Back to Journals » International Journal of General Medicine » Volume 15
Bioinformatics Analysis Identified the Hub Genes, mRNA–miRNA–lncRNA Axis, and Signaling Pathways Involved in Rheumatoid Arthritis Pathogenesis
Authors Yang M ![]() , Zheng H, Su Y, Xu K, Yuan Q, Aihaiti Y, Cai Y, Xu P
, Zheng H, Su Y, Xu K, Yuan Q, Aihaiti Y, Cai Y, Xu P ![]()
Received 16 December 2021
Accepted for publication 28 March 2022
Published 8 April 2022 Volume 2022:15 Pages 3879—3893
DOI https://doi.org/10.2147/IJGM.S353487
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Mingyi Yang,1,* Haishi Zheng,1,* Yani Su,2,* Ke Xu,1 Qiling Yuan,1 Yirixiati Aihaiti,1 Yongsong Cai,1 Peng Xu1
1Department of Joint Surgery, HongHui Hospital, Xi’an Jiaotong University, Xi’an, Shanxi, 710054, People’s Republic of China; 2Yan’an University Affiliated Hospital, Yan’an, Shanxi, 716000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Peng Xu, HongHui Hospital, Xi’an Jiaotong University, No. 555, Youyi East Road, Beilin District, Xi’an City, Shaanxi Province, 710054, People’s Republic of China, Tel +86 13772090019, Email [email protected]
Objective: Rheumatoid arthritis (RA) is a nonspecific, chronic, systemic autoimmune disease characterized by symmetric polyarticular synovitis. Bioinformatics analysis of potential biomarkers, mRNA–miRNA–lncRNA axes, and signaling pathways in the pathogenesis of RA provides potential targets and theoretical basis for further research on RA.
Methods: The GSE1919 and GSE77298 datasets were downloaded from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo). Perl was used to perform data merging, and R was used to perform batch correction. The “limma” package of R was used to screen differentially expressed genes, and the “clusterProfiler” package was used to perform enrichment analysis of the Gene Ontology and Kyoto Encyclopedia of Genes and Genomes. Search Tool for the Retrieval of Interacting Genes/Proteins was used to construct the protein–protein interaction network, Cytoscape was used for module analysis, and R was used to screen for hub genes. GraphPad Prism was used to plot the receiver operating characteristic curve of the hub genes. Gene set enrichment analysis and competitive endogenous RNA network analysis were performed on hub genes with the greatest diagnostic values. The hub gene with the greatest diagnostic value was verified using immunohistochemical staining.
Results: We obtained nine hub genes (ITGB2, VAMP8, HLA-A, PTAFR, SYK, FCER1G, HLA-DPB1, LCP2, and ACTR2) and four mRNA–miRNA–lncRNA axes (ITGB2-hsa-miR-486-3p-SNHG3, ITGB2-hsa-miR-338-5p-XIST, ITGB2-hsa-miR-5581-3p-XIST, and ITGB2-hsa-miR-1226-5p-XIST) related to the pathogenesis of RA. The nine hub genes were highly expressed, and ITGB2 had the highest diagnostic value for RA. We also identified signaling pathways related to the pathogenesis of RA: Fc epsilon Rl and chemokine signaling pathways. The immunohistochemical results showed that ITGB2 expression was significantly upregulated in RA.
Conclusion: The hub genes, mRNA–miRNA–lncRNA axes, and signaling pathways related to RA pathogenesis identified in this study provide a new research direction for the mechanism, diagnosis, and treatment of RA.
Keywords: rheumatoid arthritis, synovial, genes, lncRNA
Introduction
Rheumatoid arthritis (RA) is a chronic systemic immune inflammatory disease that involves joint swelling, stiffness, and pain, and is characterized by persistent synovitis, systemic inflammation, and autoantibodies. Although RA affects many tissues and organs, it primarily affects the synovium and joints. Synovitis can erode the joint surface and lead to deformity and loss of function, ultimately resulting in chronic disability and reduced life expectancy. Immune system abnormalities are also involved in RA pathogenesis. Multiple branches of the immune system, including T cells, B cells, antigen-presenting cells, and various cytokines, have been shown to participate in the autoimmune process of RA.1,2 RA affects approximately 1% of the population and is most common among women and the elderly. RA can occur at any age, but is most common in people 40–70 years of age, and its incidence increases with age.3 Approximately 40% of RA patients become disabled after 10 years.3 Currently, no drugs can cure RA.4 When RA develops to a late stage, surgical treatment of large joints, such as total knee and total hip arthroplasty, must be performed. Uncontrolled active RA can cause joint damage, disability, cardiovascular disease, and other comorbidities, and seriously affect the quality of life of patients. It not only imposes heavy economic and psychological burdens on patients but also has a significant influence on social stability.
RA causes persistent synovial inflammation and associated damage to the articular cartilage and underlying bones.2 Interactions between T and B lymphocytes, fibroblast-like synoviocytes (FLS), and macrophages lead to overproduction and overexpression of tumor necrosis factor (TNF). This causes excessive production of cytokines, such as interleukin-6 (IL-6), which in turn leads to persistent synovial inflammation and joint destruction.5 Joint swelling in RA results from inflammation of the synovium caused by the invasion of leukocytes into the normally sparse synovial compartment.6 The inflammatory environment of the synovial compartment is regulated by a complex network of cytokines and chemokines, in which TNF, IL-6, and granulocyte-monocyte colony-stimulating factor play important roles.7 Cytokines and chemokines aggravate the inflammatory response by activating endothelial cells and attracting immune cells to aggregate in the synovial cavity, and chondrocytes are damaged after stimulation with cytokines.6 At present, the pathogenesis of RA is not fully understood and may be related to genomic variation, gene expression, protein translation, and post-translational modification.8 Further research on the pathogenesis of RA is of great significance for patients with RA and for society.
A microarray is a high-throughput genomics technology that is used to understand complex interactions and networks in the development of diseases. Significant progress has been made in the field of medicine that has greatly promoted research on the pathogenic processes of diseases.9 In recent years, microarray technology, based on high-throughput platforms, has been widely used for gene expression profiling. Bioinformatics is an emerging discipline that combines molecular biology and information technology, and has great potential to reveal the molecular mechanisms of diseases.10 Specific proteins and compounds involved in RA have been detected using microarray technology, including fulvic acid, histone acetyltransferase, nuclear factor kappa B (NF-kB), and prostaglandin D2 synthase.11,12 In this study, bioinformatics technology was used to analyze the gene expression profile chip in RA synovial tissue to identify potential biomarkers and molecular pathways related to the pathogenesis of RA. This provides a theoretical basis and direction for mechanism-related research of RA and a potential target for the diagnosis and treatment of RA.
Materials and Methods
Microarray Data
The Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo) is a public functional genome database that stores high-throughput gene expression data, chips, and microarrays.13 Previous studies have obtained potential biomarkers in RA pathogenesis from the GSE1919 and GSE77298 datasets through bioinformatics analysis,14,15 so we combined these two datasets for our analysis.14 The GSE1919 and GSE77298 gene expression datasets were downloaded from the GEO database. GSE1919 contained five RA and five normal synovial tissue samples, and GSE77298 contained 16 RA and seven normal synovial tissue samples. The GSE128813 dataset, which contains three samples of RA synovial tissue and three samples of normal synovial tissue, was downloaded from GEO. The GSE128813 dataset used Arraystar Human LncRNA Microarray v4.0 (Arraystar Inc., Rockville, MD) to analyze the expression of long noncoding RNA (lncRNA) and messenger RNA (mRNA) in normal synovial tissue and RA synovial tissue. The differentially expressed lncRNAs and mRNAs between the two groups were further analyzed.16
Identification of Differentially Expressed Genes(DEGs)
R is a complete operating software for data processing, statistics, and graphics.17 The “limma” package of R provides a complete solution for analyzing experimental gene expression data by handling complex experimental designs and borrowing of information, thereby overcoming the problem of small sample sizes.18 Limma has become a popular choice for obtaining key genes through differential expression analysis of microarray and high-throughput data, and contains powerful tools for reading, normalizing, and exploring such data.18 Limma can also analyze expression profiles based on co-regulated gene sets or higher-order expression signatures, offering greater possibilities for the biological interpretation of gene expression differences.18 In the present study, the limma package of R was used to identify differentially expressed genes in RA synovial tissue and normal synovial tissue in GSE1919 and GSE77298 datasets.19 Screening conditions were |log(FC)| ≥ 1 and adj. p val < 0.05.
Enrichment Analysis
The Gene Ontology (GO) database is used to annotate genes and analyze their biological processes.20 The Kyoto Encyclopedia of Genes and Genomes (KEGG) database integrates information on genomes, chemistry, and system functions.21 The “clusterProfiler” package of R for gene cluster comparison can be applied to gene clusters obtained from gene expression data and from other pathways such as protein–protein interaction (PPI) modules and micro RNA (miRNA) target genes.22 The “clusterProfiler” package of R is used to analyze high-throughput data obtained via transcriptomics or proteomics, apply biological term classification and enrichment analysis to gene clustering comparisons, and contribute to a better understanding of higher-order functions of biological systems.23 The “clusterProfiler” package was used to perform GO enrichment analysis of DEGs, and the screening conditions were set as p value < 0.01 and q value < 0.01.24 The package was also used to perform KEGG enrichment analysis of DEGs, and the filtering conditions were set as p value < 0.001 and q value < 0.001.24
PPI Network Construction and Module Analysis
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) (http://string-db.org) is a public online database of gene and protein interactions that helps users easily access unique and wide-ranging experiments and predict interaction relationship information.25 In the present study, STRING was used to construct a PPI network of DEGs, and interactions with a comprehensive score > 0.4 was considered to be statistically significant.10 Cytoscape is a software that focuses on open-source network visualization and analysis, and it provides a basic functional layout and query network that are combined into a visual network based on basic data.26 Cytoscape visualizes PPI networks.19 Molecular complex detection (MCODE), a plugin for Cytoscape, detects densely connected regions in large PPI networks representing molecular complexes.27 The method employs vertex weighting based on local neighborhood densities, which allows for fine-tuning of clusters of interest without considering the rest of the network, and examines cluster interconnections associated with protein networks.27 Cytoscape’s MCODE plug-in analyzes the top-level modules.10
Hub Gene Screening and Receiver Operating Characteristic (ROC) Curve Drawing
Degree centrality is the number of interactions connecting a protein to its neighbors, and is a fundamental parameter in PPI networks used to evaluate nodes in the network.28 R screens the top ten genes with the highest degree of network connection in PPI as hub genes and visualizes them.29 The hub genes, obtained using the intersection of the hub gene and the genes contained in the top module, may play important roles in RA pathogenesis. To identify hub genes with high diagnostic value for RA, we plotted receiver operating characteristic curves using GraphPad Prism.30–32
Gene Set Enrichment Analysis (GSEA)
After the ROC curves of the hub genes were drawn, the hub gene with the largest area under the curve was determined to have the highest diagnostic value for RA. To explore the specific roles of the most valuable hub genes in the diagnosis and pathogenesis of RA, we used the GSEA method to identify the biological processes and signaling pathways related to RA.33
Construction of a Competitive Endogenous RNA (ceRNA) Network and Prediction of mRNA–miRNA–lncRNA Axis
To further study the regulatory network of the hub gene with the greatest diagnostic value for RA, we constructed a ceRNA network by mining miRNAs and lncRNAs. TargetScan, miRDB, and miRWalk databases were used to predict the target miRNA of the hub gene, and the prediction results of the three databases intersected.34 The starBase database was used to predict the target lncRNA of miRNAs,35 and Cytoscape was used to construct the ceRNA network.36 The “limma” package of R was used to perform differential analysis of the GSE128813 dataset to obtain DEGs and differentially expressed lncRNAs (DElncRNAs) of RA synovial tissue and normal synovial tissue.19 The DEGs and DElncRNAs obtained from GSE128813 intersected with the target lncRNA predicted by starBase. The mRNA–miRNA–lncRNA axis related to RA pathogenesis was determined based on the intersection of lncRNAs and their corresponding miRNAs and mRNAs.19
Immunohistochemical (IHC) and Hematoxylin–Eosin (HE) Staining
The hub gene with the greatest diagnostic value was experimentally verified using IHC staining. Human knee synovial tissues from patients with RA (n = 11) and osteoarthritis (OA) (n = 10) (Supplementary Table 1) were collected from the Department of Joint Surgery, Honghui Hospital, Xi’an Jiaotong University. Since normal synovial tissue could not be collected in the Honghui Hospital, so we used OA synovial tissue as the control group. Patients with previous synovitis were excluded from the collection of OA synovial tissue to ensure that it approximation to normal synovial tissue. All enrolled subjects met the 2010 American College of Rheumatology diagnostic criteria for RA and OA with radiographic evidence and clinical history.37 All patients signed an informed consent form and the study was approved by the hospital ethics committee.
The fresh synovial tissue samples were fixed overnight in 10% buffered formalin (pH 7.0) and embedded in paraffin. All tissue blocks were serially sectioned (5 μm) for IHC and HE staining.38
All operations were performed in accordance with the procedures of the SABC (rabbit IgG) kit (SA1022; Wuhan Boster Biological Technology, Ltd., Wuhan, China). The samples were deparaffinized with xylene, dehydrated with ethanol, and heated to retrieve antigens in 0.01 M citrate buffer (pH 6.0). Sections were treated with 3% H2O2 for 10 min at room temperature to inactivate endogenous peroxidase, and then incubated with 5% BSA blocking solution for 1 h at room temperature. To detect ITGB2 protein expression levels, sections were incubated with rabbit anti-ITGB2 antibody (anti-ITGB2, 1:200, Proteintech, Rosemont, IL, USA) overnight at 4 °C, followed by incubation with biotinylated goat anti-rabbit secondary antibody at 37 °C for 30 min. After treatment with Strept Avidin Biotin-peroxidase Complex (SABC) for 30 min, freshly prepared DAB reagent was used for color development (Boster, Wuhan, China). Slides were washed for 10 min in tap water after 10s on slides with hematoxylin (AR1180-1; Boster, Wuhan, China). After viewing and photographing these sections with a microscope, image processing software (Image-Pro Plus, Media Cybernetics, Inc., Silver Spring, MD, USA) was used to analyze the percentage of IHC signals in each photographed field. IHC staining of each tissue section was performed by two independent pathologists.
Results
Identification of DEGs
Perl was used to merge the same genes in the GSE1919 and GSE77298 datasets. The batch effect is a variable introduced by technical factors in sample processing.39 Usually, it is impossible to sequence all samples simultaneously by the same person or sequencer.39 Large technical differences may exist between samples from different batches, constituting batch effects.39 The “sva” package of R supports surrogate variable estimation with the sva function, direct adjustment for known batch effects with the ComBat function, and adjustment for batch and latent variables in prediction problems with the fsva function.39 The “sva” package of R performed batch correction on the merged data of the two data sets, thereby reducing batch effects. The “limma” package of R was used to screen for DEGs. There were 415 DEGs at a cut-off value of |log(FC)| ≥ 1 and adj. p val < 0.05, including 250 upregulated and 165 downregulated DEGs.40 R visualized the volcano plot (Figure 1A) and heatmap (Figure 1B) of the DEGs.
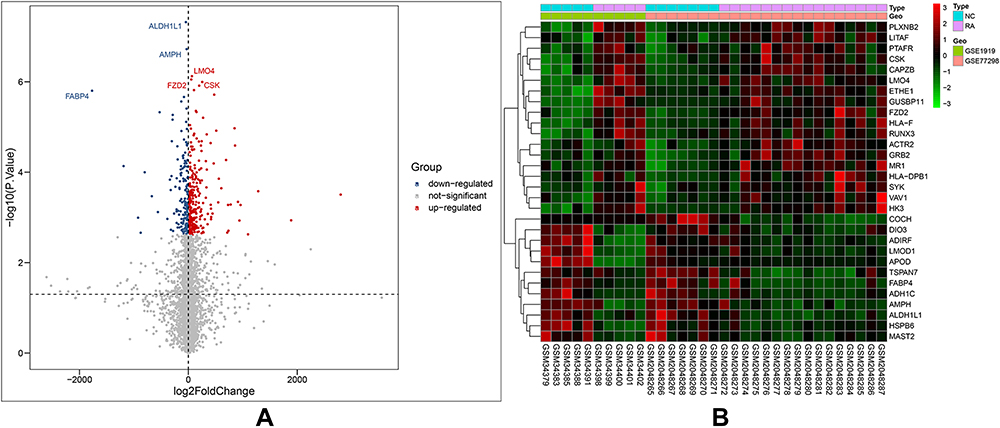 |
Figure 1 Differentially expressed genes identified in the two datasets GSE1919 and GSE77298. (A) Volcanomap of differentially expressed genes. Grey is a gene with no difference, red is an up-regulated gene, and blue is a down-regulated gene. The figure shows the names of the top three up-regulated genes and down-regulated genes. (B) Heatmap of differentially expressed genes. The abscissa is the samples of the two datasets, the ordinate is the differentially expressed genes, red is high expression, and green is low expression. |
Enrichment Analysis
R was used to perform GO and KEGG enrichment analyses of the DEGs. GO enrichment results showed that DEGs were mainly enriched in the actin filament binding, actin binding, amide binding, MHC protein complex binding, cell adhesion molecule binding, MHC class II protein complex binding, phosphotyrosine residue binding, peptide binding, protein tyrosine kinase activity, peptide antigen binding, non-membrane spanning protein tyrosine kinase activity, phosphoprotein binding, protein phosphorylated amino acid binding and antigen binding (Figure 2A). KEGG analysis showed that DEGs were primarily enriched in antigen processing and presentation, viral myocarditis, Yersinia infection, FcγR-mediated phagocytosis, natural killer cell-mediated cytotoxicity, cell adhesion molecules, Th17 cell differentiation, platelet activation, allograft rejection, chemokine signaling pathway, graft-versus-host disease, Th1 and Th2 cell differentiation, phagosomes, type I diabetes, and FcεRI signaling pathway (Figure 2B).
 |
Figure 2 Enrichment analysis of differentially expressed genes. (A) GO enrichment, the abscissa is the number of enriched differential genes, and the ordinate is the result of GO enrichment. (B) KEGG enrichment, the abscissa is the ratio of the number of enriched differential genes in the total differential genes, and the ordinate is the result of KEGG enrichment. |
PPI Network Construction and Module Analysis
STRING was used to perform PPI network analysis of DEGs, with a total of 266 nodes and 939 edges. Cytoscape was used to visualize the PPI network (Figure 3). Cytoscape’s MCODE plug-in was used for module analysis. The analysis standards were: MCODE cutoff > 5, degree cutoff = 2, node score cutoff = 0.2, maximum depth = 100, and k-core = 2. Fourteen top-level modules were displayed, and the first four top-level modules interacted most closely (Figure 4A).
 |
Figure 3 The protein interaction network of differentially expressed genes. The darker the color, the more significant the difference. The top 5 genes with the most significant differences were: leukocyte associated immunoglobulin like receptor 1(LAIR1), platelet activating factor receptor (PTAFR), integrin subunit beta 2(ITGB2), integrin subunit alpha X (ITGAX), cytochrome b-245 alpha chain (CYBA). |
 |
Figure 4 Analysis of protein interaction network. (A) Cytoscape’s MCODE plug-in obtains the top four closely interacting top-level modules. (B) R gets the top ten Hub genes with the highest network connectivity. The abscissa is the connectivity score, and the ordinate is the Hub gene. |
Screening of Hub Genes and Drawing of ROC Curves
R analyzed ten hub genes: integrin subunit beta 2 (ITGB2), vesicle-associated membrane protein 8 (VAMP8), major histocompatibility complex, class I, A (HLA-A), platelet activating factor receptor (PTAFR), spleen-associated tyrosine kinase (SYK), Fc epsilon receptor Ig (FCER1G), major histocompatibility complex, class II, DP beta 1 (HLA-DPB1), lymphocyte cytosolic protein 2 (LCP2), vav guanine nucleotide exchange factor 1 (VAV1), and actin-related protein 2 (ACTR2) (Figure 4B). We found that nine of the ten hub genes (ITGB2, VAMP8, HLA-A, PTAFR, SYK, FCER1G, HLA-DPB1, LCP2, and ACTR2) overlapped with the DEGs in the first four top-level modules. These nine hub genes are highly expressed in RA synovial tissue. ROC curves for the nine hub genes showed that ITGB2, VAMP8, HLA-A, PTAFR, SYK, FCER1G, HLA-DPB1, LCP2, and ACTR2 have diagnostic value for RA. Among the nine hub genes, ITGB2 (area under the curve = 0.972) had the highest diagnostic value for RA (Figure 5).
 |
Figure 5 ROC curves were drawn for the nine genes common to the first four top-level modules and Hub genes. The larger the area under the curve (AUC), the higher the diagnostic value of rheumatoid arthritis. The closer the AUC is to 1, the higher the diagnostic value. AUC of 0.5–0.7 indicates a lower diagnostic value. AUC of 0.7–0.9 indicates a certain diagnostic value. AUC above 0.9 has high diagnostic value. |
GSEA
GSEA of ITGB2 showed that the biological process was mainly enriched in adaptive immune response and interleukin 12 production. ITGB2 was primarily enriched in the Toll like receptor signaling pathway, B cell receptor signaling pathway, T cell receptor signaling pathway, Fc epsilon Rl signaling pathway, chemokine signaling pathway and RIG l like receptor signaling pathway (Figure 6).
 |
Figure 6 GSEA enrichment analysis was performed on the Hub gene (ITGB2) with the highest diagnostic value for rheumatoid arthritis. In terms of biological processes, ITGB2 was significantly enriched in adaptive immune response and interleukin 12 production. In terms of signaling pathways, ITGB2 was significantly enriched in Toll like receptor signaling pathway, B cell receptor signaling pathway, T cell receptor signaling pathway, Fc epsilon Rl signaling pathway pathway, chemokine signaling pathway and RIG l like receptor signaling pathway. |
Construction of ceRNA Network and Prediction of mRNA–miRNA–lncRNA Axis
ITGB2 predicted the target miRNAs in the TargetScan, miRDB, and miRWalk databases, and the intersection of the three databases yielded a total of 13 miRNAs (Figure 7A and B). Among the 13 miRNAs, five miRNAs (hsa-miR-486-3p, hsa-miR-338-5p, hsa-miR-29b-1-5p, hsa-miR-5581-3p, and hsa-miR-1226- 5p) targeting lncRNAs were predicted using the starBase database. After removing duplicate lncRNAs, five miRNAs predicted a total of 262 target lncRNAs. Cytoscape was used to construct the ceRNA network based on ITGB2, five miRNAs that could target lncRNAs in the starBase database, and 262 targeted lncRNAs predicted by five miRNAs (Figure 8). GSE128813 obtained 262 DEGs and DElncRNAs through difference analysis and R visualization of the volcano plot (Figure 9A) and heatmap (Figure 9B). The intersection of the DEGs and DElncRNAs obtained from GSE128813 and the targeted lncRNAs of ITGB2 resulted in two lncRNAs: small nucleolar RNA host gene 3 (SNHG3) and X inactive specific transcript (XIST) (Figure 10A). Based on the ceRNA network, we determined the miRNAs and mRNAs corresponding to SNHG3 and XIST, and obtained four mRNA–miRNA–lncRNA axes related to RA pathogenesis (Figure 10B).
 |
Figure 7 Target miRNAs prediction of ITGB2. (A) ITGB2 predicted a total of 13 common miRNAs in the three databases TargetScan, miRDB and miRWalk. (B) 13 miRNAs targeted by ITGB2. |
 |
Figure 8 ITGB2 predicted a total of 13 targeted miRNAs in the TargetScan, miRDB and miRWalk databases, of which 5 miRNAs obtained a total of 262 targeted lncRNAs in the starBase database. Cytoscape construct the ceRNA network. Yellow is the hub gene ITGB2 with the greatest diagnostic value for RA. Green are the five miRNAs that can predict lncRNAs in the starBase database. Pink is the 262 lncRNAs predicted by 5 miRNAs in the starBase database. |
 |
Figure 9 Differential expression analysis of GSE128813. (A) Volcanomap, black is the genes and lncRNAs with no difference, red is the genes and lncRNAs that are up-regulated, and green is the genes and lncRNAs that are down-regulated. (B) Heatmap, the abscissa is the sample, the ordinate is the differentially expressed genes and differentially expressed lncRNAs, red is high expression, and green is low expression. |
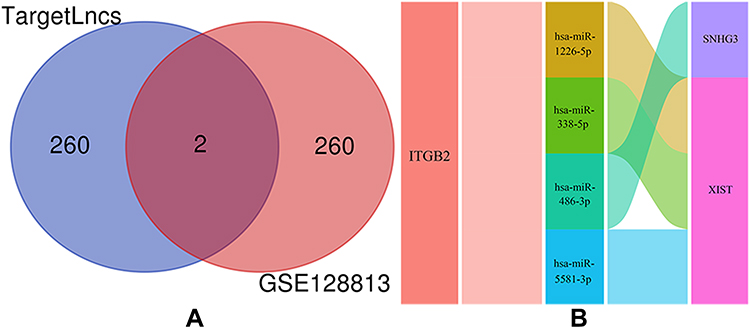 |
Figure 10 mRNA–miRNA–lncRNA axes. (A) The intersection of differentially expressed genes and differentially expressed lncRNAs obtained by GSE128813 and targeted lncRNAs in ceRNA network was obtained to obtain two lncRNAs. (B) According to two lncRNAs and their corresponding miRNAs and mRNAs, 4 mRNA–miRNA–lncRNA axes are obtained. |
IHC and HE Staining
The expression levels of ITGB2 protein in the synovial tissue of patients with RA and OA were detected using IHC with ITGB2 antibody and rabbit IgG (isotype). Compared with OA, we found more severe synovitis, including synovial hyperplasia, lymphocyte infiltration, and vascular hyperplasia, in the synovial tissue of patients with RA. The relative expression of ITGB2 was significantly up-regulated in the RA synovium (Figure 11).
 |
Figure 11 Immunohistochemistry (IHC) staining using anti-ITGB2 antibodies and hematoxylin and eosin (HE) staining of OA and RA synovial tissues. (A) OA synovial tissues, HE, 10x. (B) OA synovial tissues, HE, 20x. (C) RA synovial tissues, HE, 10x. (D) RA synovial tissues, HE, 20x. (E) OA synovial tissues, IHC (anti-ITGB2), 10x. (F) OA synovial tissues, IHC (anti-ITGB2), 20x. (G) RA synovial tissues, IHC (anti-ITGB2), 10x. (H) RA synovial tissues, IHC (anti-ITGB2), 20x. (I) IHC statistics. N(OA)=10, N(RA)=11. Data were shown as the mean ± standard deviation (SD), **p <0.01. |
Discussion
The synovium is the primary site of the RA pathological process, and as key pro-inflammatory factors, FLS are the most important target cells that participate in the occurrence and development of RA.41,42 FLS ensure the integrity of joint structure and dynamics by controlling the composition of joint synovial fluid and the extracellular matrix of the joint lining,43 but can also promote the occurrence and development of RA by producing pathogenic mediators such as cytokines and proteases.44 Studies have shown that in the peri-synovial microenvironment, RA-FLS are more invasive and specific, with aggressive inflammation, matrix regulation, and invasion. They can also enhance chondrocyte catabolism and synovial osteoclast cell generation to promote joint destruction.45,46 RA-FLS exhibit an aggressive phenotype that is hyperproliferative, anti-apoptotic, and enhances migration and invasion abilities, and the aggressive phenotype of FLS is a key factor in tissue damage.43 An increase in FLS in RA is associated with disease duration, macrophage infiltration in the synovium, and severity of cartilage erosion.47 FLS and their key signaling molecules are considered important for therapeutic strategies in RA.48 Understanding the mechanisms underlying the aggressive features of FLS provides the potential to identify novel targets for the treatment of RA, thereby reducing inflammation and bone erosion to alleviate RA progression.
We identified nine hub genes (ITGB2, VAMP8, HLA-A, PTAFR, SYK, FCER1G, HLA-DPB1, LCP2, and ACTR2) that may be potential biomarkers involved in RA pathogenesis using a series of bioinformatics analyses. These nine hub genes are highly expressed in RA synovial tissue. ROC curve analysis showed that among the nine hub genes, ITGB2 had the highest diagnostic value for RA. We then constructed a ceRNA network based on ITGB2 and combined it with the GSE128813 dataset to obtain four mRNA–miRNA–lncRNA axes (ITGB2-hsa-miR-486-3p-SNHG3, ITGB2-hsa- miR-338-5p-XIST, ITGB2-hsa-miR-5581-3p-XIST, and ITGB2-hsa-miR-1226-5p-XIST) that may be related to RA pathogenesis. We verified via IHC that ITGB2 was highly expressed in RA synovial tissue, which was consistent with our bioinformatics analysis results. Differential analysis of the GSE128813 dataset revealed that SNHG3 and XIST were DElncRNAs in RA synovial tissue. Among the four mRNA–miRNA–lncRNA axes, both mRNA and lncRNA were differentially expressed in RA synovial tissue. Therefore, these mRNA–miRNA–lncRNA axes may be involved in the regulation of RA pathogenesis, and their potential roles in RA pathogenesis need to be studied further. The enrichment results of DEGs and GSEA results of ITGB2 indicated that the Fc epsilon Rl and chemokine signaling pathways may be related to the pathogenesis of RA.
ITGB2 encodes an integrin chain that binds to multiple others to form different integrin heterodimers. Integrins are integral cell surface proteins that are involved in cell adhesion and cell surface-mediated signaling. ITGB2 plays an important role in immune responses, and deletion of this gene results in defective leukocyte adhesion.49 Mutations in ITGB2 cause type I leukocyte adhesion defects characterized by recurrent bacterial infections, pus formation, poor wound healing, and persistent neutropenia.50 Defects in ITGB2 also affect leukocyte transport mediated by the adhesion between myeloid leukocytes and inflammatory endothelial cells, which is essential for bacterial defense and wound healing.50 In this study, we found that ITGB2 is highly expressed in RA, is a potential biomarker in RA pathogenesis, and has a high diagnostic value for RA. Studies have found a bidirectional relationship between RA and depression; RA is an autoimmune disorder associated with fatigue, pain, and sleep disturbance symptoms, which are also present in depression.51 ITGB2 is highly expressed in the RA pathway in unidirectional depression,52 which is consistent with our finding that ITGB2 is highly expressed in RA. Furthermore, inflammatory pathways are the main links between RA and depression, and cytokines are the main triggers.53 Therefore, we speculate that the potential role of ITGB2 in RA pathogenesis may be related to cytokines and inflammatory pathways; however, further studies are needed to verify this.
The lncRNA SNHG3 not only regulates the proliferation and migration of laryngeal cancer by regulating the miR-384/WEE1 axis,54 but also promotes the proliferation and migration of bladder cancer through the miR-515-5p/GINS2 axis.55 Furthermore, SNHG3 acts as a ceRNA of miR-485 to upregulate autophagy related 7 (ATG7) expression to promote autophagy-induced neuronal apoptosis,56 and also acts as a ceRNA to promote the malignant development of colorectal cancer.57 It can be seen that lncRNA SNHG3 not only functions as a ceRNA but is also involved in proliferation and migration. In addition, RA-FLS have the potential to proliferate and migrate,58 and the role of lncRNA SNHG3 in RA may also be related to proliferation and migration. In this study, we found that SNHG3 may be involved in the pathogenesis of RA through the ITGB2-hsa-miR-486-3p-SNHG3 axis, and that miR-486-3p may also be involved in RA pathogenesis. Previous studies have found a correlation between miR-486-3p and RA; miR-486-3p is differentially expressed in RA, and when high-risk individuals progress to RA, the expression of miR-486-3p in serum decreases.59 This demonstrates the reliability of the results of this study to a certain extent. In summary, we speculate that SNHG3 may act as a ceRNA to regulate the proliferation and migration of RA-FLS through the ITGB2-hsa-miR-486-3p-SNHG3 axis. Further studies are needed to demonstrate its specific mechanism in the pathogenesis of RA.
The lncRNA XIST affects the proliferation and differentiation of osteoblasts by binding let-7c-5p to regulate signal transducer and activator of transcription 3 (STAT3), thereby promoting the occurrence of RA.60 Quercetin can inhibit TNF-α-induced inflammatory cytokine production and XIST expression in RA-FLS, and silencing XIST can inhibit the inflammatory response of TNF-α-treated cells through miR-485.4 The interaction between lncRNA XIST and miR-126-3p regulates the NF-κB signaling pathway in RA-FLS; downregulation of XIST can inhibit the proliferation of FLS by increasing the expression of miR-126-3p/NF-κB, thereby inhibiting the occurrence and development of RA.61 A study found that in RA, miR-338-5p was significantly downregulated and ADAM metallopeptidase with thrombospondin type 1 motif 9 (ADAMTS9) was significantly overexpressed, and miR-338-5p inhibited the proliferation and invasion of RA-FLS by inhibiting ADAMTS9, suggesting that supplementing miR-338-5p may be a potential clinical treatment for RA.62 Studies have also found that miR-338-5p is highly expressed in RA. miR-338-5p can promote the proliferation, invasion, and inflammatory response of RA-FLS by directly downregulating the expression of sprouty RTK signaling antagonist 1 (SPRY1),63 and also promotes the proliferation and migration of RA-FLS by targeting nuclear factor of activated T cells 5 (NFAT5).64 Although the current findings on the expression of miR-338-5p in RA are contrary, they also illustrate the potential role of miR-338-5p in RA pathogenesis. Although there are no reports related to miR-5581-3p and RA, one study found that miR-5581-3p promotes hemophilia A by targeting the F8 mRNA of hemophilia A without F8 mutation and inhibiting the expression of FVIII protein.65 However, related research on miR-1226-5p has not yet been reported. The results of this study suggest that the ITGB2-hsa-miR-338-5p/hsa-miR-5581-3p/hsa-miR-1226-5p-XIST axis plays a role in the pathogenesis of RA. In summary, we speculate that the regulatory role of the ITGB2-hsa-miR-338-5p/hsa-miR-5581-3p/hsa-miR-1226-5p-XIST axis in RA pathogenesis may be related to the proliferation and differentiation of osteoblasts, the inflammatory response, the NF-κB signaling pathway, and the proliferation, migration, invasion, and inflammatory response of RA-FLS.
However, this study has certain limitations. First, the sample size of this study was relatively small, and the sampling method did not eliminate the effects of sex and other diseases of the patients. Second, other potential biomarkers, apart from ITGB2, the mRNA–miRNA–lncRNA axis, and signaling pathways identified in this study, have not yet been verified experimentally; however, this will be the focus of our next study.
Conclusion
In this study, nine hub genes that may be related to RA pathogenesis were identified through microarray gene expression data and bioinformatics analysis. Nine hub genes were highly expressed in RA synovial tissue. Through ROC curve analysis, we found that among the nine hub genes, ITGB2 had the highest diagnostic value for RA. Subsequently, we analyzed ITGB2 and obtained four mRNA–miRNA–lncRNA axes that may have regulatory roles in RA pathogenesis. In addition, we identified signaling pathways that may be related to RA pathogenesis: Fc epsilon Rl and chemokine signaling pathways. Finally, we carried out experimental verification of ITGB2 via IHC, and the verification results were consistent with the results of the bioinformatics analysis. Since the results of this study have not been verified by further experiments, they can only provide direction for RA further research. We plan to further study their potential value in the mechanism, diagnosis, and treatment of RA.
Abbreviations
RA, rheumatoid arthritis; FLS, fibroblast-like synoviocytes; TNF, tumor necrosis factor; IL-6, interleukin-6; NF-kB, nuclear factor kappa B; GEO, Gene Expression Omnibus; lncRNA, long noncoding RNA; mRNA, messenger RNA; DEGs, differentially expressed genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein–protein interaction; miRNA, micro RNA; STRING, Search Tool for the Retrieval of Interacting Genes/Proteins; MCODE, Molecular complex detection; ROC, receiver operating characteristic; GSEA, Gene Set Enrichment Analysis; ceRNA, competitive endogenous RNA; DElncRNAs, differentially expressed lncRNAs; IHC, Immunohistochemical; HE, hematoxylin-eosin; OA, osteoarthritis; ITGB2, integrin subunit beta 2; VAMP8, vesicle-associated membrane protein 8; HLA-A, major histocompatibility complex, class I, A; PTAFR, platelet activating factor receptor; SYK, spleen-associated tyrosine kinase; FCER1G, Fc epsilon receptor Ig; HLA-DPB1, major histocompatibility complex, class II, DP beta 1; LCP2, lymphocyte cytosolic protein 2; VAV1, vav guanine nucleotide exchange factor 1; ACTR2, actin-related protein 2; SNHG3, small nucleolar RNA host gene 3; XIST, X inactive specific transcript; ATG7, autophagy related 7; STAT3, signal transducer and activator of transcription 3; ADAMTS9, ADAM metallopeptidase with thrombospondin type 1 motif 9; SPRY1, sprouty RTK signaling antagonist 1; NFAT5, nuclear factor of activated T cells 5.
Data Sharing Statement
The data supporting the results of the study are available from the GEO database (https://www.ncbi.nlm.nih.gov/geo/).
Ethics Approval and Informed Consent
The studies involving human participants were reviewed and approved by the Clinical Research Ethics Committee of HongHui Hospital, Xi’an Jiaotong University (No, 202203006). The patients/ participants provided their written informed consent to participate in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82072432), China Postdoctoral Science Foundation(No. 2020M673454), Foundation of the Natural Science Basic Research of Shaanxi Province of China(No. 2021JQ-924).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yamin R, Berhani O, Peleg H, et al. High percentages and activity of synovial fluid NK cells present in patients with advanced stage active Rheumatoid Arthritis. Sci Rep. 2019;9(1):1351. doi:10.1038/s41598-018-37448-z
2. Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet. 2010;376(9746):1094–1108. doi:10.1016/S0140-6736(10)60826-4
3. Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358(9285):903–911. doi:10.1016/S0140-6736(01)06075-5
4. Sun HT, Li JP, Qian WQ, Yin MF, Yin H, Huang GC. Quercetin suppresses inflammatory cytokine production in rheumatoid arthritis fibroblast-like synoviocytes. Exp Ther Med. 2021;22(5):1260. doi:10.3892/etm.2021.10695
5. Feldmann M, Brennan FM, Maini RN. Rheumatoid arthritis. Cell. 1996;85(3):307–310. doi:10.1016/S0092-8674(00)81109-5
6. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–2038. doi:10.1016/S0140-6736(16)30173-8
7. Feldmann M, Maini SRN. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev. 2008;223:7–19. doi:10.1111/j.1600-065X.2008.00626.x
8. Song X, Lin Q. Genomics, transcriptomics and proteomics to elucidate the pathogenesis of rheumatoid arthritis. Rheumatol Int. 2017;37(8):1257–1265. doi:10.1007/s00296-017-3732-3
9. Xiong Y, Mi BB, Liu MF, Xue H, Wu QP, Liu GH. Bioinformatics analysis and identification of genes and molecular pathways involved in synovial inflammation in rheumatoid arthritis. Med Sci Monit. 2019;25:2246–2256. doi:10.12659/MSM.915451
10. Cai W, Li H, Zhang Y, Han G. Identification of key biomarkers and immune infiltration in the synovial tissue of osteoarthritis by bioinformatics analysis. PeerJ. 2020;8:e8390. doi:10.7717/peerj.8390
11. Zheng F, Yu X, Huang J, Dai Y. Circular RNA expression profiles of peripheral blood mononuclear cells in rheumatoid arthritis patients, based on microarray chip technology. Mol Med Rep. 2017;16(6):8029–8036. doi:10.3892/mmr.2017.7638
12. Ouyang Q, Wu J, Jiang Z, et al. Microarray expression profile of circular RNAs in peripheral blood mononuclear cells from rheumatoid arthritis patients. Cell Physiol Biochem. 2017;42(2):651–659. doi:10.1159/000477883
13. Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol. 2016;1418:93–110. doi:10.1007/978-1-4939-3578-9_5
14. Zhang R, Zhou X, Jin Y, et al. Identification of differential key biomarkers in the synovial tissue between rheumatoid arthritis and osteoarthritis using bioinformatics analysis. Clin Rheumatol. 2021;40(12):5103–5110. doi:10.1007/s10067-021-05825-1
15. Wu R, Long L, Zhou Q, Su J, Su W, Zhu J. Identification of hub genes in rheumatoid arthritis through an integrated bioinformatics approach. J Orthop Surg Res. 2021;16(1):458. doi:10.1186/s13018-021-02583-3
16. Bi X, Guo XH, Mo BY, et al. LncRNA PICSAR promotes cell proliferation, migration and invasion of fibroblast-like synoviocytes by sponging miRNA-4701-5p in rheumatoid arthritis. EBioMedicine. 2019;50:408–420. doi:10.1016/j.ebiom.2019.11.024
17. Chan BKC. Data Analysis Using R Programming. Adv Exp Med Biol. 2018;1082:47–122. doi:10.1007/978-3-319-93791-5_2
18. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
19. Aihaiti Y, Song Cai Y, Tuerhong X, et al. Therapeutic effects of naringin in rheumatoid arthritis: network pharmacology and experimental validation. Front Pharmacol. 2021;12:672054. doi:10.3389/fphar.2021.672054
20. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–29. doi:10.1038/75556
21. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi:10.1093/nar/28.1.27
22. Yu G, He Q-Y. Functional similarity analysis of human virus-encoded miRNAs. J Clin Bioinforma. 2011;1(1):15. doi:10.1186/2043-9113-1-15
23. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
24. Tang F, Lu Z, Wang J, et al. Competitive endogenous RNA (ceRNA) regulation network of lncRNAs, miRNAs, and mRNAs in Wilms tumour. BMC Med Genomics. 2019;12(1):194. doi:10.1186/s12920-019-0644-y
25. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi:10.1093/nar/gky1131
26. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
27. Bader GD, Hogue CWV. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2. doi:10.1186/1471-2105-4-2
28. Zhu N, Hou J, Wu Y, et al. Identification of key genes in rheumatoid arthritis and osteoarthritis based on bioinformatics analysis. Medicine. 2018;97(22):e10997. doi:10.1097/MD.0000000000010997
29. Li L, Lei Q, Zhang S, Kong L, Qin B. Screening and identification of key biomarkers in hepatocellular carcinoma: evidence from bioinformatic analysis. Oncol Rep. 2017;38(5):2607–2618. doi:10.3892/or.2017.5946
30. Zhao X, Zhang L, Wang J, et al. Identification of key biomarkers and immune infiltration in systemic lupus erythematosus by integrated bioinformatics analysis. J Transl Med. 2021;19(1):35. doi:10.1186/s12967-020-02698-x
31. Wang C, Tan S, Liu WR, et al. RNA-Seq profiling of circular RNA in human lung adenocarcinoma and squamous cell carcinoma. Mol Cancer. 2019;18(1):134. doi:10.1186/s12943-019-1061-8
32. Li S, Chen L, Xu C, et al. Expression profile and bioinformatics analysis of circular RNAs in acute ischemic stroke in a South Chinese Han population. Sci Rep. 2020;10(1):10138. doi:10.1038/s41598-020-66990-y
33. Subramanian A, Tamayo P, Mootha VK. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
34. Yi XH, Zhang B, Fu YR, Yi ZJ. STAT1 and its related molecules as potential biomarkers in Mycobacterium tuberculosis infection. J Cell Mol Med. 2020;24(5):2866–2878. doi:10.1111/jcmm.14856
35. Peng S, Chen M, Yin M, Feng H. Identifying the potential therapeutic targets for atopic dermatitis through the immune infiltration analysis and construction of a ceRNA network. Clin Cosmet Investig Dermatol. 2021;14:437–453. doi:10.2147/CCID.S310426
36. Jiang J, Bi Y, Liu XP, et al. To construct a ceRNA regulatory network as prognostic biomarkers for bladder cancer. J Cell Mol Med. 2020;24(9):5375–5386. doi:10.1111/jcmm.15193
37. Aletaha D, Neogi T, Silman AJ, et al. Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–2581. doi:10.1002/art.27584
38. Du Y, Wang Q, Tian N, Lu M, Zhang XL, Dai SM. Knockdown of nrf2 exacerbates TNF-alpha-induced proliferation and invasion of rheumatoid arthritis fibroblast-like synoviocytes through activating JNK pathway. J Immunol Res. 2020;2020:6670464. doi:10.1155/2020/6670464
39. Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–883. doi:10.1093/bioinformatics/bts034
40. Lin X, Li H, Yang T, et al. Transcriptomics analysis of lens from patients with posterior subcapsular congenital cataract. Genes. 2021;12(12):1904. doi:10.3390/genes12121904
41. Brown AK, Quinn MA, Karim Z, et al. Presence of significant synovitis in rheumatoid arthritis patients with disease-modifying antirheumatic drug-induced clinical remission: evidence from an imaging study may explain structural progression. Arthritis Rheum. 2006;54(12):3761–3773. doi:10.1002/art.22190
42. Croft AP, Naylor AJ, Marshall JL, et al. Rheumatoid synovial fibroblasts differentiate into distinct subsets in the presence of cytokines and cartilage. Arthritis Res Ther. 2016;18(1):270. doi:10.1186/s13075-016-1156-1
43. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33. doi:10.1038/nrrheum.2012.190
44. Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16(6):316–333. doi:10.1038/s41584-020-0413-5
45. Gui H, Liu X, Wang ZW, He DY, Su DF, Dai SM. Expression of cannabinoid receptor 2 and its inhibitory effects on synovial fibroblasts in rheumatoid arthritis. Rheumatology. 2014;53(5):802–809. doi:10.1093/rheumatology/ket447
46. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. doi:10.1056/NEJMra1004965
47. Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov. 2012;11(3):234–250. doi:10.1038/nrd3669
48. Lefevre S, Knedla A, Tennie C, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15(12):1414–1420. doi:10.1038/nm.2050
49. Altorki T, Muller W, Brass A, Cruickshank S. The role of beta2 integrin in dendritic cell migration during infection. BMC Immunol. 2021;22(1):2. doi:10.1186/s12865-020-00394-5
50. Madkaikar M, Italia K, Gupta M, et al. Leukocyte adhesion deficiency-I with a novel intronic mutation presenting with pyoderma gangrenosum- like lesions. J Clin Immunol. 2015;35(4):431–434. doi:10.1007/s10875-015-0155-3
51. Vallerand IA, Patten SB, Barnabe C. Depression and the risk of rheumatoid arthritis. Curr Opin Rheumatol. 2019;31(3):279–284. doi:10.1097/BOR.0000000000000597
52. Dmitrzak-Weglarz M, Szczepankiewicz A, Rybakowski J, et al. Transcriptomic profiling as biological markers of depression - A pilot study in unipolar and bipolar women. World J Biol Psychiatry. 2021;22(10):744–756. doi:10.1080/15622975.2021.1907715
53. Bruce TO. Comorbid Depression in Rheumatoid Arthritis: pathophysiology and Clinical Implications. Curr Psychiatry Rep. 2008;10(3):258–264. doi:10.1007/s11920-008-0042-1
54. Wang L, Su K, Wu H, Li J, Song D. LncRNA SNHG3 regulates laryngeal carcinoma proliferation and migration by modulating the miR-384/WEE1 axis. Life Sci. 2019;232:116597. doi:10.1016/j.lfs.2019.116597
55. Dai G, Huang C, Yang J, et al. LncRNA SNHG3 promotes bladder cancer proliferation and metastasis through miR-515-5p/GINS2 axis. J Cell Mol Med. 2020;24(16):9231–9243. doi:10.1111/jcmm.15564
56. Cao Y, Pan L, Zhang X, Guo W, Huang D. LncRNA SNHG3 promotes autophagy-induced neuronal cell apoptosis by acting as a ceRNA for miR-485 to up-regulate ATG7 expression. Metab Brain Dis. 2020;35(8):1361–1369. doi:10.1007/s11011-020-00607-1
57. Huang W, Tian Y, Dong S, et al. The long non-coding RNA SNHG3 functions as a competing endogenous RNA to promote malignant development of colorectal cancer. Oncol Rep. 2017;38(3):1402–1410. doi:10.3892/or.2017.5837
58. Zhu S, Ye Y, Shi Y, et al. Sonic hedgehog regulates proliferation, migration and invasion of synoviocytes in rheumatoid arthritis via JNK signaling. Front Immunol. 2020;11:1300. doi:10.3389/fimmu.2020.01300
59. Heinicke F, Zhong X, Flam ST, et al. MicroRNA expression differences in blood-derived CD19+ B cells of methotrexate treated rheumatoid arthritis patients. Front Immunol. 2021;12:663736. doi:10.3389/fimmu.2021.663736
60. Wang ZQ, Xiu DH, Jiang JL, Liu GF. Long non-coding RNA XIST binding to let-7c-5p contributes to rheumatoid arthritis through its effects on proliferation and differentiation of osteoblasts via regulation of STAT3. J Clin Lab Anal. 2020;34(11):e23496. doi:10.1002/jcla.23496
61. Liu W, Song J, Feng X, Yang H, Zhong W. LncRNA XIST is involved in rheumatoid arthritis fibroblast-like synoviocytes by sponging miR-126-3p via the NF-kappaB pathway. Autoimmunity. 2021;54(6):326–335. doi:10.1080/08916934.2021.1937608
62. Sun Y, Sun X, Liu Z, Wang X, Li Y. MiR-338-5p suppresses rheumatoid arthritis synovial fibroblast proliferation and invasion by targeting ADAMTS-9. Clin Exp Rheumatol. 2018;36(2):195–202.
63. Yang Y, Wang Y, Liang Q, Yao L, Gu S, Bai X. MiR-338-5p promotes inflammatory response of fibroblast-like synoviocytes in rheumatoid arthritis via targeting SPRY1. J Cell Biochem. 2017;118(8):2295–2301. doi:10.1002/jcb.25883
64. Guo T, Ding H, Jiang H, Bao N, Zhou L, Zhao J. miR-338-5p regulates the viability, proliferation, apoptosis and migration of rheumatoid arthritis fibroblast-like synoviocytes by targeting NFAT5. Cell Physiol Biochem. 2018;49(3):899–910. doi:10.1159/000493222
65. Meng F. Hsa-miR-5581-3p and Hsa-miR-542-3p target the F8 gene in hemophilia A without F8 mutations. Mediterr J Hematol Infect Dis. 2021;13(1):e2021041. doi:10.4084/MJHID.2021.041
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.