (100 nM, Figure 3A), BF and fluorescence microscopy reveal very strong signals of N-TER peptide–FluoNTC complexes, which are extensively distributed over the surface of 3T3-L1 preadipocytes. In parallel, AFM height images show strongly agglomerated complexes built up by smaller particles. These agglomerates mostly adhere to the cell surface but also to the glass bottom of the dish. In contrast to high nucleic acid concentrations, the detection of N-TER peptide–FluoNTC complexes at low concentrations (5 nM, Figure 3B) reveals a decreased number of agglomerates as expected following results from previous studies.41 In general, the formation of particle agglomerates is characterized by a two-step process. The process starts with the self-assembly of primary particles based on electrostatic interactions between the negatively charged nucleic acids and the positively charged peptide domain. Their size is typically in the range from 100 to 300 nm. This is followed by the second step, the agglomeration of these primary particles due to additional hydrophobic peptide–peptide interactions. The propensity to form agglomerates is therefore specific to the N-TER peptide-based DDS, which cancels long-time stability as discussed previously with the agglomerates exceeding even several µm.41
The concentration-dependent increase of agglomeration has also been observed for N-TER peptide complexes from fluorescence microscopic images (Figure 4). The images show randomly distributed N-TER peptide–FluoNTC complexes (red) being adhered to the layer of 3T3-L1 cells, which are stained with fluorescent dyes to clearly distinguish between the cytoskeleton (green) and the nuclei (blue). As already described before, low nucleic acid concentrations (Figure 4, left column) reveal decreased numbers of agglomerates in contrast to higher ones (Figure 4, middle column). To ensure that the red fluorescent signal is specific to N-TER peptide–FluoNTC complexes, non-transfected cells are prepared in parallel as controls (Figure 4, cells only, right column).
As tendency for agglomeration increases with increasing concentrations of the complexes at constant transfection volumes, the system-specific properties to form agglomerates might be also responsible for the limiting effect on adipocyte differentiation at higher concentrations of free N-TER peptide as well as the complexed form with NTC, which is already indicated in Figure 2A. Earlier studies have demonstrated that agglomeration of N-TER peptide–nucleic acid complexes occurs immediately after preparation. This agglomeration effect is even more enhanced after dilution with the cell culture medium DMEM because of the additional charged components (eg, amino acids, vitamins and inorganic salts).41 Consequently, these agglomerates adhere to the cell surface (see Figures 3 and 4) and possibly induce a mechanical stress response. This response could potentially cause physical deformation of the cytoskeleton and thus, affect adipocyte differentiation. These considerations are in agreement with other studies describing decreased lipid droplet accumulation during adipocyte differentiation of 3T3-L1 cells as a response to mechanical stress.56,57 Hampered adipocyte differentiation could also result just from a blocked receptor access due to the presence of agglomerates. The number or affinity of receptors is regulated in response to a ligand, such as insulin, dexamethasone and IBMX, which are responsible for receptor-mediated signals inducing the adipogenic transcription cascade.58,59 However, increased numbers of agglomerates might block these docking sites of receptors resulting in reduced ligand-receptor interactions and, consequently, affecting the adipogenic differentiation program. The function of membrane ion channels, which are essential for proliferation and the physiological homeostasis, could also be disturbed similarly by the presence of agglomerates being adhered to the cell surface.60
Besides the intrinsic properties, the smaller particles occurring at low nucleic acid concentrations seem to be slightly embedded in the cytoskeleton (marked in AFM height image, Figure 3B). Unlike the strongly agglomerated complexes increasingly occurring at high nucleic acid concentrations, which appear more likely to be located onto the cell surface (marked in AFM height image, Figure 3A). The presented results further reveal polydisperse properties of the N-TER peptide-based DDS. The smaller particles exhibit heights of about 100–150 nm as well as larger agglomerates of about 600 nm marked as size profile 1 and 2, respectively (see supplementary materials, Figure S1). In general, cellular uptake mechanisms are known to be size-dependent,61,62 which has been also described by Simeoni et al for N-TER peptide complexes.36 From the results above it can be concluded that the DDS is able to transport the miRNAs into the 3T3-L1 cells. If this is achieved by small primary particles or by agglomerated ones cannot be identified by the methods at hand, but it might even be possible that several uptake mechanisms (passive or active) occur simultaneously: primary particle uptake, removal of individual particles from the agglomerates at the cell surface, or others.
 |
Figure 3 N-TER peptide–FluoNTC complexes (blue colored) at high (100 nM, image A, above) and low (5 nM, image B, below) nucleic acid concentrations on formaldehyde-fixated 3T3-L1 preadipocytes. Topography and cellular adhesion of the complexes are visualized by using a triple combination of AFM, BF and fluorescence microscopy at the same sample area. Heights of the particles/agglomerates are estimated by using particle size profiles (marked as 1 and 2; for further information about the respective particle size profiles, see supplementary materials Figure S1). |
As already mentioned, adhesion of N-TER peptide–nucleic acid complexes takes place on the cell surface (Figures 3 and 4). The adhesion forces between the complexes and the cell surface as well as the glass bottom are concluded to be strong. This we can derive from the fact that the attachment required withstands the forces occurring during PBS washing, which is used to remove unbound sample material followed by formaldehyde cell fixation. N-TER peptides exhibit amphipathic properties containing hydrophobic as well as positively charged amino acids (lysine and arginine), which could be advantageous regarding cellular uptake by allowing interactions with both hydrophilic (eg, negatively charged cell surface compounds) and hydrophobic (eg, lipid membranes) surfaces.4,63,64 The initial cell surface contact of the peptide-based DDS is most probably taking place through electrostatic interactions between the hydrophilic amino acid sequence carrying positive charges and the negatively charged heparan sulfate proteoglycans as one of the main components in the extracellular matrix.40,63,65
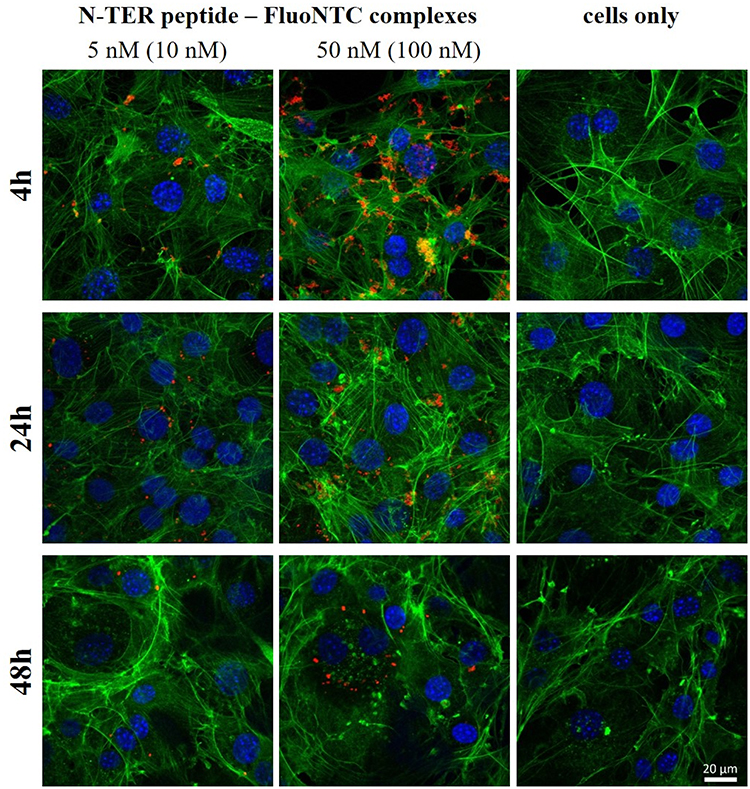 |
Figure 4 Fluorescence microscopic images of 3T3-L1 cells at 37°C transfected with N-TER peptide–FluoNTC complexes (red) at 10 nM or 100 nM (4-hr post-transfection) and at 5 nM or 50 nM (24-hr and 48-hr post-transfection). Cells only represent the non-transfected cells. Hoechst 33342 (blue) and Alexa Fluor 488 Phalloidin (green) are used to stain cell nuclei and cytoskeleton, respectively. |
Extracellular administration of miRNA-27a using N-TER peptide-based DDS affects the degree of adipocyte differentiation indicated by reduced formation of lipid droplets. This biological response to miRNA-27a treatment is detected by using light microscopy images (Figure 1) and absorbance measurements (Figure 2) of in lipid droplets accumulated ORO in mature adipocytes at d6 of differentiation. Based on these results, it is therefore assumed that the observed anti-adipogenic effect must correlate with cellular uptake and cytoplasmic release of miRNA-27a after in-vitro incubation of 3T3-L1 preadipocytes using the peptide-based DDS. For verification purposes, z-stacks of cLSM studies are aimed at confirming cellular uptake by tracing fluorescent-labelled NTC within the first 48 hrs after transfection in 3T3-L1 cells. Z-stack cross sections are prepared providing three-dimensional data set to distinguish between cellular adhesion and actual cellular uptake (see Figure 5). Non-transfected cells are presented again to truly assign the red fluorescence to the N-TER peptide–FluoNTC complexes after continuous observations over 4 hrs, 24 hrs and 48 hrs (Figure 5, cells only, panel a). Regarding N-TER peptide–FluoNTC complexes 4-hr post-transfection, most of the fluorescent pattern appears to be located onto the cell surface partly stuck into the cytoskeleton, which matches for both nucleic acid concentrations of 10 nM (upper panel b) and 100 nM (upper panel c), exemplarily marked as dashed circles in Figure 5. After an incubation period of 24 hrs and 48 hrs, however, the z-stacks reveal red fluorescent signals mainly appearing within the cytoskeleton and thus, highly indicating the cellular uptake of the fluorescent-labelled N-TER peptide complexes, exemplarily marked as dashed circles in Figure 5 (middle and bottom panel b/c for 5 nM and 50 nM, respectively).
As frequently highlighted in the literature, CPP, such as N-TER peptide, possess the ability for delivering hydrophilic nucleic acid molecules across biological membranes, which consist of lipid bilayers as basic structural units. The present studies have investigated the ability of N-TER peptides to function as suitable carrier system for miRNA-27a across 3T3-L1 cell membranes. The peptides belong to a category of CPP termed as “primary amphipathic peptides” that are characterized by a sequential assembly of hydrophilic and hydrophobic amino acid residues.63 This amphipathic structure might provide some benefits to act as successful DDS for miRNAs, which will be discussed in the following. First, the positively charged lysine residues in the hydrophilic domain of N-TER peptides induce non-covalent complexation with negatively charged nucleic acids. On the one hand, these electrostatic interactions represent a technically simple approach to manufacture a self-assembled DDS. On the other hand, the initial cell contact of this peptide-based DDS is promoted by electrostatic interactions between the partially positively charged N-TER peptide complexes and the negatively charged proteoglycans being present on the cell surface membrane.63,65 Second, it is furthermore important to enable hydrophobic interactions with the lipid components of cell membranes. As already discussed in the section before, the translocation of amphipathic N-TER peptides across cell membranes is often reported as membrane perturbation mechanisms via, eg, pore formation or membrane thinning. During these proposed uptake mechanisms, the hydrophobic amino acid domain of amphipathic peptides tends to interact with membrane phospholipids by adopting secondary structures, preferably beta-sheets, within the cell membrane that affect its integrity as well as cytoskeleton organization leading to temporary membrane leakage and thus, enabling drug delivery across cell membranes.39,40,65,66 After exceeding a certain concentration threshold, however, this amphipathic character might induce a cytotoxic response, which can be noticed via impaired adipocyte differentiation as demonstrated before. If these observed effects are provoked by N-TER peptide-mediated membrane disruption and/or agglomeration-related mechanical stress cannot be identified by the methods at hand. It is highly probable that both phenomena contribute to the impaired cell viability in a concentration-dependent manner. Beyond that, it is also very likely for amphipathic peptides that different uptake mechanisms, such as membrane translocation and endocytosis, might occur simultaneously.63,65 This versatility could be related to an interplay of various parameters not only depending on the amino acid sequence of the peptide but also on particle sizes of the DDS, miRNA concentrations, cell types as well as experimental conditions.36,61,62,67,68 Altogether, the described discoveries support the appropriateness of the presented peptide-based DDS for miRNAs, which is confirmed by the miRNA-27a-specific effect in mature adipocytes.
 |
Figure 5 Fluorescence microscopic z-stacks of 3T3-L1 cells at 37°C. The panels show non-treated cells (panel a) or cells transfected with N-TER peptide–FluoNTC complexes (red). Panel b represents the low nucleic acid concentrations (5 nM) whereas panel c represents the high nucleic acid concentrations (50 nM). Hoechst 33342 (blue) and Alexa Fluor 488 Phalloidin (green) are used to stain cell nuclei and cytoskeleton, respectively. The dashed circles exemplarily indicate adhesion/uptake of fluorescent-labelled NTC according to the particular incubation period (4 hrs, 24 hrs, 48 hrs). |
Conclusion
In-vitro studies are performed using 3T3-L1 cells as cell culture model to investigate the degree of lipid droplet formation during adipocyte differentiation after exposure to peptide-based DDS for miRNAs. This proof of principle has been demonstrated that N-TER peptide-based DDS are capable of inducing the anti-adipogenic effect of miRNA-27a by reducing lipid droplet formation in mature adipocytes. It can be concluded that this amphipathic peptide represents a suitable DDS for miRNAs by promoting non-covalent complexation through electrostatic interactions between both components as well as cellular adhesion of the N-TER peptide–nucleic acid complexes followed by uptake across cell membranes and intracellular miRNA release to induce its biological activity. In-depth investigations are required to elucidate the mechanisms of internalization of N-TER peptide-based DDS followed by intracellular trafficking the cargo release to further improve the anti-adipogenic potential of miRNA-27a during adipocyte development. With respect to miRNA mimic replacement therapies, this approach might provide new therapeutic strategies to prevent or treat obesity and obesity-related disorders.
Abbreviations
AFM, atomic force microscopy; BF, brightfield; cLSM, confocal laser scanning microscopy; CPP, cell penetrating peptides; DDS, drug delivery systems; DM, differentiation medium; miRNA, microRNA; IBMX, isobutylmethylxanthine; IM, induction medium; NTC, non-targeting control; ORO, Oil-Red-O; PM, proliferation medium; PPARɣ, peroxisome proliferator-activated receptor ɣ; RISC, RNA-induced silencing complex.
Acknowledgment
The work was supported by grants from the University of Graz.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172(3):962–974. doi:10.1016/j.jconrel.2013.09.015
2. Guay C, Regazzi R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat Rev Endocrinol. 2013;9:513–521. doi:doi:10.1038/nrendo.2013.86
3. Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev. 2005;15(5):563–568. doi:10.1016/j.gde.2005.08.005
4. Meade BR, Dowdy SF. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv Drug Deliv Rev. 2008;60(4–5):530–536. doi:10.1016/j.addr.2007.10.004
5. Laufer SD, Detzer A, Sczakiel G, Restle T. RNA Technologies and Their Applications.
6. MacFarlane L, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics. 2010;11:537–561. doi:10.2174/138920210793175895
7. Guzman-Villanueva D, El-Sherbiny IM, Herrera-Ruiz D, Vlassov AV, Smyth HDC. Formulation approaches to short interfering RNA and MicroRNA: challenges and implications. J Pharm Sci. 2012;101(11):4046–4066. doi:10.1002/jps.23300
8. Firoozabadi AD, Shojaeii S, Hadinedoushan H. Physiological and pathological roles for MicroRNAs: implications for immunity complications. Int J Med Lab. 2014;1(1):61–75.
9. Nana-Sinkam SP, Croce CM. Clinical applications for microRNAs in cancer. Clin Pharmacol Ther. 2013;93(1):98–104. doi:10.1038/clpt.2012.192
10. Rottiers V, Näär AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. 2014;13(4):239–250. doi:10.1038/nrm3313
11. Peng Y, Yu S, Li H, Xiang H, Peng J, Jiang S. MicroRNAs: emerging roles in adipogenesis and obesity. Cell Signal. 2014;26(9):1888–1896. doi:10.1016/j.cellsig.2014.05.006
12. Iacomino G, Siani A. Role of microRNAs in obesity and obesity-related diseases. Genes Nutr. 2017;12(1):1–16. doi:10.1186/s12263-017-0577-z
13. Klaus S. Adipose tissue as a regulator of energy balance. Curr Drug Targets. 2004;5:241–250.
14. Sun L, Trajkovski M. MiR-27 orchestrates the transcriptional regulation of brown adipogenesis. Metabolism. 2014;63(2):272–282. doi:10.1016/j.metabol.2013.10.004
15. Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z. A role of miR-27 in the regulation of adipogenesis. Febs J. 2009;276(8):2348–2358. doi:10.1111/j.1742-4658.2009.06967.x
16. Kim SY, Kim AY, Lee HW, et al. miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARγ expression. Biochem Biophys Res Commun. 2010;392(3):323–328. doi:10.1016/j.bbrc.2010.01.012
17. Karbiener M, Fischer C, Nowitsch S, et al. microRNA miR-27b impairs human adipocyte differentiation and targets PPARγ. Biochem Biophys Res Commun. 2009;390(2):247–251. doi:10.1016/j.bbrc.2009.09.098
18. Dorrani M, Garbuzenko OB, Minko T, Michniak-Kohn B. Development of edge-activated liposomes for siRNA delivery to human basal epidermis for melanoma therapy. J Control Release. 2016;228:150–158. doi:10.1016/j.jconrel.2016.03.010
19. Kim HK, Davaa E, Myung CS, Park JS. Enhanced siRNA delivery using cationic liposomes with new polyarginine-conjugated PEG-lipid. Int J Pharm. 2010;392(1–2):141–147. doi:10.1016/j.ijpharm.2010.03.047
20. Shim G, Kim MG, Park JY, Oh YK. Application of cationic liposomes for delivery of nucleic acids. Asian J Pharm Sci. 2013;8(2):72–80. doi:10.1016/j.ajps.2013.07.009
21. Ghatak S, Li J, Chan YC, et al. AntihypoxamiR functionalized gramicidin lipid nanoparticles rescue against ischemic memory improving cutaneous wound healing. Nanomed Nanotechnol Biol Med. 2016;12(7):1827–1831. doi:10.1016/j.nano.2016.03.004
22. McLendon JM, Joshi SR, Sparks J, et al. Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J Control Release. 2015;210:67–75. doi:10.1016/j.jconrel.2015.05.261
23. Jain A, Barve A, Zhao Z, Jin W, Cheng K. Comparison of avidin, neutravidin, and streptavidin as nanocarriers for efficient siRNA delivery. Mol Pharm. 2017;14(5):1517–1527. doi:10.1021/acs.molpharmaceut.6b00933
24. Choi KM, Choi SH, Jeon H, Kim IS, Ahn HJ. Chimeric capsid protein as a nanocarrier for siRNA delivery: stability and cellular uptake of encapsulated siRNA. ACS Nano. 2011;5(11):8690–8699. doi:10.1021/nn202597c
25. Zhang X, Li Y, Chen YE, Chen J, Ma PX. Cell-free 3D scaffold with two-stage delivery of miRNA-26a to regenerate critical-sized bone defects. Nat Commun. 2016;7:1–15. doi:10.1038/ncomms10376
26. Chen M, Gao S, Dong M, et al. Chitosan/siRNA nanoparticles encapsulated in PLGA nanofibers for siRNA delivery. ACS Nano. 2012;6(6):4835–4844. doi:10.1021/nn300106t
27. You X, Gu Z, Huang J, Kang Y, Chu CC, Wu J. Arginine-based poly(ester amide) nanoparticle platform: from structure–property relationship to nucleic acid delivery. Acta Biomater. 2018;74:180–191. doi:10.1016/j.actbio.2018.05.040
28. Bolhassani A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim Biophys Acta Rev Cancer. 2011;1816(2):232–246. doi:10.1016/j.bbcan.2011.07.006
29. Majumder P, Bhunia S, Chaudhuri A. A lipid-based cell penetrating nano-assembly for RNAi-mediated anti-angiogenic cancer therapy. Chem Commun. 2018;54(12):1489–1492. doi:10.1039/c7cc08517f
30. Suh JS, Lee JY, Choi YS, Chong PC, Park YJ. Peptide-mediated intracellular delivery of miRNA-29b for osteogenic stem cell differentiation. Biomaterials. 2013;34(17):4347–4359. doi:10.1016/j.biomaterials.2013.02.039
31. Zhao Z, Li Y, Jain A, et al. Development of a peptide-modified siRNA nanocomplex for hepatic stellate cells. Nanomed Nanotechnol Biol Med. 2018;14(1):51–61. doi:10.1016/j.nano.2017.08.017
32. Shukla RS, Jain A, Zhao Z, Cheng K. Intracellular trafficking and exocytosis of a multi-component siRNA nanocomplex. Nanomed Nanotechnol Biol Med. 2016;12(5):1323–1334. doi:10.1016/j.nano.2016.02.003
33. Choi YS, Lee JY, Suh JS, et al. The systemic delivery of siRNAs by a cell penetrating peptide, low molecular weight protamine. Biomaterials. 2010;31(6):1429–1443. doi:10.1016/j.biomaterials.2009.11.001
34. Scheicher B, Schachner-Nedherer AL, Zimmer A. Protamine-oligonucleotide-nanoparticles: recent advances in drug delivery and drug targeting. Eur J Pharm Sci. 2015;75:54–59. doi:10.1016/j.ejps.2015.04.009
35. Ndodo ND. Nanoparticle (MPG) -mediated delivery of small RNAs into human mesenchymal stem cells. African J Biotechnol. 2015;14(37):2703–2714. doi:10.5897/AJB2015.14417
36. Simeoni F, Morris MC, Heitz F, Divita G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003;31(11):2717–2724. doi:10.1093/nar/gkg385
37. Deshayes S, Gerbal-Chaloin S, Morris MC, et al. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim Biophys Acta Biomembr. 2004;1667(2):141–147. doi:10.1016/j.bbamem.2004.09.010
38. Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell. 2008;100(4):201–217. doi:10.1042/BC20070116
39. Crombez L, Charnet A, Morris MC, Aldrian-Herrada G, Heitz F, Divita G. A non-covalent peptide-based strategy for siRNA delivery. Biochem Soc Trans. 2007;35(1):44–46. doi:10.1042/BST0350044
40. Gerbal-Chaloin S, Gondeau C, Aldrian-Herrada G, Heitz F, Gauthier-Rouvière C, Divita G. First step of the cell-penetrating peptide mechanism involves Rac1 GTPase-dependent actin-network remodelling. Biol Cell. 2007;99(4):223–238. doi:10.1042/BC20060123
41. Schachner-Nedherer A-L, Werzer O, Zimmer A. A protocol to characterize peptide-based drug delivery systems for miRNAs. ACS Omega. 2019;4(4):7014–7022. doi:10.1021/acsomega.8b03562
42. Sigma-Aldrich. N-TER Nanoparticle siRNA Transfection System - Product Information. 1–4. Available from: https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Bulletin/n2913bul.pdf. Accessed September 18, 2019.
43. Ramírez-Zacarías JL, Castro-Muñozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with oil red o. Histochem Cell Biol. 1992;97(6):493–497. doi:10.1007/BF00316069
44. Nečas D, Klapetek P. Gwyddion: an open-source software for SPM data analysis. Cent Eur J Phys. 2012;10(1):181–188. doi:10.2478/s11534-011-0096-2
45. Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4(4):263–273. doi:10.1016/j.cmet.2006.07.001
46. Kawada T, Takahashi N, Fushiki T. Biochemical and physiological characteristics of fat cell. J Nutr Sci Vitaminol. 2001;47:1–12. doi:10.3177/jnsv.47.1
47. Drechsler S, Andrä J. Online monitoring of metabolism and morphology of peptide-treated neuroblastoma cancer cells and keratinocytes. J Bioenerg Biomembr. 2011;43(3):275–285. doi:10.1007/s10863-011-9350-y
48. Orr BG, Baker JR, Leroueil PR, et al. Interaction of polycationic polymers with supported lipid bilayers and cells: nanoscale hole formation and enhanced membrane permeability. Bioconjug Chem. 2006;17(3):728–734. doi:10.1021/bc060077y
49. Saar K, Lindgren M, Hansen M, et al. Cell-penetrating peptides: a comparative membrane toxicity study. Anal Biochem. 2005;345(1):55–65. doi:10.1016/j.ab.2005.07.033
50. Schubert D, Behl C, Lesley R, et al. Amyloid peptides are toxic via a common oxidative mechanism. Proc Natl Acad Sci USA. 1995;92(6):1989–1993. doi:10.1073/pnas.92.6.1989
51. Gooding M, Browne LP, Quinteiro FM, Selwood DL. siRNA delivery: from lipids to cell-penetrating peptides and their mimics. Chem Biol Drug Des. 2012;80(6):787–809. doi:10.1111/cbdd.12052
52. Siedlecka-Kroplewska K, Kogut-Wierzbicka M, Mucha P, Wierzbicki PM, Rekowski P, Ruczynski J. Cell-penetrating peptides as a promising tool for delivery of various molecules into the cells. Folia Histochem Cytobiol. 2014;52(4):257–269. doi:10.5603/fhc.a2014.0034
53. Reissmann S. Cell penetration: scope and limitations by the application of cell-penetrating peptides. J Pept Sci. 2014;20(10):760–784. doi:10.1002/psc.2672
54. Creusot N, Gruppen H, van Koningsveld GA, de Kruif CG, Voragen AGJ. Peptide-peptide and protein-peptide interactions in mixtures of whey protein isolate and whey protein isolate hydrolysates. Int Dairy J. 2006;16(8):840–849. doi:10.1016/j.idairyj.2005.06.010
55. Cui H, Webber MJ, Stupp SI. Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials. Biopolymers. 2010;94(1):1–18. doi:10.1002/bip.21328
56. Tanabe Y. Inhibition of adipocyte differentiation by mechanical stretching through ERK-mediated downregulation of PPAR 2. J Cell Sci. 2004;117(16):3605–3614. doi:10.1242/jcs.01207
57. Shoham N, Gefen A. Mechanotransduction in adipocytes. J Biomech. 2012;45(1):1–8. doi:10.1016/j.jbiomech.2011.10.023
58. Reed BC, Lane MD. Insulin receptor synthesis and turnover in differentiating 3T3-L1 preadipocytes. Proc Natl Acad Sci. 1980;77(1):285–289. doi:10.1073/pnas.77.1.285
59. Crooke T. Glucocorticoid pread ipocytes regulation of fl-Adrenergic receptors in 3T3-L1. Mol Pharmacol. 1987;31(4):337–384.
60. Zhang XH, Zhang YY, Sun HY, Jin MW, Li GR. Functional ion channels and cell proliferation in 3T3-L1 preadipocytes. J Cell Physiol. 2012;227(5):1972–1979. doi:10.1002/jcp.22925
61. Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomedicine. 2014;9:51–63. doi:10.2147/IJN.S26592
62. Shang L, Nienhaus K, Nienhaus GU. Engineered nanoparticles interacting with cells: size matters. J Nanobiotechnology. 2014;12(1):1–11. doi:10.1186/1477-3155-12-5
63. Heitz F, Morris MC, Divita G. Themed section : vector design and drug delivery review Twenty years of cell-penetrating peptides : from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195–206. doi:10.1111/j.1476-5381.2008.00057.x
64. Phillips DC, York RL, Mermut O, McCrea KR, Ward RS, Somorjai GA. Side chain, chain length, and sequence effects on amphiphilic peptide adsorption at hydrophobic and hydrophilic surfaces studied by sum-frequency generation vibrational spectroscopy and quartz crystal microbalance. J Phys Chem C. 2007;111(1):255–261. doi:10.1021/jp0645263
65. Copolovici DM, Langel K, Eriste E, Langel Ü. Cell-penetrating peptides: design, synthesis, and applications. ACS Nano. 2014;8(3):1972–1994. doi:10.1021/nn4057269
66. Deshayes S, Morris MC, Divita G, Heitz F. Cellular and molecular life sciences cell-penetrating peptides : tools for intracellular delivery of therapeutics. Cell Mol Life Sci. 2005;62:1839–1849. doi:10.1007/s00018-005-5109-0
67. Zhao F, Zhao Y, Liu Y, Chang X, Chen C. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small. 2011;7(10):1322–1337. doi:10.1002/smll.201100001
68. Verma A, Stellacci F. Effect of surface properties on nanoparticle-cell interactions. Small. 2010;6(1):12–21. doi:10.1002/smll.200901158
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.