Back to Journals » International Journal of Nanomedicine » Volume 11
Biodegradable mixed MPEG-SS-2SA/TPGS micelles for triggered intracellular release of paclitaxel and reversing multidrug resistance
Authors Dong K, Yan Y, Wang PC, Shi XP, Zhang L, Wang K, Xing JF, Dong YL ![]()
Received 3 May 2016
Accepted for publication 5 September 2016
Published 6 October 2016 Volume 2016:11 Pages 5109—5123
DOI https://doi.org/10.2147/IJN.S111930
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Kai Dong,1 Yan Yan,2 Pengchong Wang,2 Xianpeng Shi,2 Lu Zhang,2 Ke Wang,2 Jianfeng Xing,2 Yalin Dong1
1Department of Pharmacy, The First Affiliated Hospital of Xi’an Jiaotong University, 2School of Pharmacy, Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China
Abstract: In this study, a type of multifunctional mixed micelles were prepared by a novel biodegradable amphiphilic polymer (MPEG-SS-2SA) and a multidrug resistance (MDR) reversal agent (D-α-tocopheryl polyethylene glycol succinate, TPGS). The mixed micelles could achieve rapid intracellular drug release and reversal of MDR. First, the amphiphilic polymer, MPEG-SS-2SA, was synthesized through disulfide bonds between poly (ethylene glycol) monomethyl ether (MPEG) and stearic acid (SA). The structure of the obtained polymer was similar to poly (ethylene glycol)-phosphatidylethanolamine (PEG-PE). Then the mixed micelles, MPEG-SS-2SA/TPGS, were prepared by MPEG-SS-2SA and TPGS through the thin film hydration method and loaded paclitaxel (PTX) as the model drug. The in vitro release study revealed that the mixed micelles could rapidly release PTX within 24 h under a reductive environment because of the breaking of disulfide bonds. In cell experiments, the mixed micelles significantly inhibited the activity of mitochondrial respiratory complex II, also reduced the mitochondrial membrane potential, and the content of adenosine triphosphate, thus effectively inhibiting the efflux of PTX from cells. Moreover, in the confocal laser scanning microscopy, cellular uptake and 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide assays, the MPEG-SS-2SA/TPGS micelles achieved faster release and more uptake of PTX in Michigan Cancer Foundation-7/PTX cells and showed better antitumor effects as compared with the insensitive control. In conclusion, the biodegradable mixed micelles, MPEG-SS-2SA/TPGS, could be potential vehicles for delivering hydrophobic chemotherapeutic drugs in MDR cancer therapy.
Keywords: reversal of multidrug resistance, reduction-sensitive, disulfide bond, mixed micelles, MCF-7/PTX cells
Introduction
Nowadays, chemotherapy has become the major treatment modality for cancers. However, the effect of chemotherapy has been seriously affected by the development of multidrug resistance (MDR).1–3 MDR arises from the course of chemotherapy treatment; the increased use of chemotherapeutic drugs will aggravate MDR and make drugs less effective. Although many efforts have been made to overcome the MDR cancer, limited successful therapeutic regimens and antineoplastic agents have been applied.4–9 The MDR of cancer cells results from many factors; one of the most important reasons is the reduction of drug accumulation caused by the superfamily of adenosine triphosphate (ATP)-binding cassette proteins, such as the P-glycoprotein (P-gp), MDR-associated protein (MRP), and breast cancer resistance protein (BCRP).10 In cancer cells, the overexpression of these proteins would pump out the chemotherapy drugs and would make the intracellular drug concentration less than the threshold value of cell lethal dose, thus reducing the cytotoxicity of drugs and developing MDR.
Currently, paclitaxel (PTX) is the most successful and widely used anticancer drug. It has a broad antitumor spectrum, such as lung cancer, refractory ovarian cancer, and metastatic breast cancer.11–14 However, the long-term use of PTX causes serious MDR, especially in advanced and metastatic tumors.15 Moreover, the low water solubility of PTX limits its clinical application.16 Therefore, research studies have focused on the nano-polymer drug delivery system in order to improve the water solubility of PTX and reverse MDR.17–20 For example, Wang et al and Zhang et al developed a series of mixed micelles based on Pluronic copolymers (Pluronic P105/L101 micelles and Pluronic F127/P123 micelles) to overcome MDR.21–23 They reported that the mixed micelles composed of different kinds of Pluronic copolymers could enhance the PTX accumulation in resistant cancer cells, such as resistant breast cancer Michigan Cancer Foundation-7/adriamycin (MCF-7/ADR) cells and resistant ovarian cancer SKOV-3/PTX cells, thus enhancing the cytotoxicity of PTX and reversing MDR. However, the self-assembled Pluronic micelles are easily diluted in the blood stream because of their high critical micelle concentration (CMC) value, thereby reducing their stability. Moreover, poor micellization and solubilization capacities of hydrophobic drugs are also major limitations of Pluronic copolymers. Therefore, this study aimed to prepare a kind of mixed micelles composed of two different polymers, one has a low CMC value in order to maintain the stability of micelles and the other one can reverse MDR. Poly (ethylene glycol)-phosphatidylethanolamine (PEG-PE) is a US Food and Drug Administration (FDA)-approved diblock copolymer with good biocompatibility and safety.24 The micelles composed of PEG-PE have a low CMC value (1×10−5 mol/L) and present as a 9 nm radius of sphere, including a distearoyl phosphoethanolamine core (2 nm radius) and distributed phosphorus atoms (2.5–3.5 nm radius) with negative charge.25–27 In this study, a novel amphiphilic polymer, MPEG-SS-2SA, was synthesized by replacing the phosphorus atoms with disulfide bonds, and the MPEG-SS-2SA had a similar structure to PEG-PE and a low CMC value. The micelles composed of MPEG-SS-2SA could release drugs quickly through the break of disulfide bonds in response to the reducing agent in cells (eg, glutathione, GSH).28,29 D-α-Tocopheryl polyethylene glycol succinate (TPGS) was used as the MDR reversal agent, which is the combination of polyethylene glycol (PEG) and α-tocopheryl succinate (α-TOS) and is an FDA-approved pharmaceutical excipient.30 Recent studies showed that TPGS was an effective P-gp efflux inhibitor and could selectively kill cancer cells.31 TPGS could affect the synthesis of ATP through inhibiting the functions of mitochondria, and in turn inhibit the ATP-dependent P-gp efflux.32,33 Therefore, a reduction-sensitive biodegradable mixed micelles, MPEG-SS-2SA/TPGS, were developed to achieve effective intracellular release of PTX and reverse MDR. The reduction-triggered drug release, in vitro cytotoxicity, mitochondrial respiratory complex II activity assay, mitochondrial membrane potential and ATP assay, and intracellular drug accumulation were performed. Compared with the reduction-insensitive micelles (MPEG-2SA/TPGS micelles, non-MDR reversal MPEG-SS-2SA micelles) and free PTX, the MPEG-SS-2SA/TPGS micelles showed desired reduction-sensitivity and effective reverse of MDR.
Materials and methods
Materials
PTX was purchased from Xi’an Sanjiang Bio-Engineering Co. Ltd, People’s Republic of China. Poly (ethylene glycol) monomethyl ether (MPEG, Mn =2,000), p-nitrophenyl chloroformate (p-NPC, 97%), and triethylamine (TEA) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Cystamine dihydrochloride (cystamine·2HCl), 3-amino-1,2-propanediol (ADP), stearic acid (SA), dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), N, N′-carbonyldiimidazole (CDI), and pyridine (Py, 99.5%) were obtained from Aladdin Chemistry (Shanghai, People’s Republic of China). Dithiothreitol (DTT, 99%) was purchased from Merck (Darmstadt, Germany). 3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was purchased from Sigma-Aldrich Co. Mito Tox™ OXPHOS Complex II Activity Kit was purchased from Novagen (Darmstadt, Germany). Mitochondrial membrane potential assay kit with JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyllenzimidazolycarbocyanine iodide), ATP assay kit, rhodamine123 (R123), Hoechst 33342, and BCA protein assay kit were purchased from Beyotime Biotechnology Co. Ltd (Nantong, People’s Republic of China). All other reagents were of analytical grade and were obtained from commercially available sources.
The MCF-7 human breast cancer cell line was obtained from the Cell Bank of Shanghai, Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, People’s Republic of China). The PTX-resistant MCF-7 (MCF-7/PTX) cell line was established as previously described.34 Culture plates and dishes were purchased from Corning Incorporated (Corning, NY, USA). Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium, supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, and 100 mg/mL streptomycin sulfate. All the cells were grown at 37°C in a humidified incubator containing 5% CO2 and routinely tested negative for mycoplasma. All the experiments were performed on cells during the logarithmic phase of growth. The study protocol was approved by the Xi’an Jiaotong University Medical and Biological Research Ethics Committee.
Synthesis and characterization of MPEG-SS-2SA
Synthesis of MPEG-ADP
First, the terminal hydroxyl group of MPEG was activated by p-NPC to obtain MPEG-p-NPC. Briefly, MPEG (12 g, 6 mmol) and p-NPC (3.63 g, 18 mmol) were dissolved in dichloromethane (DCM), respectively. TEA (2.6 mL, 18 mmol) was added to the MPEG solution under stirring, and then the mixed solution was added dropwise in p-NPC solution with continuous stirring for 12 h. The MPEG-p-NPC was purified by precipitation in cold diethylether, filtration, and drying in vacuum at 30°C (yield: 90.1%).
Second, the MPEG-p-NPC was reacted with ADP to obtain the MPEG-ADP. MPEG-p-NPC (6.45 g, 3.0 mmol) was dissolved in DCM, and then a thin film was formed after the solvent was removed under reduced pressure (vacuum degree: 0.09 Mpa). ADP (0.816 g, 9 mmol) was dissolved in hydrochloric acid (12 mL, 0.01 mol/L); then the solution was added to the film and the mixture was dispersed by ultrasound. Na2CO3-NaHCO3 solution (60 mL, 0.2 mol/L, pH 9.0) was added in the mixture, and the reaction was allowed to proceed under stirring for 12 h. The MPEG-ADP was purified by dialysis (MWCO 1000) against deionized water and was finally obtained by lyophilization (yield: 46.3%).
Synthesis of MPEG-SS-2SA
The hydroxyl groups of MPEG-ADP were activated by p-NPC again to obtain MPEG-ADP-p-NPC. Briefly, MPEG-ADP (2.12 g, 1.0 mmol) and p-NPC (1.22 g, 6 mmol) were dissolved in DCM, respectively. The TEA (0.84 mL, 6 mmol) was added to the MPEG-ADP solution dropwise in p-NPC solution and stirred for 12 h. The MPEG-ADP-p-NPC was purified by precipitation, filtration, and drying in vacuum (yield: 91.5%).
The obtained MPEG-ADP-p-NPC (1.94 g, 0.8 mmol) and cystamine·2HCl (2.52 g, 16.8 mmol) were dissolved in dimethyl sulfoxide (DMSO), respectively. Then the MPEG-ADP-p-NPC solution was added dropwise to the cystamine·2HCl solution, followed by adding TEA (3.16 mL, 22.8 mmol). The reaction was proceeded under N2 atmosphere at room temperature for 24 h. The MPEG-ADP-Cys was purified by dialysis (MWCO 1000) against deionized water and was obtained by lyophilization (yield: 41.4%).
SA (0.2 g, 0.7 mmol) was first dissolved in DCM. Then DCC (0.22 g, 1.05 mmol) and NHS (0.12 g, 1.05 mmol) were added to the solution and reacted under N2 atmosphere at 25°C for 4 h. MPEG-ADP-Cys (0.99 g, 0.4 mmol) was added to the SA solution and was allowed to react continuously for 12 h under N2 atmosphere. The resulting MPEG-SS-2SA was purified through precipitated in cold diethylether twice and dried in vacuum at room temperature (yield: 85.8%).
The reduction insensitive control polymer, MPEG-2SA, was synthesized by MPEG-ADP and SA under the catalysis of CDI. Briefly, SA (0.31 g, 1.08 mmol) was first dissolved in DCM. Then CDI (0.2 g, 1.2 mmol) was added to the solution and reacted under N2 atmosphere at 25°C for 24 h. Then MPEG-ADP (1.31 g, 0.6 mmol) was added to the SA solution and was allowed to react continuously for 12 h under N2 protection. The resulting MPEG-2SA was separated by precipitation and drying in vacuum at room temperature (yield: 87.2%).
1H nuclear magnetic resonance (NMR) spectra were recorded on a 300 MHz 1H NMR spectrometer (Bruker, Karlsruhe, Germany) using deuterated chloroform (CDCl3) as the solvent. Chemical shifts (δ) were measured in ppm downfield from the internal standard tetramethylsilane. The CMC value was determined using pyrene as fluorescence probe. The concentrations of MPEG-SS-2SA and MPEG-2SA varied from 1×10−6 to 1×10−4 mol/L, and the concentration of pyrene was 0.6 μM. The fluorescence spectra were recorded by RF-5301PC fluorescence spectrometer (Shimadzu, Kyoto, Japan), the excitation wavelength was 330 nm, and the emission fluorescence was monitored at 372 and 383 nm. The CMC value was determined as the cross-point when the intensity ratio I372/I383 was extrapolated at low and high concentration regions.
Preparation and characterization of PTX-loaded mixed micelles
The MPEG-SS-2SA/TPGS/PTX micelles were prepared through thin film hydration method. Briefly, PTX and a mixture (100 mg) of MPEG-SS-2SA and TPGS (molar ratio 1:1) were dissolved in 10 mL acetonitrile and sonicated for 10 min. The solvent was evaporated by rotary evaporation at 50°C for 30 min in order to obtain a PTX/copolymer thin film. Residual acetonitrile remaining in the film was removed under vacuum overnight at room temperature. The resultant thin film was hydrated with 5 mL water at 60°C for 30 min in order to obtain the micellar solution. Then the solution was filtrated through a 0.22-μm membrane filter to remove the unencapsulated PTX aggregate. Also, the MPEG-2SA/TPGS/PTX and MPEG-SS-2SA/PTX micelles were prepared as described earlier.
The three types of micelles were characterized by Malvern Zetasizer Naso ZS (Malvern Instruments, Malvern, UK) and transmission electron microscopy (TEM) on a Hitachi H 600 (Hitachi Ltd., Tokyo, Japan) instrument. The drug-loading coefficient (DL%) and entrapment efficiency (EE%) were calculated by using the following equations, respectively:
|
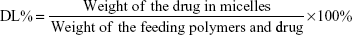
|

Reduction-triggered release of PTX from mixed micelles
The release behavior of PTX from these mixed micelles was investigated by dialysis tubes (MWCO 1000) at 37°C in two different media, that is, phosphate-buffered saline (PBS) (50 mM, pH 7.4) with or without 10 mM DTT. In order to acquire sink conditions, both the media contained 1 mol/L sodium salicylate, and 5 mL of micelle solution was dialyzed against 50 mL of the same medium.35 At predetermined time intervals, 1 mL release media were taken out and replenished with an equal volume of fresh media. The samples were filtered through 0.22-μm membrane filter and the amount of released PTX was determined by a reverse phase high-performance liquid chromatography method, which used a C18 BDS column (5 μm, 250×4.6 mm, Thermo Fisher Scientific, Waltham, MA, USA) and a mobile phase consisting of acetonitrile and water (55:45, v/v) at 1.0 mL/min. The linearity of PTX concentration was assessed by a calibration that was obtained by plotting the peak area of each analyte versus PTX concentration. The detected concentrations were in the range of 0.1–100 μg/mL. Samples were monitored at 227 nm and quantified by comparing the peak areas with the standard curve. The release experiments were conducted in triplicate, and the results at each time point were divided by the initial value in order to obtain relative values.
In vitro cytotoxicity
The MTT assay was used to investigate the cytotoxicity of the synthesized polymers (MPEG-SS-2SA and MPEG-2SA), PTX-loaded and blank micelles on MCF-7/PTX cells. Briefly, MCF-7/PTX cells were seeded in 96-well plates at a density of 5×103 cells per well and incubated overnight. After 24 h, the old growth medium was removed, and the following operations were performed: for the cytotoxicity of synthesized polymers, cells were incubated with MPEG-SS-2SA, MPEG-2SA, and TPGS growth medium solution at designated concentrations for 24 and 48 h; for the cytotoxicity of PTX-loaded micelles, cells were incubated with MPEG-SS-2SA/TPGS/PTX, MPEG-2SA/TPGS/PTX, MPEG-SS-2SA/PTX, and free PTX of various concentrations (from 0.001 to 10 μg/mL, diluted by the growth medium) for 48 h. Moreover, cells were also incubated with three blank and mixed micelles (diluted by the growth medium with the same dilution as the 10 μg/mL PTX-loaded micelles solution) for 48 h.
At the scheduled time, the wells were rinsed with PBS and then replaced with MTT solution. The plates were further incubated for 4 h at 37°C, allowing the viable cells to reduce the yellow MTT into purple formazan crystals. The supernatant was then carefully removed from each well, and 150 mL of DMSO was added to dissolve the crystals after further incubation. An enzyme-linked immunosorbent assay microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA) was used to record the absorbance at 570 nm. The cell cytotoxicity of these PTX formulations was quantified as cell viability, calculated according to the following equation:
|

Mitochondrial respiratory complex II activity assay
The activity of mitochondrial respiratory complex II was measured by a Mito Tox™ OXPHOS Complex II Activity Kit. Specifically, MCF-7/PTX cells were seeded in 12-well plates at a density of 5×105 cells per well and incubated overnight. Then the old growth medium was removed, and the cells were treated with different blank formulations (MPEG-SS-2SA/TPGS, MPEG-2SA/TPGS, MPEG-SS-2SA, and blank medium) for 0, 0.5, 1, 2, 4, and 8 h at 37°C. At predetermined time intervals, the tested medium was withdrawn, and the cells were washed three times with cold PBS, followed by adding 150 μL radio immunoprecipitation assay (RIPA) lysis buffer and being incubated for 10 min at 37°C. These samples were measured by an enzyme-linked immunosorbent assay microplate reader (Bio-Rad Laboratories Inc.) at the absorbance of 600 nm. The activity of mitochondrial respiratory complex II was normalized by the protein content in each sample, using a BCA kit. The blank medium was used as control. The relative activity of mitochondrial respiratory complex II was calculated by using the following equation:
|
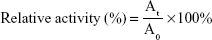
where At was the activity of mitochondrial respiratory complex II at time t and A0 was the activity of mitochondrial respiratory complex II before incubation.
Mitochondrial membrane potential assay
The change of mitochondrial membrane potential was assessed by using the lipophilic cationic membrane potential-sensitive dye JC-1.36 Briefly, the confluent MCF-7/PTX cells were treated with different blank formulations (MPEG-SS-2SA/TPGS, MPEG-2SA/TPGS, MPEG-SS-2SA, and blank medium) for 12 h and then washed three times with cold PBS. The trypsinized cells (5×105) were suspended in 500 μL of diluted JC-1 staining solution for 20 min. Then the cells were rinsed with physiological saline, subsequently, suspended in 500 μL of JC-1 staining buffer, and immediately the fluorescence intensities of FL1 (green fluorescence) and FL2 (red fluorescence) channels were measured by flow cytometry (BD FACSCalibur, Franklin Lakes, NJ, USA). The obtained values were expressed as average JC-1 FL2/FL1 (red/green) signal intensity ratio.
ATP content assay
To investigate the effects of blank micelles on intracellular ATP levels, the confluent MCF-7/PTX cells were treated with different blank micelles (MPEG-SS-2SA/TPGS, MPEG-2SA/TPGS, and MPEG-SS-2SA) for 2 h. Then the cells were washed twice with ice-cold PBS and were solubilized in cell lysates, followed by immediate centrifugation (12,000× g) at 4°C for 10 min. The supernatant was collected for ATP quantification by the luciferin/luciferase assay. The light emission of each sample was measured with an Ultra-Weak Luminescence Analyzer (BPCL, Beijing, People’s Republic of China). Raw data were converted to ATP concentration according to the standard calibration curve. ATP contents were normalized by protein content in each sample, using a BCA kit. The blank medium was used as control.
In vitro cellular uptake
Qualitative uptake
In this part, R123, a substrate for P-gp, was used instead of PTX to perform the cellular qualitative uptake assay.37–39 Specifically, MCF-7/PTX cells (1×105 per well) were seeded into a 12-well plate in RPMI-1640 complete medium containing one glass coverslip/well and incubated for 24 h. After that, the medium was removed and 2 mL growth medium (containing R123-loaded MPEG-SS-2SA/TPGS, MPEG-2SA/TPGS, MPEG-SS-2SA, and free R123) at a R123 concentration of 100 μg/mL was added into each well, respectively, and incubated for 4 h at 37°C. After incubation, coverslips were taken off, washed with PBS for three times, placed in empty wells, and treated with 1 mL of 4% paraformaldehyde for 15 min. After that, the cells were washed with PBS, and then cell nucleus was counterstained by Hoechst 33342 for 5 min. The cells were washed again by PBS and were analyzed by confocal laser scanning microscope (CLSM).
Quantitative uptake
The cellular quantitative uptake of PTX-loaded micelles was conducted by using liquid chromatography-tandem mass spectrometry (LC-MS/MS) method. Specifically, MCF-7/PTX cells, which were in the logarithmic growth phase, were seeded in 12-well plates at 1×105 per well. After 24 h, the old growth medium was removed, and the cells were exposed to MPEG-SS-2SA/TPGS/PTX, MPEG-2SA/TPGS/PTX, MPEG-SS-2SA/PTX, and PTX growth medium solution, respectively, at a PTX concentration of 5 μg/mL for 4 h at 37°C. At predetermined time intervals, cells were washed three times with cold PBS in order to terminate the uptake and remove the PTX-loaded micelles absorbed on the cell membrane. Then the cells were lysed in 200 μL RIPA lysis buffer for 10 min at 37°C. After incubation, 100 μL of cell lysate was withdrawn and extracted with acetonitrile (200 μL per sample). Then the mixture was treated with ultrasound in ice bath for further PTX extraction. After centrifuged at 12,000× g for 5 min, the supernatant of the extracts was analyzed by LC-MS/MS, and the protein content was determined using a BCA protein assay kit. The LC-MS/MS method was performed on a Hypersil GOLD C18 column, using a mixture solution of 0.1% formic acid (A) and acetonitrile containing 0.1% formic acid (B) as the mobile phase. The flow rate was 0.4 mL/min, and the run time was 5.5 min. The gradient elution was delivered as follows: at the start of run 80/20% of A/B; from 1.5 to 5.5 min, the gradient starts at 20% A and ramps to 80% A. Approximately 10 mL of the sample was injected into the system by an auto-sampler, and the column temperature was 20°C. The mass spectrometer was operated in an electronic spray positive ion mode. The selected reaction monitoring transitions used for quantification were performed at m/z 854.25/509.12 (collision energy: 15 eV) for PTX and m/z 830/527.14 (collision energy: 25 eV) for docetaxel (internal standard). The linearity was assessed by weighted (1/X2) least-squares linear regression of calibration curves based on peak area ratios of PTX to docetaxel versus actual concentrations. The detected concentrations were in the range of 5–5,000 ng/mL. The cellular accumulation of PTX was normalized with the total protein content. The uptake index (UI) was calculated by using the following equation:
UI = C/P |
where C is the concentration of PTX in the cell lysis solution. P is the protein concentration in the cell lysis solution.
Statistical analysis
Statistical analyses were performed with SPSS version 13.0 for Windows (SPSS Inc., Chicago, IL, USA). All the results were expressed as mean ± standard deviation. The statistical significance was determined by analysis of variance among ≥3 groups or Student’s t-test between the two groups. P<0.05 was considered to be statistically significant. All statistical tests were two-sided, and the confidence intervals were within 95%.
Results and discussion
Synthesis and characterization of MPEG-SS-2SA
During synthesis, MPEG first reacted with ADP after its terminal hydroxyl group was activated by p-NPC. Then, the MPEG-ADP was reacted with cystamine·2HCl under the activation of p-NPC again, in order to introduce disulfide bonds. Last, SA, the hydrophobic portion, was bound to the MPEG-ADP-Cys under the catalysis of DCC and NHS in order to obtain the final product, MPEG-SS-2SA (Figure 1). The 1H NMR results of these products in each step are illustrated in Figure 2. Specifically, during the synthesis of MPEG-ADP, p-NPC was employed to activate the terminal hydroxyl groups of MPEG, and then MPEG-ADP was formed through amide groups synthesized by the amino group of ADP. The peaks appeared at 8.28–8.34 and 7.30–7.52 ppm which belonged to the benzene ring of p-NPC (Figure 2A). However, these peaks disappeared when connected to ADP and new peaks appeared at 2.23 ppm, which were referred to the ortho-dihydroxy groups of ADP (Figure 2B).
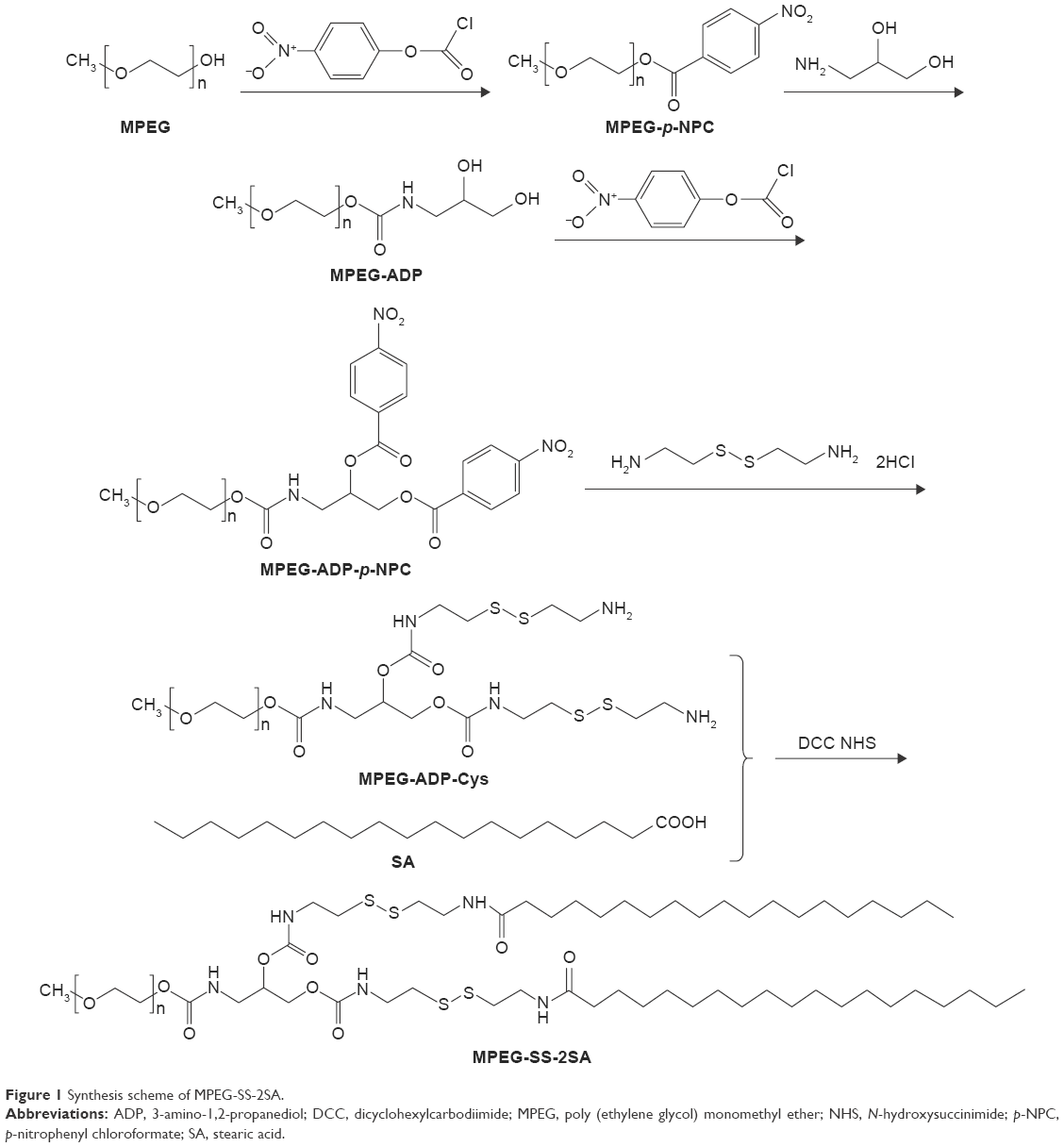 | Figure 1 Synthesis scheme of MPEG-SS-2SA. |
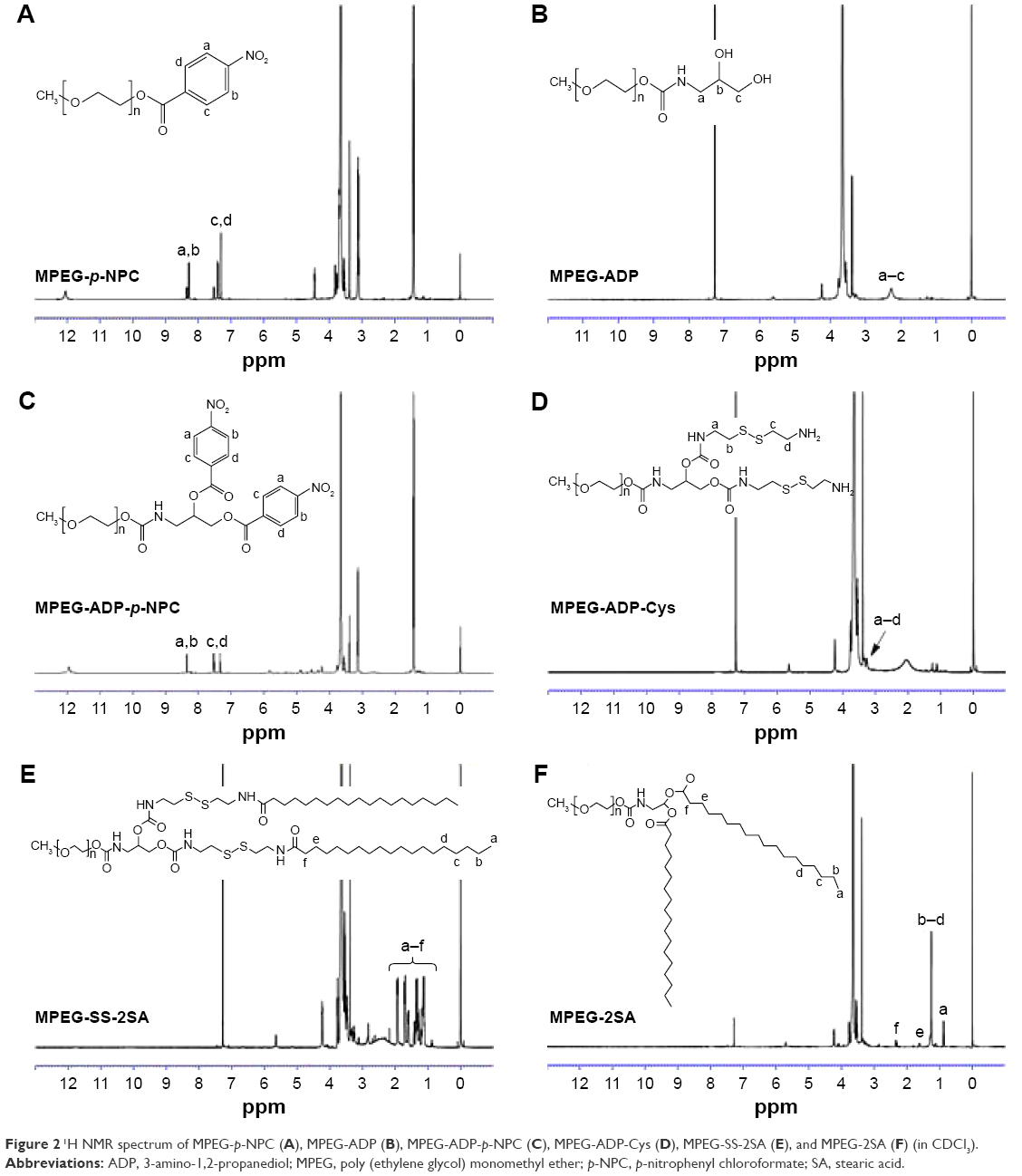 | Figure 2 1H NMR spectrum of MPEG-p-NPC (A), MPEG-ADP (B), MPEG-ADP-p-NPC (C), MPEG-ADP-Cys (D), MPEG-SS-2SA (E), and MPEG-2SA (F) (in CDCl3). |
MPEG-ADP-Cys was synthesized by activating the ortho-dihydroxy groups of ADP with p-NPC, then reacting with excessive cystamine 2HCl in the presence of TEA. From Figure 2C, the characteristic peaks of benzene ring appeared at 8.34–8.36 and 7.34–7.53 ppm after being activated by p-NPC. After the reaction with cystamine 2HCl, the amide bonds were formed. Thereby, the peaks of the benzene ring disappeared and new peaks appeared at 3.20–3.30 ppm, which were attributed to the methylene protons near the disulfide bonds (Figure 2D). The final product, MPEG-SS-2SA, was synthesized through amide bonds between MPEG-ADP-Cys and SA under the catalysis of DCC and NHS. A series of peaks appeared at 0.88–1.94 ppm, which belonged to the protons of methyl and methylene groups of SA (Figure 2E).
Moreover, the 1H NMR spectra of MPEG-2SA were similar to that of MPEG-SS-2SA (Figure 2F). A series of peaks also appeared at 0.89–2.23 ppm which belonged to the protons of SA. However, the peaks that belonged to the methylene protons near the disulfide bond did not appear in the spectra of MPEG-2SA. The CMC value was determined using pyrene as probe. MPEG-SS-2SA showed a CMC value of 2.2×10−5 mol/L, and the value of MPEG-2SA was 1.9×10−5 mol/L. Both of them were slightly higher than that of PEG-PE (1×10−5 mol/L).
Characterization of PTX-loaded mixed micelles
The physical characterization and drug-loading parameters of these mixed micelles were summarized in Table 1. The characterization information showed that the size of MPEG-SS-2SA/PTX micelles was ~30 nm, with relatively low polydispersity indexes. After combination with a designated amount of TPGS, the size of MPEG-SS-2SA/TPGS/PTX micelles increased to ~60 nm. This was because the two polymers had different structures, namely, the proportion of hydrophilic and hydrophobic segments in each amphiphilic polymer (MPEG-SS-2SA and TPGS) was different, thus affecting the morphology of each mixed micelle. The size of MPEG-2SA/TPGS micelles was slightly smaller than that of MPEG-SS-2SA/TPGS micelles (43 nm), which was probably due to the lack of disulfide bonds in MPEG-2SA. Moreover, as depicted in Figure 3, all of the three micelles were spherical and homogeneous in shape and correlated well with the results of laser diffraction particle size. In many solid tumors, the pore size of permeable vasculature is >50 nm. Therefore, these micelles could be able to selectively accumulate in solid tumors by the enhanced permeability and retention effect to achieve passive targeting.40,41
 | Table 1 Physical characteristics of PTX-loaded mixed micelles (mean ± SD, n=3) |
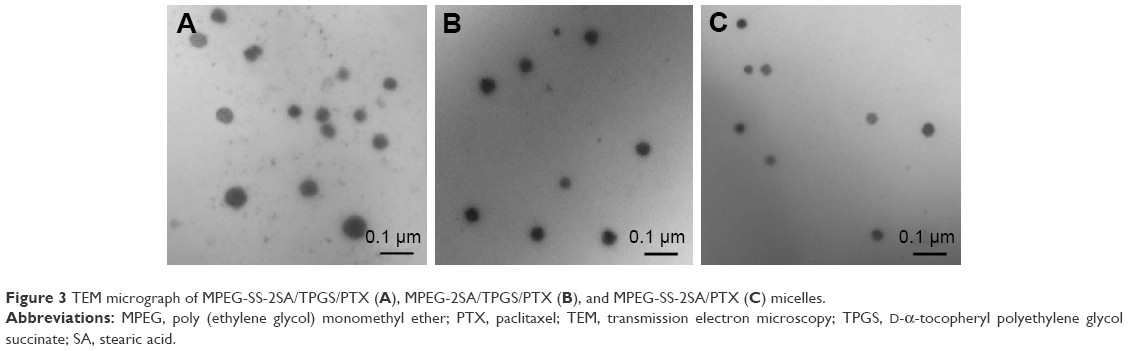 | Figure 3 TEM micrograph of MPEG-SS-2SA/TPGS/PTX (A), MPEG-2SA/TPGS/PTX (B), and MPEG-SS-2SA/PTX (C) micelles. |
Reduction-triggered release of PTX
The release of PTX from the mixed micelles was investigated by dialysis tubes (MWCO 1000) in pH 7.4 PBS with or without 10 mM DTT at 37°C. Furthermore, in order to acquire sink conditions, both the media contained 1 mol/L sodium salicylate, since PTX has high solubility in sodium salicylate solution (28.1 μg/mL), and 5 mL of micelle solution was dialyzed against 50 mL of the same medium. DTT is a kind of small molecule reducing agent, which can degrade disulfide bonds into thiol groups. Moreover, the concentration of GSH in cancer cells is higher than that in normal cells; this reductive environment of cancer cells will promote breaking of the disulfide bonds.42–44 Therefore, the media with 10 mM DTT imitate a reductive environment analogous to that of the intracellular compartments of cancer cells, such as cytosol and nucleus.45 Results showed that micelles contained disulfide bonds that released PTX rapidly in the media with DTT. Specifically, as shown in Figure 4A, MPEG-SS-2SA micelles showed the fastest release of PTX (almost all PTX released after 12 h), followed by the MPEG-SS-2SA/TPGS micelles (>70% PTX released after 24 h), the MPEG-2SA/TPGS micelles without disulfide bonds released minimum amount of PTX (<40%). However, in the media without DTT, these three kinds of micelles exhibited similar release behaviors (<45% PTX released after 24 h, Figure 4B). Therefore, it was obvious that the cleavage of disulfide bonds caused by DTT could degrade micelles and release drugs quickly. Moreover, due to the nondegradable TPGS, the MPEG-SS-2SA/TPGS mixed micelles could not degrade only by the cleavage of disulfide bonds; therefore, they showed slower degradation and drug release than MPEG-SS-2SA micelles. For MPEG-2SA/TPGS micelles, they could only release drugs by diffusion because of the lack of disulfide bonds, thus achieving the minimal drug release. In the media without DTT, these three kinds of micelles all released PTX by diffusion and showed similar release behaviors.
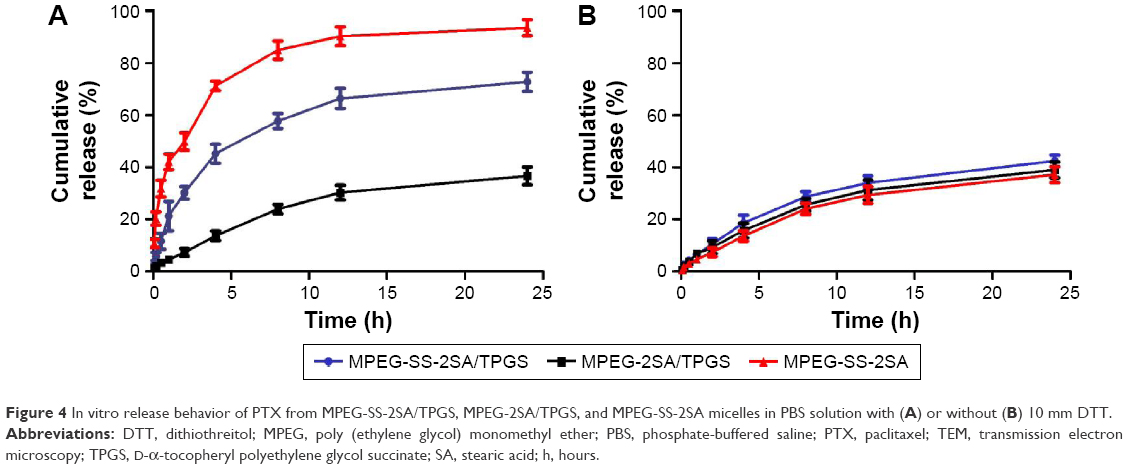 | Figure 4 In vitro release behavior of PTX from MPEG-SS-2SA/TPGS, MPEG-2SA/TPGS, and MPEG-SS-2SA micelles in PBS solution with (A) or without (B) 10 mm DTT. |
In vitro cytotoxicity
As shown in Figure 5A and B, MPEG-SS-2SA and MPEG-2SA did not present any cytotoxicity in 24 or 48 h with the increase of polymer concentrations, and the cell viabilities were all >85%. However, the cytotoxicity of TPGS on MCF-7/PTX cells increased significantly with the increase in concentration, indicating that TPGS could obviously induce cell apoptosis, which was consistent with related references.46–49 Figure 5C showed that when compared with free PTX, the drug-loaded micelles significantly reduced the cell viability. The IC50 value of MCF-7/PTX cell line was 10.68±2.14 μg/mL for free PTX, 2.35±0.26 μg/mL for MPEG-SS-2SA/PTX, 0.32±0.04 μg/mL for MPEG-2SA/TPGS/PTX, and 0.11±0.02 μg/mL for MPEG-SS-2SA/TPGS/PTX. As compared with micelles without TPGS and the free PTX, the mixed micelles with TPGS exhibited much higher cytotoxicity against MCF-7/PTX cells. The MPEG-SS-2SA/TPGS/PTX micelles showed higher cytotoxicity than MPEG-2SA/TPGS/PTX, suggesting that breaking of disulfide bonds caused the degradation of micelles and the release of PTX and TPGS more quickly, thereby achieving greater toxicity. Compared with the free PTX, the MPEG-SS-2SA micelles could transport more PTX into cells relying on their amphiphilic structure and passive targeting ability. However, due to the lack of TPGS, the MPEG-SS-2SA micelles were unable to effectively suppress drug efflux; hence, their toxicity was less than that of the two TPGS micelles. From Figure 5D, the two blank TPGS micelles also expressed certain cytotoxicity (cell viability was <45% for both). Moreover, the MPEG-SS-2SA/TPGS exhibited a slightly higher cytotoxicity than MPEG-2SA/TPGS, which was due to the rapid degradation of micelles and the release of TPGS caused by disulfide bonds. However, the cytotoxicity of the two blank micelles was both weaker than the PTX-loaded micelles. The blank MPEG-SS-2SA micelles barely expressed cytotoxicity, indicating that the introduction of TPGS significantly enhanced the cytotoxicity of micelles and also promoted the absorption and effects of PTX.
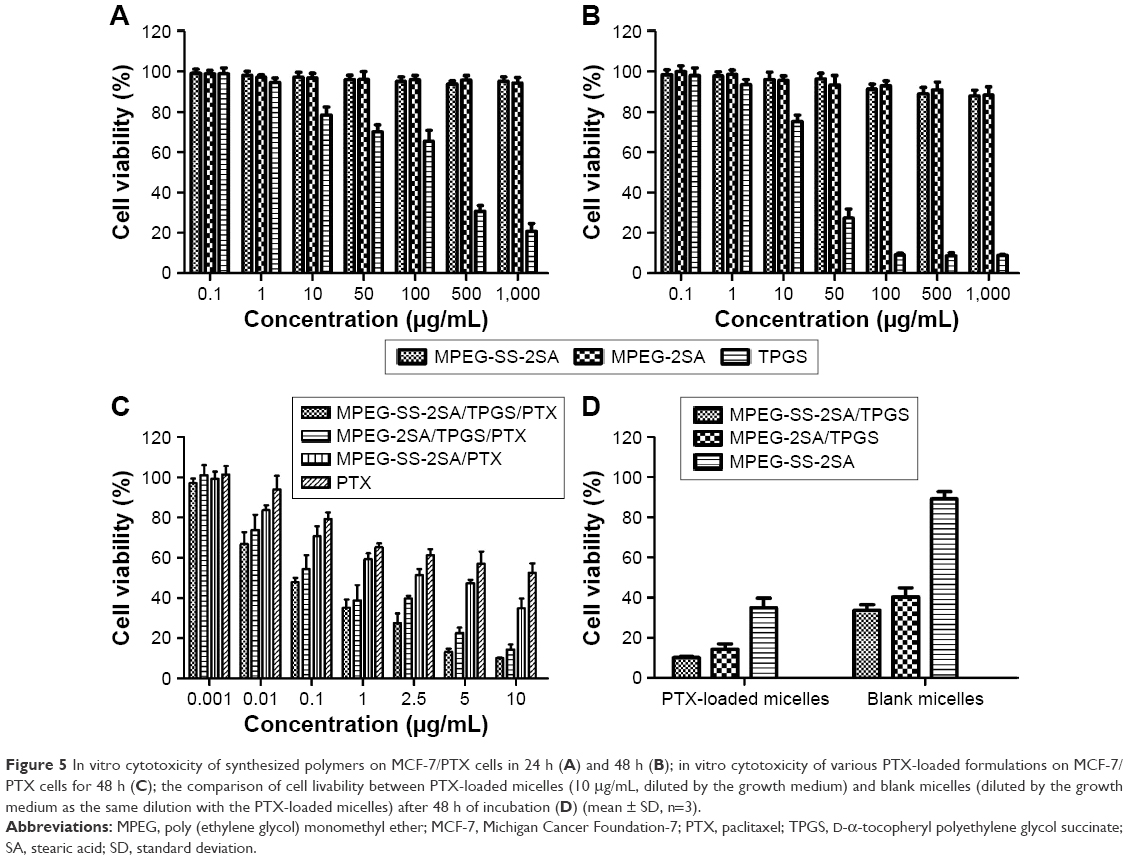 | Figure 5 In vitro cytotoxicity of synthesized polymers on MCF-7/PTX cells in 24 h (A) and 48 h (B); in vitro cytotoxicity of various PTX-loaded formulations on MCF-7/PTX cells for 48 h (C); the comparison of cell livability between PTX-loaded micelles (10 μg/mL, diluted by the growth medium) and blank micelles (diluted by the growth medium as the same dilution with the PTX-loaded micelles) after 48 h of incubation (D) (mean ± SD, n=3). |
Effect of mixed micelles on the activity of mitochondrial respiratory complex II
Mitochondrial respiratory complex II, also known as the succinate-coenzyme Q reductase or succinate dehydrogenase, is an important part of the mitochondrial electron transport, which links the tricarboxylic acid cycle to mitochondrial electron transport chain.50 Current research studies have shown that the mechanism of cell apoptosis induced by TPGS was mainly due to the destruction and inhibition of mitochondrial respiratory complex II.51–53 The dysfunction of complex II can cause the leakage of electrons during the transfer in the complex II and the activation of calcium channel, resulting in the mitochondria and intracellular overload of calcium and eventually leading to the disappearance of the mitochondrial membrane potential, which is one of the mechanisms of mitochondrial dysfunction.54,55 Micelles containing TPGS significantly inhibited the activity of mitochondrial respiratory complex II, and the inhibition effect gradually increased over the incubation time (Figure 6). However, the MPEG-SS-2A micelles barely affected the activity of complex II. Moreover, MPEG-SS-2SA/TPGS showed stronger inhibition effect than MPEG-2SA/TPGS, indicating that, because of the breaking of disulfide bonds, the TPGS was quickly released and resulted in stronger inhibition. However, the MPEG-2SA/TPGS micelles showed relatively weak inhibition due to the lack of disulfide bonds.
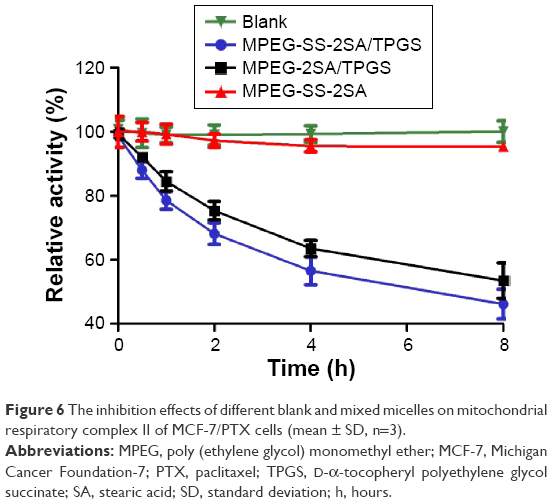 | Figure 6 The inhibition effects of different blank and mixed micelles on mitochondrial respiratory complex II of MCF-7/PTX cells (mean ± SD, n=3). |
Effect of mixed micelles on mitochondrial function and ATP level
Recently, many studies have demonstrated that TPGS can damage the structure of mitochondria and the energy of cells, by disrupting the electron transport of the mitochondrial respiratory chain as discussed earlier, thus reducing the drug efflux caused by energy-dependent efflux pumps.56,57 In order to further investigate the mechanism of resistance reversal of the mixed micelles, the present study investigated the effects of these micelles on mitochondria and ATP level.
The effects on mitochondrial membrane potential (MP) and ATP level of MCF-7/PTX cells were assessed with different formulations (MPEG-SS-2SA/TPGS, MPEG-2SA/TPGS, and MPEG-SS-2SA). The MP was determined by JC-1, which was able to reversibly transform from a monomer form (green florescence) to an aggregate form (red florescence) when it was bound with high MP. Mitochondrial depolarization (nonfunctional mitochondria) was indicated by a decrease in the ratio of red/green fluorescence intensity. The MPEG-SS-2SA/TPGS micelles significantly decreased the average JC-1 red/green fluorescence intensity in MCF-7/PTX cells (R/G =1.15±0.16, P<0.01; Figure 7A). Moreover, the MPEG-2SA/TPGS micelles achieved a similar effect (R/G =1.57±0.26, P<0.01). The subtle difference between the two micelles was caused by the disulfide bonds which made the MPEG-SS-2SA/TPGS micelles release TPGS quickly, thereby significantly reducing the MP. However, the MPEG-SS-2SA micelles showed no effect on the MP (R/G =3.69±0.17) due to the lack of TPGS. The intracellular ATP level was determined by luciferin/luciferase assay. The cells treated with blank medium were used as control, and the ATP level was normalized as 100%. As depicted in Figure 7B, the MPEG-SS-2SA/TPGS micelles significantly decreased the ATP level of MCF-7/PTX cells (49% of the normal level, P<0.01). However, because of the absence of disulfide bonds, the MPEG-2SA/TPGS micelles were unable to quickly release TPGS or reduce the cellular energy supply (67% of the normal level, P<0.01). Compared with the former two micelles, the MPEG-SS-2SA micelles slightly dropped the ATP level to 93% of the normal level.
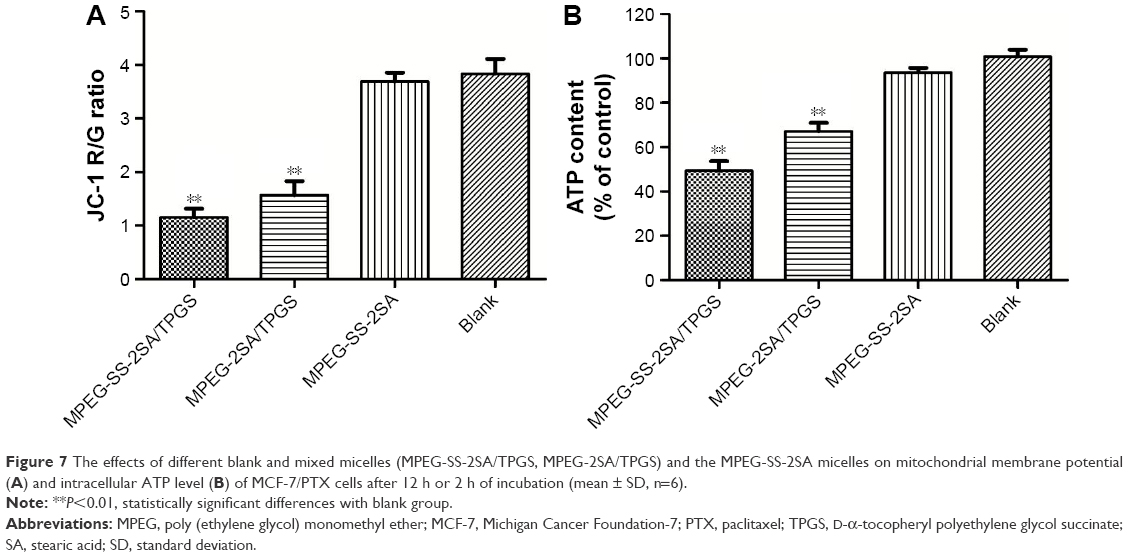 | Figure 7 The effects of different blank and mixed micelles (MPEG-SS-2SA/TPGS, MPEG-2SA/TPGS) and the MPEG-SS-2SA micelles on mitochondrial membrane potential (A) and intracellular ATP level (B) of MCF-7/PTX cells after 12 h or 2 h of incubation (mean ± SD, n=6). |
The depletion of ATP is extremely important for MDR reversal since many efflux transporters are ATP-dependent proteins, such as P-gp, MRP, and BCRP.58 They need to consume energy to maintain their function in MDR cells. In this work, the MPEG-SS-2SA/TPGS micelles decreased both MP and ATP levels with the aid of TPGS. It indicated that the disulfide bonds of MPEG-SS-2SA were quickly broken in tumor cells with high level of GSH. Therefore, the micelles composed of TPGS and MPEG-SS-2SA could be rapidly degraded and then could release TPGS to inhibit the mitochondria and reduce ATP level, thus preventing the efflux of PTX. The MPEG-2SA/TPGS micelles showed less reduction of MP and ATP level than MPEG-SS-2SA/TPGS. It suggested that the lack of disulfide bonds limited the degradation and drug release of micelles. Moreover, the MPEG-SS-2SA micelles scarcely decreased the level of MP and ATP, suggesting that the micelles without TPGS were unable to inhibit the energy supply of cells, thus hardly reversing MDR.
In vitro cellular uptake
The in vitro cellular uptake behaviors of these mixed micelles were determined by the qualitative and quantitative uptake assays by loading R123 or PTX, respectively. In the qualitative uptake assay, CLSM was employed to visualize cellular uptake of the R123-loaded micelles (also denoted by MPEG-SS-2SA/TPGS, MPEG-2SA/TPGS, and MPEG-SS-2SA) and the free R123 solution after 4 h of incubation. Strong fluorescence was observed in cells with R123-loaded MPEG-SS-2SA/TPGS micelles, then followed by the MPEG-2SA/TPGS micelles, and MPEG-SS-2SA micelles only showed weak fluorescence (Figure 8). Moreover, the weakest fluorescence was observed in cells incubated with free R123 solution. The results of the quantitative uptake showed that, after 4 h of incubation, the UI of free PTX was only 175.75±4.29 ng/mg protein (Figure 9). However, the UI of MPEG-SS-2SA/TPGS and MPEG-2SA/TPGS micelles was 1,012.06±44.65 and 741.19±28.27 ng/mg protein, respectively. The UI of MPEG-SS-2SA micelles was much lower (443.15±35.02 ng/mg protein). Moreover, compared with the free PTX, the three micelles showed an obviously time-dependent cellular uptake behavior, which meant that the cellular uptake of these micelles increased over time and that the MPEG-SS-2SA/TPGS micelles could significantly improve the uptake of PTX.
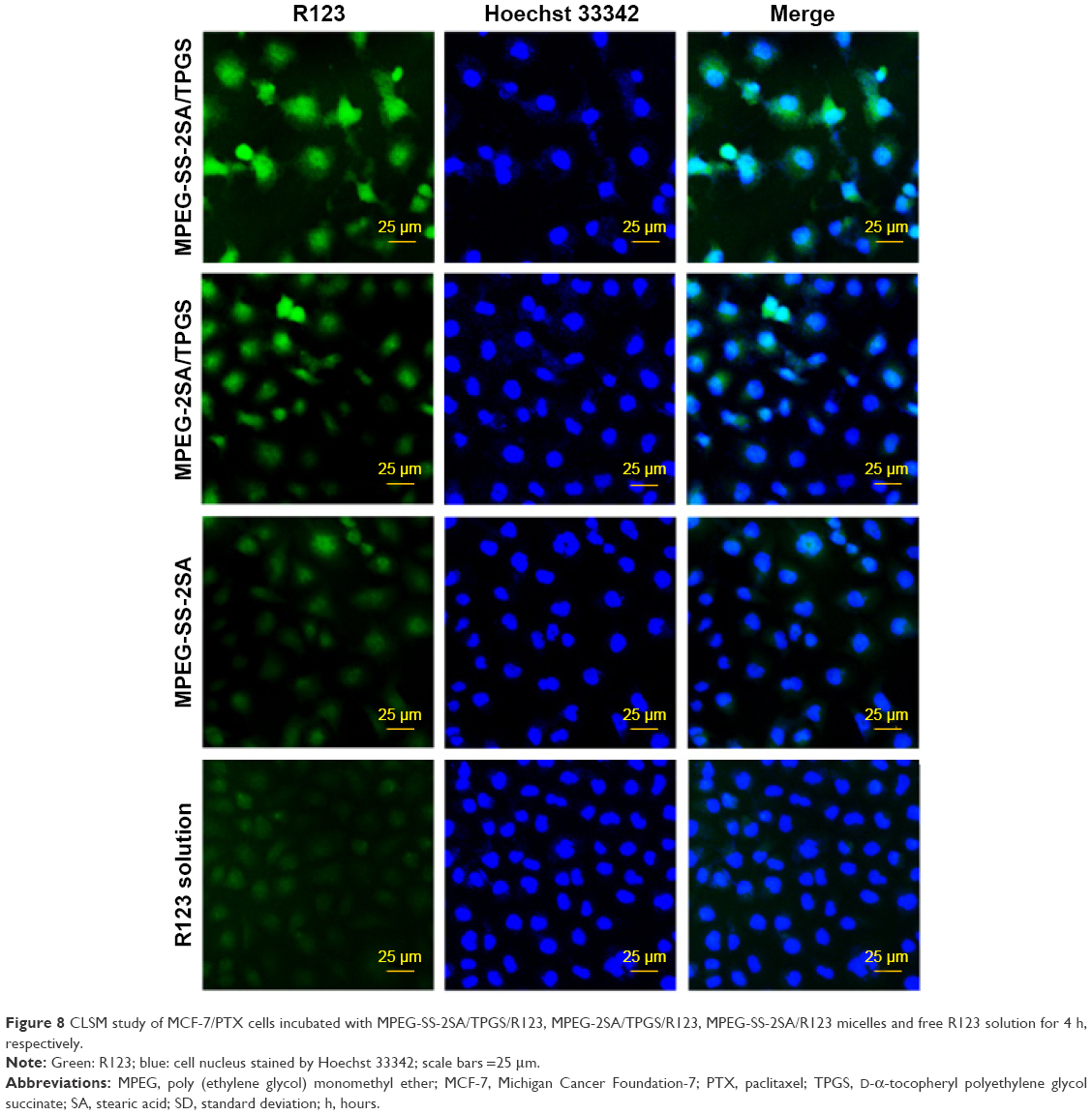 | Figure 8 CLSM study of MCF-7/PTX cells incubated with MPEG-SS-2SA/TPGS/R123, MPEG-2SA/TPGS/R123, MPEG-SS-2SA/R123 micelles and free R123 solution for 4 h, respectively. |
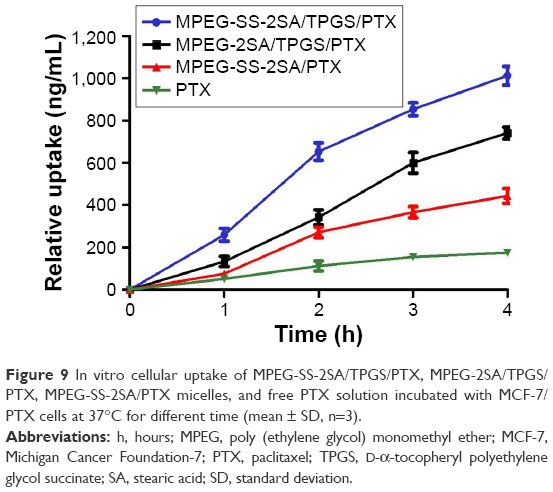 | Figure 9 In vitro cellular uptake of MPEG-SS-2SA/TPGS/PTX, MPEG-2SA/TPGS/PTX, MPEG-SS-2SA/PTX micelles, and free PTX solution incubated with MCF-7/PTX cells at 37°C for different time (mean ± SD, n=3). |
The increase of the R123 fluorescence and the UI of PTX could be attributed to two aspects: first, the reduction-sensitive disulfide bonds could be cleaved in the intracellular environment with high level of GSH. Then the micelles could be degraded quickly and could release R123 or PTX, thus improving the uptake of R123 or PTX. Second, TPGS could be released simultaneously after the breaking of disulfide bonds. TPGS acted on the mitochondria and significantly inhibited the cell energy supply, thereby inhibiting the energy-dependent drug efflux and increasing the intracellular amount of R123 or PTX (Figure 10). Compared with the MPEG-SS-2SA/TPGS micelles, the MPEG-2SA/TPGS micelles did not possess disulfide bonds hence, their degradation and drug release were relatively slow. Moreover, although the MPEG-SS-2SA micelles could rapidly release R123 or PTX, they were unable to suppress the drug efflux due to the lack of TPGS. Hence, their cellular uptake was much lower than the former two micelles. These results indicated that the MPEG-SS-2SA/TPGS mixed micelles could quickly release drugs in cancer cells and effectively inhibit the drug efflux, finally reversing drug resistance.
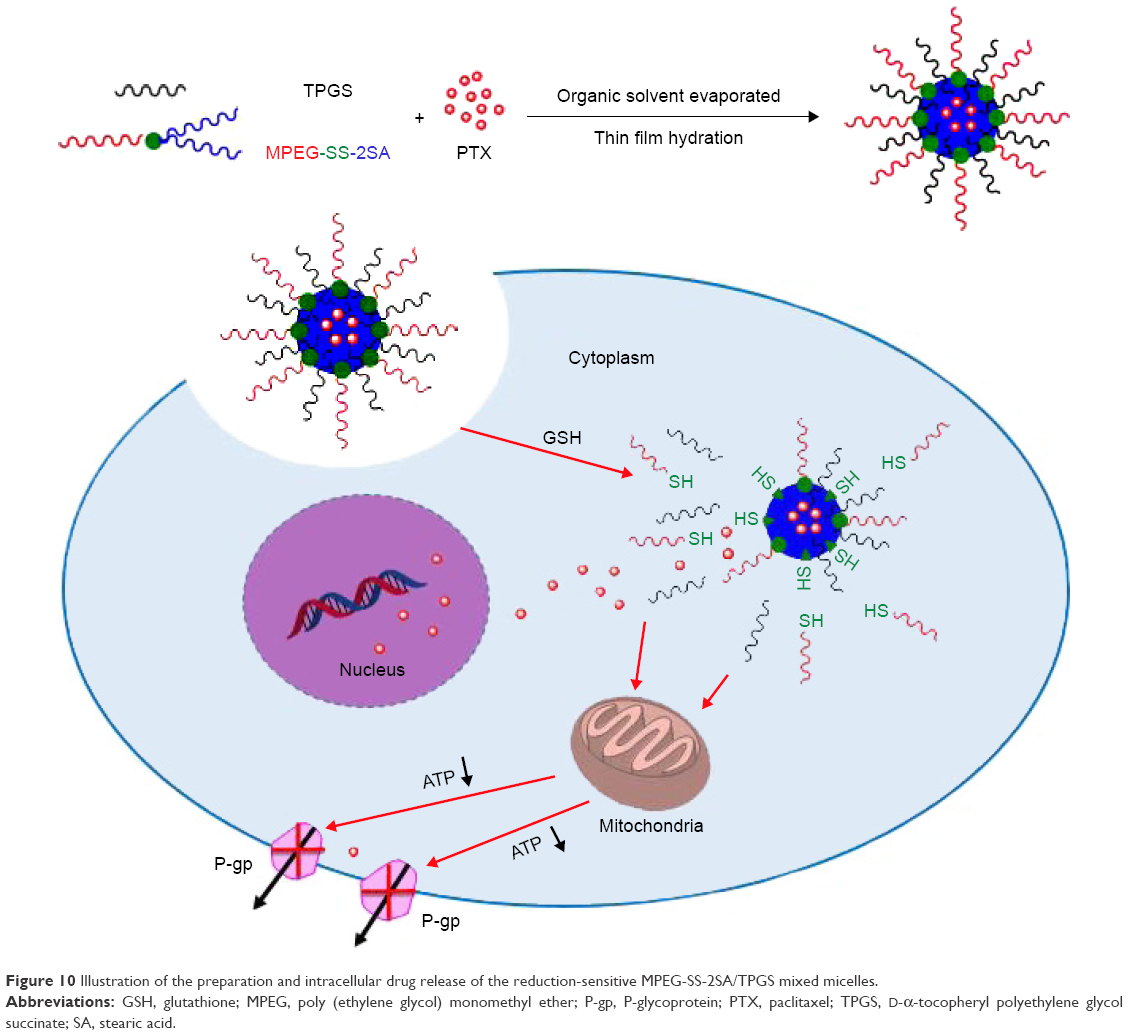 | Figure 10 Illustration of the preparation and intracellular drug release of the reduction-sensitive MPEG-SS-2SA/TPGS mixed micelles. |
Conclusion
In this study, a new amphiphilic polymer, MPEG-SS-2SA, was successfully synthesized. Then the mixed micelles, MPEG-SS-2SA/TPGS, were prepared by combining the polymer with TPGS. The in vitro release study demonstrated that the mixed micelles could rapidly release PTX under the reductive condition due to the breaking of the disulfide bonds. Moreover, the MPEG-SS-2SA/TPGS micelles could effectively inhibit the function of mitochondria and decrease the level of ATP, thus increasing the cellular accumulation of PTX and finally reversing the MDR. The combined effects of reduction-responsive degradation and the inhibition of drug efflux will make the multifunctional mixed micelles be used in a distinct way for MDR cancer treatment.
Acknowledgments
This work was supported financially by Natural Science Foundation of China (No 81473177) and Natural Science Foundation of Shaanxi Province (Nos 2016JM8020 and 2016JM8015).
Disclosure
The authors report no conflicts of interest in this work.
References
Kroger N, Achterrath W, Hegewisch-Becker S, Mross K, Zander AR. Current options in treatment of anthracycline-resistant breast cancer. Cancer Treat Rev. 1999;25(5):279–291. | ||
Naito S, Yokomizo A, Koga H. Mechanisms of drug resistance in chemotherapy for urogenital carcinoma. Int J Urol. 1999;6(9):427–439. | ||
Gillet J-P, Gottesman MM. Mechanisms of multidrug resistance in cancer. In: Zhou J, editor. Multi-Drug Resistance in Cancer. Vol. 596. Totowa (NJ): Humana Press Inc; 2010:47–76. | ||
Fields A, Hochster H, Runowicz C, et al. PSC833: initial clinical results in refractory ovarian cancer patients. Curr Opin Oncol. 1998;10(Suppl 1):S21. | ||
Wigler PW. PSC833, cyclosporin A, and dexniguldipine effects on cellular calcein retention and inhibition of the multidrug resistance pump in human leukemic lymphoblasts. Biochem Biophys Res Commun. 1999;257(2):410–413. | ||
Verschraegen CF, Gupta F, Loyer E, et al. A phase II clinical and pharmacological study of oral 9-nitrocamptothecin in patients with refractory epithelial ovarian, tubal or peritoneal cancer. Anti-Cancer Drugs. 1999;10(4):375–383. | ||
Cruz F, Wolf A. Effects of the novel cyclosporine derivative PSC833 on glucose metabolism in rat primary cultures of neuronal and glial cells. Biochem Pharmacol. 2001;62(1):129–139. | ||
Baekelandt M, Lehne G, Trope CG, et al. Phase I/II trial of the multidrug-resistance modulator valspodar combined with cisplatin and doxorubicin in refractory ovarian cancer. J Clin Oncol. 2001;19(12):2983–2993. | ||
Chico I, Kang MH, Bergan R, et al. Phase I study of infusional paclitaxel in combination with the P-glycoprotein antagonist PSC 833. J Clin Oncol. 2001;19(3):832–842. | ||
Baguley BC. Multiple drug resistance mechanisms in cancer. Mol Biotechnol. 2010;46(3):308–316. | ||
Adler LM, Herzog TJ, Williams S, Rader JS, Mutch DG. Analysis of exposure times and dose-escalation of paclitaxel in ovarian-cancer cell-lines. Cancer. 1994;74(7):1891–1898. | ||
Guastalla JP, Lhomme C, Dauplat J, et al. Taxol (paclitaxel) safety in patients with platinum pretreated ovarian carcinoma: an interim analysis of a phase II multicenter study. Ann Oncol. 1994;5(Suppl 6):S33–S38. | ||
Kubota T, Matsuzaki SW, Hoshiya Y, et al. Antitumor activity of paclitaxel against human breast carcinoma xenografts serially transplanted into nude mice. J Surg Oncol. 1997;64(2):115–121. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1–2):179–192. | ||
Liu Y, Huang L, Liu F. Paclitaxel nanocrystals for overcoming multidrug resistance in cancer. Mol Pharm. 2010;7(3):863–869. | ||
Mathew AE, Mejillano MR, Nath JP, Himes RH, Stella VJ. Synthesis and evaluation of some water-soluble prodrugs and derivatives of taxol with antitumor-activity. J Med Chem. 1992;35(1):145–151. | ||
Wang YZ, Yu L, Han LM, Sha XY, Fang XL. Difunctional Pluronic copolymer micelles for paclitaxel delivery: synergistic effect of folate-mediated targeting and pluronic-mediated overcoming multidrug resistance in tumor cell lines. Int J Pharm. 2007;337(1–2):63–73. | ||
Van Vlerken LE, Duan ZF, Little SR, Seiden MV, Amiji MM. Augmentation of therapeutic efficacy in drug-resistant tumor models using ceramide coadministration in temporal-controlled polymer-blend nanoparticle delivery systems. AAPS J. 2010;12(2):171–180. | ||
Punfa W, Suzuki S, Pitchakarn P, et al. Curcumin-loaded PLGA nanoparticles conjugated with anti-P-glycoprotein antibody to overcome multidrug resistance. Asian Pac J Cancer Prev. 2014;15(21): 9249–9258. | ||
Wang N, He T, Shen YM, et al. Paclitaxel and tacrolimus coencapsulated polymeric micelles that enhance the therapeutic effect of drug-resistant ovarian cancer. ACS Appl Mater Interfaces. 2016;8(7):4368–4377. | ||
Wang YZ, Li YJ, Wang QS, Wu J, Fang XL. Pharmacokinetics and biodistribution of paclitaxel-loaded Pluronic P105/L101 mixed polymeric micelles. Yakugaku Zasshi. 2008;128(6):941–950. | ||
Zhang W, Shi YA, Chen YZ, Ye JA, Sha XY, Fang XL. Multifunctional Pluronic P123/F127 mixed polymeric micelles loaded with paclitaxel for the treatment of multidrug resistant tumors. Biomaterials. 2011;32(11):2894–2906. | ||
Wang YZ, Hao JG, Li YJ, et al. Poly (caprolactone)-modified Pluronic P105 micelles for reversal of paclitaxcel-resistance in SKOV-3 tumors. Biomaterials. 2012;33(18):4741–4751. | ||
Wei XL, Liang W. Nanostructrue and function of PEGylated phospholipid micelles encapsulation drugs. Prog Biochem Biophys. 2013;40(10):948–954. | ||
Tang N, Du GJ, Wang N, Liu CC, Hang HY, Liang W. Improving penetration in tumors with nanoassemblies of phospholipids and doxorubicin. J Natl Cancer Inst. 2007;99(13):1004–1015. | ||
Wang J, Xing XQ, Fang XC, et al. Cationic amphiphilic drugs self-assemble to the core-shell interface of PEGylated phospholipid micelles and stabilize micellar structure. Philos Trans A Math Phys Eng Sci. 2013;371(2000):17. | ||
Fang XC, Yang T, Wang LY, et al. Nano-cage-mediated refolding of insulin by PEG-PE micelle. Biomaterials. 2016;77:139–148. | ||
Son S, Singha K, Kim WJ. Bioreducible BPEI-SS-PEG-cNGR polymer as a tumor targeted nonviral gene carrier. Biomaterials. 2010;31(24):6344–6354. | ||
Oumzil K, Khiati S, Grinstaff MW, Barthelemy P. Reduction-triggered delivery using nucleoside-lipid based carriers possessing a cleavable PEG coating. J Control Release. 2011;151(2):123–130. | ||
Zhang ZP, Tan SW, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Collnot EM, Baldes C, Wempe MF, et al. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: influence on ATPase activity and membrane fluidity. Mol Pharm. 2007;4(3):465–474. | ||
Shieh MJ, Hsu CY, Huang LY, Chen HY, Huang FH, Lai PS. Reversal of doxorubicin-resistance by multifunctional nanoparticles in MCF-7/ADR cells. J Control Release. 2011;152(3):418–425. | ||
Guo YY, Chu M, Tan SW, et al. Chitosan-g-TPGS nanoparticles for anticancer drug delivery and overcoming multidrug resistance. Mol Pharm. 2014;11(1):59–70. | ||
Chen SY, Dong Q, Hu SS, et al. Proteomic analysis of the proteins that are associated with the resistance to paclitaxel in human breast cancer cells. Mol Biosyst. 2014;10(2):294–303. | ||
Lee JW, Lee SC, Acharya G, Chang CJ, Park K. Hydrotropic solubilization of paclitaxel: analysis of chemical structures for hydrotropic property. Pharm Res. 2003;20(7):1022–1030. | ||
Simon L, Szilagyi G, Bori Z, Telek G, Magyar K, Nagy Z. Low dose (−)deprenyl is cytoprotective: it maintains mitochondrial membrane potential and eliminates oxygen radicals. Life Sci. 2005;78(3):225–231. | ||
Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS). Pharm Res. 1999;16(10):1550–1556. | ||
Yumoto R, Murakami T, Nakamoto Y, Hasegawa R, Nagai J, Takano M. Transport of rhodamine 123, a P-glycoprotein substrate, across rat intestine and Caco-2 cell monolayers in the presence of cytochrome P-450 3A-related compounds. J Pharmacol Exp Ther. 1999;289(1):149–155. | ||
Mistry P, Stewart AJ, Dangerfield W, et al. In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576. Cancer Res. 2001;61(2):749–758. | ||
Feng SS, Chien S. Chemotherapeutic engineering: application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem Eng Sci. 2003;58(18):4087–4114. | ||
Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457(7228):426–433. | ||
Wu GY, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134(3):489–492. | ||
Cheng R, Feng F, Meng FH, Deng C, Feijen J, Zhong ZY. Glutathione-responsive nano-vehicles as a promising platform for targeted intracellular drug and gene delivery. J Control Release. 2011;152(1):2–12. | ||
Wen HY, Dong HQ, Xie WJ, et al. Rapidly disassembling nanomicelles with disulfide-linked PEG shells for glutathione-mediated intracellular drug delivery. Chem Commun. 2011;47(12):3550–3552. | ||
Sun HL, Guo BN, Cheng R, Meng FH, Liu HY, Zhong ZY. Biodegradable micelles with sheddable poly (ethylene glycol) shells for triggered intracellular release of doxorubicin. Biomaterials. 2009;30(31):6358–6366. | ||
Zhang ZP, Feng SS. The drug encapsulation efficiency, in vitro drug release, cellular uptake and cytotoxicity of paclitaxel-loaded poly (lactide)-tocopheryl polyethylene glycol succinate nanoparticles. Biomaterials. 2006;27(21):4025–4033. | ||
Pan J, Feng SS. Targeted delivery of paclitaxel using folate-decorated poly (lactide) – vitamin E TPGS nanoparticles. Biomaterials. 2008;29(17):2663–2672. | ||
Na C, Si-Shen F. Doxorubicin conjugated to d-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS): conjugation chemistry, characterization, in vitro and in vivo evaluation. Biomaterials. 2008;29(28):3856–3865. | ||
Chee Wee G, Si-Shen F. Transferrin-conjugated nanoparticles of poly (lactide)-d-alpha-tocopheryl polyethylene glycol succinate diblock copolymer for targeted drug delivery across the blood-brain barrier. Biomaterials. 2010;31(30):7748–7757. | ||
Sun F, Huo X, Zhai YJ, et al. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell. 2005;121(7):1043–1057. | ||
Su ZG, Chen ML, Xiao YY, et al. ROS-triggered and regenerating anticancer nanosystem: an effective strategy to subdue tumor’s multidrug resistance. J Control Release. 2014;196:370–383. | ||
Almeida J, Ball BA. Effect of alpha-tocopherol and tocopherol succinate on lipid peroxidation in equine spermatozoa. Anim Reprod Sci. 2005;87(3–4):321–337. | ||
Youk HJ, Lee E, Choi MK, et al. Enhanced anticancer efficacy of alpha-tocopheryl succinate by conjugation with polyethylene glycol. J Control Release. 2005;107(1):43–52. | ||
Kolker S, Schwab M, Horster F, et al. Methylmalonic acid, a biochemical hallmark of methylmalonic acidurias but no inhibitor of mitochondrial respiratory chain. J Biol Chem. 2003;278(48):47388–47393. | ||
Mbaya E, Oules B, Caspersen C, et al. Calcium signalling-dependent mitochondrial dysfunction and bioenergetics regulation in respiratory chain Complex II deficiency. Cell Death Differ. 2010;17(12):1855–1866. | ||
Wang Y, Guo M, Lu Y, et al. Alpha-tocopheryl polyethylene glycol succinate-emulsified poly(lactic-co-glycolic acid) nanoparticles for reversal of multidrug resistance in vitro. Nanotechnology. 2012;23(49):14. | ||
Assanhou AG, Li WY, Zhang L, et al. Reversal of multidrug resistance by co-delivery of paclitaxel and lonidamine using a TPGS and hyaluronic acid dual-functionalized liposome for cancer treatment. Biomaterials. 2015;73:284–295. | ||
Perez-Tomas R. Multidrug resistance: retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13(16):1859–1876. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.