Back to Journals » International Journal of Nanomedicine » Volume 12
Bioglass implant-coating interactions in synthetic physiological fluids with varying degrees of biomimicry
Authors Popa AC, Stan GE, Husanu MA, Mercioniu I, Santos LF, Fernandes HR, Ferreira JMF ![]()
Received 26 September 2016
Accepted for publication 3 December 2016
Published 21 January 2017 Volume 2017:12 Pages 683—707
DOI https://doi.org/10.2147/IJN.S123236
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
AC Popa,1,2 GE Stan,1 MA Husanu,1 I Mercioniu,1 LF Santos,3 HR Fernandes,4 JMF Ferreira4
1National Institute of Materials Physics, Măgurele, 2Army Centre for Medical Research, Bucharest, Romania; 3Centro de Química Estrutural, Instituto Superior Técnico (CQE-IST), University of Lisbon, Lisbon, 4Department of Materials and Ceramics Engineering, Centre for Research in Ceramics and Composite Materials (CICECO), University of Aveiro, Aveiro, Portugal
Abstract: Synthetic physiological fluids are currently used as a first in vitro bioactivity assessment for bone grafts. Our understanding about the interactions taking place at the fluid–implant interface has evolved remarkably during the last decade, and does not comply with the traditional International Organization for Standardization/final draft International Standard 23317 protocol in purely inorganic simulated body fluid. The advances in our knowledge point to the need of a true paradigm shift toward testing physiological fluids with enhanced biomimicry and a better understanding of the materials’ structure-dissolution behavior. This will contribute to “upgrade” our vision of entire cascades of events taking place at the implant surfaces upon immersion in the testing media or after implantation. Starting from an osteoinductive bioglass composition with the ability to alleviate the oxidative stress, thin bioglass films with different degrees of polymerization were deposited onto titanium substrates. Their biomineralization activity in simulated body fluid and in a series of new inorganic–organic media with increasing biomimicry that more closely simulated the human intercellular environment was compared. A comprehensive range of advanced characterization tools (scanning electron microscopy; grazing-incidence X-ray diffraction; Fourier-transform infrared, micro-Raman, energy-dispersive, X-ray photoelectron, and surface-enhanced laser desorption/ionization time-of-flight mass spectroscopies; and cytocompatibility assays using mesenchymal stem cells) were used. The information gathered is very useful to biologists, biophysicists, clinicians, and material scientists with special interest in teaching and research. By combining all the analyses, we propose herein a step forward toward establishing an improved unified protocol for testing the bioactivity of implant materials.
Keywords: biomaterials, bioglass, in vitro biomimetic assays, proteins
Introduction
The ability of certain silica-based glass formulations to bond hard and soft tissues was discovered in the late sixties by Hench et al.1 Since then, new glass formulations have been proposed to expand the functionalities of bioactive glass (BG), including nanostructured implant coatings synthesized by chemical and physical methods, low-dimensional objects for drug delivery, or regenerative bone scaffolds fabricated by additive manufacturing, robocasting, or 3-D printing.2–8
Currently, the bioactivity of BGs is routinely assessed through their immersion in simulated body fluid (SBF) solution,9 a simplistic method that does not account for the complex biological processes that take place after implantation. The synthetic SBF solution introduced to the scientific community in 1990 by Kokubo et al reproduces only the inorganic composition of physiological media (ie, blood plasma).9 The remarkable simplicity of both the chemical preparation of SBF and the in vitro testing protocol were quickly perceived by the scientific biomaterial community. Meanwhile, several modifications of the base formula were proposed by the Oyane et al,10,11 as detailed in a summary article.12 Such initiatives culminated in an internationally standardized in vitro assay dating from 2007: International Organization for Standardization (ISO)/final draft International Standard (FDIS) 23317 (last revised in 2014).13 Its predicting ability was soon after questioned by Bohner and Lemaitre.14 Their Scopus (www.scopus.com) search using the keywords “bioactivity” and “simulated body fluid” (in all fields) led to 1,975 hits in 2009, and a similar search in 2016 led to 7,475 hits (6,123 since 2007)! This means that in spite of the criticisms raised, this simple in vitro SBF testing method had become very popular and classically adopted for assessing the bioactivity/bioreactivity of various materials (eg, glasses, apatites).
The ISO/FDIS 2331713 protocol proposes immersing testing samples (discs or squares of diameter/size of 10±2 mm and thickness of 2±1 mm) in SBF at pH 7.4 and 36.5°C for different periods during 4 weeks, without any specific indication about the atmosphere. Although disc or rectangular plate samples are preferred for their easiness in setting the recommended Vs:Sa ratio at 100 mm (Vs is the volume of SBF solution [mm3] and Sa the apparent surface area of the specimen [mm2]), specimens of any configuration and size can be used. However, can the ability of a material to induce the formation of a hydroxyapatite (HA) surface layer when immersed in SBF solution be viewed as an obvious predictor of bioactivity and its capacity to induce bone-tissue rehabilitation? As SBF is just a collection of electrolytes, SBF testing has some obvious shortcomings:
- As to its difference from true intercellular fluid, SBF does not contain a series of important organic substances, eg, glucose, amino acids, oligopeptides, proteins, vitamins, lactic acids, urea, creatinine, metabolic products, lipids, and hormones.
- An inability to describe the complex processes (ie, bleeding/clotting, inflammation, and tissue repair) taking place at the implantation site.
- The internal medium has a steady pH of 7.35–7.4, with a partial pressure of CO2 dissolved in the intercellular medium of about 5 kPa, not considered in the ISO standard.13 SBF tests are often performed in normal atmosphere, and the pH of SBF is known to rise during incubation to as high as ~9–9.5, which can accelerate the nucleation of calcium phosphate layers.14 Such extreme alkaline conditions do not naturally occur in the human body, which would otherwise lead to ionization of proteins, change their functions, and induce cell necrosis.
- Coherent directives concerning the characteristics of the testing vials are lacking. In our opinion, supported by others,15 the liquid must be filtered to sterilization through 0.22 μm filters, in order to ensure sterility against most bacteria and fungi. The contamination of sterile samples with bacteria and fungi could lead to biofilm formation, pH variation, and ionic modification of the solution, yielding faulty results.
- The use of a normal-atmosphere incubator could lead to pronounced evaporation of the liquid in order to achieve the saturating H2O pressure at 37°C. Gaseous communication between the vial and the normal-atmosphere incubator can gradually lead to SBF alkalinization. The SBF solution, already rich in Ca and P ions to the point of saturation, will concentrate even more and favor spontaneous crystallization, hence leading to altered results. Some researchers are trying to circumvent this by periodically replenishing the solution. However, opening the vial periodically makes it prone to contamination. The SBF should be used in an incubator with humidified atmosphere at 37°C (as in the case of in vitro tests in cell cultures) for an opened testing vial or in closed vials. Therefore, a closed vial with minimum air volume above the liquid should be favored when using a simple incubator, as it limits the evaporation and pH modification of solution, thus offering a stabler environment. Nonetheless, a more proper test should be done in a 5% CO2 humidified-atmosphere incubator at 37°C.
- Another important concern of the SBF protocol is the lack of equity in testing conditions for specimens of different forms (bulk materials, thin films, powders, scaffolds) while adopting the Vs:Sa ratio of 100 mm defined for bulk specimens.12,13 For example, a thin film of BG (ρ=2.7 g/cm3) with 1 μm thickness, surface area of 100 mm2, and mass of 0.27 mg is likely to be completely solubilized in the equivalent volume of SBF testing medium (10 mL). Under those conditions, the pinnacle of HA formation (the supersaturation of BG sample:liquid interface) would hardly be achieved, explaining the longer times required for the biomineralization phenomena to occur in thin BG films. This means that Vs is overestimated for thin films.
- For porous materials, ISO/FDIS 2331713 ambiguously states that the volume of SBF should be greater than the calculated Vs. In other words, the ISO/FDIS 23317 protocol cannot be regarded as a unifying procedure for bioactivity assessment applicable to bulk specimens, powders, or other samples with less defined shapes. Inexplicably, this discrepancy has not been fully stressed yet.
Most literature studies have focused on testing bulk materials. When mentioned, the quantity of BG material per SBF volume varies between ~0.005 and 30 mg/mL (see Table 1), meaning a rather heterogeneous interpretation of data gathered by this type of in vitro assay, precluding comparative interstudy analyses.
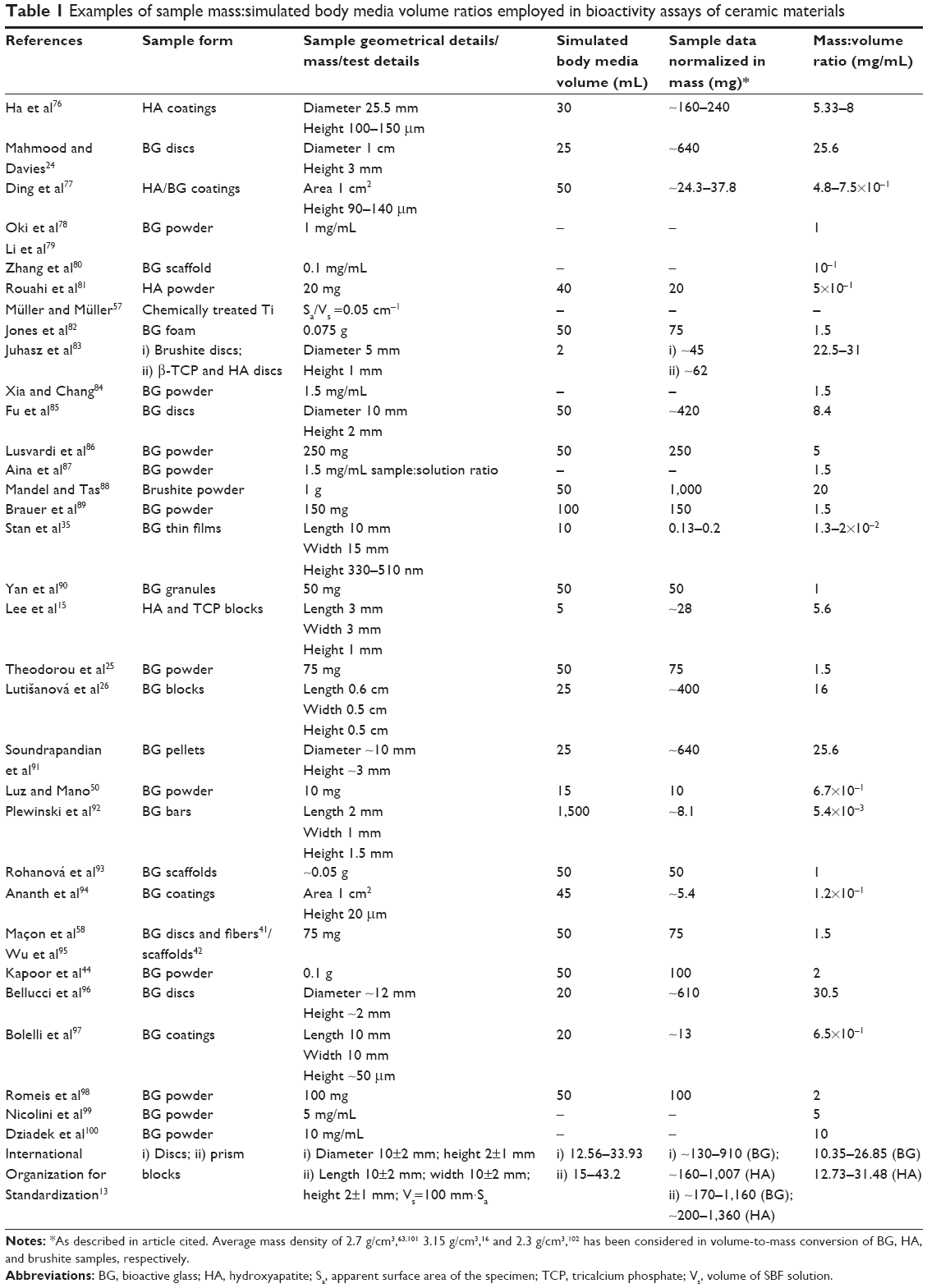 | Table 1 Examples of sample mass:simulated body media volume ratios employed in bioactivity assays of ceramic materials |
Another simplification is the synthetic and scholastic description of the bone-healing process (presently known as the Hench mechanism),16–19 which states that only after mineralization (ie, HA chemical growth and crystallization) can the process of chemoattraction and adhesion of stem cells to the surface occur. Cell adhesion on surfaces can occur in 2–4 hours, and subsequently proliferation starts within the first 12 hours,20–22 thus likely before the HA layer starts to develop onto those surfaces in true biomimetic conditions. Moreover, during healing, the bone is subjected to a remodeling phase in which osteoclasts take out HA from the bone matrix and change the proteinaceous 3-D architecture of the bone matrix at microscopic level. Therefore, the entire bone-healing process around an implant proves to be far more complex than presented in Hench’s mechanism,16–19 being thus hard to anticipate by a simple SBF test.
All these aspects raise a wall of doubts around the classical methodology of testing bioactivity by immersion materials in SBF. The German philosopher Arthur Schopenhauer once said: “The discovery of truth is prevented more effectively, not by the false appearance things present and which mislead into error, not directly by weakness of the reasoning powers, but by preconceived opinion, by prejudice”.23 The search for the truth is the driving force that equally motivates researchers, scientists, and philosophers. In this process, the researcher should first question their own ideas, being the accuser, the defender, and the judge altogether.
Nevertheless, a simple, inexpensive yet scientifically more accurate in vitro bioactivity-screening test is still (unequivocally) necessary, and would be highly beneficial for the scientific community. The use of synthetic body fluid with improved biomimicry would allow not only extrapolation of the behavior of a given material in a living organism but also serve as a trustworthy selection method for materials, prior to the expensive in vitro tests in cell cultures and/or in vivo tests in animal models. We are not allowed by reason, ethics, laws, or economics to subject any number of animals to painful procedures in the absence of solid proof of potential biofunctionality.
Little attention toward addressing this specific subject has been given during the years. To the best of our knowledge, alternative proposals to SBF date back to the year 2000.24 Similar yet incomplete questions have been raised by a small number of researchers.14,15,24–30 Our studies are among the pioneering works on complex biomimetic testing solutions containing organic/inorganic components.31,32 The subject gained more traction after the interrogative work of two leading scientists.14 The gradually increased awareness of the research community of the shortcomings of SBF testing14,15,33 has pointed to the need for more reliable bioactivity testing protocols with enhanced prediction ability of in vivo behavior.
The present study aims to contribute to this important target. The bioactivity of BG films was investigated using different testing solutions and protocols: 1) according to ISO/FDIS 23317 in SBF,13 and 2) using inorganic–organic solutions (with and without proteins) that more closely mimic the human physiological intercellular medium (eg, Dulbecco’s Modified Eagle’s Medium [DMEM]) used for the in vitro culturing of cells. In some cases, DMEM was further supplemented with proteins found in human plasma, as they allow harmonious in vitro growth of cells. The aim was to evaluate if such amino acids and/or proteins get adsorbed on the surface of the tested specimens and understand the nature of such an organic layer, and how it interferes with the biomineralization process. BG coatings with different silica contents were deposited onto titanium substrates. The role of the glass network connectivity (NC) in governing the dissolution rate of BG in aqueous media and thereby its biomineralization capacity is well known, and offers a solid and trusty basis to sustain the findings and their interpretation here reported. A unified protocol is also proposed for testing the bioactivity of implant materials, irrespective of the composition of the testing medium or the physical shape of specimens.
Materials and methods
Bioglass-powder synthesis
The source material for film preparation consisted of a bioactive and cytocompatible alkali-free BG powder with ZnO additive34 with the following nominal composition (mol%): SiO2 38.5, CaO 36.1, P2O5 5.6, MgO 15.2, ZnO 4, and CaF2 0.6. High-purity powders of SiO2 (>99.5%; Merck, Darmstadt, Germany), CaCO3 (>99.5%; Merck), MgCO3 (>99%; Merck), ZnO (>99%; Sigma-Aldrich, St Louis, MO, USA), NH4H2PO4 (>99%; Sigma-Aldrich), and CaF2 (>99.9%, 325 mesh; Sigma Aldrich) were used for preparing the BG. Homogeneous mixtures of batches (~100 g) obtained by dry ball milling were preheated at 900°C for 1 hour for decarbonization and then melted in Pt crucibles at 1,550°C for 1 hour. The melt was poured into cold water to obtain a glass frit, which was dried and then milled in a high-speed agate mill, resulting in fine glass powders with mean particle size of ~10–20 μm (determined by light-scattering technique, Coulter LS 230, Fraunhofer optical model; Beckman Coulter, Brea, CA, USA).
Target and substrate preparation
BG cathode targets with 110 mm diameter and 3 mm thickness were consolidated from the powdered frit by mild pressing at room temperature in a tantalum port target. This procedure has proven to assure good target consistency during the prolonged sputtering processes.32,35 Here, we introduce a simple route of producing BG films with different contents of silica/degrees of polymerization, starting from the same base material. High-purity fused silica plates (Präzisions Glas & Optik, Iserlohn, Germany) with a surface area of 100 mm2 (this case) were fixed in the “racetrack” region (area where the most intensive sputtering phenomena occur, due to the high density of magnetron plasma, consequently leading to maximal erosion) of the BG target (Figure 1). This allowed tailoring of the composition and structure of the prepared BG coatings, as is shown henceforth.
 | Figure 1 Schematics of the magnetron cathode targets employed in the framework of this study. |
Pure titanium (commercially pure, grade 1; Mateck GmbH, Jülich, Germany) plates (10×10 mm2) were used as substrates. All substrates were cleaned successively by ultrasonication in acetone, ethanol, and distilled water, and dried by argon-gas purging. Silicon <100> wafers had previously been used as substrates to determine by optical measurements the films’ deposition rates under the sputtering conditions detailed herein.
Magnetron sputtering deposition
The BG films were prepared using a UVN-75R1 sputtering-deposition system with a magnetron planar cathode with plasma ring of ~57 mm outside diameter and ~43 mm inside diameter, operating at a radio frequency of 1.78 MHz. The vacuum chamber was initially evacuated down to a pressure of ~2×10−3 Pa. The substrates were plasma etched for 10 minutes to increase the film’s adherence.36 The cathode target surface was first presputtered in an argon atmosphere for ~60 minutes at a working pressure of 0.4 Pa, in order to remove any possible remnant superficial impurities and equilibrate the surface composition.
For all types of targets, the deposition of BG films was carried out at the same argon pressure (0.4 Pa). A target–substrate distance of 35 mm was used. The substrate reached a maximum temperature of ~150°C (in the case of the longest deposition time [~3 hours]), as indicated by a built-in temperature controller. The typically low deposition temperature inside the magnetron sputtering chamber allowed the preservation in the deposited films of the amorphous state.
BG coatings with different silica contents were prepared using three types of targets: simple (BG-O), and with three (BG-3S) and five (BG-5S) silica plates attached in the region of the target racetrack (Figure 1). BG coatings of equal thickness (~1 μm) were synthesized based on deposition rates previously determined.
Physicochemical characterization
The envisaged quality of the source material for the cathode targets was checked prior to the films’ preparation by structural analysis. Prior to and/or after the in vitro testing, the composition and structure of the BG films were assessed by a plethora of characterization techniques, in order to gather detailed and consistent information to support the comparative biological behavior of the as-designed BG coatings in simulated body media with increased complexity and biomimetic accuracy.
The bonding configuration of the BG target and as-sputtered coatings, prior to and after the in vitro testing, was analyzed by Fourier-transform infrared (FTIR) spectroscopy (Spectrum BX II; PerkinElmer, Waltham, MA, USA). Measurements were performed in attenuated total reflectance mode (using a Pike Miracle diamond head) within a range of 570–4,000 cm−1 with 4 cm−1 resolution and a total of 128 scans per experiment.
The short-range-order structural modifications emerging in the case of BG coatings tested in vitro in body fluids were further gauged locally by micro-Raman spectroscopy employing a LabRam HR 800 Evolution (Horiba, Kyoto, Japan) confocal Raman microscopy system. The measurements were carried out with a 532 nm excitation laser, using a 1,800 g/mm grating and 100× objective lens. The incident laser power on the samples was ~10 mW and the spot diameter ~1 μm. For each sample, 128 measurements of 20 seconds’ integration were performed.
The surface morphology of the films was examined by scanning electron microscopy (SEM) under secondary electron mode (acceleration voltage 15 kV) with Tescan Lyra III and Hitachi SU-70 microscopes. Thin conductive carbon films were sputtered on the samples’ surface to avoid electrical charge accumulation.
The amorphous status of the as-manufactured BG powder was verified by X-ray diffraction (XRD) with a Bruker D8 Advance diffractometer (CuKα [λ=1.5418 Å] radiation), using a high-efficiency LynxEye linear detector. The crystallographic structure of the soaked BG films was investigated by grazing-incidence XRD (GIXRD; grazing angle 2°) using the Bruker D8 in parallel-beam setting equipped with a copper-anode X-ray tube. Measurements were carried out in the 2θ range from 20° to 60°, with a step size of 0.04° and 50 seconds’ acquisition per step.
The elemental composition of the BG films was determined by energy-dispersive X-ray spectroscopy (EDS) measurements, performed with a Bruker Quantax 400. The EDS analyses were performed on five different regions of 50×50 μm2 located on the sample surface, the results being presented further as means ± standard deviation (n=5).
X-ray photoelectron spectroscopy (XPS) measurements were conducted in a dedicated chamber (Specs Surface Nano Analysis GmbH, Wedding, Germany) equipped with a monochromatized X-ray source (Al Kα: hν =1,486.7 eV). Photoelectrons were recorded using a Phoibos 150 hemispherical analyzer and a flood gun operating at 1 eV using a 0.1 mA current, in order to avoid sample charging during measurements. The analyzer was operated in fixed-transmission mode with a pass energy of 20 eV, leading to an overall resolution of the system (source + analyzer) of 0.75 eV. During measurements, the pressure was kept below 3×10−9 mbar. The energy scale was calibrated using the C–C component of the C1s peak at 284.5 eV. Core-level spectra were fitted with Voigt lines and associated integrals of the Voigt profile,37 each component with its own inelastic background associated with the inelastic scattering of photoelectrons on their way out of the sample.38
In vitro biological testing of sputtered glass films in physiological fluids with various degrees of biomimicry
The in vitro bioactivity of BG samples (ie, ~1 μm-thick BG coatings with surface area of 1 cm2, deposited on 1 mm-thick Ti coupons), reflected by their capability of inducing calcium phosphate formation onto their surfaces, was investigated by immersion in the following.
Simulated body fluid
We complied with all the indications included in the ISO standard: solution composition (in millimoles: Na+ 142, K+ 5, Mg2+ 1.5, Ca2+ 2.5, Cl− 147.8, HCO3− 4.2, HPO42− 1, SO42− 0.5), volume of solution calculated with formula Vs=100 mm·Sa, temperature 36.5°C, pH 7.4, normal pressure. Also, even if there was no specific recommendation in the ISO standard, the SBF was filtered through sterilized filters (cameo 25 AS-MSI, pore size 0.22 μm) to avoid microbial infestation of the initial medium. The samples were kept in a humidified atmosphere, to prevent SBF from evaporation.
Dulbecco’s Modified Eagle’s Medium
Type D8437 (Sigma-Aldrich) solution, currently used as medium for cell cultures (its complex composition can be found on the manufacturer’s website). All the aforementioned ISO 23317 standard requirements (imposed in the case of SBF testing) were abided by (including the normal-atmosphere and pressure-condition norms). This testing medium shall be denoted henceforth as “DS”.
DMEM supplemented with 10% fetal bovine serum in normal atmosphere
All the ISO 23317 standard requirements (imposed in the case of SBF testing) were abided by (including the normal-atmosphere and pressure-condition norms). This testing medium shall be denoted further as “DC”.
DMEM supplemented with 10% fetal bovine serum in homeostatic atmosphere
This testing medium (5 kPa CO2) shall be denoted henceforth as “DCC”.
The samples were soaked in 15 mL sterile polyethylene tubes with the given synthetic body media for 28 days. All tests were performed in duplicate. Precautions were taken to assure sterility of the samples and vials (by dry-heat procedure at 180°C for 1 hour), as well as of the testing media. All operations were carried out in a biosafety cabinet with laminar flow used for cell cultures. Each sample batch was placed in a humidified atmosphere incubator with or without 5% CO2 in the atmosphere. After their extraction, the samples were rinsed gently with deionized water and left to dry at ambient temperature in a desiccator.
Surface-enhanced laser desorption/ionization time-of-flight mass spectroscopy analyses
The adsorption profiles of proteins onto the surface of BG films during in vitro testing in complex inorganic–organic media were assayed by a state-of-the-art biological analysis method: surface-enhanced laser desorption/ionization time-of-flight (SELDI-ToF) mass spectroscopy. CM10 ProteinChip® (Bio-Rad Laboratories, Hercules, CA, USA) arrays were abraded and polished to remove the original special surface, and then prepared for their coating with BG films following the protocol described in the “Magnetron sputtering deposition” section. The chips were sterilized by dry heating for 1 hour at 180°C. Subsequently, the as-prepared samples were mounted in the bioreactor, sterilized by autoclaving, and incubated with cell-culture DMEM containing various concentrations of fetal bovine serum (FBS; 5%, 10%, and 25%) or in pure FBS (100%). After different soaking times (6, 12, 24, and 96 hours), the chips were washed with deionized water and the spots subsequently incubated with 2 μL energy-absorbing molecule buffer (5 mg sinapic acid dissolved in 400 μL solvent composed of 50% acetonitrile, 0.5% trifluoroacetic acid, 49.5% H2O, high-performance liquid-chromatography purity grade; Sigma Aldrich) and dried in a laminar flow hood.
Samples were analyzed with the ProteinChip SELDI-ToF platform and ProteinChip Data Manager software. All materials and reagents used in this experiment were purchased from Bio-Rad. Parameters were: mass range 6,000–200,000, focus mass 25,000, matrix attenuation 6,000, sampling rate 400, calibration method PCS4000 instrument default, acquisition method SELDI quantization, warming energy 4,400 (one shot), data energy 4,000 (ten shots).
In vitro testing in mesenchymal stem cell cultures
Biocompatibility assays were performed in mesenchymal stem cell (MSC) cultures (Lonza, Basel, Switzerland). The selection of this specific cell line was justified by the higher sensitiveness of MSCs toward cytotoxic factors, from the group of cells (eg, fibroblasts, osteoblasts, osteoclasts, MSCs) that can interact with a given implant surface.
MTS assay
Cell proliferation was investigated by a classic MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) test (Promega Corporation, Madison, WI, USA). After the MSC cultures reached a sufficient degree of proliferation and nucleation in order to conduct triplicate experiments, cells were detached and seeded onto control surfaces (polycarbonate cell-culture control) and BG coatings with the highest (ie, BG-O) and lowest (ie, BG-5S) concentrations of silica. All samples had a designed surface area of 100 mm2.
MSCs were maintained in culture with complete media (DMEMα modification, with 15% FBS, GlutaMax producer-recommended dilution, and L-ascorbic acid), and at every subculturing procedure they were dissociated to a unicellular suspension. On each type of surface (controls and implant coatings) 104 cells were seeded in 100 μL of complete culture media. After 5 hours, 1 mL of complete cell-culture media was added. After 24 hours of culturing, mitochondrial activity was assessed by MTS. The culture medium was removed, and fresh 400 mL cell-culture medium without phenol red was added. After 30 minutes, 80 μL of MTS substrate (Promega) was added and the plates reintroduced in the incubator for 1 hour. Volumes of 200 μL of the cell-culture media were transferred to microplates, and absorbance was read at 490 nm.
Immunofluorescence assays
Cell proliferation, morphology, and degree of differentiation were assessed by investigating expression of the CD90 marker by indirect immunofluorescence assay. Cells were fixed with a 2% paraformaldehyde solution for 30 minutes, and then washed and permeabilized by incubation for 30 minutes with 0.075% saponin in phosphate-buffered saline (PBS; Sigma-Aldrich). Cells were further washed three times for 10 minutes with PBS. Blocking of unspecific coupling sites was realized by incubation for 30 minutes with 2% bovine serum albumin (Sigma-Aldrich) in PBS. The samples were then incubated with a 1:50 dilution of CD90 primary antibody (Santa Cruz Biotechnology, Dallas, TX, USA). After two PBS washes of 15 minutes and one of 30 minutes, the samples were incubated at room temperature for 1 hour with the appropriate secondary antibody (antirabbit produced in goat coupled with CruzFluor™ 488; Santa Cruz Biotechnology). After another set of PBS washes – two of 15 minutes and one of 30 minutes – samples were incubated for 10 minutes with 0.3 μg/mL (4′,6-diamidino-2-phenylindole dihydrochloride) (DAPI) (Sigma-Aldrich). Samples were washed again three times for 10 minutes, and mounted with 170 μm-thick cover glass and fluorescence mounting medium (DakoCytomation, Glostrup, Denmark). For the negative controls of experiments, we used an incubation of samples with PBS with 2% bovine serum albumin, instead of the primary antibody.
Statistics
Statistical significance analysis was performed using the unpaired Student’s t-test. Differences were considered significant when P<0.05.
Results
Characterization of starting BG powder and as-sputtered BG coatings
XRD analyses performed in symmetric (θ-θ) geometry confirmed the amorphous status (within the experimental sensitivity limit) of the BG-O target material (Figure 2). The pattern was dominated by a pronounced halo centered at 2θ≈29°–30°, characteristic of a vitreous silica-based compound.39
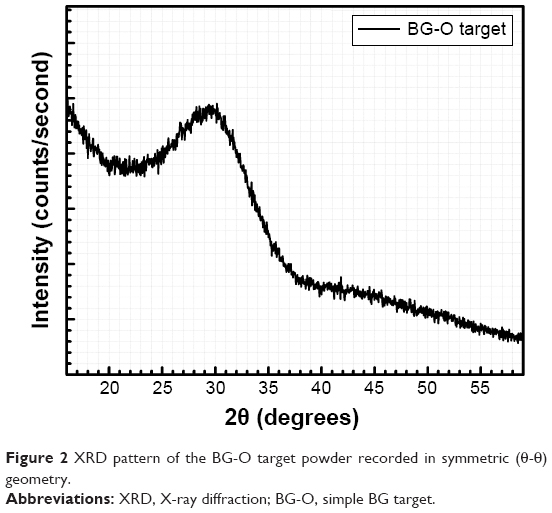 | Figure 2 XRD pattern of the BG-O target powder recorded in symmetric (θ-θ) geometry. |
Micro-Raman analysis supported the XRD findings, further evidencing the strongly depolymerized structure of the BG-O powder (Figure 3), dominated by the tetrahedral structural units (QSi) with two nonbridging oxygen atoms 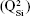 The prominent broad Raman band of the spectrum, positioned in the wave-number region of 900–1,100 cm−1, had its maximum centered at 956 cm−1, pertaining to the
The prominent broad Raman band of the spectrum, positioned in the wave-number region of 900–1,100 cm−1, had its maximum centered at 956 cm−1, pertaining to the 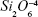 rings and chains and two weaker shoulders, peaking at ~869 cm−1 and ~1,040 cm−1 (ascribed to the contribution of monomers
rings and chains and two weaker shoulders, peaking at ~869 cm−1 and ~1,040 cm−1 (ascribed to the contribution of monomers 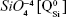 and to the bidimensional
and to the bidimensional 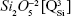 units, respectively).40–43 Typical bands of rocking (ρ) motion of bridging oxygen in structural units containing nonbridging oxygen (~623 cm−1) and symmetric oxygen stretching of Si–O–Si and symmetric O–Si–O angular deformation of the coupled silicate groups (470–420 cm−1 region) have also been shown.40,43
units, respectively).40–43 Typical bands of rocking (ρ) motion of bridging oxygen in structural units containing nonbridging oxygen (~623 cm−1) and symmetric oxygen stretching of Si–O–Si and symmetric O–Si–O angular deformation of the coupled silicate groups (470–420 cm−1 region) have also been shown.40,43
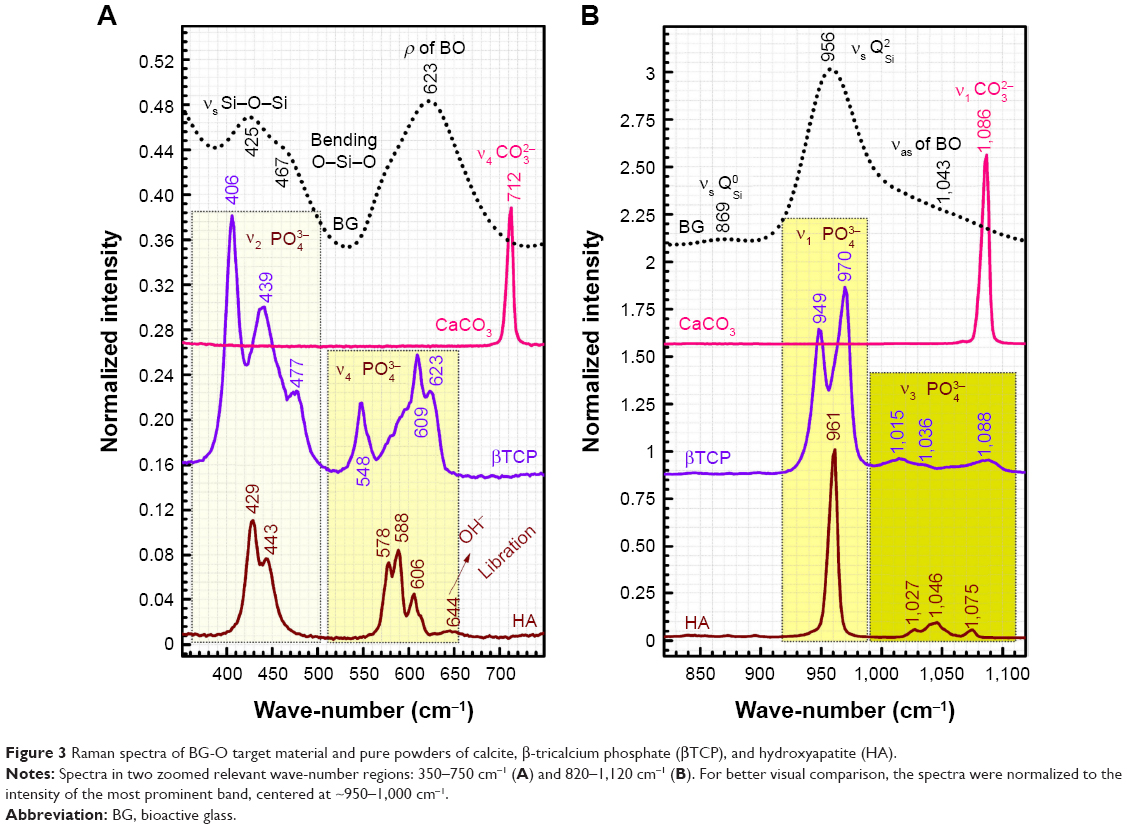 | Figure 3 Raman spectra of BG-O target material and pure powders of calcite, β-tricalcium phosphate (βTCP), and hydroxyapatite (HA). |
Table 2 presents the comparative oxide compositions of the BG powder and BG-O, BG-3S, and BG-5S sputtered films, together with the corresponding NC values calculated with the formula proposed in Kapoor et al.44 Progressive silica enrichment of the BG films was observed: BG-O < BG-3S < BG-5S. An antagonistic relationship between SiO2 and CaO was depicted by the EDS results. When the SiO2:BG ratio was increased in the target racetrack region, the SiO2 content was augmented progressively, with the CaO concentration reducing proportionally. No substantial P2O5, MgO, or ZnO concentration modifications were noticed among BG films. However, the P2O5 and ZnO contents were found to be lower in the case of all as-sputtered BG films in comparison to the parent-target composition. This is determined by the high volatility of these species.45,46
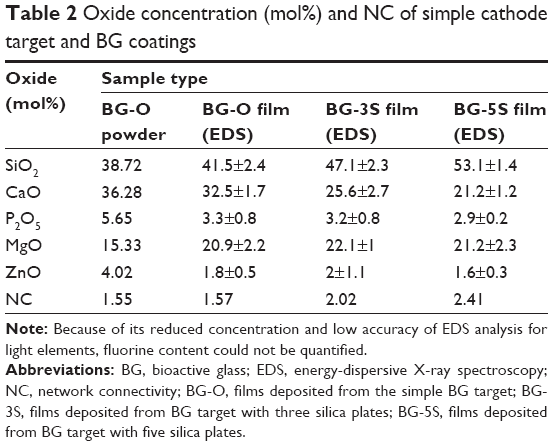 | Table 2 Oxide concentration (mol%) and NC of simple cathode target and BG coatings |
The BG-O target and films presented analogous infrared envelopes denoting their close structural similarity, featuring a highly depolymerized glass structure dominated by the silicate tetrahedral units with various numbers of nonbridging oxygen atoms 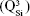 . Overlapped IR vibrational bands of the
. Overlapped IR vibrational bands of the 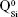 ,
, 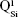 ,
, 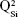 and
and 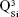 units, emerging in the 850–1,010 cm−1 wave-number region,40,47–49 constituted a solid testimony of the low NC of these BG structures. The contribution of P–O bond vibrations (elicited by the orthophosphate functional groups,
units, emerging in the 850–1,010 cm−1 wave-number region,40,47–49 constituted a solid testimony of the low NC of these BG structures. The contribution of P–O bond vibrations (elicited by the orthophosphate functional groups, 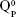 , which are typically the dominant phosphate environment in such glass compositions),50 is hard to deduce, due to the low P content of the films and to the superimposition of the highest-intensity IR bands of phosphate with those of silicate.40
, which are typically the dominant phosphate environment in such glass compositions),50 is hard to deduce, due to the low P content of the films and to the superimposition of the highest-intensity IR bands of phosphate with those of silicate.40
As expected, the gradual increase in the silica content of the BG-3S and BG-5S films was reflected in an augmentation of the polymerization degree of the films’ glass network. FTIR spectra (Figure 4) indicated an intensity enhancement of the band positioned at ~1,030–1,050 cm−1, associated with the stretching vibrations of bridging oxygen atoms (Si–O–Si) in all 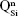 silicate species. The blue shift of these vibration bands with silica content (to 1,036 cm−1 for BG-3S and to 1,044 cm−1 for BG-5S) denoted a progressive augmentation of the bonds’ strength in this type of film. Furthermore, in the case of the BG-5S films, the presence of fully polymerized silica domains was hinted by the appearance of a new band (centered at ~1,160 cm−1), determined by the
silicate species. The blue shift of these vibration bands with silica content (to 1,036 cm−1 for BG-3S and to 1,044 cm−1 for BG-5S) denoted a progressive augmentation of the bonds’ strength in this type of film. Furthermore, in the case of the BG-5S films, the presence of fully polymerized silica domains was hinted by the appearance of a new band (centered at ~1,160 cm−1), determined by the 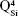 silicate units.
silicate units.
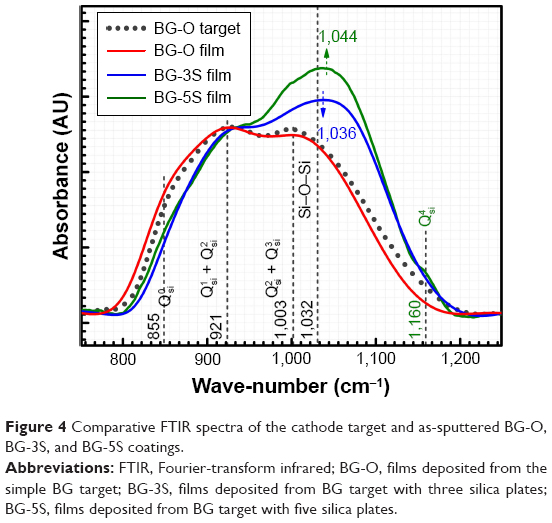 | Figure 4 Comparative FTIR spectra of the cathode target and as-sputtered BG-O, BG-3S, and BG-5S coatings. |
SEM analysis of BG-coating surfaces (Figure 5) revealed morphologies that were typical of amorphous structures deposited by radio-frequency magnetron sputtering,51–53 consisting of fairly smooth film matrices. However, one can notice that more uniform coatings were obtained with increasing silica contents.
 | Figure 5 Comparative surface morphology as evidenced by SEM for (A) BG-O, (B) BG-3S, and (C) BG-5S coatings. |
Characterization of in vitro-tested BG films
FTIR and GIXRD
FTIR (Figure 6) and GIXRD (Figure 7) analyses revealed that all BG coatings soaked in SBF for 28 days under normal atmosphere induced the formation of a thick but poorly crystallized HA layer (Figure 6A vs 6D, Figure 7), as ubiquitously reported in the literature. On the other hand, the BG coatings tested in vitro in SBF but incubated at 37°C in a 5% CO2 atmosphere led to the growth of a thinner and less structured layer of HA (data not presented). The presence of the distinctive IR vibrational bands of HA is emphasized (Figure 6A): the asymmetric ν4 bending mode of (PO4)3− groups (~561 and 602 cm−1), νL libration of hydroxyl groups (~624 cm−1), symmetric ν1 stretching of (PO4)3− groups (~963 cm−1), and asymmetric ν3 stretching modes of (PO4)3− groups (~1,025 and 1,097 cm−1).54 The slight deviation in the bands’ maxima (Figure 6A) with respect to a pure highly crystalline HA (Figure 6D) indicated the biomineralization of an HA layer with structural faults (due to ionic substitutions with species or functional groups present in the SBF solution).55 The occurrence of supplemental low-intensity bands was also signaled (Figure 6A), and can be ascribed to the juxtaposition of the ν2 bending mode of carbonate and vibrations typical of labile (HPO4)2− ions present in nonapatitic domains (peaking at 873 cm−1) and to the symmetric stretching vibrations of Si-O bonds elicited in various environments (original BG film, silica-rich gel layer formed in the previous stages of bioactivity17,56) centered at 819, 808, and 737 cm−1.
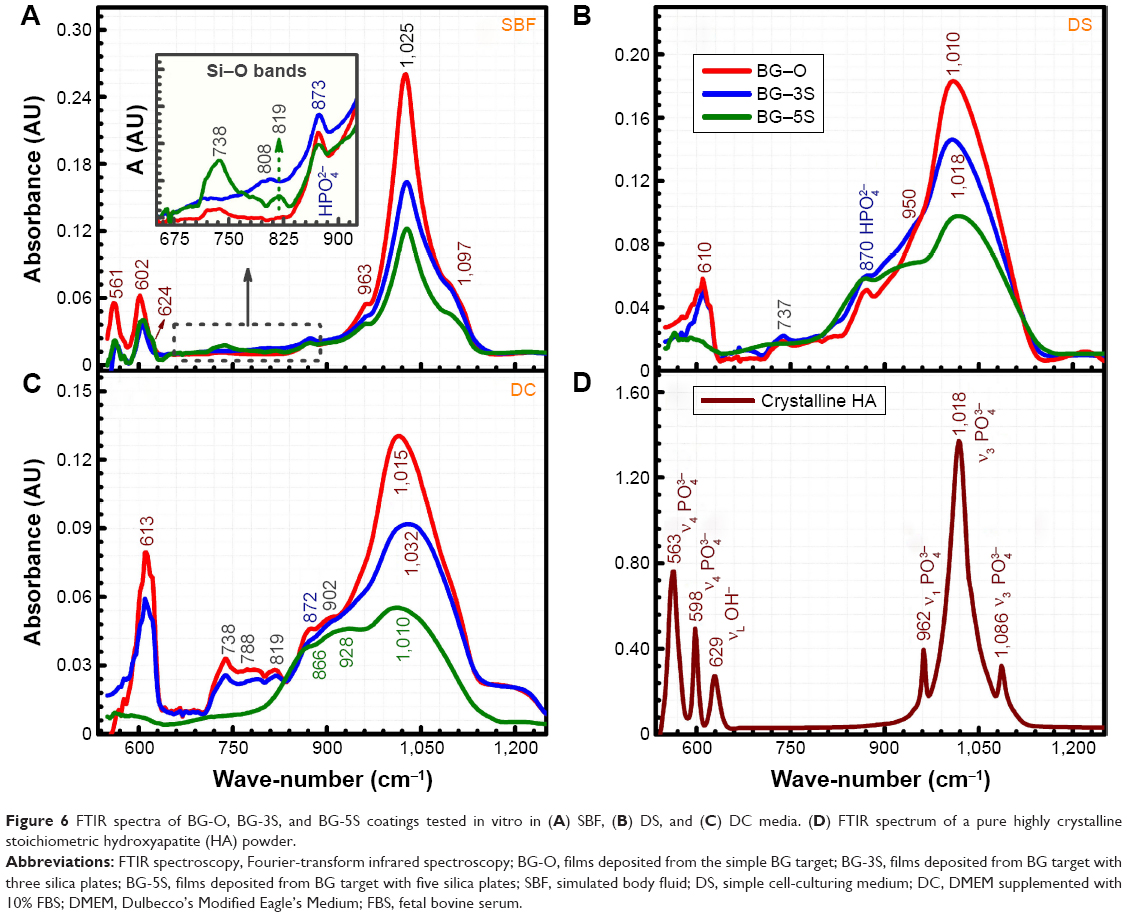 | Figure 6 FTIR spectra of BG-O, BG-3S, and BG-5S coatings tested in vitro in (A) SBF, (B) DS, and (C) DC media. (D) FTIR spectrum of a pure highly crystalline stoichiometric hydroxyapatite (HA) powder. |
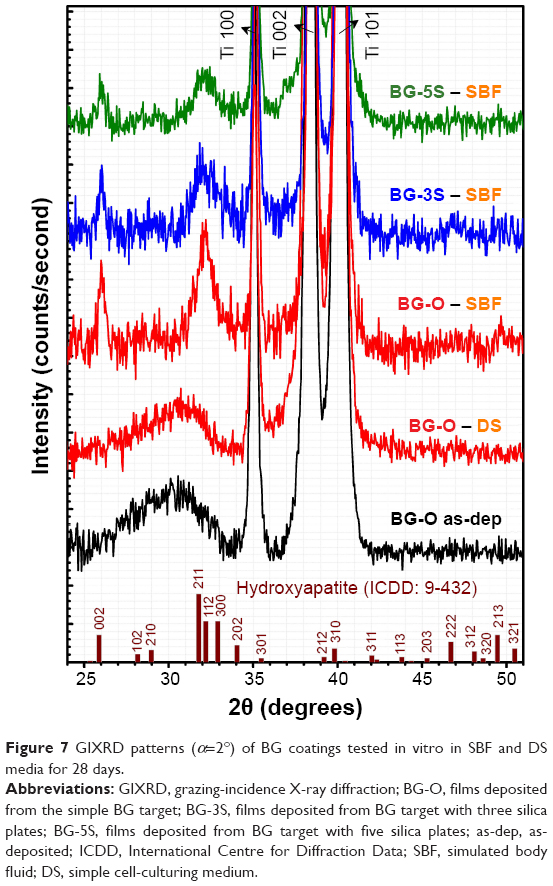 | Figure 7 GIXRD patterns (α=2°) of BG coatings tested in vitro in SBF and DS media for 28 days. |
The integral area of the prominent phosphate asymmetric stretching band can be approximated with the thickness of HA films (Beer–Lambert law) chemically grown in vitro in simulated body media, and thus can be associated with one sample capacity of biomineralization.55 As expected, one can notice that a lower concentration of silica and thus a greater degree of depolymerization is reflected in a thicker calcium phosphate (CaP) layer (Figure 6A). This phenomenon was more acute in the case of tests conducted in the classical SBF solution, where BG-O area > BG-3S area > BG-5S area.
BG coatings immersed in DS and DC exhibited sluggish formation and crystallization of biomimetic HA layers, suggesting that the organic components in the testing media slowed the biomineralization processes. After 28 days of immersion, the samples featured a layer of amorphous calcium phosphate, as revealed by FTIR analysis (Figure 6B and C). The IR spectra also revealed that a higher degree of NC endowed a lower dissolution rate to BG films in both DMEM-based media, one being able to discriminate more easily the vibration modes originating from BG coatings when their silica content was larger (Figure 6B and C). Therefore, the BG-5S films exhibited the lowest in vitro bioreactivity. Their immersion in DC (with the highest compositional complexity) originated the smallest structural changes compared to the initial structure of the as-sputtered films (see Figure 4 vs Figure 6C). Samples tested in DCC did not present noticeable structural changes with respect to the as-deposited samples (data not shown).
GIXRD-analysis results (Figure 7) were in agreement with the FTIR spectra. The broad diffraction maxima recorded in the case of films tested in SBF indicated the formation of a nanocrystalline HA layer with c-axis preferential orientation, typical of apatitic layers grown in vitro.55,57,58 For samples tested in vitro in inorganic–organic DS, only a halo was emphasized (slightly shifted toward higher angles with respect to the hump observed for the as-deposited layer), which denoted the amorphous status of the CaP deposits (at the sensitivity limit of the employed XRD machine). No significant long-range structural changes occurred in tested DC or DCC samples (data not presented). GIXRD patterns indicated a similar biomineralization evolutionary ladder, as did the aforementioned FTIR results. The less intense HA peaks from BG-3S and BG-5S films in comparison to BG-O ones indicated less extensive biomineralization with increasing NC.
SEM-EDS and micro-Raman spectroscopy
SEM investigations further strengthened the latency of the biomineralization processes with increased complexity of the testing medium. The morphology of all types of samples had been dramatically modified after 28 days of immersion in SBF (Figure 8). The fairly smooth appearance of the BG coatings was converted into a rough matrix composed of intertwining fine needle-like crystals with EDS-estimated Ca:P ratios within 1.4–1.5, on the top of which had grown spheroid particulates (1.5–2.5 μm diameter) with Ca:P ratios close to that of stoichiometric HA (1.6–1.75). This morphology of spheroid aggregates made of nanosize elongated crystals is characteristic of well-developed HA coatings precipitated from SBF.12,55,57
 | Figure 8 SEM images of BG-O (A–C), BG-3S (D–F), and BG-5S (G–I) coatings tested in vitro for 28 days in SBF medium. |
BG coatings tested in vitro for 28 days in DS (Figure 9) and DC (Figure 10) exhibited surface morphologies that were radically different from those obtained in SBF (Figure 8). Spherical nodules (0.4–0.75 μm diameter) forming “fractal”-like aggregate chains were observed for samples tested in DS (Figure 9). The biomineralization processes of samples tested in DC (Figure 10) were at an even earlier stage. Only smaller and lower-density (in comparison to DS samples) spheroidal nodules (0.1–0.2 μm diameter) were observed. Such formations resemble very well those obtained for BG films soaked in SBF for 7 days (Figure 10G), thus confirming the latency of biomineralization in DC. The size of the nodules and their inorganic nature revealed by the Raman spectroscopy (as further detailed) and absence of medium turbidity and/or of any biofilm on the surface of BG coatings after 28 days excluded the possibility of attributing these nodules to any bacterial contamination. In the case of DCC tests, only the less polymerized BG-O films exhibited the presence of scarce and very fine (0.035–0.09 μm diameter) nodules (Figure 10H), explaining why no significant changes could be discerned by FTIR spectroscopy or GIXRD.
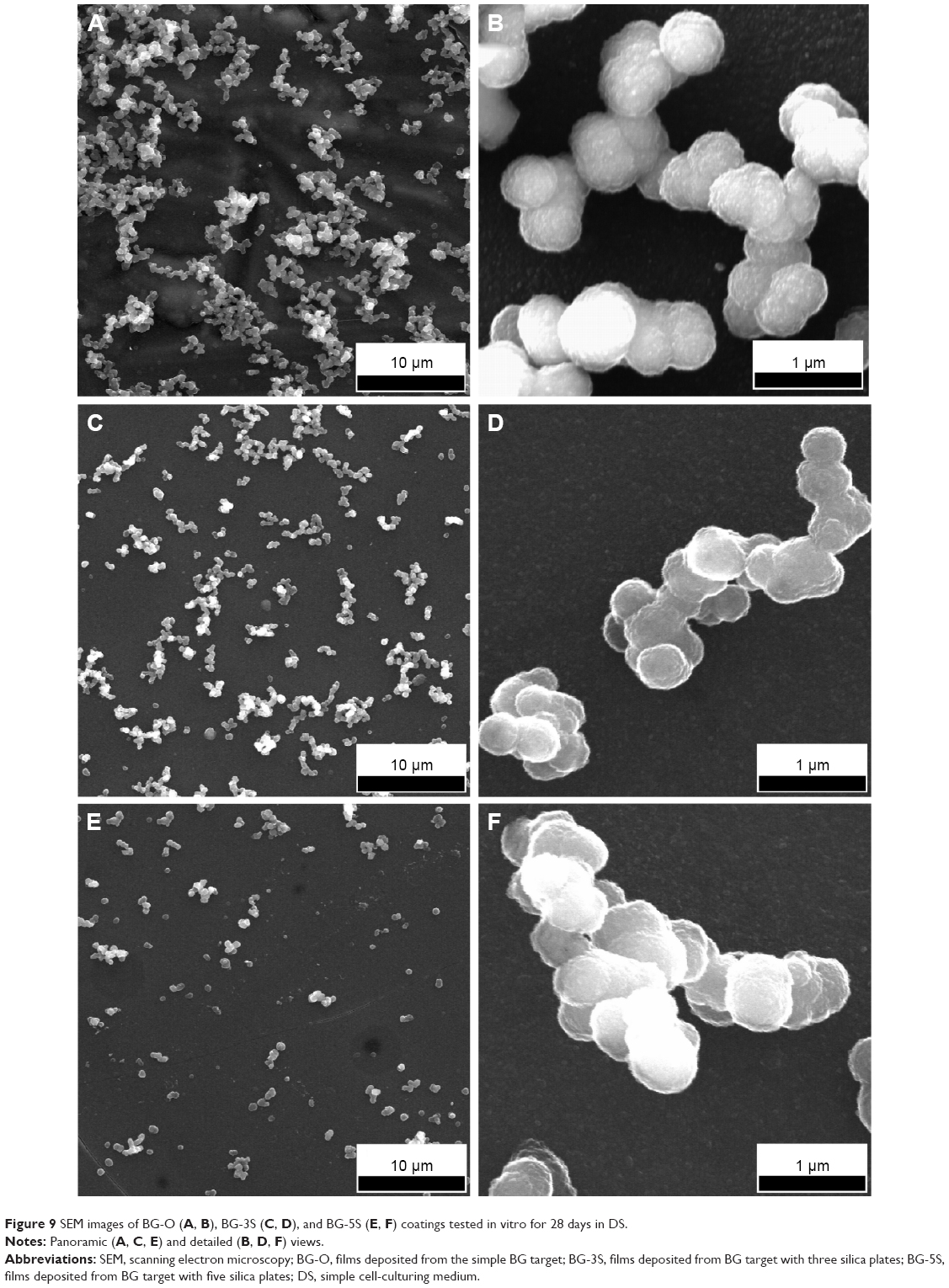 | Figure 9 SEM images of BG-O (A, B), BG-3S (C, D), and BG-5S (E, F) coatings tested in vitro for 28 days in DS. |
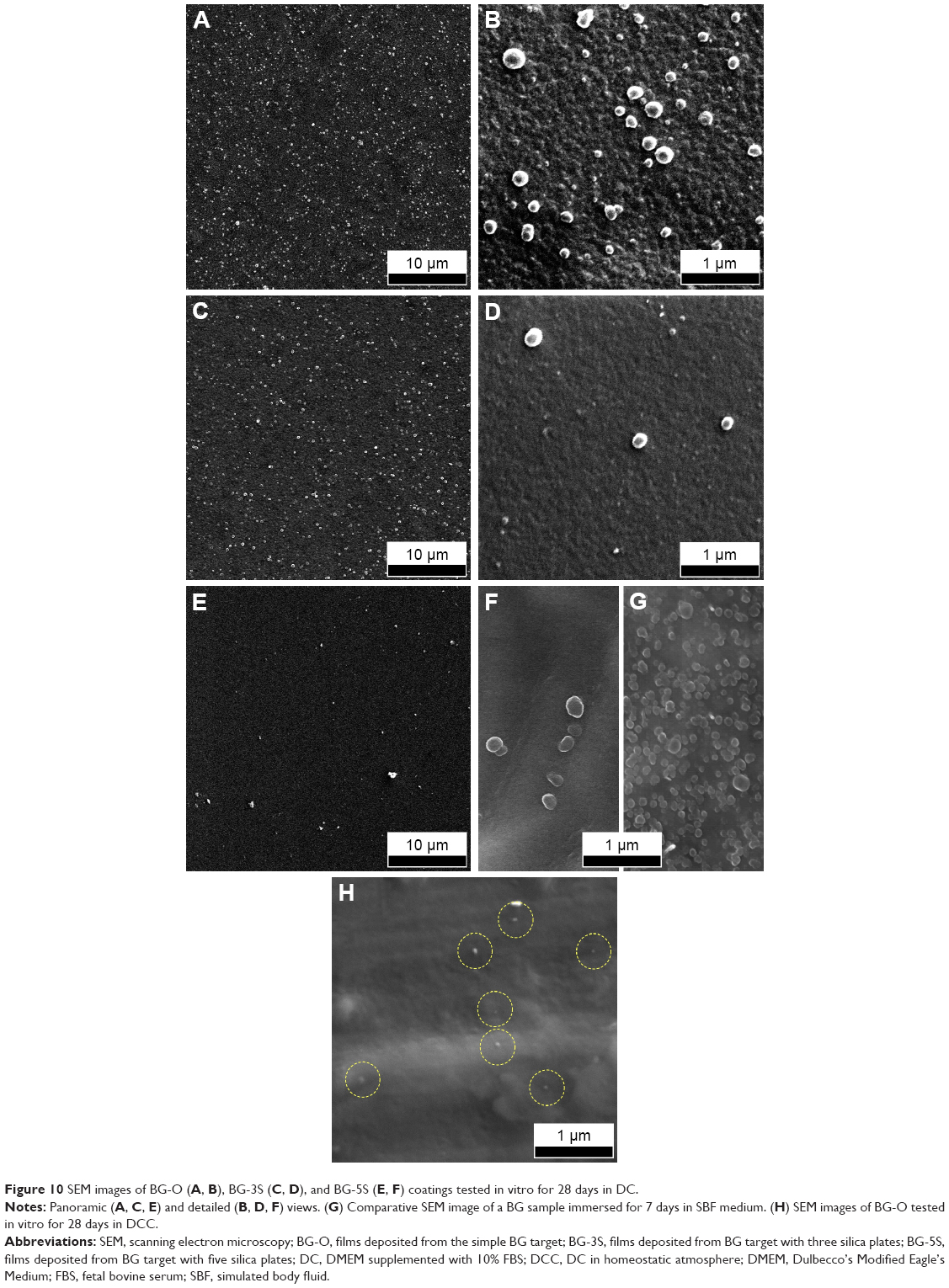 | Figure 10 SEM images of BG-O (A, B), BG-3S (C, D), and BG-5S (E, F) coatings tested in vitro for 28 days in DC. |
The population of spheroid particulates tended to decrease (Figures 8–10) with increasing silica contents in BG films (Table 2). This qualitative observation further suggests the sluggish biomineralization processes with increasing NC, in good agreement with the FTIR-spectroscopy results (Figure 7).
To shed further light on the influence of the in vitro testing composition on the extent of biomineralization, more systematic microscopy studies were performed. The area occupied by the spheroid aggregates/nodules on a given surface was quantified on the basis of at least ten SEM images (at least 500 particles have been numbered) per type of sample (ie, BG-O, BG-3S, and BG-5S). The results plotted in Figure 11 confirm the gradual deceleration of biomineralization in the presence of amino acids and/or proteins found in the human intercellular fluid. Moreover, the increase in NC systematically decreased the reactivity of the BG films, regardless of the type of testing medium.
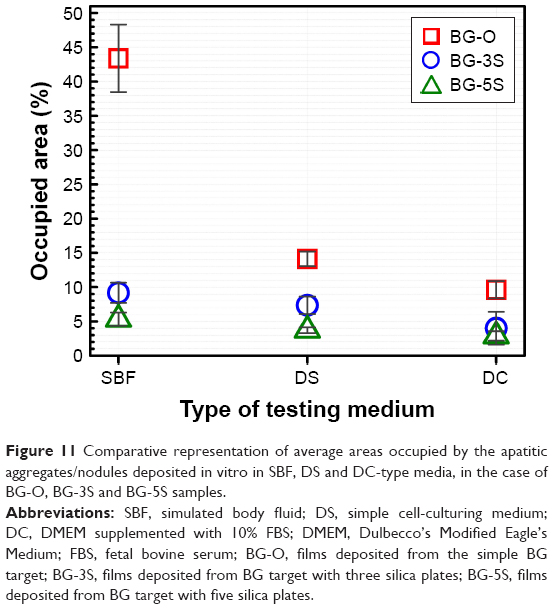 | Figure 11 Comparative representation of average areas occupied by the apatitic aggregates/nodules deposited in vitro in SBF, DS and DC-type media, in the case of BG-O, BG-3S and BG-5S samples. |
Micro-Raman spectroscopy (spot size ≈1 μm) was used to shed further light on the local structure of spheroid aggregates observed by SEM (Figures 8–10). The spectra of the BG films tested in vitro in all simulated body media (Figure 12) can be compared to the spectra recorded for BG-O target material, and pure crystalline powders (Sigma-Aldrich) of calcite (CaCO3), β-tricalcium phosphate [β-TCP; β-Ca3(PO4)2] and HA [Ca10(PO4)6(OH)2] (Figure 3). Two zoomed wave-number windows (350–680 cm−1 and 820–1,120 cm−1) relevant to both glass and calcium phosphate structures are shown in order to facilitate a better view of the bands. Only the spectra of the least reactive samples (BG-5S) collected in the regions marked with crosshairs in the optical images are shown in Figure 12, as a demonstration of spheroid aggregates nature developed under different in vitro conditions.
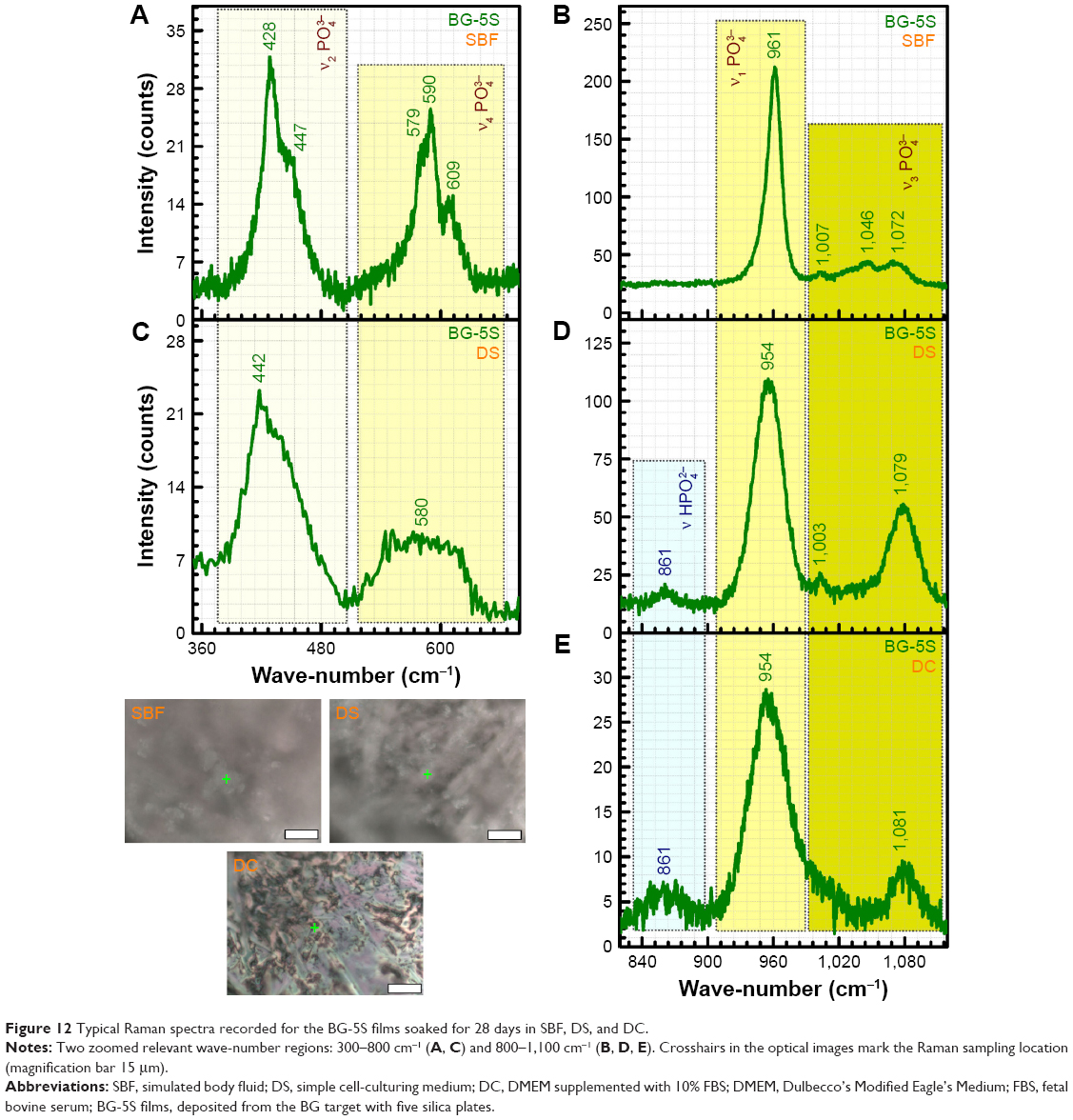 | Figure 12 Typical Raman spectra recorded for the BG-5S films soaked for 28 days in SBF, DS, and DC. |
The spheroid aggregates formed on BG films immersed in SBF clearly consisted of HA, eliciting all the typical active Raman bands (Figure 12 vs Figure 3) of tetrahedral (PO4)3− at similar wave-numbers as crystalline HA powder: the strong symmetric stretching mode (ν1) peak (P-O bond) at ~961 cm−1, triply degenerated asymmetric stretching mode (ν3) (P-O bond) at ~1,007, 1,046, and 1,072 cm−1, triply degenerated bending mode (ν4) (O−P−O bond) at ~579, 590, and 609 cm−1, and doubly degenerated bending mode (ν2) (O−P−O bond) at ~428 and 447 cm−1.54,59
Spheroid nodules formed on BG films immersed in DS (Figure 12C and D) and DC (Figure 12E) also consisted of an apatite-like phase, but with a lower degree of ordering (eg, amorphous calcium phosphate), as suggested by the less defined (separated) and broader allure of the Raman bands. The structural ordering of the in vitro-grown deposits was decreasing in the order of SBF > DS > DC. In the case of the inorganic–organic media (ie, DS and DC), one can also notice the higher intensity of the band positioned at ~861 cm−1, which can be ascribed to the P-OH stretching mode of the (HPO4)2− group.54,59 The occurrence of this band suggests that labile ions, ie, (HPO4)2−, were present and occupied nonapatitic lattice sites. Therefore, the Raman analysis suggests that the material chemically deposited onto BG films in DS and DC is still in an intermediary structural development stage, in good agreement with the SEM results already presented.
Cytocompatibility tests in MSCs
MTS assays
Adhesion and proliferation of MSCs on BG coatings with the lowest (BG-O) and the highest (BG-5S) degrees of polymerization were studied at 24 hours after stem cells (SCs) were seeded. The results showed good biocompatibility of the BG coatings (Figure 13) compared with polycarbonate-treated surfaces dedicated to cell cultures. Differences between samples were found to be small, with no statistical significance (P>0.05).
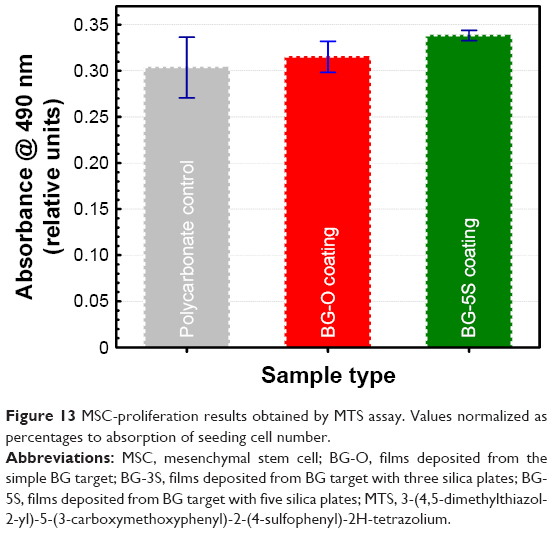 | Figure 13 MSC-proliferation results obtained by MTS assay. Values normalized as percentages to absorption of seeding cell number. |
Fluorescence microscopy
The morphology of MSCs grown on BG coatings was examined by epifluorescence microscopy. The cells were stained for the CD90 marker (specific for MSCs), and nuclei were counterstained with DAPI. All BG coatings showed very good biocompatibility, since the cells adhered in great numbers and retained normal morphology (Figure 14). Moreover, on BG-5S coatings, SCs began to organize in aggregates and spheres (Figure 14A and D). Usually, normal (nontumoral) cells grow in monolayers. Because they have a different growing-regulation mechanism, the SCs can develop spheroid colonies, where they replicate a complex system of SCs and their niche. Such behavior is usually observed in MSC cultures that retain their stemness properties.60
 | Figure 14 Immunofluorescence for CD90 expression in mesenchymal stem cells. |
The biocompatibility results (Figures 13 and 14) could be interpreted as proof that BG coatings alone enabled the adhesion and proliferation of SCs in the absence of an HA interlayer. This allows further asserting that the complex process of bone–implant healing somehow differs from the postulated time-dependent sequence of interfacial reactions, involving the formation of an HA-surface layer necessary for implant–cell interactions and bonding to living tissues. The inorganic and organic chemical interactions along with cellular development are rather interlaced processes.
Discussion
The morphostructural and compositional results presented suggest that the presence of amino acids and/or proteins in the in vitro testing media determined a slower biomineralization process and calcium phosphate deposits that were still in an incipient structuring stage after 28 days of immersion. This contrasts with the thick, rough, and fairly crystallized HA layer grown in SBF for the same incubation period.
Hindered nucleation of apatite in the absence of amino acids and/or organic compounds has also been reported in bioactivity tests performed only on bulk samples.15,25,28,30 However, no systematic and insightful assessments have been performed yet to shed light on the complex processes involved and their causality.
One possible explanation lies in the lower pH values attained by the DMEM solutions with respect to the SBF. In the case of SBF tests performed under normal atmosphere, after 28 days the solution reached pH values of 9–9.5, while for SBF tests performed in 5% CO2 atmosphere, after the same soaking period, the pH rose to ~7.8. A pH value in the range of 8–10 is known to be propitious for HA nucleation.14,57,61,62 However, such pH values are detrimental to biological equilibrium. This hints that testing the biological potential of a material by immersing it in SBF under a normal atmosphere is not a reliable assay, because the conditions found in vivo are not faithfully reproduced. The simple cell-culturing medium (DS) behaved analogously to SBF in a normal atmosphere, slowly turning to a very alkaline environment, with the pH reaching ~9, after 28 days. For DC kept in a normal atmosphere, pH reached 8–8.5. The supplemented DMEM kept in the incubator under homeostatic conditions had throughout the testing period a constant pH value of ~7.3–7.4, similar to that found in the human body.
On the other hand, the presence of amino acids24 and other serum proteins (ie, DC and DCC) could determine the formation of an organic protective layer by adsorption processes of such molecules onto the implant surface. Such a “screening” layer could play an important role in the retardation of bioactivity processes, by halting an earlier dissolution of the BG coating in the testing media, and thus consequently partially or even totally impede the ionic exchanges that govern the biomineralization mechanism.16,17,63
This explains why reduced structural changes were seen in DMEM for BG coatings with high silica content, which by the nature of their bonding architecture (strongly polymerized) already imprint to the material lower rates of solubilization in aqueous media. However, up to this point, no clues regarding the nature of such an organic layer have been provided.
To confirm the formation of the organic protective layer by adsorption processes of amino acids and/or serum proteins on the BG coatings, their surface chemistry was surveyed by XPS, a powerful tool whose sensitivity can discern differences in the chemical environment of relevant elements. Our XPS studies focused on the analysis of the N1s (Figure 15A) and C1s (Figure 15B) core-electron levels (nitrogen being a chemical element specific to the structure of both amino acids and proteins) in the case of BG-O coatings tested in various simulated body media. While no N1s signal could be detected at the limit of experimental sensitivity for BG-O coatings tested in SBF, the films tested in DS, DC, and DCC featured two or three component maxima (Table 3). The peaks situated at lower binding energy (ie, ~398.9 eV and 399.9–400.7 eV) were associated with neutral amine and carbon-bonded nitrogen in an amidic bond, respectively.64–72 The peak situated at higher binding energy (~400.3–402.2 eV) pertained to the protonated amine groups.64–72
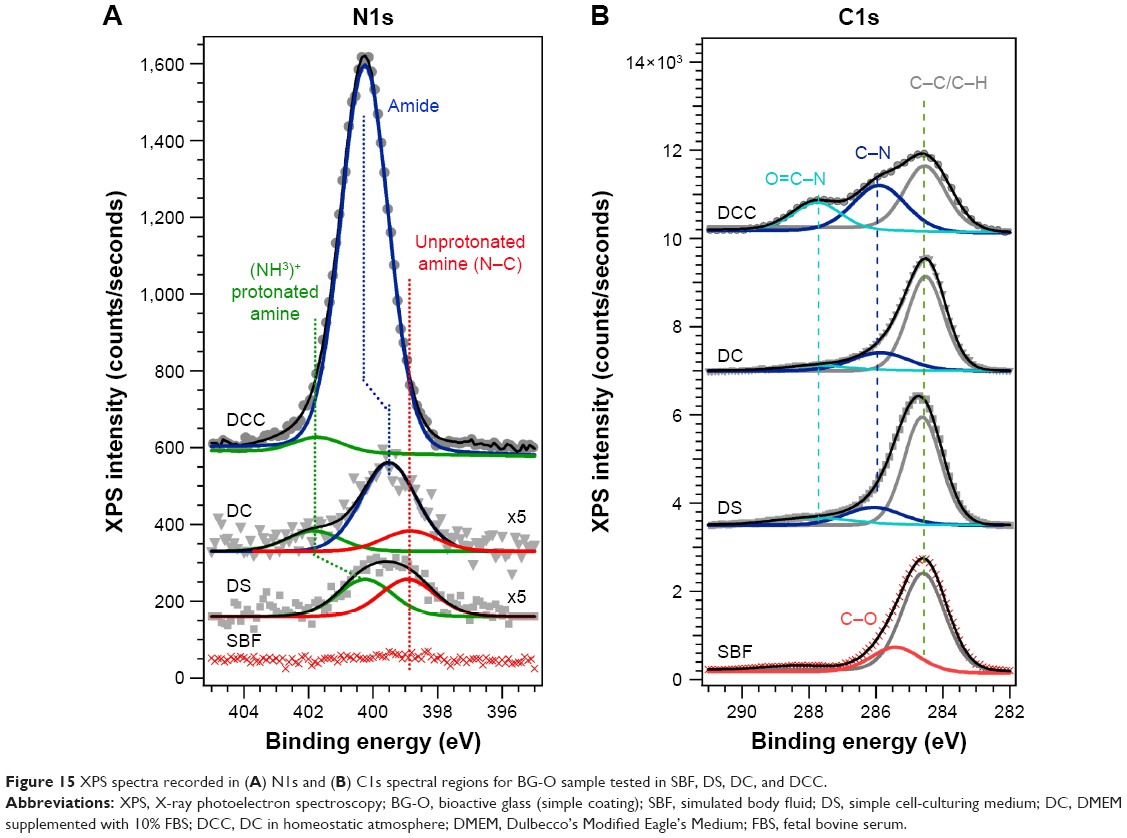 | Figure 15 XPS spectra recorded in (A) N1s and (B) C1s spectral regions for BG-O sample tested in SBF, DS, DC, and DCC. |
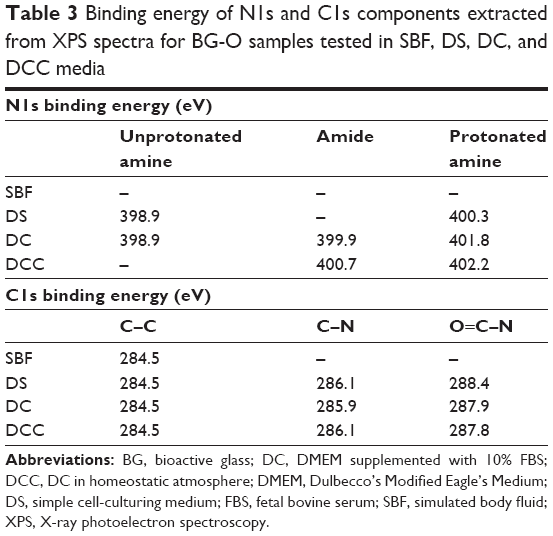 | Table 3 Binding energy of N1s and C1s components extracted from XPS spectra for BG-O samples tested in SBF, DS, DC, and DCC media |
In the case of samples tested in DS (which contained only amino acids), the spectra exhibited only the maxima of amino groups: protonated amino groups at higher binding energy (~400.3 eV), and unprotonated (neutral) amino groups at lower binding energies (~398.9 eV) (Figure 15A). The addition of fetal serum (for DC experiments), corroborated with a lower pH (thus a less reducing environment) induced a robust shift in the protonated amino groups component toward higher binding energies (401.8 eV), accompanied by the appearance of a central component: the signature of amides from the proteins (at ~399.9 eV). In the homeostatic regime (DCC tests), an even less reducing environment, the protonated group almost vanished, and the only component, a broad and intense peak, was associated to amidic formations/groups. We cannot exclude the presence of unprotonated amino groups, but their detection was beyond the resolution of the experimental setup. This behavior was to be expected, since in such conditions the pH is at physiologic values (~7.3–7.4), where many blood proteins are in the neutral state and can be adsorbed on the BG surface. In contrast, in the DC experiments (under normal air atmosphere), the pH increased and the proteins likely became more electronegative. For every molecule, the adsorption process can occur when from all driving forces (ie, chemical, electrostatic, Van der Waals, hydrogen bonds) the proadsorption ones overcome the ones against. The adsorption occurs until a new equilibrium state is reached. An elevated pH determines the formation of a great number of silanol groups at the BG–solution interface that create an electronegative surface, at which the electronegative proteins might be electrostatically impeded to adsorb in high quantities.
All C1s spectra (Figure 15B), regardless of medium type, featured a common component peaking at ~284.5 eV, assigned to C–C/C–H bonds of adventitious hydrocarbon contamination and/or aliphatic carbon of the amino acid-pending groups.65,68,69 When amino acids and proteins are present in the environment (DS, DC, DCC), a new component appears at ~285.9–286.1 eV, associated with C–N bonds.64,66,67,70–72 Its position is rather robust, irrespective of changes in the testing conditions (Table 3). For tested SBF samples, a second component was elicited at ~285.5 eV, ascribed to C–O bonds. The third component, signaled in the case of tested DS, DC, and DCC samples at higher binding energies, was assigned to O=C–N bonds, characteristic of amide groups.64,66–68,70–72 In the case of the DCC samples, one can notice a significant increase in the intensity of this component, in good agreement with the N1s findings (Figure 15A). Therefore, the XPS results confirmed the hypothesis of the formation of an organic screening layer (of amino acid and/or protein nature) that induced latency in the bioactivity process.
The XPS results encouraged us to continue the research to clarify this phenomenology further, in an attempt to unveil for the first time the nature of the organic screening layer formed under homeostatic conditions. With this purpose, the samples’ surface was assayed by a state-of-the-art biological analysis method – SELDI-ToF.
SELDI-ToF results showed an increase in protein adsorption on the surface of BG coatings with increasing amounts of FBS added to the testing medium (Figure 16A) and a positive time-dependent evolutionary trend (Figure 16B). The protein assignment to each SELDI peak is given in Table 4. The concentration of adsorbed proteins increased from 6 to 12 hours, and saturation seemed to be achieved after ~24 hours. The adsorbed level of proteins was likely conditioned by the timely evolution of the electrochemical properties of the BG surface. Interestingly, even if the albumin was present in high concentrations (>50% from the total serum proteins → 3 g of albumin per liter of serum), it was adsorbed in lower amounts than other proteins (eg, apolipoprotein D, haptoglobin, transferrin, plasminogen, ceruloplasmin) that are found in lower quantities in the testing medium (Table 4, Figure 17).
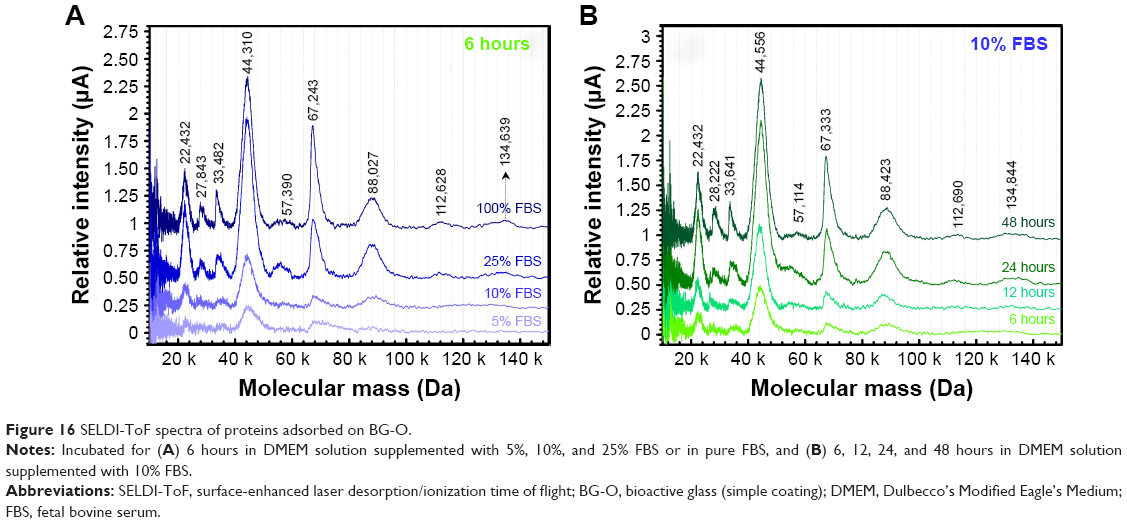 | Figure 16 SELDI-ToF spectra of proteins adsorbed on BG-O. |
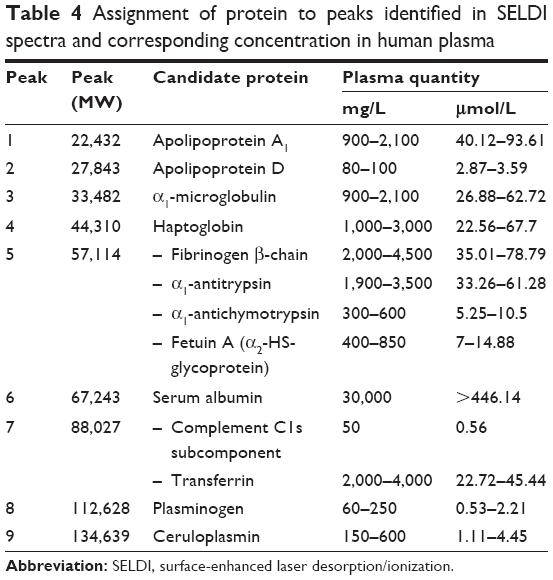 | Table 4 Assignment of protein to peaks identified in SELDI spectra and corresponding concentration in human plasma |
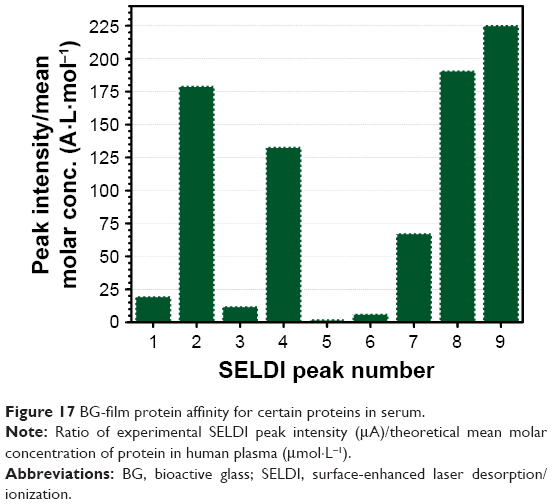 | Figure 17 BG-film protein affinity for certain proteins in serum. |
The affinity of radio-frequency magnetron sputtering BG coatings for certain proteins with pronounced hydrophilic character may be one of the explanations for their excellent cell biocompatibility. On the other hand, it also warrants the retarded formation and/or structuring of HA on the surface of BG films immersed in complex inorganic–organic environments that more closely mimic the composition of intercellular fluid. The formation of silanol (Si-OH) groups at the BG coating–testing medium interface in the early stages of incubation, as a consequence of the ionic exchange process and the breakage of Si–O–Si bonds followed by hydrolysis and condensation,16–19 could thus lead to more complex interactions than predicted: 1) with the inorganic part of the testing medium (as is only the case with SBF) determining the chemical growth of HA (largely acknowledged), and 2) with the organic component of the intercellular fluid, which in light of the findings presented here tends to delay the biomineralization processes at the implant surface.
However, similar evolutionary reduction trends in bioactivity were recorded (Figure 11) with increasing silica contents in BG films. High concentrations of network formers are known to enhance the structural polymerization of BG and decrease its bioreactivity,17 regardless of the type of in vitro testing conditions. Therefore, it is suggested that the biomineralization processes theorized in the past two decades on the basis of tests performed in SBF can be considered (somewhat) valid, but the speed with which these may occur must be seriously questioned. This needs to be emphasized, as the therapeutic efficiency of a BG for osseous reconstruction is appreciated precisely by the temporal interval required to achieve the bonding with the neighboring living tissue.16–19,73
Our results suggest that tests performed in DMEM-like media supplemented with proteins under correct homeostatic conditions are more appropriate and truthful assays to assess in vitro biomineralization capacity of a given glass coating than classical bioactivity tests in SBF. The classical approach can often force the biomineralization phenomena by unnaturally increasing the pH value. Moreover, the SBF solution is already supersaturated with respect to HA components,14 and even the lowest local increases in Ca2+ and (PO4)3− concentrations are likely to induce spontaneous precipitation of calcium phosphates and even calcite on the surfaces of the testing samples. Literature studies are available reporting on the inexplicable growth of HA layers on surfaces of otherwise-renowned bioinert materials (eg, zirconia, polymeric fibers).74,75 Tests performed in biomimetic media could eliminate such hazards and avoid false-positive events.
As highlighted in Table 1, research approaches are nowadays rather inhomogeneous, and hinder a reliable comparison of the mineralization capacity in simulated body media of an ever-growing range of BG compositions and formulations, as well pointed out in a recent round-robin SBF-testing article.58 Therefore, a further important issue is devising a unified SBF-testing procedure that can be applied in a more integrated and rational manner in the evaluation of bioactivity of bulk structures (eg, pellets, powders, scaffolds) and low-dimensional objects (eg, thin films).
As stated at the end of Introduction, the Vs:Sa ratio proposed in the ISO standard can no longer yield a reliable response concerning the multitudes of forms and dimensions of testing specimens. The modified method proposed in the round-robin SBF-testing article58 does not satisfactorily resolve this limitation. Irrespective of the composition and mimicry of the in vitro bioactivity-testing media, a meaningful unifying procedure still needs to be better equated. Considering that interfacial reactions take place at the solid–liquid interface, this desideration could be coherently solved based on the following premises:
- the Vs:Sa ratio needs to be constant for all types of samples (bulk, thin films, powders, scaffolds)
- the centimeter is a more convenient linear unit, as cubed centimeters directly translate in the unit volume (milliliters) commonly employed in SBF tests
- adopting the following standard shapes and dimensions (in centimeters) of bulk samples, which comply with ISO/FDIS 23317:201413 and have total surface areas of 3 cm2 (3×10−4 m2): 1) square shape of 1×1×0.25 cm, volume 0.25 cm3; 2) disc shape diameter 1.2 cm, thickness 0.2 cm, volume 0.23 cm3; or 3) any nonstandard shape/dimensions, provided the same Vs:Sa ratio is assured
- since Vs in ISO/FDIS 23317:201413 is clearly overestimated for thin films and underestimated for powders, a unifying value of Vs:Sa =20 mm (2 cm) is proposed for all types of samples, irrespective of their morphological/structural features, which is five times less than recommended in the ISO standard13
- accordingly, such bulk samples with Sa =3 cm2 should be immersed in 6 mL of SBF solution, corresponding to 0.5 cm2/mL, which could be designated as the unifying standard area (SA) per milliliter of SBF solution.
For spherical particles of uniform size, the specific surface area (SSA) is given by: SSA =6/ρd (ρ = specific density in kg·m−3 or in Mg·m−3), and d = diameter of the particles (meters). For powdered samples, the mass would be strictly dependent on density and average size of the particles and volume of simulated body media solution, as given by the equation:
|
|
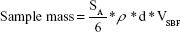
Table 5 reports examples of estimated mass for powders of different average particle sizes and densities covering the common range for BG. It can be seen that reasonable mass values are obtained for particles with average diameters within the 30–50 μm range, meaning that there are no obstacles to extending the same standard area (0.5 cm2/mL or 5×10−5 m2·mL−1) proposed for bulk samples to powdered samples. The same criterion can easily be applied to thin films. For example, a square thin film with an area of 1 cm2 (10−4 m2), a third of that of the standard bulk sample (3 cm2 or 3×10−4 m2), could be immersed in 2 mL of simulated body media solution (Vs:Sa =2 cm). Extending the same standard procedures to porous scaffolds just requires determining/estimating the area of contact between the testing material and the simulated body media solution. While this might be relatively easy for scaffolds fabricated by additive manufacturing based on geometrical considerations, more random structures will require complementary characterization techniques, such as microscopy coupled with image analysis. However, the most important output from this discussion is the demonstration that a unified method for bioactivity testing in simulated body media is feasible and desirable.
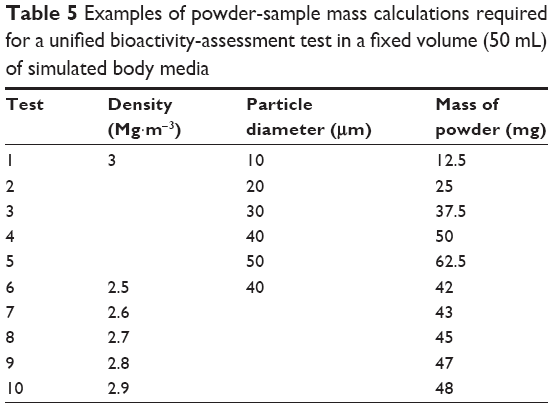 | Table 5 Examples of powder-sample mass calculations required for a unified bioactivity-assessment test in a fixed volume (50 mL) of simulated body media |
Conclusion
Insightful discussions about bioactivity assessment require the recognition of SBF-testing limitations and devising improved protocols for further rigorous consensual validation by the scientific community. This study aimed to contribute to this important target. The results presented and discussed enable the conclusion that bioactivity patterns of thin BG films in SBF differ strongly from those observed in inorganic–organic solutions that better mimic the internal medium under homeostatic conditions. Although biomineralization occurred under all studied conditions, the speed of the process was quite different. If for SBF assays the speed was high, for tests taking place in solutions that tend to mimic the internal medium, the speed decreased dramatically. When getting closer to biomimicry (amino acids, glucose and/or serum addition, CO2 partial pressure), the biomineralization process was slower.
The initial adsorption of organic molecules onto the surface of testing films within the first few hours of incubation was demonstrated by XPS and SELDI-ToF, which tend to slow the process by “screening” the BG matrix from the medium. Although the in vivo conditions can hardly be matched in vitro, it is our belief that only an exigent and homogeneous testing of BGs can filter the best formulations and boost their translation to clinical applications.
Irrespective of the medium used, it is of paramount importance to unify bioactivity-testing protocols for samples with different forms and dimensions (bulk objects, thin films, powders, scaffolds). Our discussion demonstrated that this target can easily be achieved (especially for bulk samples, thin films, and powders) by assuming the same standard area per milliliter (SA =0.5 cm2/mL or 5×10−5 m2·mL−1), which was derived from the area exposed by the bulk samples proposed in ISO/FDIS 23317:2014 and a testing volume of 6 mL SBF solution. This test protocol has a rational basis, and could easily be adopted by most research laboratories around the world to allow comparison of dissolution and apatite-nucleation rates. Whether the system could be static or kept under certain agitation conditions to get reproducible and comparable results is something that needs to be further investigated by the scientific community.
Acknowledgments
ACP, GES, and MAH are grateful for the financial support of the Romanian National Authority for Scientific Research and Innovation, CNCS-UEFISCDI, in the framework of projects PN-II-RU-TE-2011-4-0164 (contract 49/2011), PN-II-RU-TE-2014-4-0180 (contract 73/2015), and Core Programme PN 16 48-3/2016. The support of CICECO – Aveiro Institute of Materials (Ref UID/CTM/50011/2013), funded by FEDER funds through the Operational Programme Competitiveness Factors (COMPETE 2020), and the Portuguese Foundation for Science and Technology (FCT) is acknowledged. The authors thank Dr B Almeida, Dr M Enculescu, and Dr F Miculescu for the help provided with some of the SEM analyses.
Disclosure
The authors report no conflicts of interest in this work.
References
Hench LL, Splinter RJ, Allen WC, Greenlee TK. Bonding mechanisms at the interface of ceramic prosthetic materials. J Biomed Mater Res. 1971;5:117–141. | ||
Lee JH, Kang MS, Mahapatra C, Kim HW. Effect of aminated mesoporous bioactive glass nanoparticles on the differentiation of dental pulp stem cells. PLoS One. 2016;11:e0150727. | ||
Cai Y, Guo L, Shen H, et al. Degradability, bioactivity, and osteogenesis of biocomposite scaffolds of lithium-containing mesoporous bioglass and mPEG-PLGA-b-PLL copolymer. Int J Nanomedicine. 2015;10:4125–4136. | ||
Stuart BW, Titman JJ, Gimeno-Fabra M, Ahmeda I, Grant DM. Insights into structural characterisation and thermal properties of compositionally equivalent vapour-condensed and melt quenched glasses. Mater Des. 2016;111:174–184. | ||
Rath SN, Brandl A, Hiller D, et al. Bioactive copper-doped glass scaffolds can stimulate endothelial cells in co-culture in combination with mesenchymal stem cells. PLoS One. 2014;9:e113319. | ||
Chen XH, Zhao YB, Geng SN, et al. In vivo experimental study on bone regeneration in critical bone defects using PIB nanogels/boron-containing mesoporous bioactive glass composite scaffold. Int J Nanomedicine. 2015;10:839–846. | ||
Stuart BW, Gimeno-Fabra M, Segal J, Ahmed I, Grant DM. Degradation and characterization of resorbable phosphate-based glass thin-film coatings applied by radio-frequency magnetron sputtering. ACS Appl Mater Interfaces. 2015;7:27362–27372. | ||
Chen SJ, Jian ZY, Huang LS, et al. Mesoporous bioactive glass surface modified poly(lactic-co-glycolic acid) electrospun fibrous scaffold for bone regeneration. Int J Nanomedicine. 2015;10:3815–3827. | ||
Kokubo T, Kushitani H, Sakka S, Kitsugi T, Yamamuro T. Solutions able to reproduce in vivo surface-structure change in bioactive glass-ceramic A-W. J Biomed Mater Res. 1990;24:721–734. | ||
Oyane A, Onuma K, Ito A, Kim HM, Kokubo T, Nakamura T. Formation and growth of clusters in conventional and new kinds of simulated body fluids. J Biomed Mater Res A. 2003;64:339–348. | ||
Oyane A, Kim HM, Furuya T, Kokubo T, Miyazaki T, Nakamura T. Preparation and assessment of revised simulated body fluids. J Biomed Mater Res A. 2003;65:188–195. | ||
Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 2006;27:2907–2915. | ||
International Organization for Standardization. ISO 23317:2014 – Implants for Surgery: In Vitro Evaluation for Apatite-Forming Ability of Implant Materials. 3rd ed. Geneva: ISO; 2014. | ||
Bohner M, Lemaitre J. Can bioactivity be tested in vitro with SBF solution? Biomaterials. 2009;30:2175–2179. | ||
Lee JTY, Leng Y, Chow KL, et al. Cell culture medium as an alternative to conventional simulated body fluid. Acta Biomater. 2011;7:2615–2622. | ||
Hench LL, Best S. Ceramics, glasses, and glass-ceramics. In: Ratner BD, editor. Biomaterials Science: An Introduction to Materials in Medicine. New York: Elsevier; 2004:153–169. | ||
Hench LL. Bioceramics: from concept to clinic. J Am Ceram Soc. 1991;74:1487–1510. | ||
Hench LL. Chronology of bioactive glass development and clinical applications. New J Glass Ceram. 2013;3:67–73. | ||
Jones JA. Review of bioactive glass: from Hench to hybrids. Acta Biomater. 2013;9:4457–4486. | ||
Murphy MB, Suzuki RK, Sand TT, Chaput CD, Gregory CA. Short term culture of human mesenchymal stem cells with commercial osteoconductive carriers provides unique insights into biocompatibility. J Clin Med. 2013;2:49–66. | ||
Mader EK, Butler G, Dowdy SC, et al. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J Transl Med. 2013;11:20. | ||
Kim JH, Jekar DW, Kim M, et al. Effects of ECM protein mimetics on adhesion and proliferation of chorion derived mesenchymal stem cells. Int J Med Sci. 2014;11:298–308. | ||
Schopenhauer A. Aphorismenzur Lebensweisheit/The Wisdom of Life and Counsels and Maxims. Translated by TB Saunders. Amherst, NY: Prometheus Books;1995. | ||
Mahmood TA, Davies JE. Incorporation of amino acids within the surface reactive layers if bioactive glass in vitro: an XPS study. J Mater Sci Mater Med. 2000;11:19–23. | ||
Theodorou G, Goudouri OM, Kontonasaki E, et al. Comparative bioactivity study of 45S5 and 58S bioglasses in organic and inorganic environment. Bioceram Dev Appl. 2011;1:D110154. | ||
Lutišanová G, Palou MT, Kozánková J. Comparison of bioactivity in vitro of glass and glass ceramic materials during soaking in SBF and DMEM medium. Ceram Silikaty. 2011;55:199–207. | ||
Liu P, Tao JH, Cai YR, Pan HH, Xu XR, Tang RK. Role of fetal bovine serum in the prevention of calcification in biological fluids. J Cryst Growth. 2008;310:4672–4675. | ||
Shah FA, Brauer DS, Wilson RM, Hill RG, Hing KA. Influence of cell culture medium composition on in vitro dissolution behavior of a fluoride-containing bioactive glass. J Biomed Mater Res A. 2014;102:647–654. | ||
Tas AC. The use of physiological solutions or media in calcium phosphate synthesis and processing. Acta Biomater. 2014;10:1771–1792. | ||
Shah FA, Brauer DS, Hill RG, Jing KA. Apatite formation of bioactive glasses is enhanced by low additions of fluoride but delayed in the presence of serum proteins. Mater Lett. 2015;153:143–147. | ||
Faure J, Balamurugan A, Benhayoune H, Torres P, Balossier G, Ferreira JM. Morphological and chemical characterisation of biomimetic bone like apatite formation on alkali treated Ti6Al4V titanium alloy. Mater Sci Eng C Mater Biol Appl. 2009;29:1252–1257. | ||
Socol G, Macovei AM, Miroiu F, et al. Hydroxyapatite thin films synthesized by pulsed laser deposition and magnetron sputtering on PMMA substrates for medical applications. Mater Sci Eng B Adv Funct Solid State Mater. 2010;169:159–168. | ||
Pan H, Zhao X, Darvell BW, Lu WW. Apatite-formation ability: predictor of “bioactivity”? Acta Biomater. 2010;6:4181–4188. | ||
Kapoor S, Goel A, Correia AF, et al. Influence of ZnO/MgO substitution on sintering, crystallisation, and bio-activity of alkali-free glass-ceramics. Mater Sci Eng C Mater Biol Appl. 2015;53:252–261. | ||
Stan GE, Pina S, Tulyaganov DU, Ferreira JM, Pasuk I, Morosanu CO. Biomineralization capability of adherent bio-glass films prepared by magnetron sputtering. J Mater Sci Mater Med. 2010;21:1047–1055. | ||
Galca AC, Stan GE, Trinca LM, Negrila CC, Nistor LC. Structural and optical properties of c-axis oriented aluminum nitride thin films prepared at low temperature by reactive radio-frequency magnetron sputtering. Thin Solid Films. 2012;524:328–333. | ||
Teodorescu CM, Esteva JM, Karnatak RC, El Afif A. An approximation of the Voigt I profile for the fitting of experimental X-ray absorption data. Nucl Instrum Methods Phys Res A. 1994;345:141–147. | ||
Mardare D, Luca D, Teodorescu CM, Macovei D. On the hydrophilicity of nitrogen-doped TiO2 thin films. Surf Sci. 2007;601:4515–4520. | ||
Kapoor S, Goel A, Tilocca A, et al. Role of glass structure in defining the chemical dissolution behavior, bioactivity and antioxidant properties of zinc and strontium co-doped alkali-free phosphosilicate glasses. Acta Biomater. 2014;10:3264–3278. | ||
Agathopoulos S, Tulyaganov DU, Ventura JM, Kannan S, Karakassides MA, Ferreira JM. Formation of hydroxyapatite onto glasses of the CaO-MgO-SiO2 system with B2O3, Na2O, CaF2 and P2O5 additives. Biomaterials. 2006;27:1832–1840. | ||
Ziemath EC, Aegerter MA. Raman and infrared investigations of glass and glass-ceramics with composition 2Na2O-1CaO-3SiO2. J Mater Res. 1994;1:216–225. | ||
Bellucci D, Bolelli G, Cannillo V, Cattini A, Sola A. In situ Raman spectroscopy investigation of bioactive glass reactivity: simulated body fluid solution vs Tris-buffered solution. Mater Charact. 2011;62:1021–1028. | ||
Lin CC, Huang LC, Shen P. Na2CaSi2O6-P2O5 based bioactive glasses – part 1: elasticity and structure. J Non Cryst Solids. 2005;351:3195–3203. | ||
Kapoor S, Semitela A, Goel A, et al. Understanding the composition-structure-bioactivity relationships in diopside (CaO·MgO·2SiO2)-tricalcium phosphate (3CaO·P2O5) glass system. Acta Biomater. 2015;15:210–226. | ||
Surmenev RA. A review of plasma-assisted methods for calcium phosphate-based coatings fabrication. Surf Coat Technol. 2012;206:2035–2056. | ||
Verbist K, Van Tendeloo G, Ye M, Schroeder J, Mehbod M, Deltour R. Inclusions in magnetron sputtered YBa2Cu3–xMxO7–δ thin films: a study by means of electron microscopy. Microsc Microanal Microstruct. 1996;7:17–25. | ||
Aguiar H, Solla EL, Serra J, et al. Orthophosphate nanostructures in SiO2-P2O5-CaO-Na2O-MgO bioactive glasses. J Non Cryst Solids. 2008;354:4075–4080. | ||
Ma J, Chen CZ, Wang DG, Meng XG, Shi JZ. Influence of the sintering temperature on the structural feature and bioactivity of sol-gel derived SiO2-CaO-P2O5 bioglass. Ceram Int. 2010;36:1911–1916. | ||
Lin CC, Chen SF, Leung KS, Shen P. Effects of CaO/P2O5 ratio on the structure and elastic properties of SiO2-CaO-Na2O-P2O5 bioglasses. J Mater Sci Mater Med. 2012;23:245–258. | ||
Luz GM, Mano JF. Preparation and characterization of bioactive glass nanoparticles prepared by sol-gel for biomedical applications. Nanotechnology. 2011;22:494014. | ||
Stan GE, Popa AC, Galca AC, Aldica G, Ferreira JM. Strong bonding between sputtered bioglass-ceramic films and Ti-substrate implants induced by atomic inter-diffusion post-deposition heat-treatments. Appl Surf Sci. 2013;280:530–538. | ||
Stuart B, Gimeno-Fabra M, Segal J, Ahmed I, Grant DM. Preferential sputtering in phosphate glass systems for the processing of bioactive coatings. Thin Solid Films. 2015;589:534–542. | ||
Popa AC, Stan GE, Besleaga C, et al. Submicrometer hollow bioglass cones deposited by radio frequency magnetron sputtering: formation mechanism, properties, and prospective biomedical applications. ACS Appl Mater Interfaces. 2016;8:4357–4367. | ||
Markovic M, Fowler BO, Tung MS. Preparation and comprehensive characterization of a calcium hydroxyapatite reference material. J Res Natl Inst Stand Technol. 2004;109:553–568. | ||
Stan GE, Pasuk I, Husanu MA, et al. Highly adherent bioactive glass thin films synthetized by magnetron sputtering at low temperature. J Mater Sci Mater Med. 2011;22:2693–2710. | ||
Berbecaru C, Stan GE, Pina S, Tulyaganov DU, Ferreira JM. The bioactivity mechanism of magnetron sputtered bioglass thin films. Appl Surf Sci. 2012;258:9840–9848. | ||
Müller L, Müller FA. Preparation of SBF with different HCO3− content and its influence on the composition of biomimetic apatites. Acta Biomater. 2006;2:181–189. | ||
Maçon AL, Kim TB, Valliant EM, et al. A unified in vitro evaluation for apatite forming ability of bioactive glasses and their variants. J Mater Sci Mater Med. 2015;26:115. | ||
Koutsopoulos S. Synthesis and characterization of hydroxyapatite crystals: a review study on the analytical methods. J Biomed Mater Res. 2002;62:600–612. | ||
Popa AC, Stan GE, Enculescu M, Tanase C, Tulyaganov DU, Ferreira JM. Superior biofunctionality of dental implant fixtures uniformly coated with durable bioglass films by magnetron sputtering. J Mech Behav Biomed Mater. 2015;51:313–327. | ||
Zhang D, Hupa M, Hupa L. In situ pH within particle beds of bioactive glasses. Acta Biomater. 2008;4:1498–1505. | ||
Cerruti M, Greenspan D, Powers K. Effect of pH and ionic strength on the reactivity of Bioglass 45S5. Biomaterials. 2005;26:1665–1674. | ||
O’Donnell MD. Melt-derived bioactive glasses. In: Jones J, Clare A, editors. Bio-Glasses: An Introduction. New York: Wiley; 2012:13–27. | ||
Shchukarev A, Mladenovic Z, Ransjö M. Surface characterization of bone graft substitute materials conditioned in cell culture medium – 2: protein adsorption. Surf Interface Anal. 2012;44:919–923. | ||
Vanea E, Simon V. XPS study of protein adsorption onto nanocrystalline aluminosilicate microparticles. Appl Surf Sci. 2011;257:2346–2352. | ||
Deligianni DD, Katsala N, Ladas S, Sotiropoulou D, Amedee J, Missirlis YF. Effect of surface roughness of the titanium alloy Ti-6Al-4V on human bone marrow cell response and on protein adsorption. Biomaterials. 2001;22:1241–1251. | ||
Carvalho I, Galindo RE, Henriques M, Palacio C, Carvalho S. Influence of culture media on the physical and chemical properties of Ag-TiCN coatings. J Phys D Appl Phys. 2014;47:335401. | ||
Gruian C, Vanea E, Simon S, Simon V. FTIR and XPS studies of protein adsorption onto functionalized bioactive glass. Biochim Biophys Acta. 2012;1824:873–881. | ||
Barazzouk S, Daneault C. Amino acid and peptide immobilization on oxidized nanocellulose: spectroscopic characterization. Nanomaterials (Basel). 2012;2:187–205. | ||
Bartiaux S, Lhoest JB, Genet MJ, Bertrand P, Rouxhet PG. Poly(amino-acids) by XPS: analysis of poly(L-serine). Surf Sci Spectra. 1994;3:342–347. | ||
Hurisso BB, Lovelock KRJ, Licence P. Amino acid-based ionic liquids: using XPS to probe the electronic environment via binding energies. Phys Chem Chem Phys. 2011;13:17737–17748. | ||
Stevens JS, de Luca AC, Pelendritis M, Terenghi G, Downes S, Schroeder SL. Quantitative analysis of complex amino acids and RGD peptides by X-ray photoelectron spectroscopy (XPS). Surf Interface Anal. 2013;45:1238–1246. | ||
Karlsson KH, Hupa L. Thirty-five years of guided tissue engineering. J Non Cryst Solids. 2008;354:717–721. | ||
Thaveedeetrakul A, Witit-Anun N, Boonamnuayvitaya V. The role of target-to-substrate distance on the DC magnetron sputtered zirconia thin films’ bioactivity. Appl Surf Sci. 2012;258:2612–2619. | ||
Renke-Gluszko M, El Fray M. The effect of simulated body fluid on the mechanical properties of multiblock poly(aliphatic/aromatic-ester) copolymers. Biomaterials. 2004;25:5191–5198. | ||
Ha SW, Reber R, Eckert KL, et al. Chemical and morphological changes of vacuum-plasma-sprayed hydroxyapatite coatings during immersion in simulated physiological solutions. J Am Ceram Soc. 1998;81:81–88. | ||
Ding SJ, Ju CP, Lin JH. Morphology and immersion behavior of plasma sprayed hydroxyapatite/bioactive glass coatings. J Mater Sci Mater Med. 2000;11:183–190. | ||
Oki A, Parveen B, Hossain S, Adeniji S, Donahue H. Preparation and in vitro bioactivity of zinc containing sol-gel-derived bioglass materials. J Biomed Mater Res A. 2004;69:216–221. | ||
Li X, Wang XP, Hua ZL, Shi JL. One-pot synthesis of magnetic and mesoporous bioactive glass composites and their sustained drug release property. Acta Mater. 2008;56:3260–3265. | ||
Zhang K, Washburn NR, Simon CG Jr. Cytotoxicity of three-dimensionally ordered macroporous sol-gel bioactive glass (3DOM-BG). Biomaterials. 2005;26:4532–4539. | ||
Rouahi M, Champion E, Gallet O, Jada A, Anselme K. Physico-chemical characteristics and protein adsorption potential of hydroxyapatite particles: Influence on in vitro biocompatibility of ceramics after sintering. Colloids Surf B Biointerfaces. 2006;47:10–19. | ||
Jones JR, Ehrenfried LM, Saravanapavan P, Hench LL. Controlling ion release from bioactive glass foam scaffolds with antibacterial properties. J Mater Sci Mater Med. 2006;17:989–996. | ||
Juhasz JA, Best SM, Auffret AD, Bonfield W. Biological control of apatite growth in simulated body fluid and human blood serum. J Mater Sci Mater Med. 2008;19:1823–1829. | ||
Xia W, Chang J. Preparation, in vitro bioactivity and drug release property of well-ordered mesoporous 58S bioactive glass. J Non Cryst Solids. 2008;354:1338–1341. | ||
Fu Q, Rahaman MN, Sonny Bal B, Brown RF, Day DE. Mechanical and in vitro performance of 13–93 bioactive glass scaffolds prepared by a polymer foam replication technique. Acta Biomater. 2008;4:1854–1864. | ||
Lusvardi G, Malavasi G, Menabue L, Aina V, Morterra C. Fluoride-containing bioactive glasses: surface reactivity in simulated body fluids solutions. Acta Biomater. 2009;5:3548–3562. | ||
Aina V, Malavasi G, Pla AF, Munaron L, Morterra C. Zinc-containing bioactive glasses: surface reactivity and behaviour towards endothelial cells. Acta Biomater. 2009;5:1211–1222. | ||
Mandel S, Tas AC. Brushite (CaHPO4·2H2O) to octacalcium phosphate (Ca8(HPO4)2(PO4)4·5H2O) transformation in DMEM solutions at 36.5°C. Mater Sci Eng C Mater Biol Appl. 2010;30:245–254. | ||
Brauer DS, Karpukhina N, O’Donnell MD, Law RV, Hill RG. Fluoride-containing bioactive glasses: effect of glass design and structure on degradation, pH and apatite formation in simulated body fluid. Acta Biomater. 2010;6:3275–3282. | ||
Yan PH, Wang JQ, Ou JF, Li ZP, Lei ZQ, Yang SR. Synthesis and characterization of three-dimensional ordered mesoporous-macroporous bioactive glass. Mater Lett. 2010;64:2544–2547. | ||
Soundrapandian C, Bharati S, Basu D, Datta S. Studies on novel bioactive glasses and bioactive glass-nano-Hap composites suitable for coating on metallic implants. Ceram Int. 2011;37:759–769. | ||
Plewinski M, Schickle K, Lindner M, Kirsten A, Weber M, Fischer H. The effect of crystallization of bioactive bioglass 45S5 on apatite formation and degradation. Dent Mater. 2013;29:1256–1264. | ||
Rohanová D, Boccaccini AR, Horkavcová D, Bozděchová P, Bezdičkac P, Castorálová M. Is non-buffered DMEM solution a suitable medium for in vitro bioactivity tests? J Mater Chem B Mater Biol Med. 2014;2:5068–5076. | ||
Ananth KP, Suganya S, Mangalaraj D, Ferreira JM, Balamurugan A. Electrophoretic bilayer deposition of zirconia and reinforced bioglass system on Ti6Al4V for implant applications: an in vitro investigation. Mater Sci Eng C Mater Biol Appl. 2013;33:4160–4166. | ||
Wu ZY, Hill RG, Yue S, Nightingale D, Lee PD, Jones JR. Melt-derived bioactive glass scaffolds produced by a gel-cast foaming technique. Acta Biomater. 2011;7:1807–1816. | ||
Bellucci D, Sola A, Anesi A, Salvatori R, Chiarini L, Cannillo V. Bioactive glass/hydroxyapatite composites: mechanical properties and biological evaluation. Mater Sci Eng C Mater Biol Appl. 2015;51:196–205. | ||
Bolelli G, Bellucci D, Cannillo V, et al. Comparison between suspension plasma sprayed and high velocity suspension flame sprayed bioactive coatings. Surf Coat Technol. 2015;280:232–249. | ||
Romeis S, Hoppe A, Detsch R, Boccaccini AR, Schmidt J, Peukert W. Top-down processing of submicron 45S5 bioglass for enhanced in vitro bioactivity and biocompatibility. Procedia Eng. 2015;102:534–541. | ||
Nicolini V, Varini E, Malavasi G, et al. The effect of composition on structural, thermal, redox and bioactive properties of Ce-containing glasses. Mater Des. 2016;97:73–85. | ||
Dziadek M, Zagrajczuk B, Menaszek E, Wegrzynowicz A, Pawlik J, Cholewa-Kowalska K. Gel-derived SiO2-CaO-P2O5 bioactive glasses and glass-ceramics modified by SrO addition. Ceram Int. 2016;42:5842–5857. | ||
Lopez-Esteban S, Saiz E, Fujino S, Oku T, Suganuma K, Tomsia AP. Bioactive glass coatings for orthopedic metallic implants. J Eur Ceram Soc. 2003;23:2921–2930. | ||
Mindat [website on the Internet]. Brushite. Available from: http://www.mindat.org/min-793.html. Accessed September 20, 2016. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.