Back to Journals » Cancer Management and Research » Volume 17

Bioinformatics Analysis and Experimental Validation of Lactylation Related Genes in Lung Adenocarcinoma
Authors Gao L, Zhang Y ![]() , Wu F
, Wu F ![]() , Liu B, Xie X
, Liu B, Xie X
Received 20 May 2025
Accepted for publication 8 September 2025
Published 12 September 2025 Volume 2025:17 Pages 1947—1960
DOI https://doi.org/10.2147/CMAR.S533289
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bilikere Dwarakanath
Li Gao,1,* Yadi Zhang,2,3,* Fengrui Wu,2,3 Bin Liu,1 Xin Xie2,3
1Department of Respiratory and Critical Care Medicine, Fuyang People’s Hospital, Fuyang, Anhui, 236012, People’s Republic of China; 2Anhui Province Key Laboratory of Pollution Damage and Biological Control for Huaihe River Basin, Fuyang Normal University, Fuyang, Anhui, 236041, People’s Republic of China; 3Anhui Province Key Laboratory of Embryo Development and Reproductive Regulation, Fuyang Normal University, Fuyang, Anhui, 236041, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Bin Liu, Department of Respiratory and Critical Care Medicine, Fuyang People’s Hospital, 501 Sanqing Road, Fuyang, Anhui, 236012, People’s Republic of China, Email [email protected] Xin Xie, Anhui Province Key Laboratory of Pollution Damage and Biological Control for Huaihe River Basin, Fuyang Normal University, Fuyang Normal University, 100 Qinghe West Road, Fuyang, Anhui, 236041, People’s Republic of China, Email [email protected]
Purpose: Lactylation, a novel post-translational modification, is dysregulated in various tumors and influences lung cancer progression. However, its role in lung adenocarcinoma (LUAD) remains unclear. Based on multi-omics analysis results, this study investigated lactylation levels in LUAD tissues and explored the dual research positioning of lactylation as a prognostic marker and therapeutic target.
Methods: Lactylation levels in LUAD tissue microarrays were assessed using immunohistochemistry and immunofluorescence. Western blot analysis validated these findings. Differential expression analysis of lactylation-related genes was conducted using The Cancer Genome Atlas (TCGA, n=365), based on |log2 fold-change (FC)|≥ 2. KEGG pathway analysis identified key biological pathways, and COX regression analysis pinpointed prognostic genes. Single-cell RNA sequencing data from the GEO database validated these genes, with mitochondrial gene threshold < 20%.
Results: Lactylation levels were significantly elevated in LUAD tissues compared to adjacent non-cancerous tissues, as shown by immunohistochemistry and confirmed by Western blot analysis. Differential analysis identified 17 lactylation-related genes enriched in pathways such as AMPK signaling and cellular senescence. COX regression analysis identified five risk genes: KIF2C, MKI67, HMGA1, PFKP, and CCNA2. Validation with single-cell RNA sequencing data revealed high expression levels of these genes in LUAD tissues and the LUAD cell line H1299. Functional validation revealed that the 5 genes panel significantly regulates global lactylation modification in vitro.
Conclusion: LUAD tissues exhibit elevated lactylation levels, suggesting their potential as prognostic biomarkers. The identified genes—KIF2C, MKI67, HMGA1, PFKP, and CCNA2—are highly expressed in cancerous tissues and correlate with LUAD prognosis. These findings highlight their value as tumor biomarkers and therapeutic targets, offering new opportunities for targeted LUAD treatments.
Keywords: lactylation, LUAD, single-cell sequencing, prognostic signature
Introduction
Lung cancer is among the leading causes of cancer-related mortality worldwide, with non-small cell lung cancer (NSCLC) accounting for approximately 80–85% of cases based on pathological classification.1 According to global cancer statistics, over 2 million new cases of lung cancer are diagnosed annually.2,3 Among NSCLC subtypes, lung adenocarcinoma (LUAD) represents approximately 55–60%.4 Early-stage LUAD is often asymptomatic, leading to late diagnoses and poor prognoses. Common clinical symptoms include hemoptysis, chest tightness, and dyspnea. Despite significant advances in the treatment of LUAD due to innovations in surgery, chemotherapy, targeted and immunotherapy, the prognosis of LUAD patients remains poor, with 5-year survival rates below 25%.5 Therefore, identifying novel biomarkers and therapeutic targets is crucial for improving early diagnosis and prognosis in LUAD patients.
Proteins, the functional units of cells, are regulated through genomic and epigenetic mechanisms, as well as post-translational modifications (PTMs), which involve covalent addition of functional groups, proteolytic cleavage of subunits, or protein degradation. Lactic acid is traditionally considered as a metabolic waste product produced during glycolysis in normal cells under hypoxic conditions. In the 1920s, Otto Warburg’s observations revealed that tumor cells exhibit enhanced glucose uptake and prefer glycolysis even in the absence of hypoxia, resulting in increased lactic acid production through aerobic glycolysis, known as the Warburg effect.6 Lactylation, a PTM identified in 2019, involves lysine lactylation of histones in response to hypoxia, is thought to regulate gene transcription through epigenetic mechanisms,7 interferon stimulation, or bacterial invasion. This modification occurs in the promoter regions of specific genes, modulating inflammatory gene expression and facilitating the transition of M1 macrophages to M2 macrophages.7,8
In addition to histones, non-histone proteins also undergo lactylation, which influences the progression of diseases such as inflammation, fibrosis, and tumor progression.9,10 For instance, in the tumor microenvironment, lactate promotes the lactylation of N-myc downstream-regulated gene 3 (NDRG3) under hypoxic conditions, inhibiting proteasome-mediated degradation.11 Yan et al found that hypoxia-induced lactylation of SOX9 enhances stemness, migration, and invasion in NSCLC cells.12 These findings suggest that targeting protein lactylation could serve as a promising therapeutic strategy for cancer treatment.
In this study, we analyzed lactylation levels in LUAD tissues using immunohistochemistry, immunofluorescence, and Western blot techniques. The results showed a strong association between elevated lactylation levels and LUAD progression, establishing lactylation as an independent prognostic factor. Additionally, differentially expressed lactylation-related genes were identified using data from The Cancer Genome Atlas (TCGA) and analyzed through KEGG enrichment analysis, due to the large sample size and comprehensive clinical data of TCGA. Five lactylation-associated risk genes (KIF2C, MKI67, HMGA1, PFKP, and CCNA) were identified using COX regression analysis. These genes were further validated using single-cell RNA sequencing data from the GEO database, due to the abundance of GEO database datasets with strong selectivity and their mRNA expression levels were analyzed in LUAD tissues and corresponding cell lines. The flowchart is shown in Figure 1.
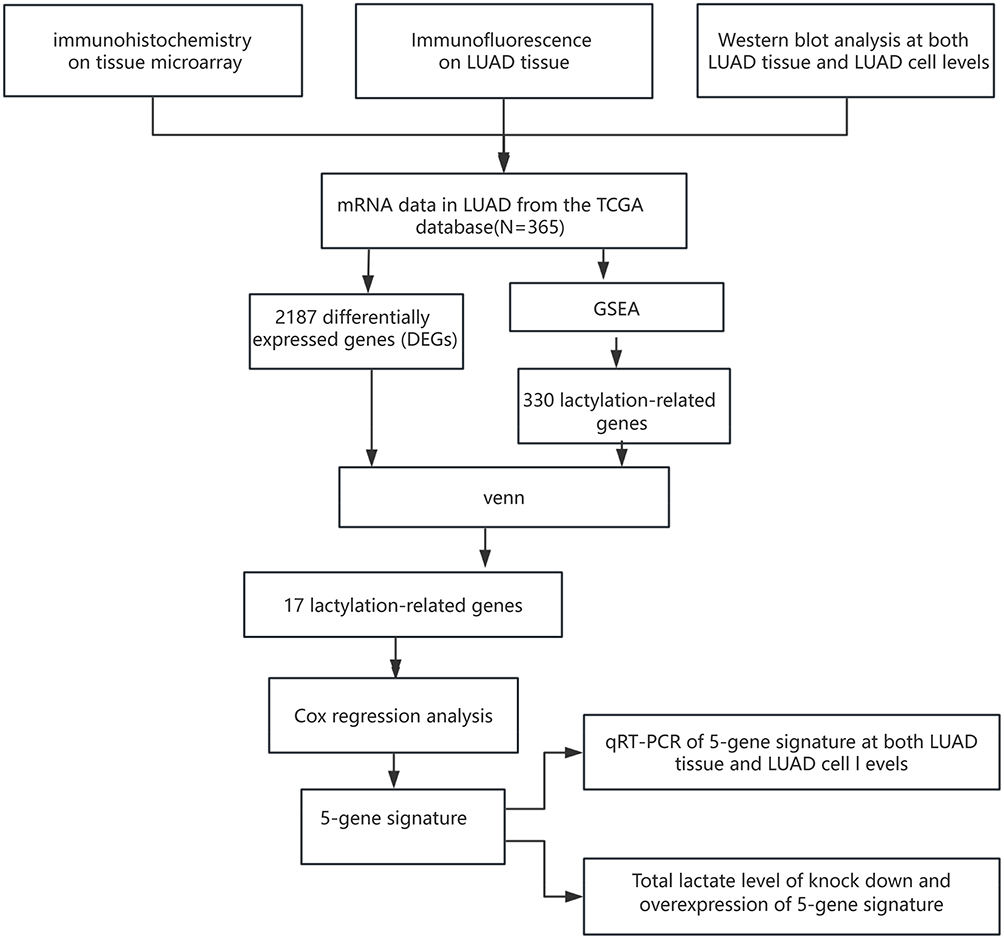 |
Figure 1 The flowchart of this study. Abbreviations: LUAD, lung adenocarcinoma; TCGA, The Cancer Genome Atlas; DEGs, differentially expressed genes; GSEA, Gene Set Enrichment Analysis; RT-qPCR, Real-time quantitative polymerase chain reaction. |
The integration of multi-omics analysis is the innovation of this study, filling the gap in this field. Our findings indicate that LUAD tissues exhibit high levels of lactylation modifications, underscoring the potential of targeting lactylation and its associated genes as viable therapeutic strategies. Furthermore, the five identified risk genes (KIF2C, MKI67, HMGA1, PFKP, and CCNA) are highly expressed in cancerous tissues, serving as tumor biomarkers and potential therapeutic targets. Given the heightened global lactylation levels in adenocarcinoma, targeting lactylation and its related mechanisms emerges as a promising therapeutic, Our research also has shortcomings, such as the lack of systematic studies on lactylation modification in LUAD.
Materials and Methods
Data Collection and Processing
This study included a training cohort of 365 LUAD patients, comprising RNA expression profiles and corresponding clinical data retrieved from TCGA database (https://portal.gdc.cancer.gov/). Single-cell RNA sequencing (scRNA-seq) data for LUAD were retrieved from the GSE143423 dataset (https://www.ncbi.nlm.nih.gov/geo/), which included three lung cancer samples.
Differential Expression Analysis
Differential expression analysis of mRNA in LUAD patients was conducted using the “DESeq2” R package with data from the TCGA database. Differentially expressed genes (DEGs) were identified based on a false discovery rate (FDR) < 0.05 and |log2 fold-change (FC)| ≥ 2. Genes whose expression levels in tumor tissues exceeded fourfold those in adjacent non-cancerous tissues were selected. Heatmaps and volcano plots of DEGs were created using the “pheatmap” R package. A Venn diagram was generated to identify intersecting DEGs with lactylation-associated genes.
Identification of Lactylation-Associated Prognostic Genes
Univariate Cox regression analysis of intersecting DEGs was performed using the “survival” R package. Genes with a significant p-value of < 0.05 were identified as prognostic genes. Forest plots were generated, and LUAD patients were stratified into high- and low-expression groups based on the median expression of each gene. Survival differences between groups were analyzed using the Log rank test, and Kaplan-Meier (K-M) survival curves were plotted.
Protein-Protein Interaction (PPI) Network of Lactylation-Associated Prognostic Genes
A PPI network of prognostic lactylation-associated genes was constructed using the STRING database (https://www.stringdb.org) with a medium confidence score threshold of > 0.4. The network was visualized in Cytoscape software, and gene correlations were assessed with the CytoNCA plugin (v3.10.2). Gradient color mapping was applied to the network graph based on correlation scores to highlight key relationships.
Functional Enrichment Analysis
Gene Ontology (GO) and KEGG enrichment analyses were conducted on lactylation-associated prognostic genes to explore their biological functions and specific mechanisms. A significant threshold of P < 0.05 was used to identify meaningful results.
Survival Analysis of High- and Low-Lactylation Groups
The ssGSEA algorithm was applied to score lactylation-associated prognostic genes for each patient. Based on the median score, patients were categorized into high-lactylation and low-lactylation groups. Survival differences between these groups were analyzed using the Log rank test, and Kaplan-Meier (K-M) survival curves were generated to illustrate the findings.
Single-Cell Validation
The “Seurat” R package was used to analyze scRNA-seq data from the GSE143423 dataset and evaluate the expression of lactylation-associated risk genes across various lung cell populations. Quality control procedures included filtering out cells with fewer than three expressed genes, retaining cells with 200–7000 expressed genes, and excluding cells with mitochondrial gene expression exceeding 20%. After normalization, 12,144 cells were identified. Data integration, identification of highly variable genes (HVGs), and principal component analysis (PCA) were performed. Clustering was performed using the “FindClusters” and “FindNeighbors” functions, and cell annotation was conducted with the “SingleR” Package. The results were visualized using the “ggplot2” R package.
Cell Culture
The human bronchial epithelial cell line BEAS-2B and LUAD cell lines H1299, H1975, PC9, and A549 were obtained from the National Collection of Authenticated Cell Cultures.
BEAS-2B cells were cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum (FBS, Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin-streptomycin (MedChemExpress, Monmouth, NJ, USA). LUAD cell lines H1299, H1975, and PC9 were maintained in RPMI 1640 with 10% FBS and 1% penicillin-streptomycin, while A549 cells were cultured in F-12K medium (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS and 1% penicillin-streptomycin. All cell cultures were maintained at 37°C in humidified atmosphere with 5% CO2.
Plasmids and RNA Interference
The coding sequence of human MKI67 was cloned and inserted into the eukaryotic plasmid vector H6919 pCDNA3.1–3FLAG (Shanghai OBiO Technology Corp., Ltd.). The coding sequences of human CCNA2, KIF2C, PFKP, and HMGA1 were cloned and inserted into the eukaryotic plasmid vector pcDNA3.1(+) (Shanghai GenePharma Co., Ltd.) to generate MKI67, CCNA2, KIF2C, PFKP, and HMGA1 expression plasmids. Subsequently, lung adenocarcinoma cells (A549) were infected using plasmid. For shRNA, the pSLenti-U6-shRNA-CMV- EGFP-F2A-Puro-WPRE vector was used to generate MKI67 knockdown A549 cells. The pGPU6-GFP-Neo vector was used to generate CCNA2, KIF2C, PFKP, and HMGA1 knockdown A549 cells. The sequences of shRNAs were as follows: shMKI67#1: 5′-GCA GCA ATA GAC GGC TAC AAA-3′; shMKI67#2: 5′-GCC GAA GTC AAC ATG ATA TTT-3′; shMKI67#3: 5′-CCA TGA GCA GGA GGC AAT ATT-3′. shCCNA2#1: 5′-AGC TGG CCT GAA TCA TTA ATA-3′; shCCNA2#2: 5′-CCT TAA GGA TCT TCC TGT AAA-3′; shCCNA2#3: 5′-GAA GTA GGA GAA GAA TAT AAA-3′. shKIF2C#1: 5′-GCA TAA GCT CCT GTG AAT ATA-3′; shKIF2C#2: 5′-AGC AGG CTA GCA GAC AAA TAA-3′; shKIF2C#3: 5′-CCC ACG CGT GCT TCC AAA TTA-3′. shPFKP#1: 5′-ACT TCA TTT ACC AGC TGT ATT-3′; shPFKP#2: 5′-CAG GAA CGG CCA GAT CGA TAA-3; shPFKP#3: 5′-ATC AGA TCC CAA AGA CCA ATT-3′. shHMGA1#1: 5′-AGC GAA GTG CCA ACA CCT AAG-3′; shHMGA1#2: 5′-TGA GTG AGT CGA GCT CGA AGT-3′; shHMGA1#3: 5′-GGC ATC TCG CAG GAG TCC TC-3′. Plasmid DNA was transfected into A549 cells by Lipo8000™ Transfection Reagent (Beyotime, China) according to the manufacturer’s protocol.
Clinical Sample Collection
This study complies with the Declaration of Helsinki. Clinical tissue samples were collected from twenty-eight LUAD patients at Fuyang People’s Hospital. None of the patients had received preoperative treatments. The study was approved by the hospital’s ethics committee (Medical Ethics Review [2024] 103) and informed consent was obtained from all participants. Pathological diagnoses were confirmed by at least two pathologists in accordance with the American Joint Committee on Cancer (AJCC) guidelines.
Western Blot Analysis
The collected 28 pairs of adjacent non-tumor and tumor tissues from lung cancer patients were used for Western blot experiments. Total proteins were extracted from LUAD cell lines and clinical tissue samples using RIPA buffer (Sigma-Aldrich) on ice. Proteins were separated via SDS-PAGE and transferred onto PVDF membranes (Millipore, CAS#: 24937-79-9). The membranes were blocked with 5% non-fat milk for 2 hours at room temperature, followed by overnight incubation at 4°C with a pan-Kla antibody (PTM Bio, Shanghai, China, Cat# PTM-1401, 1:1000). After three washes with TBST (10 minutes each), the membranes were incubated with Goat Anti-Rabbit IgG H&L (HRP) (Abcam, Cambridge, UK, Cat# ab97051, 1:10000) at room temperature for 2 hours and washed again. Protein bands were visualized using an enhanced chemiluminescence (ECL) kit (Beyotime) and visualized with a gel imaging system.
Quantitative Real-Time PCR (qRT-PCR)
The collected 18 pairs of adjacent non-tumor and tumor tissues from lung cancer patients were used for Western blot experiments. Total RNA was extracted from LUAD cells and tissue samples using Trizol reagent (Invitrogen, USA). RNA was reverse-transcribed into complementary DNA (cDNA) using a synthesis kit (Nanjing, Jiangsu Province, China). qRT-PCR analysis was conducted with TB Green Premix Ex Taq II (TaKaRa, Japan) on a Roche LightCycler®480 system, with GAPDH as the internal control. The thermal cycling protocol was as follows: initial denaturation at 95°C for 15 min, followed by 40 cycles of denaturation at 95°C for 10s and annealing/extension at 60°C for 30s. Relative gene expression levels were calculated using the 2−ΔΔCt method. The primer sequences used are listed in Table 1.
 |
Table 1 The Primer Sequences of qRT-PCR |
Immunohistochemical Staining
LUAD and adjacent non-cancerous tissues were embedded in paraffin and sectioned into 4-μm slices. After deparaffinization, sections were incubated overnight at 4°C with a pan-Kla antibody (PTM BIO, PTM-1404, China), followed by a 2-hour incubation with goat anti-rabbit HRP conjugate (Affinity Biosciences, Jiangsu, China, Cat#: S0001, 1:200) at room temperature. Staining was analyzed using a light microscope. LUAD tissue microarrays (HLugA150CS04) were obtained from Shanghai Outdo Biotech Co., Ltd., and the protein level of pan-Kla was evaluated using an Aperio scanner (Leica Biosystems).
Immunofluorescence Staining
The procedures for deparaffinization, rehydration, and antigen retrieval were identical to those in IHC protocol. Tissue sections were incubated overnight at 4°C with a pan-Kla antibody (PTM BIO, PTM-1404, China), followed by a 50-minute incubation with Goat Anti-Rabbit IgG (H+L) Fluor594-conjugated (Affinity Biosciences, Jiangsu, China, Cat#: S0006, 1: 200) in the dark at room temperature. Nuclei were counterstained with DAPI (Beyotime, C1006), and the slices were examined using a confocal microscope.
Statistical Analysis
For normally distributed variables between two groups, statistical significance was assessed using unpaired Student’s t-tests. Mann–Whitney U-tests were used for non-normally distributed variables. Kaplan-Meier survival analysis and Log rank tests were conducted to compare overall survival rates between high-risk and low-risk groups. Receiver operating characteristic (ROC) curves and area under the curve (AUC) values were generated to evaluate the predictive performance of risk scores. All statistical analyses were performed using R software (version 4.3.1), with a significant threshold of P < 0.05.
Results
Lactylation Levels Are Elevated in LUAD and Positively Correlated with Tumor Progression
Previous studies have demonstrated that hyperglycolysis in LUAD leads to increased lactate production, which serves as a key substrate for protein lactylation. Although lactylation has been implicated in tumorigenesis and tumor progression, its specific role in LUAD remains unclear. This study examined lactylation levels in LUAD tissues and their clinical significance.
To assess overall lactylation levels in LUAD, immunohistochemical staining was performed on LUAD tissue microarrays. The results revealed significantly higher lactylation levels in cancerous tissues compared to adjacent non-cancerous tissues (Figure 2A). Compare with adjacent non-cancerous tissues, the lactylation levels in cancerous tissues cytoplasm and nuclear increased 2.46-fold 29.4-fold subsequently (Figure 2B). Similar findings were observed in paired cancerous and adjacent tissues from twenty-eight LUAD patients at Fuyang People’s Hospital, where tumor tissues exhibited consistently elevated lactylation levels (Figure 2C and Supplemental Figure 1). Meanwhile, the lactylation levels were also measured in the alveolar epithelial cell line BEAS-2B and four LUAD cell lines. The results demonstrated significantly higher lactylation levels in the LUAD cell lines H1299 and A549 compared to BEAS-2B (Figure 2D). To assess the prognostic significance of lactylation, the ssGSEA algorithm was applied to score lactylation-related prognostic genes in each patient. Patients were categorized into high- and low-lactylation groups based on median scores, and K-M survival curves and ROC curves were plotted. K-M analysis showed that patients in the high-lactylation group have significantly worse survival outcomes (P < 0.001) (Figure 2E). ROC curve analysis demonstrated that the AUCs for 1, 3, and 5 years exceed 0.6 (Figure 2F), highlighting lactylation as a potential prognostic biomarker for LUAD. Additionally, Immunofluorescence analysis further confirmed these findings, showing significantly higher lactylation levels in LUAD tissues compared to adjacent non-cancerous tissues (Figure 2G). Collectively, these findings indicate that elevated lactylation levels are positively correlated with LUAD progression.
 |
Figure 2 Increased lactylation levels in LUAD are positively correlated with tumor progression. (A) Immunohistochemical staining of tissue microarrays comparing lactylation levels in adjacent non-cancerous tissues (n = 75) and LUAD tissues (n = 75). Scale bars: left panel, 600 μm; right panel, 50 μm. ****P<0.0001 compared with Normal groups; (B) The pan-kla expression score in non-cancerous tissues (n = 75) and LUAD tissues (n = 75). (C) Western blot analysis of lactylation levels in adjacent non-cancerous tissues (N) and cancerous tissues (T). (D) Western blot analysis comparing lactylation levels in the alveolar epithelial cell line, BEAS-2B, and four LUAD cell lines (A549, H1299, H1975, and PC9). (E) K-M survival analysis showing the association between lactylation levels and OS in LUAD patients. (F) Time-dependent ROC curves illustrating the prognostic accuracy of lactylation levels. (G) Immunofluorescence staining visualizing lactylation levels in normal and cancerous tissues. |
Identification of Prognostic Genes Associated with Lactylation in LUAD
Using the LUAD patient cohort from TCGA database, 2187 differentially expressed genes (DEGs) were identified, including 1636 upregulated and 551 downregulated genes (|logFC| ≥ 2, FDR < 0.05) (Figure 3A and B). Heatmaps highlighted the top 100 DEGs between normal and LUAD tissues. Gene set enrichment analysis (GSEA) identified 330 lactylation-related genes (P < 0.001, R > 0.2). A Venn diagram was used to intersect these genes with the 2187 DEGs yielding 17 overlapping genes (Figure 3C). Volcano plots and heatmaps visualized these 17 genes (Figure 3E and F).
 |
Figure 3 Identification of prognostic lactylation-related genes in LUAD. (A) Volcano plot highlighting DEGs between normal tissues and LUAD patients. (B) Heatmap showing the expression patterns of DEGs in normal and LUAD tissues. (C) Venn diagram showing the overlap between lactylation-related genes and DEGs. (D) GO enrichment analysis of lactylation-related genes. (E) Visualization of 17 lactylation-related genes on the volcano plot. (F) Heatmap displaying the expression of 17 lactylation-related genes. (G) KEGG pathway enrichment analysis of lactylation-related genes. (H) PPI network depicting interactions among the 17 lactylation-related genes. (I) Molecular docking model of CCNA2 interacting with domain of MKI67. (J) Molecular docking model of CCNA2 interacting with domain of KIF2C. |
Functional enrichment analysis of lactylation-associated genes was conducted. GO analysis revealed involvement in biological processes such as fructose-6-phosphate kinase activity, monocarboxylic acid metabolism, fatty acid metabolism, and nucleosome formation. These genes were associated with cellular components such as the cyclin-CDK2 complex, fructose-6-phosphate kinase complex, and presynaptic intermediate filament cytoskeleton, with key biological processes including mitochondrial localization, negative regulation of DNA recombination, and chromatin condensation (Figure 3D). KEGG analysis indicated significant enrichment in pathways such as AMPK signaling, cellular senescence, and the pentose phosphate pathway (Figure 3G).
A PPI network for the lactylation-related genes was constructed using the STRING database. Gene correlations were analyzed using the CytoNCA plugin in Cytoscape (v3.10.2), with genes of higher importance visualized in deeper colors (Figure 3H). Meanwhile, using PRISM tool exhibited the interacting structural motifs of CCNA2 with MKI67 and CCNA2 with KIF2C, suggesting a protein-protein interaction interface (Figure 3I and J).
Correlation Between Expression of Lactylation-Related Genes and Prognosis
Univariate Cox regression analysis revealed that six of the 17 lactylation-related genes are significantly associated with LUAD survival outcomes (Figure 4A). Five genes (KIF2C, MKI67, CCNA2, HMGA1, and PFKP) were negatively correlated with survival, while PRAM1 was positively correlated. Forest plots depicted these associations (Figure 4B).
 |
Figure 4 Correlation between lactylation-related gene expression and prognosis. (A) Correlation analysis of the 17 DEGs with LUAD prognosis. (B) Forest plot illustrating the association between six lactylation-related genes and prognosis. (C) qRT-PCR analysis of the expression of lactylation risk genes (KIF2C, MKI67, HMGA1, PFKP, and CCNA2) in adjacent non-cancerous tissues (N) and cancerous tissues (T). (D) qRT-PCR analysis comparing the expression of lactylation risk genes (KIF2C, MKI67, HMGA1, PFKP, and CCNA2) in the alveolar epithelial cell line, BEAS-2B, and LUAD cell lines (A549, H1299, PC9, and H1975). (E) The lactylation levels of A549 cell were measured after overexpression KIF2C, MKI67, HMGA1, PFKP, and CCNA2. (F) The lactylation levels of A549 cell were measured after knockdown KIF2C, MKI67, HMGA1, PFKP, and CCNA2. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. |
To validate these findings, mRNA expression levels of KIF2C, MKI67, HMGA1, PFKP, and CCNA2 were measured in cancerous and adjacent non-cancerous tissues using qRT-PCR. All five genes were significantly overexpressed in cancerous tissues (Figure 4C). Meanwhile, RNA expression in the alveolar epithelial cell line BEAS-2B and LUAD cell lines revealed elevated RNA expression levels of these genes in the LUAD cell line H1299 (Figure 4D). Additionally, we measured lactylation levels following the overexpression or knockdown of KIF2C, MKI67, HMGA1, PFKP, and CCNA2. The results demonstrated that individual overexpression of these five lactylation-related genes significantly elevated global protein lactylation in A549 cells (Figure 4E), whereas their knockdown led to a reduction in lactylation levels (Figure 4F). These results suggest that these five genes are associated with poor prognosis in LUAD.
Single-Cell Transcriptomic Analysis of Lactylation-Associated Genes
To examine the expression patterns of lactylation risk genes across different cell types in LUAD tissues, single-cell transcriptomic analysis was conducted using GSE143423 dataset. The cells were clustered into 12 groups (Figure 5A), and t-SNE visualization identified seven distinct cell clusters (Figure 5B). The distribution of the five prognostic genes (KIF2C, MKI67, HMGA1, PFKP, and CCNA2) was analyzed within these clusters (Figure 5C–H). CCNA2 and MKI67 were predominantly expressed in epithelial cells and monocytes. HMGA1 was mainly expressed in epithelial cells, macrophages, and monocytes, while PFKP was enriched in monocytes, macrophages, and epithelial cells. KIF2C displayed low expression across all cell clusters.
 |
Figure 5 Single-cell transcriptome analysis of lactylation-related genes. (A and B) Single-cell transcriptome analysis categorizing LUAD tissues into seven distinct cell clusters. (C–H) Expression patterns of lactylation risk genes (KIF2C, MKI67, HMGA1, PFKP, and CCNA2) across the seven cell clusters identified in LUAD tissues. |
Discussion
LUAD often presents without obvious symptoms in its early stages, leading to late diagnoses in most patients. Although advances in immunotherapy and targeted therapies have provided new hope for improving outcomes in advanced LUAD,13,14 the five-year survival rate remains rather low. In 2022, lung cancer accounted for 1.817 million deaths globally, ranking as the leading cause of cancer-related mortality (GLOBOCAN data). Lactate, a key metabolite of glycolysis, not only supports cell growth but also plays a regulatory role in biological functions by serving as a donor of lactyl groups for covalent modification of lysine residues on proteins. This process, known as lactylation, modulates protein activity and function, influencing disease onset and progression. For example, Wang et al reported a positive correlation between total lysine lactylation (Kla) levels and the progression of anaplastic thyroid carcinoma (ATC). Elevated Kla levels, driven by lactate from aerobic glycolysis, activated the transcription of genes essential for ATC proliferation total Kla levels, such as histone H4K12 lactylation.15 Similarly, Yu et al demonstrated that lactate accumulation in uveal melanoma results in elevated overall Kla levels compared to normal melanocyte tissues. In this context, histone lactylation promoted tumor progression by regulating YTHDF2 transcription.8 Jin et al found that lactylated CCNE2 promotes hepatocellular carcinoma (HCC) cell growth.16 Yang et al reported higher Kla levels in gastric cancer tissues than in adjacent non-cancerous tissues, with elevated Kla levels correlating with poor prognosis.17 Yang et al linked histone lactylation to unfavorable outcomes in patients with clear cell renal cell carcinoma (ccRCC).18 Despite these findings, the specific relationship between lactylation and LUAD pathogenesis and prognosis remains poorly understood.
In this study, we first evaluated the clinical significance of overall lactylation levels in LUAD. Compared with adjacent non-cancerous tissues, LUAD tumor tissues exhibited elevated overall lactylation levels, a trend that was observed in LUAD cell lines compared with normal epithelial cells (BEAS-2B). Survival analysis using Kaplan-Meier and ROC curves revealed that patients in the high-lactylation group have significantly poor overall survival, establishing lactylation as an independent prognostic factor negatively correlated with LUAD outcomes. Transcriptomic analysis identified five lactylation risk genes (KIF2C, MKI67, HMGA1, PFKP, and CCNA2) that are differentially expressed in LUAD. The mRNA expression levels of these genes were consistently overexpressed in LUAD tissues and cell lines. Single-cell transcriptomic analysis further showed that these five risk genes are predominately expressed in epithelial cell clusters within LUAD tissues, with MKI67, CCNA2, HMGA1, and PFKP being particularly prominent.
Previous studies have investigated the roles of these five lactylation risk genes in various cancers. KIF2C promotes cervical cancer growth by inhibiting the p53 signaling pathway and activating the mTORC1 pathway.19 It also promotes prostate cancer progression via p65 regulation20 and has been identified as a key hub gene in lung cancer progression,21,22 with high expression levels correlating with reduced overall survival (OS). Functionally, KIF2C enhances proliferation, migration, and invasion while reducing apoptosis in NSCLC cell lines.23 MKI67, which encodes the Ki-67 protein, is active during the G1, S, G2, and M phases of the cell cycle.24 It is widely recognized as a primary marker of cell proliferation25,26 and a hallmark of tumorigenesis. HMGA1 has been implicated in several tumors, where it is associated with poor prognosis. For instance, ST8SIA6-AS1 promotes hepatocellular carcinoma progression by targeting the miR-142-3p/HMGA1 axis,27 while the long coding RNA MYU promotes ovarian cancer cell proliferation by inhibiting miR-6827-5p and upregulating HMGA1.28 In lung cancer, high HMGA1 expression is associated with poor outcomes in lung cancer29 and is linked to glycolysis regulation in LUAD via the PI3K/AKT pathway.30 PFKP, a glycolytic enzyme, plays a key role in cancer cell metabolism. Its high expression in primary LUAD tissues correlates with poor prognosis and cancer cell proliferation.31 Beyond its glycolytic role, PFKP enhances ATP and NADPH production through PFKP/AMPK/ACC2-mediated long-chain fatty acid oxidation, promoting cancer cell survival.32 Cyclin A2 (CCNA2), a member of the cyclin family, regulates the G1/S and G2/M transitions by binding and activating cyclin-dependent kinase 1 (CDK1) and cyclin-dependent kinase 2 (CDK2).33 High CCNA2 expression is associated with poor prognosis in LUAD.34,35 However, further research is needed to clarify how these genes influence lactylation-mediated LUAD progression.
Conclusion
This study demonstrates a negative correlation between overall lactylation levels in cancerous tissues and LUAD prognosis, suggesting that overall lactylation could serve as a valuable prognostic indicator. Moreover, five lactylation-associated risk genes (KIF2C, MKI67, HMGA1, PFKP, and CCNA2) were identified as key drivers for LUAD progression. These genes were overexpressed in both LUAD cell lines and tissues and were strongly negatively correlated with poor patient outcomes. Single-cell transcriptomic analysis demonstrated the expression patterns of these five genes across seven specific cell clusters in LUAD tissues. CCNA2 and MKI67 were predominately expressed in epithelial cells and monocytes, while HMGA1 was enriched in epithelial cells, macrophages, and monocytes. PFKP exhibited high expression in monocytes, macrophages, and epithelial cells, whereas KIF2C showed low expression across all cell clusters. These findings highlight the potential of lactylation-associated genes as tumor biomarkers and therapeutic targets, future research should focus on mechanism investigations and functional validation experiments, as well asexploring the potential of targeting LRGs in clinical trials, such as CCNA2, MKI67, etc.
Data Sharing Statement
Data is provided within the manuscript or supplementary information files.
Acknowledgments
The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by grants from Bengbu Medical University Science and Technology Project (2023byzd223).
Disclosure
The authors declare no competing interests.
References
1. Cao W, Chen HD, Yu YW, Li N, Chen WQ. Changing profiles of cancer burden worldwide and in China: a secondary analysis of the global cancer statistics 2020. Chin Med J. 2021;134(7):783–791. doi:10.1097/Cm9.0000000000001474
2. Pine SR. Rethinking gamma-secretase inhibitors for treatment of non-small-cell lung cancer: is notch the target? Clin Cancer Res. 2018;24(24):6136–6141. doi:10.1158/1078-0432.Ccr-18-1635
3. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
4. Wang N, Chen WQ, Zhu WX, Xing XM, Lu AP, Yang L. Incidence trends and pathological characteristics of lung cancer in urban Beijing during period of 1998–2007. Zhonghua Yu Fang Yi Xue Za Zhi. 2011;45(3):249–254.
5. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12–49. doi:10.3322/caac.21820
6. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–337. doi:10.1038/nrc3038
7. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. doi:10.1038/s41586-019-1678-1
8. Yu J, Chai P, Xie M, et al. Histone lactylation drives oncogenesis by facilitating m(6)A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22(1):85. doi:10.1186/s13059-021-02308-z
9. Zhang Y, Zhai Z, Duan J, et al. Lactate: the mediator of metabolism and immunosuppression. Front Endocrinol. 2022;13:901495. doi:10.3389/fendo.2022.901495
10. Zhou J, Xu W, Wu Y, et al. GPR37 promotes colorectal cancer liver metastases by enhancing the glycolysis and histone lactylation via Hippo pathway. Oncogene. 2023;42(45):3319–3330. doi:10.1038/s41388-023-02841-0
11. Lee DC, Sohn HA, Park ZY, et al. A lactate-induced response to hypoxia. Cell. 2015;161(3):595–609. doi:10.1016/j.cell.2015.03.011
12. Yan F, Teng Y, Li X, et al. Hypoxia promotes non-small cell lung cancer cell stemness, migration, and invasion via promoting glycolysis by lactylation of SOX9. Cancer Biol Ther. 2024;25(1):2304161. doi:10.1080/15384047.2024.2304161
13. Lee JB, Kim HR, Ha SJ. Immune checkpoint inhibitors in 10 years: contribution of basic research and clinical application in cancer immunotherapy. Immune Netw. 2022;22(1):e2. doi:10.4110/in.2022.22.e2
14. de Mello RA, Neves NM, Tadokoro H, Amaral GA, Castelo-Branco P, Zia VAA. New target therapies in advanced non-small cell lung cancer: a review of the literature and future perspectives. J Clin Med. 2020;9(11):3543. doi:10.3390/jcm9113543
15. Wang X, Ying T, Yuan J, et al. BRAFV600E restructures cellular lactylation to promote anaplastic thyroid cancer proliferation. Endocr Relat Cancer. 2023;30(8). doi:10.1530/erc-22-0344
16. Jin J, Bai L, Wang D, et al. SIRT3-dependent delactylation of cyclin E2 prevents hepatocellular carcinoma growth. EMBO Rep. 2023;24(5):e56052. doi:10.15252/embr.202256052
17. Yang D, Yin J, Shan L, Yi X, Zhang W, Ding Y. Identification of lysine-lactylated substrates in gastric cancer cells. iScience. 2022;25(7):104630. doi:10.1016/j.isci.2022.104630
18. Yang J, Luo L, Zhao C, et al. A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRβ signaling drives clear cell renal cell carcinoma progression. Int J Biol Sci. 2022;18(8):3470–3483. doi:10.7150/ijbs.73398
19. Yang J, Wu Z, Yang L, et al. Characterization of kinesin family member 2C as a proto-oncogene in cervical cancer. Front Pharmacol. 2021;12:785981. doi:10.3389/fphar.2021.785981
20. Liu X, Lin Y, Long W, et al. The kinesin-14 family motor protein KIFC2 promotes prostate cancer progression by regulating p65. J Biol Chem. 2023;299(11):105253. doi:10.1016/j.jbc.2023.105253
21. Abdel-Maksoud MA, Hassan F, Mubarik U, et al. An in-silico approach leads to explore six genes as a molecular signatures of lung adenocarcinoma. Am J Cancer Res. 2023;13(3):727–757.
22. Chen B, Xie X, Lan F, Liu W. Identification of prognostic markers by weighted gene co-expression network analysis in non-small cell lung cancer. Bioengineered. 2021;12(1):4924–4935. doi:10.1080/21655979.2021.1960764
23. Guo J, Zhang W, Sun L, et al. KIF2C accelerates the development of non-small cell lung cancer and is suppressed by miR-186-3p via the AKT-GSK3β-β-catenin pathway. Sci Rep. 2023;13(1):7288. doi:10.1038/s41598-023-30073-5
24. Gerlach C, Sakkab DY, Scholzen T, Dassler R, Alison MR, Gerdes J. Ki-67 expression during rat liver regeneration after partial hepatectomy. Hepatology. 1997;26(3):573–578. doi:10.1002/hep.510260307
25. Shirendeb U, Hishikawa Y, Moriyama S, et al. Human papillomavirus infection and its possible correlation with p63 expression in cervical cancer in Japan, Mongolia, and Myanmar. Acta Histochem Cytochem. 2009;42(6):181–190. doi:10.1267/ahc.09030
26. Hooghe B, Hulpiau P, van Roy F, De Bleser P. ConTra: a promoter alignment analysis tool for identification of transcription factor binding sites across species. Nucleic Acids Res. 2008;36(Web Server issue):W128–32. doi:10.1093/nar/gkn195
27. Feng T, Yao Y, Luo L, et al. ST8SIA6-AS1 contributes to hepatocellular carcinoma progression by targeting miR-142-3p/HMGA1 axis. Sci Rep. 2023;13(1):650. doi:10.1038/s41598-022-26643-8
28. Wang S, Zheng Q, Wang J, Chen S, Chen L. Long non-coding RNA MYU promotes ovarian cancer cell proliferation by sponging miR-6827-5p and upregulating HMGA1. Pathol Oncol Res. 2023;29:1610870. doi:10.3389/pore.2023.1610870
29. Saed L, Jeleń A, Mirowski M, Sałagacka-Kubiak A. Prognostic significance of HMGA1 expression in lung cancer based on bioinformatics analysis. Int J Mol Sci. 2022;23(13):6933. doi:10.3390/ijms23136933
30. Ma Y, Ma X, Du B, Li X, Li Y. HMGA1 is a prognostic biomarker and correlated with glycolysis in lung adenocarcinoma. J Cancer. 2024;15(10):2913–2927. doi:10.7150/jca.89056
31. Shen J, Jin Z, Lv H, et al. PFKP is highly expressed in lung cancer and regulates glucose metabolism. Cell Oncol. 2020;43(4):617–629. doi:10.1007/s13402-020-00508-6
32. Chen J, Zou L, Lu G, et al. PFKP alleviates glucose starvation-induced metabolic stress in lung cancer cells via AMPK-ACC2 dependent fatty acid oxidation. Cell Discov. 2022;8(1):52. doi:10.1038/s41421-022-00406-1
33. Baumann K. Genome stability: cyclin’ on mRNA. Nat Rev Mol Cell Biol. 2016;17(11):676–677. doi:10.1038/nrm.2016.142
34. Yang X, Zhou Y, Ge H, Tian Z, Li P, Zhao X. Identification of a transcription factor‑cyclin family genes network in lung adenocarcinoma through bioinformatics analysis and validation through RT‑qPCR. Exp Ther Med. 2023;25(1):63. doi:10.3892/etm.2022.11762
35. Chen S, Zhao Z, Wang X, Zhang Q, Lyu L, Tang B. The predictive competing endogenous RNA regulatory networks and potential prognostic and immunological roles of cyclin A2 in pan-cancer analysis. Front Mol Biosci. 2022;9:809509.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.