Back to Journals » International Journal of Nanomedicine » Volume 12
Biodegradable mesoporous delivery system for biomineralization precursors
Authors Yang HY, Niu LN, Sun JL, Huang XQ, Pei DD, Huang C ![]() , Tay FR
, Tay FR
Received 26 November 2016
Accepted for publication 30 December 2016
Published 25 January 2017 Volume 2017:12 Pages 839—854
DOI https://doi.org/10.2147/IJN.S128792
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Hong-ye Yang,1 Li-na Niu,2 Jin-long Sun,2 Xue-qing Huang,3 Dan-dan Pei,4 Cui Huang,1 Franklin R Tay5
1The State Key Laboratory Breeding Base of Basic Science of Stomatology, Key Laboratory for Oral Biomedicine Ministry of Education, School and Hospital of Stomatology, Wuhan University, Wuhan, Hubei, People’s Republic of China; 2State Key Laboratory of Military Stomatology, National Clinical Research Center for Oral Diseases, Shaanxi Key Laboratory of Oral Diseases, Department of Prosthodontics, School of Stomatology, The Fourth Military Medical University, Xi’an, Shaanxi, People’s Republic of China; 3Department of Prosthodontics, Guanghua School and Hospital of Stomatology, Guangdong Key Laboratory of Stomatology, Yat-sen University, Guangzhou, Guangdong, People’s Republic of China; 4Department of Prosthodontics, College of Stomatology, Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China; 5Department of Endodontics, College of Dental Medicine, Augusta University, Augusta, GA, USA
Abstract: Scaffold supplements such as nanoparticles, components of the extracellular matrix, or growth factors have been incorporated in conventional scaffold materials to produce smart scaffolds for tissue engineering of damaged hard tissues. Due to increasing concerns on the clinical side effects of using large doses of recombinant bone-morphogenetic protein-2 in bone surgery, it is desirable to develop an alternative nanoscale scaffold supplement that is not only osteoinductive, but is also multifunctional in that it can perform other significant bone regenerative roles apart from stimulation of osteogenic differentiation. Because both amorphous calcium phosphate (ACP) and silica are osteoinductive, a biodegradable, nonfunctionalized, expanded-pore mesoporous silica nanoparticle carrier was developed for loading, storage, and sustained release of a novel, biosilicification-inspired, polyamine-stabilized liquid precursor phase of ACP for collagen biomineralization and for release of orthosilicic acid, both of which are conducive to bone growth. Positively charged poly(allylamine)-stabilized ACP (PAH-ACP) could be effectively loaded and released from nonfunctionalized expanded-pore mesoporous silica nanoparticles (pMSN). The PAH-ACP released from loaded pMSN still retained its ability to infiltrate and mineralize collagen fibrils. Complete degradation of pMSN occurred following unloading of their PAH-ACP cargo. Because PAH-ACP loaded pMSN possesses relatively low cytotoxicity to human bone marrow-derived mesenchymal stem cells, these nanoparticles may be blended with any osteoconductive scaffold with macro- and microporosities as a versatile scaffold supplement to enhance bone regeneration.
Keywords: amorphous calcium phosphate, biomineralization, collagen, mesoporous silica, osteoinductive, poly(allylamine)
Introduction
Although bone is one of the most frequently damaged organs in the human body, it is one of the few tissues that can regenerate completely without scar tissue formation. Between 1992 and 2007, autogenous and artificial bone grafts were placed in close to 2 million patients in the US alone.1 As life expectancy and social demands of the elderly continue to increase, there is likely to be an upsurge in the global demand for bone grafting materials. Contemporary regenerative medicine principles, as epitomized by the “diamond concept”, require the utilization of scaffolds, mesenchymal stem cells, growth factors, and the establishment of mechanical competence to optimize bone regeneration in non-self-healing large bone defects.2 From a tissue engineering perspective, scaffolds provide the 3-D environment for filling of bone defects, the matrix for cell seeding and proliferation, the spaces for angiogenesis, and the framework to achieve mechanical competency during bone regeneration.3
Calcium phosphate-based ceramics have been intensively studied over the past four decades as load-bearing bone graft substitutes.4 Nevertheless, they fall short of completely satisfying the three essential requisites for an ideal hard tissue scaffold: strength and toughness combination approximating those of cortical bone, complete biodegradability, and osteoinductivity.5 To circumvent at least some of these limitations, scaffold supplements such as nanoparticles, components of the extracellular matrix or growth factors have been incorporated into conventional scaffold materials to produce smart scaffolds that are more conducive to bone regeneration.6 Although bone morphogenetic protein-2 is critical for osteogenesis and bone regeneration,7 there have been recent concerns regarding the clinical side effects associated with the use of large doses of this recombinant growth factor supplement.8 Thus, it is desirable to develop a multifunctional scaffold supplement that is osteoinductive, biodegradable, and performs other significant bone regenerative roles apart from stimulation of osteogenic differentiation.
Bone morphogenetic proteins are not the only agents that induce heterotrophic bone deposition in surgical wounds. Osteoinductivity has also been demonstrated in several calcium phosphate phases,9 including amorphous calcium phosphate (ACP).10 Apart from its osteoinductivity, ACP is the raw material for biomineralization of collagen fibrils, a process that accounts for the second level of structural hierarchy in bone.11 The existence of a transient ACP phase in developing hard tissues has been definitively confirmed.12 The ACP involved in osteogenesis is not necessarily produced by bone-forming cells adjacent to the site of active bone growth and may be transported as raw materials to the bone-forming site via blood vessels.13 This suggests that bone regeneration may also be enhanced by supplementation of ACP derived from an extrinsic source. Although ACP nanoparticles have previously been incorporated as scaffold supplements,14 this solid type of ACP is not the form that can be readily utilized for intrafibrillar mineralization of collagen. The latter is generated from solution via a cascade of events, beginning with the aggregation of charged calcium trihydrogen phosphate complexes [Ca(HPO4)34−] as prenucleation clusters. The latter eventually lose their hydrogen ions and accumulate calcium ions to produce ACP.15 In the presence of polyanionic analogs of matrix proteins involved in biomineralization, ACP is stabilized in a nanoparticulate, moldable status that has been coined the polymer-induced liquid precursor phase (PILP).16 This PILP phase infiltrates the intrafibrillar spaces via the gap zones and is eventually transformed into apatite crystallites within the mineralized collagen fibrils.17 In the context of osteogenesis, there are advantages in having mineralized collagen matrices available in a surgical wound. Human mesenchymal stem cells (hMSCs) grown ex vivo on mineralized collagen matrices underwent osteogenic differentiation in an adipogenic medium.18 Mineralized collagen scaffolds induced bone regeneration of critical-sized defects in vivo without addition of exogenous growth factors.19 Thus, it is envisaged that osteogenesis will be promoted by the controlled delivery of polymer-stabilized ACP in a bioactive form that can be readily utilized for collagen biomineralization.
Because the PILP phases of ACP are produced by liquid–liquid phase separation from supersaturated calcium and phosphate-containing solutions,20 it is impossible for them to be retained in sufficient quantity within a surgical site when a dispersion of these nanoparticles is applied in solution form. Thus, it is necessary to design a delivery system for polymer-stabilized ACP to be collected, transferred to the bony crypt, and eventually be released. Mesoporous silica nanoparticle (MSN) is a popular scaffold supplement due to its biocompatibility and biodegradability,21 with the mesopores serving as reservoirs for sustained drug release.22 The MSN gradually decomposes in the physiological pH range through hydrolysis of the Si–O bonds, releasing orthosilicic acid, the biologically-relevant, water-soluble form of Si. As early as 1970, Carlisle described the importance of Si in the initiation of bone mineralization and regeneration,23 which has been confirmed by numerous works published over the past 40 years.24 In particular, orthosilicic acid released from silica is osteoinductive in that it stimulates type I collagen synthesis and osteoblastic differentiation in human osteoblast-like cells or hMSCs.25,26 Similarly, Si ions released from MSN stimulated osteogenic differentiation of hMSCs.27 Because the osteostimulatory effect of Si spans over different stages in osteogenesis, continuous release of orthosilicic acid by MSN after depletion of the CaP cargo load would be highly desirable.
Some technical hurdles have to be overcome in the design of an MSN delivery system for polymer-stabilized ACP. Although MSN has large specific surface areas and large pore volumes, they are limited by their small pore sizes, which restrict the dimension of the cargo molecule to be loaded. Nanoparticles with larger pores also permit more effective release of the cargo molecule and induce faster degradation of the silica.28 Since pore size is tunable during MSN synthesis, it is enticing to use currently available pore-swelling techniques to create expanded-pore MSNs (pMSN) for more effective accommodation of the polymer-stabilized ACP.29 Due to the presence of silanol groups in pMSN, the latter have to be amine-functionalized to create positively-charged surfaces for loading of ACP that are traditionally stabilized by polyanionic nucleation inhibitors such as poly(aspartic) acid.16,17 Such a strategy, however, defeats the purpose of creating pMSN because of the potential reduction in pore size after pore surface functionalization with aminosilane30 and the potential adverse effect of an organosilane passivation layer on silica dissolution.31 Because of the necessity to use pMSN without surface modification for optimizing the dual function of ACP storage and Si dissolution, a new positively charged PILP phase was developed by Niu et al to maintain the electrostatic attraction between the bare pMSN and polymer-stabilized ACP precursors. Inspired by the effect of long-chain polyamines on stabilization of orthosilicic acid during biosilicification of silica-containing organisms, Niu et al have developed poly(allylamine)-stabilized ACP (PAH-ACP) for intrafibrillar mineralization of collagen.32 However, it is not known whether this novel PILP phase remains effective for biomineralization following its loading, storage, and release from the pMSN. Accordingly, three hypotheses were tested in the present study: 1) positively charged PAH-ACP may be effectively loaded and released from nonfunctionalized pMSN; 2) PAH-ACP released from loaded pMSN still retains its ability to infiltrate and mineralize collagen fibrils; and 3) complete degradation of pMSN occurs following unloading of their PAH-ACP cargo.
Methods
All chemical reagents were purchased from MilliporeSigma (St Louis, MO, USA), including cetyltrimethyl ammonium bromide (CTAB, molecular weight 364.45), tetraethyl orthosilicate (TEOS), mesitylene (TMB), poly(allylamine) hydrochloride (PAH, molecular weight 17,500), calcium chloride (CaCl2), potassium phosphate dibasic (K2HPO4), tris(hydroxymethyl)aminomethane (Tris)-buffered saline (TBS), bovine skin-derived type I collagen lyophilized powder, hydroxyethyl methacrylate, and triethylene glycol methacrylate. All chemicals were used as received.
Synthesis of MSN with enlarged pores
pMSN was synthesized using a previously reported technique with modification.33 Briefly, 1 g of CTAB and 7 mL TMB were dissolved in a solution containing 480 mL water and 3.5 mL of 2N NaOH. The mixture was stirred vigorously at 80°C for 4 h. Five mL of TEOS was added quickly to the solution and the mixture was stirred for 2 h. The white precipitate was centrifuged and washed with deionized water and ethanol three times each and oven-dried at 60°C overnight. The as-synthesized powders were calcined in air at 550°C for 5 h to remove the structural template (CTAB) and pore swelling agent (TMB).
Preparation of PAH-ACP loaded pMSN
PAH-ACP precursors were prepared at room temperature by rapid mixing of equal volumes of 9 mM CaCl·2H2O and 4.2 mM Na2HPO4. The PAH was added to the calcium ion-containing solution before mixing to achieve a final PAH concentration of 500 μg/mL. For loading of mineralization precursors, 200 mg of pMSN was dispersed in 50 mL of PAH-ACP-containing solution and stirred at room temperature for 24 h to establish equilibrium. The PAH-ACP-loaded pMSN (PAH-ACP@pMSN) was centrifuged to generate a white precipitate, which was washed with deionized water and stored wet at −20°C until use.
Characterization
The mesostructural characteristics of pMSN and PAH-ACP@pMSN were examined using transmission electron microscopy (TEM; JEM-1230; JEOL, Tokyo, Japan) at 110 kV and atomic force microscopy (AFM; DI Nanoscope IV, Veeco, NY, USA) in the tapping mode. Elemental mapping was evaluated using a Tecnai G2 scanning TEM (FEI, Hillsboro, OR, USA) at 200 kV. X-ray diffraction (XRD) of pMSN was performed with a diffractometer (X’Pert PRO, PANalytical, Almelo, the Netherlands) with Ni-filtrated Cu Kα radiation (40 kV, 40 mA). Fourier transform-infrared spectra (FT-IR) were evaluated by a Nicolet 6700 attenuated total reflection spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Surface analysis was performed by recording nitrogen adsorption–desorption isotherms using porosimetry (ASAP2020; Micromeritics Corp., Norcross, GA, USA). The Brunauer-Emmett-Teller (BET) method was used to calculate the surface areas, and the Barrett-Joyner-Halenda (BJH) analysis was used to calculate pore size distributions. Thermogravimetric analysis (TGA) was performed with a Q500 analyzer (TA Instruments, New Castle, DE, USA) at a rate of 10°C/min in nitrogen atmosphere. The zeta potential of nanoparticles was measured with a Zetasizer Nano ZS system (Malvern Instruments Ltd., Westborough, MA, USA). Deionized water (pH =7.0) was used to suspend the nanoparticles and the zeta potential was measured immediately after ultrasonication for 15 min.
Calcium, phosphorus, and silicon release
TBS containing no Ca and P ions was used to evaluate the amount of Ca, P, and Si released from the PAH-ACP@pMSN. Specifically, 100 mg of PAH-ACP@pMSN was immersed in 10 mL of TBS (pH 7.4). Elements released from the PAH-ACP@pMSN were measured at 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, and 30 days. At each time point, the mixture was centrifuged at 4,000 rpm for 4 min. A 1 mL aliquot of the supernatant were removed and replaced with fresh solution. The aliquots were analyzed for Ca, P, and Si content using inductively coupled plasma–atomic emission spectroscopy (ICP-AES; Perkin Elmer, MA, USA).
Mineralization of reconstituted collagen
A single-layer fibrillar collagen model was used to examine the effect of PAH-ACP release from the mesoporous delivery system on intrafibrillar mineralization. For collagen reconstitution, 200-mesh Ni TEM grids were placed in 0.2 mg/mL collagen/acetic acid; the latter was prepared by dissolving bovine skin-derived type I collagen powder in acetic acid. The collagen solution was adjusted with ammonia vapor to pH 8 and left to gel at room temperature for 5 days. After removing excess solution and air-drying, self-assembled collagen fibrils were deposited on the TEM grids.
Fifty milligrams of PAH-ACP@pMSN was immersed in 1.5 mL of TBS buffer (pH =7.4) inside each well of a 24-well plate. A Ni TEM grid covered with reconstituted collagen fibrils was floated on the top of the suspension. The assembly was incubated in a 100% relative humidity chamber at 37°C. Grids were retrieved after 1, 2, or 3 days for TEM.
Concomitant collagen matrix mineralization and pMSN degradation
Although intrafibrillar mineralization of a 3-D collagen scaffold occurred after blending it with PAH-ACP@pMSN, such a procedure did not provide evidence of pMSN dissolution that occurred slowly over time. Resorption of pMSN occurred after confinement of PAH-ACP@pMSN within a polymerized hydrophilic resin matrix; efflux of PAH-ACP and solubilized silica caused by water sorption created pMSN-free voids within the resin matrix. Nevertheless, such a protocol did not permit observation of collagen mineralization. To circumvent this dilemma, a hybrid procedure was adopted based on what dentists do for bonding tooth-colored fillings to dentin. In this procedure, pieces of bovine dentin were partially demineralized with 15% phosphoric acid to create a 5 μm thick bed of mineral-free collagen fibrils on the surface of the dentin block. The PAH-ACP@pMSN powder was sprinkled onto the dentin surface and embedded with a hydrophilic resin blend (50/50 wt% of 2-hydroxyethyl methacrylate and triethylene glycol dimethacrylate containing tertiary amine catalyst and camphorquinone photoinitiator) solvated in ethanol. Application of the resin blend to demineralized dentin resulted in partial infiltration of the resinous material into the collagen matrix; the basal portion of the matrix was resin-sparse and contained denuded collagen fibrils that are susceptible to remineralization by the released PAH-ACP. Using this protocol, dissolution of the pMSN and remineralization of the collagen matrix in areas not completely infiltrated by resin could be observed simultaneously.
Specimens to be mineralized were stored in TBS at 37°C for 24 h or 3 months. The retrieved specimens were longitudinally sectioned to produce slabs for TEM observation. The specimens were dehydrated in an ascending ethanol series (50%–100%), immersed in propylene oxide, and embedded in epoxy resin. Ninety millimeter-thick sections were prepared with an ultramicrotome and examined unstained by TEM at 110 kV.
Effect of PAH-ACP@pMSN on cytotoxicity and osteogenic differentiation of hMSCs
Detailed methods are included in the Supplementary materials.
Results and discussion
The pMSN was synthesized by adding a pore-expanding agent (TMB) to CTAB-templated, base-catalyzed sol-gel condensation reaction of silica.33 Powder XRD (Figure 1A) shows one strong diffraction peak, 2θ =1.76°, and three weak diffraction peaks, 2θ =3.09°, 3.62°, and 4.81°. Based on the Bragg formula λ =2d sinθ, where λ is the wavelength of the probing radiation, d is the constant of the planar crystal between atoms, and θ is the incident angle, the corresponding crystal plane d-spacings of pMSN were found to be 5.01, 2.85, 2.44, and 1.84 nm, respectively. These d-spacings are attributed to the 100, 110, 200, and 210 planes of 2D hexagonal reflection in the P6 mm space group, indicating that pMSN has a hexagonal mesoporous structure.34 Infrared spectra (Figure 1B) provided evidence for successful preparation of PAH-ACP@pMSN; pMSN-CTAB displayed strong absorbance at 2,942 and 2,871 cm−1 for C–H stretching vibrations and 1,478 cm−1 for C–H deformation.35 These CTAB-derived signals disappeared after the templating agent was completely removed by calcination. Compared with calcined pMSN, PAH-ACP@pMSN displayed two additional adsorption peaks at 1,568 and 1,635 cm−1 that were assigned to the N–H bending and C=O stretching vibrations of PAH, respectively.36
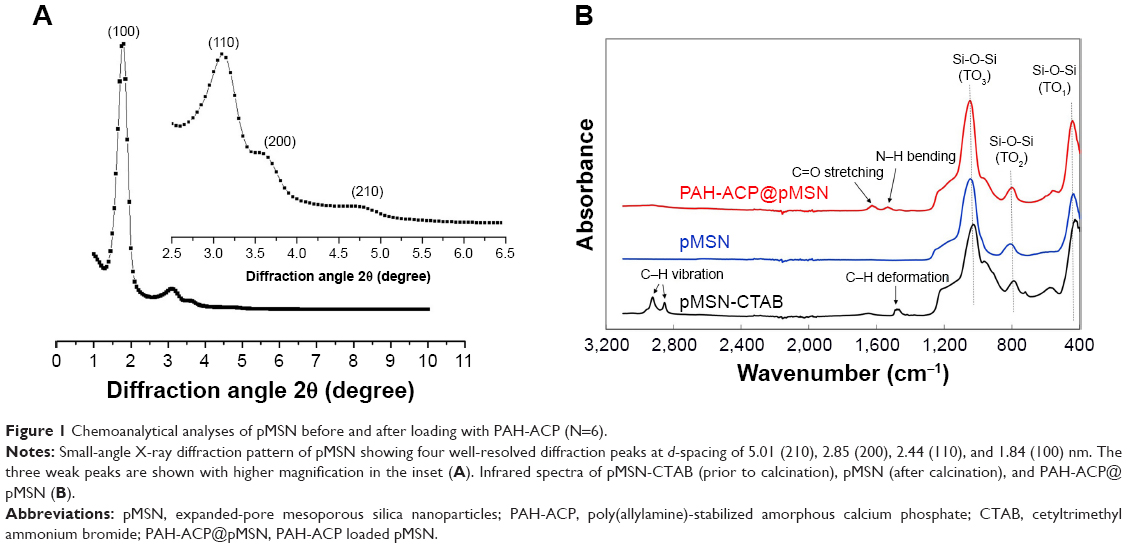 | Figure 1 Chemoanalytical analyses of pMSN before and after loading with PAH-ACP (N=6). |
When examined with TEM (Figure 2A-left), pMSN were predominantly spherical, between 50–100 nm in diameter, and with occasional olive-shaped configurations.33 High magnification of a spherical pMSN (Figure 2A-middle) revealed highly regular mesoporosity comprising a hexagonal array typical of MCM-41-type MSN. Mesoporosities became less clearly defined after PAH-ACP loading (Figure 2B-left, middle). To observe the internal morphologic changes in pMSN, the nanoparticles were embedded in epoxy resin for ultramicrotomy. Examination of the epoxy resin-embedded PAH-ACP@pMSN sections confirmed the presence of amorphous materials in the mesopores and in the periphery of the sectioned nanoparticles (Figure 2B-right). Scanning TEM–energy dispersive X-ray elemental mapping (Figure 2C) of the sectioned PAH-ACP@pMSN revealed strong intraparticulate Ca and P signals that are indicative of the infiltration of ACP precursors into the mesopores. Height and phase AFM images of pMSN (Figure 2D, top) showed a relatively flat surface with needle-like columns that are representative of the surface mesopores. By contrast, AFM of PAH-ACP@pMSN (Figure 2D, bottom) demonstrated a rough appearance with large surface granules.
 | Figure 2 Ultrastructure of pMSN before and after loading with PAH-ACP. |
Nitrogen sorption by pMSN demonstrated a type IV isotherm with well-defined steps at 0.4–0.6P/P0 (Figure 3A), which is indicative of the presence of uniform mesoporous channels and narrow pore size distribution. Unloaded pMSN have a specific surface area of 983.6 m2/g, pore volume of 0.99 cm3/g, and mean pore size of 5.0 nm (Figure 3B). The well-defined large pore structure and high specific surface area of pMSN are demonstrations of potential for loading molecules.37 Loading of amorphous materials reduced the adsorbed nitrogen volume of PAH-ACP@MSN but exerted no effect on the shape of the hysteresis loop (Figure 3A). The BET specific surface area, pore volume, and pore size of PAH-ACP@pMSN were reduced to 641.8 m2/g, 0.71 cm3/g, and 2.3 nm (Figure 3B), respectively. These findings indicate that amorphous precursors have infiltrated the internal mesoporosities of the pMSN. The increase in mean particle size from 67.1 nm for pMSN to 93.4 nm for PAH-ACP@pMSN suggests that additional PAH-ACP accumulates on the nanoparticle surface.
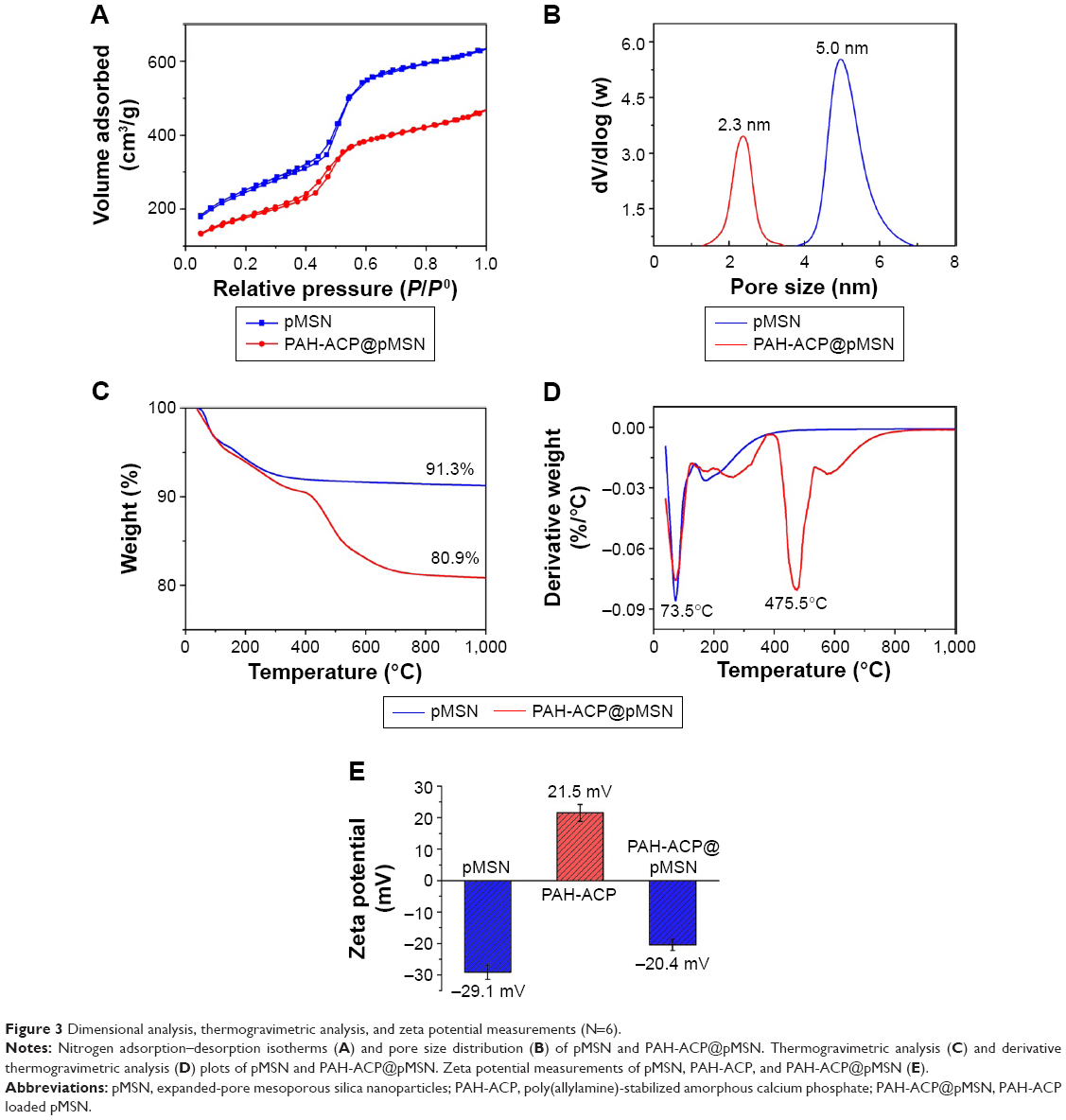 | Figure 3 Dimensional analysis, thermogravimetric analysis, and zeta potential measurements (N=6). |
The amount of precursors adsorbed on pMSN was estimated through TGA. After heating to 1,000°C, unloaded pMSN displayed a total weight loss of 8.7 wt% (Figure 3C), which was mainly attributed to the removal of physisorbed water (73.5°C; Figure 3D). The PAH-ACP@pMSN showed a total weight loss of 19.1 wt% with an additional discernible derivative weight-loss peak at 475.5°C, which may be attributed to the decomposition of organic substances (PAH). The amount of adsorbed PAH was approximately 10.4 wt%, suggesting that the amount of PAH-ACP adsorbed on pMSN was higher than 10.4 wt%.
Measurement of the zeta (ζ) potentials of pMSN, PAH-ACP, and PAH-ACP@pMSN (Figure 3E) revealed the driving force behind the loading of amorphous precursors. The mean ζ potential of pMSN was −29.1 mV because of the presence of silanol groups, whereas that of PAH-ACP was +21.5 mV. The negative ζ potential of PAH-ACP@pMSN (−20.4 mV) is suggestive of electrostatic interaction as a mechanism for adsorption of PAH-ACP by the mesoporous delivery system.
Cumulative release profiles of Ca, P, and Si at pH 7.4 are presented in Figure 4A. Both Ca and P were rapidly released from PAH-ACP@pMSN during the first 10 days; release of these elements declined from 10 to 30 days. These power law-related release profiles are indicative of the depletion of the loaded calcium phosphate phase. The Ca/P ratio of the releasing solution gradually dropped to close to 1.5 after 10 days (Figure 4B), close to the Ca/P ratio of ACP at neutral pH.38 By contrast, Si continued to be released linearly up to the 30-day cut-off time (Figure 4C), indicating that the pMSN gradually dissolved during long-term storage following release of the mineralization precursors. Thus, the first hypothesis that positively charged PAH-ACP may be effectively loaded and released from nonfunctionalized pMSN was validated.
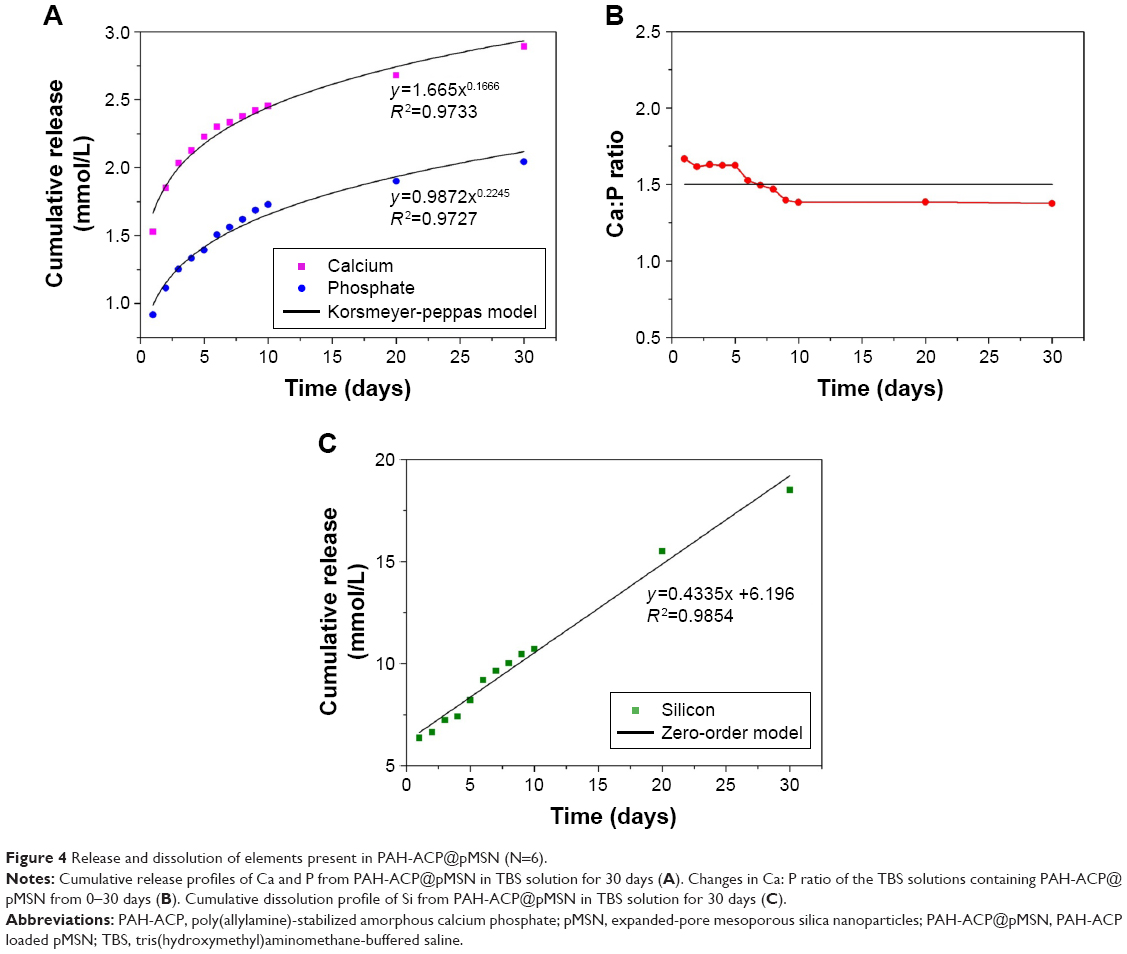 | Figure 4 Release and dissolution of elements present in PAH-ACP@pMSN (N=6). |
Intrafibrillar mineralization of collagen was examined initially with a single-layer reconstituted collagen model. After 24 h, mineralization precursors were released from the pMSN, some of which had infiltrated the unstained collagen fibrils resulting in their partial mineralization (Figure 5A). After 3 days, the electron density of the unstained collagen became higher after complete intrafibrillar mineralization of the fibrils (Figure 5B). High magnification of a highly mineralized, unstained fibril shows mineral strands filling the spirally-arranged microfibrillar spaces (Figure 5C). Selected area electron diffraction revealed arc-shaped diffraction pattern along the 002 plane of apatite that is indicative of the deposition of minerals along the c-axis of the fibril (Figure 5C inset). A 2-day specimen stained with uranyl acetate showed attachment of released PAH-ACP precursors to the collagen surface, initial state of apatite crystallization in a partially mineralized fibril, and collagen banding in a highly mineralized fibril (Figure 5D). Based on these results, the second hypothesis that PAH-ACP released from loaded pMSN still retains its ability to infiltrate and mineralize collagen fibrils was validated. The positively charged PAH-ACP precursors were released from the pMSN probably because of competitive displacement by zwitterions derived from Tris-buffered saline, in which the PAH-ACP@pMSN were suspended.39 Gradual dissolution of silica probably also promoted the release of the mineralization precursors. Several theories explain the driving forces behind the influx of mineralization precursors into collagen. The first theory involves capillary infiltration of liquid-like status ACP produced by liquid–liquid phase separation into the intrafibrillar water compartments of the collagen fibril.40 Although the liquid-like status of polymer-stabilized amorphous calcium carbonate has been demonstrated,41,42 a similar status has never been demonstrated for ACP. Likewise, the proposed capillary action of a liquid phase within a liquid has only been demonstrated in macroscopic systems using capillary tubing and bulk collagen scaffold.43 The second theory involves electrostatic interactions between net positive charges along the gap zones of a collagen fibril and the negatively charged polyelectrolyte-stabilized ACP.17,44 This theory, however, cannot account for the attraction and infiltration of positive-charged PAH-ACP into the collagen fibril, which results in heavily mineralized fibrils that are similar in appearance to those infiltrated by polyanion-stabilized mineral precursors.17,45 Based on the size exclusion mechanism of intrafibrillar mineralization,46 PAH is excluded from entering the intrafibrillar compartments of the collagen fibril; depletion of the nucleation inhibitor would result in the formation of crystalline calcium phosphate phases inside the collagen fibril. Because nucleation inhibitors do not need to enter collagen fibrils to induce mineralization,47 a third theory was recently proposed in which the negatively charged or positively charged polyelectrolytes reside in the extrafibrillar region to establish a balance between electroneutrality and osmotic equilibrium in the vicinity of the collagen fibrils. Establishment of Gibbs–Donnan equilibrium enables the influx of prenucleation clusters or ACP into the internal milieu of the fibrils to result in intrafibrillar mineralization.32
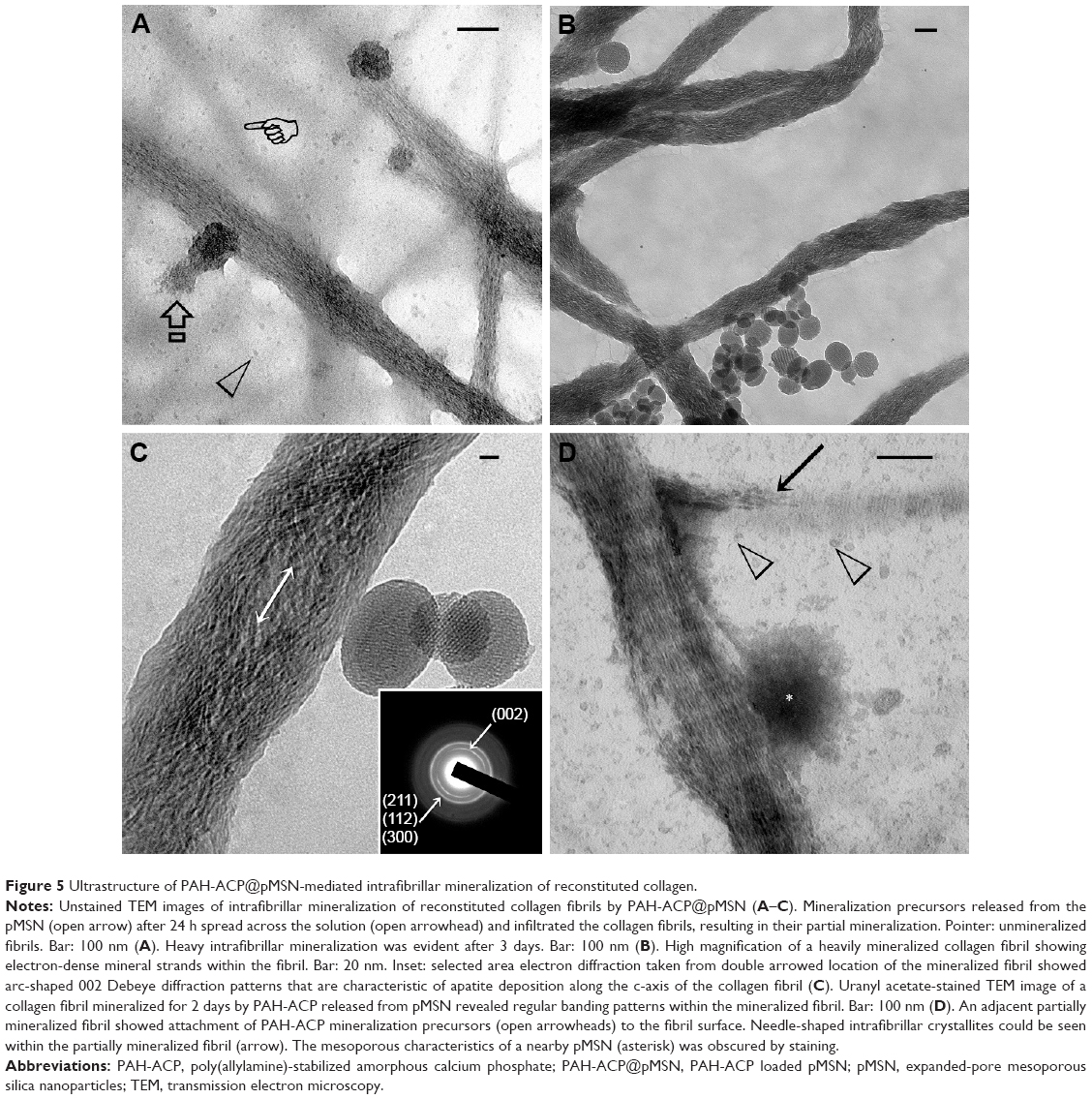 | Figure 5 Ultrastructure of PAH-ACP@pMSN-mediated intrafibrillar mineralization of reconstituted collagen. |
Biodegradation of the delivery vehicles and mineralization of thicker 3-D collagen scaffolds, both of which required a longer observation period, were concurrently accomplished via a tooth filling-inspired approach.48 The PAH-ACP@pMSN was sandwiched between a layer of polymerized hydrophilic methacrylate resin and a 5 μm thick layer of completely demineralized collagen matrix. The latter was prepared by applying 15% phosphoric acid to the surface of a small piece of bovine dentin for 15 sec, followed by vigorous water rinsing. Incomplete infiltration of the methacrylate resin into the water-filled collagen scaffold produced denuded collagen fibrils at the base of the scaffold that are susceptible to intrafibrillar mineralization (Figure 6A). After 24 h, release of mineralization precursors from the pMSN was apparent along the resin–scaffold interface (Figure 6B). After 3 months of storage in deionized water, collagen fibrils along the base of the demineralized collagen scaffold were heavily remineralized (Figure 6C), while dissolution of the pMSN left behind electron-lucent voids along the resin–scaffold interface (Figure 6D). This facile confinement model enables observation of complete degradation of the pMSN after water aging. Hence, the third hypothesis that complete degradation of pMSN occurs following unloading of their PAH-ACP cargo was also validated. It has been shown that covalent attachment of a layer of polyethylene glycol to the MSN surface resulted in passivation of the degradation kinetics of the mesoporous silicon structures.49 Although electrostatic interaction is not as strong as covalent attachment, there is concern that binding of free PAH (available from size exclusion of PAH during infiltration of ACP into collagen) to the pMSN may dampen the degradation of silica. This concern was alleviated by the results of the confinement experiment.
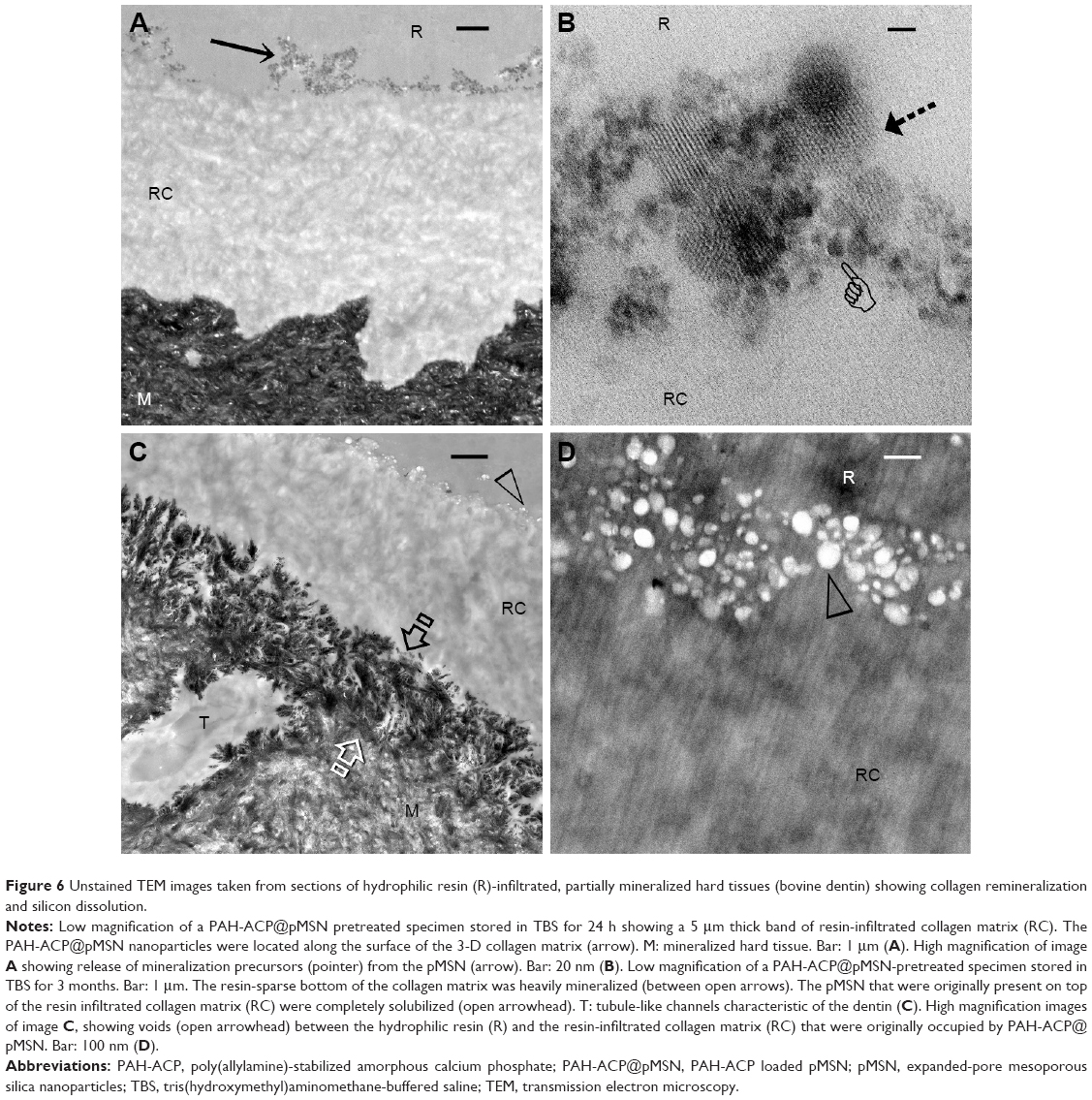 | Figure 6 Unstained TEM images taken from sections of hydrophilic resin (R)-infiltrated, partially mineralized hard tissues (bovine dentin) showing collagen remineralization and silicon dissolution. |
It has been shown that MSN used as carriers of therapeutic agents had negligible effects on cell mitosis even upon phagocytosis by vascular endothelial cells.50 Because the biocompatibility of PAH-ACP@pMSN is unknown, proliferation of hMSCs was performed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. This was achieved by incubating the stem cells with different concentrations of PAH-ACP@pMSN suspension (0–1,280 μg/mL) for 24 h (Figure S1). Significant difference in relative mitochondrial dehydrogenase activity was identified among the concentration groups (P<0.05). Viability of hMSCs remained at approximately 90% even with the highest PAH-ACP@pMSN concentration (1,280 μg/mL). These results demonstrate that PAH-ACP@pMSN possess relatively low cytotoxicity to cells associated with anabolic activities within a surgical site.
The effects of PAH-ACP@pMSN on osteogenic differentiation of hMSCs were further evaluated by comparisons to unexposed hMSCs (control) and hMSCs exposed to unloaded pMSN. The osteogenic target markers employed for quantitative reverse transcriptase-polymerase chain reaction were Runt-related transcription factor 2 (Runx2), alkaline phosphatase (ALP), bone sialoprotein (BSP), dentin matrix protein-1 (DMP-1) osteocalcin (OCN), and osterix (OSX), with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the housekeeping gene. Using Dunn–Sidak correction of α =0.0085 to control familywise error rate in multiple comparison procedures (see “Supplementary materials” – Methods), there were no differences among the three groups for Runx2, BSPIJN-128792 and OCN. For ALP and DMP-1, gene expressions were significantly upregulated, in the order: PAH-ACP@pMSN > pMSN > control. For OSX, gene expressions were upregulated in the order: PAH-ACP@pMSN = pMSN > control (Figure S2). Upregulation of ALP expression at the gene level was also observed at the enzyme level, with PAH-ACP@pMSN > pMSN > control (P<0.05; Figure S3). Taken together, the data indicate that PAH-ACP@pMSN has a promotional effect on osteogenesis of hMSCs.
Figure 7 is a schematic summarizing the development of a biodegradable mesoporous carrier for the delivery of ACP mineralization precursors. Biodegradation of the delivery vehicles in a surgical site represents a biomimetic approach in regenerating natural bony tissues without leaving behind nonresorbable foreign bodies or resorting to cell-mediated mechanisms for their removal. Release of orthosilicic acid from the degrading pMSN promotes healthier bone formation via enhanced deposition of collagen25 as well as stimulating the proliferation and osteogenic differentiation of hMSCs via specific cell signaling pathways of those cells.26 In addition, the CaP phase released from the pMSN is in the same form (ie, ACP) as those produced within the intracellular vesicles of hMSCs that are transported to the extracellular matrix for mineralization.51,52 Thus, the PAH-ACP provides additional raw materials in a form that is readily available to the newly deposited collagen fibrils within the reorganizing extracellular matrix of the surgical site for collagen biomineralization and bone regeneration.
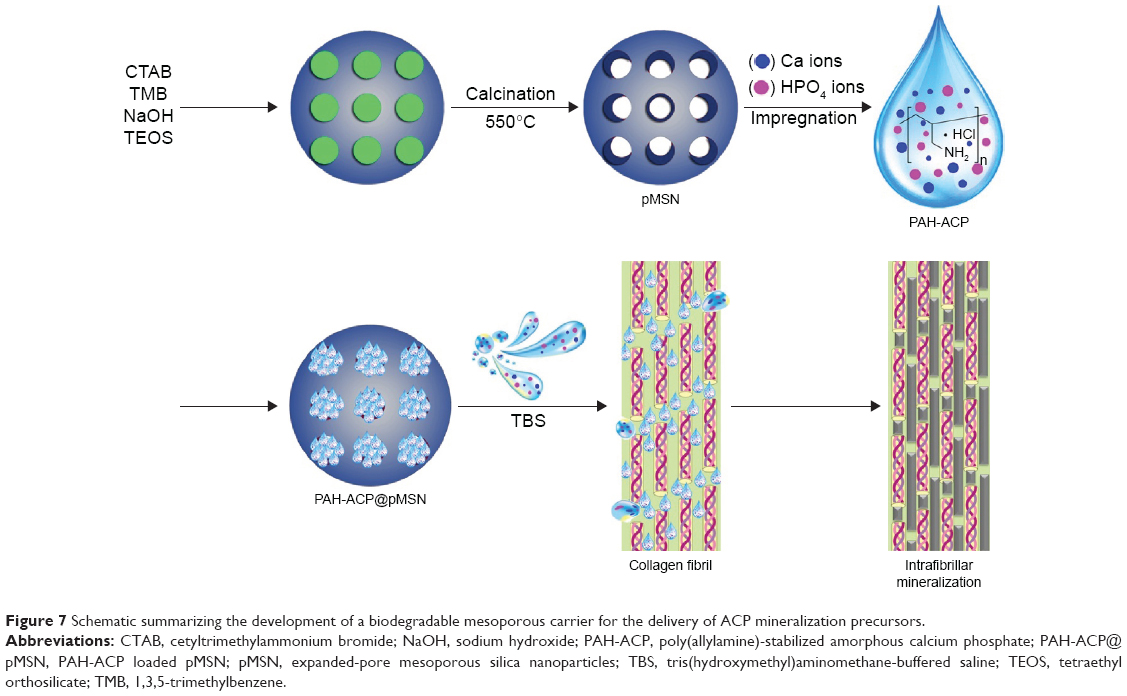 | Figure 7 Schematic summarizing the development of a biodegradable mesoporous carrier for the delivery of ACP mineralization precursors. |
Conclusion
The PAH-ACP@pMSN may be blended with any osteoconductive scaffold with macro- and microporosities as a versatile scaffold supplement. While blending may be achieved in different ways, these nanoparticles may be incorporated in “inks” for 3-D of personalized scaffolds as long as the particles are prevented from dehydration to preserve the liquid-like, moldable characteristics of the PAH-ACP. Such a requirement should not be considered a restriction in the use of the nanoparticles since live cells can be similarly printed using contemporary 3-D printing technology.53 From a biosafety perspective, this may circumvent the introduction of potent osteoinductive agents such as recombinant human bone morphogenetic protein-2 to a surgical site.54 Because orthosilicic acid release has recently been shown to promote angiogenesis and cell homing via modulation of monocytes,55 these properties need to be evaluated in future studies using the present mineralization precursor-loaded biodegradable delivery system. In addition, the potential for PAH-ACP@pMSN to be used as a nanoparticulate supplement for commonly employed tissue engineering scaffolds requires further validation in in vivo small animal models containing critical-sized surgical bone defects.
Acknowledgments
This work was supported by the National Nature Science Foundation of China grant 81371191, grant 2015AA020942 from the National High Technology Research and Development Program of China, and grant 81400555 from the National Nature Science Foundation of China and program IRT13051 from Changjiang Scholars and Innovative Research Team in University.
Author contributions
HYY performed the synthesis and characterization of the pMSN and PAH-ACP@pMSN materials and the in vitro mineralization experiments; LNN and JLS performed the cell culture experiments; XQH and DDP reconstituted collagen and collected and prepared bovine teeth. CH and FRT conceived and designed the experiments and wrote the paper. All authors contributed toward data analysis, drafting and revising the paper, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Kinaci A, Neuhaus V, Ring DC. Trends in bone graft use in the United States. Orthopedics. 2014;37(9):e783–e788. | ||
Dimitriou R, Jones E, McGonagle D, Giannoudis PV. Bone regeneration: current concepts and future directions. BMC Med. 2011;9:66. | ||
Szpalski C, Wetterau M, Barr J, Warren SM. Bone tissue engineering: current strategies and techniques – Part I: Scaffolds. Tissue Eng Part B Rev. 2012;18(4):242–257. | ||
Habraken W, Habibovic P, Epple M, Bohner M. Calcium phosphates in biomedical applications: materials for the future? Mater Today. 2016;19(2):69–87. | ||
Bohner M, Galea L, Doebelin N. Calcium phosphate bone graft substitutes: failures and hopes. J Eur Ceram Soc. 2012;32(11):2663–2671. | ||
Motamedian SR, Hosseinpour S, Ahsaie MG, Khojasteh A. Smart scaffolds in bone tissue engineering: a systematic review of literature. World J Stem Cells. 2015;7(3):657–668. | ||
Bessa PC, Casal M, Reis RL. Bone morphogenetic proteins in tissue engineering: the road from laboratory to clinic, part II (BMP delivery). J Tissue Eng Regen Med. 2008;2(2–3):81–96. | ||
James AW, LaChaud G, Shen J, et al. A review of the clinical side effects of bone morphogenetic protein-2. Tissue Eng Part B Rev. 2016;22(4):284–297. | ||
Chai YC, Carlier A, Bolander J, et al. Current views on calcium phosphate osteogenicity and the translation into effective bone regeneration strategies. Acta Biomater. 2012;8(11):3876–3887. | ||
Syed-Picard FN, Jayaraman T, Lam RS, Beniash E, Sfeir C. Osteoinductivity of calcium phosphate mediated by connexin 43. Biomaterials. 2013;34(15):3763–3774. | ||
Weiner S, Wagner HD. The material bone: structure mechanical function relations. Ann Rev Mater Sci. 1998;28:271–298. | ||
Mahamid J, Aichmayer B, Shimoni E, et al. Mapping amorphous calcium phosphate transformation into crystalline mineral from the cell to the bone in zebrafish fin rays. Proc Natl Acad Sci U S A. 2010;107(14):6316–6321. | ||
Akiva A, Malkinson G, Masic A, et al. On the pathway of mineral deposition in larval zebrafish caudal fin bone. Bone. 2015;75(6):192–200. | ||
Buschmann J, Härter L, Gao S, et al. Tissue engineered bone grafts based on biomimetic nanocomposite PLGA/amorphous calcium phosphate scaffold and human adipose-derived stem cells. Injury. 2012;43(10):1689–1697. | ||
Habraken WJ, Tao J, Brylka LJ, et al. Ion-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat Commun. 2013;4:1507. | ||
Olszta MJ, Cheng X, Jee SS, et al. Bone structure and formation: a new perspective. Mater Sci Eng R: Reports. 2007;58(3–5):77–116. | ||
Nudelman F, Pieterse K, George A, et al. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat Mater. 2010;9(12):1004–1009. | ||
Kang H, Shih YR, Varghese S. Biomineralized matrices dominate soluble cues to direct osteogenic differentiation of human mesenchymal stem cells through adenosine signaling. Biomacromolecules. 2015;16(3):1050–1061. | ||
Ren X, Tu V, Bischoff D, et al. Nanoparticulate mineralized collagen scaffolds induce in vivo bone regeneration independent of progenitor cell loading or exogenous growth factor stimulation. Biomaterials. 2016;89:67–78. | ||
DiMasi E, Liu T, Olszta MJ, Gower LB. Laser light scattering observations of liquid–liquid phase separation in a polymer-induced liquid-precursor (PILP) mineralization process. MRS Proceedings. 2005;873:K10.6. | ||
Shadjou N, Hasanzadeh M. Silica-based mesoporous nanobiomaterials as promoter of bone regeneration process. J Biomed Mater Res A. 2015;103(11):3703–3716. | ||
Wang Y, Zhao Q, Han N, et al. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomedicine. 2015;11(2):313–327. | ||
Carlisle EM. Silicon: a possible factor in bone calcification. Science. 1970;167(3916):279–280. | ||
Rodella LF, Bonazza V, Labanca M, Lonati C, Rezzani R. A review of the effects of dietary silicon intake on bone homeostasis and regeneration. J Nutr Health Aging. 2014;18(9):820–826. | ||
Reffitt DM, Ogston N, Jugdaohsingh R, et al. Orthosilicic acid stimulates collagen type 1 synthesis and osteoblastic differentiation in human osteoblast-like cells in vitro. Bone. 2003;32(2):127–135. | ||
Han P, Wu C, Xiao Y. The effect of silicate ions on proliferation, osteogenic differentiation and cell signalling pathways (WNT and SHH) of bone marrow stromal cells. Biomater Sci. 2013;1:379–392. | ||
Shi M, Zhou Y, Shao J, et al. Stimulation of osteogenesis and angiogenesis of hBMSCs by delivering Si ions and functional drug from mesoporous silica nanospheres. Acta Biomater. 2015;21(7):178–189. | ||
Douroumis D, Onyesom I, Maniruzzaman M, Mitchell J. Mesoporous silica nanoparticles in nanotechnology. Crit Rev Biotechnol. 2013;33(3):229–245. | ||
Kruk M. Access to ultralarge-pore ordered mesoporous materials through selection of surfactant/swelling-agent micellar templates. Acc Chem Res. 2012;45(10):1678–1687. | ||
Kamarudin NHN, Jalil AA, Triwahyono S, et al. Role of 3-aminopropyltriethoxysilane in the preparation of mesoporous silica nanoparticles for ibuprofen delivery: effect on physicochemical properties. Microporous Mesoporous Mater. 2013;180:235–241. | ||
Dekeyser CM, Buron CC, Derclaye SR, Jonas AM, Marchand-Brynaert J, Rouxhet PG. Degradation of bare and silanized silicon wafer surfaces by constituents of biological fluids. J Colloid Interface Sci. 2012;378(1):77–82. | ||
Niu LN, Jee SE, Jiao K, et al. Collagen intrafibrillar mineralization as a result of the balance between osmotic equilibrium and electroneutrality. Nat Mater. Epub 2016 Nov 7. | ||
Slowing II, Trewyn BG, Lin VS. Mesoporous silica nanoparticles for intracellular delivery of membrane-impermeable proteins. J Am Chem Soc. 2007;129(28):8845–8849. | ||
Ishii Y, Nishiwaki Y, Al-Zubaidi A, Kawasaki S. Pore size determination in ordered mesoporous materials using powder X-ray diffraction. J Phys Chem C. 2013;117(35):18120–18130. | ||
Moreira AF, Gaspar VM, Costa EC, et al. Preparation of end-capped pH-sensitive mesoporous silica nanocarriers for on-demand drug delivery. Eur J Pharm Biopharm. 2014;88(3):1012–1025. | ||
Wang Y, Yu A, Caruso F. Nanoporous polyelectrolyte spheres prepared by sequentially coating sacrificial mesoporous silica spheres. Angew Chem Int Ed Engl. 2005;44(19):2888–2892. | ||
Argyo C, Weiss V, Bräuchle C, Bein T. Multifunctional mesoporous silica nanoparticles as a universal platform for drug delivery. Chem Mater. 2014;26(1):435–451. | ||
Dorozhkin SV. Amorphous calcium (ortho) phosphates. Acta Biomater. 2010;6(12):4457–4475. | ||
Rachas I, Tadros ThF, Taylor P. The displacement of adsorbed polymer from silica surfaces by the addition of a nonionic surfactant. Colloids Surf A. 2000;161:307–319. | ||
Gower LB. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem Rev. 2008;108(11):4551–4627. | ||
Gower LB, Odom DJ. Deposition of calcium carbonate films by a polymer-induced liquid-precursor (PILP) process. J Crystal Growth. 2000;210(4):719–734. | ||
Wolf SE, Leiterer J, Pipich V, Barrea R, Emmerling F, Tremel W. Strong stabilization of amorphous calcium carbonate emulsion by ovalbumin: gaining insight into the mechanism of ‘polymer-induced liquid precursor’ processes. J Am Chem Soc. 2011;133(32):12642–12649. | ||
Gower LB. Biomimetic mineralization of collagen. In: Aparicio C, Ginebra MP, editors. Biomineralization and Biomaterials: Fundamentals and Applications, 1st ed. Cambridge, CA: Woodhead Publishing, Elsevier Ltd; 2016:187–232. | ||
Xu Z, Yang Y, Zhao W, et al. Molecular mechanisms for intrafibrillar collagen mineralization in skeletal tissues. Biomaterials. 2015;39(1):59–66. | ||
Liu Y, Kim YK, Dai L, et al. Hierarchical and non-hierarchical mineralisation of collagen. Biomaterials. 2011;32(5):1291–1300. | ||
Price PA, Toroian D, Lim JE. Mineralization by inhibitor exclusion: the calcification of collagen with fetuin. J Biol Chem. 2009;284(25):17092–17101. | ||
Nudelman F, Lausch AJ, Sommerdijk NA, Sone ED. In vitro models of collagen biomineralization. J Struct Biol. 2013;183(2):258–269. | ||
Pashley DH, Tay FR, Breschi L, et al. State of the art etch-and-rinse adhesives. Dent Mater. 2011;27(1):1–16. | ||
Godin B, Gu J, Serda RE, et al. Tailoring the degradation kinetics of mesoporous silicon structures through PEGylation. J Biomed Mater Res A. 2010;94(4):1236–1243. | ||
Serda RE, Ferrati S, Godin B, Tasciotti E, Liu X, Ferrari M. Mitotic trafficking of silicon microparticles. Nanoscale. 2009;1(2):250–259. | ||
Mahamid J, Sharir A, Gur D, Zelzer E, Addadi L, Weiner S. Bone mineralization proceeds through intracellular calcium phosphate loaded vesicles: a cryo-electron microscopy study. J Struct Biol. 2011;174(3):527–535. | ||
Boonrungsiman S, Gentleman E, Carzaniga R, et al. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc Natl Acad Sci U S A. 2012;109(35):14170–14175. | ||
O’Brien CM, Holmes B, Faucett S, Zhang LG. Three-dimensional printing of nanomaterial scaffolds for complex tissue regeneration. Tissue Eng Part B Rev. 2015;21(1):103–114. | ||
Fu R, Selph S, McDonagh M, et al. Effectiveness and harms of recombinant human bone morphogenetic protein-2 in spine fusion: a systematic review and meta-analysis. Ann Intern Med. 2013;158(12):890–902. | ||
Sun JL, Jiao K, Niu LN, et al. Intrafibrillar silicified collagen scaffold modulates monocyte to promote cell homing, angiogenesis and bone regeneration. Biomaterials. 2017;113(1):203–216. |
Supplementary materials
Cytotoxicity assay
Human bone marrow-derived human mesenchymal stem cells (hMSCs; FC-0057; Lifeline® Cell Technology, Frederick, MD, USA) were used to evaluate the cytotoxicity of poly(allylamine)-stabilized amorphous calcium phosphate-loaded expanded-pore mesoporous silica nanoparticles (PAH-ACP@pMSN) using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, the hMSCs were seeded at 5,000 cells/well in 96-well plates and cultured for 24 h to achieve 80%–85% confluency. The cells were then exposed to a series of increasing concentrations of PAH-ACP@pMSN (0, 10, 20, 40, 80, 160, 320, 640, 1,280 μg/mL). After 24 h of incubation, 10 μL of MTT solution (5 mg/mL) was added to each well and the plate was incubated for 4 h. The medium was replaced with 150 μL of dimethyl sulfoxide for dissolution of the MTT formazan produced by intracellular mitochondrial dehydrogenases. The optical density of formazan salt at 490 nm was measured with a plate reader (Synergy H1; Biotek Instruments Inc., Winooski, VT, USA). Background absorbance of media was subtracted from the readings. Experiments were performed in sextuplicate.
Effect of PAH-ACP@pMSN on osteogenic differentiation of hMSCs
Cell culture
The hMSCs were plated in a 24-well format at a concentration of 3×105/well and cultured in 77% Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum, 10% horse serum, 1% L-glutamine, and 2% 100 U/mL penicillin, and 100 mg/mL streptomycin. The cells were cultured for 4 days to enable them to become established. The expansion medium was replaced with osteogenic medium (complete expansion medium supplemented with 50 μg/mL ascorbic acid, 10 mmol/L β-glycerophosphate, and 100 nmol/L dexamethasone; MilliporeSigma, St Louis, MO, USA). Differentiated hMSCs were exposed to pMSN (1,000 μg/mL) or PAH-ACP@pMSN (1,280 μg/mL) for 7 days. Differentiated cells not exposed to materials were used as control.
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)
Expression of osteogenic differentiation markers by hMSCs was examined in sextuplicate. The total RNA present in the hMSCs derived from each experimental or control group was isolated and purified using RNeasy Mini Kit (QIAGEN, Valencia, CA, USA). Quality control of the extracted RNA samples was performed by measuring their UV absorbance with the NanoDrop ND-1000 UV-visible spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). The quantity of extracted RNA was determined from the RNA absorbance at 260 nm; RNA purity was determined from the ratio of the absorbance (A@260 nm/A@280 nm and A@260/A@230 >1.8 and <2.0). Equal amounts of total RNA (0.1 mg/mL) were reverse-transcribed into single-stranded complementary DNA (cDNA) using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) in a 7,300 real-time PCR system (Applied Biosystems). Reverse transcription was performed at 25°C for 10 min, 37°C for 120 min, and 85°C for 5 min. The cDNA was stored at −20°C until use.
Osteogenic target markers employed for qRT-PCR were Runt-related transcription factor 2 (RUNX2), alkaline phosphatase (ALP), bone sialoprotein (BSP), dentin matrix protein-1 (DMP-1), osteocalcin (OCN), and osterix (OSX). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the housekeeping gene. A FastStart Universal SYBR Green Master Kit (Roche Applied Science, Indianapolis, IN, USA) was employed for qRT-PCR, using forward and reverse primers for the aforementioned genes (Table S1) in the 7,300 qRT-PCR system. Plates were loaded in the thermocycler and run in regular cycles. Each cycle consisted of 50°C for 2 min, 95°C for 10 min, 40 cycles of denaturing/annealing at 95°C for 15 sec, and 60°C for 1 min. The comparative threshold cycle method (ΔΔCt) was employed for relative quantification of gene expression. The expression level of each target cDNA marker was normalized to the GAPDH endogenous control. Data output was expressed as fold regulations of mRNA expression levels, given by 2−ΔΔCt. Gene expressions of hMSCs exposed to the pMSN and PAH-ACP@pMSN were compared with unexposed hMSCs (control), with the latter taken to be unity (value =1).
Alkaline phosphatase (ALP) activity
Intracellular release of ALP enzyme was measured using the Quantichrom ALP assay kit (Bioassay Systems, Hayward, CA, USA). Colorimetric determination was based on hydrolysis of p-nitrophenyl phosphate by ALP into inorganic phosphate and p-nitrophenol, a yellow-colored product. After exposure of the pMSN (1,000 μg/mL) or PAH-ACP@pMSN (1,280 μg/mL) to hMSCs cultured in osteogenic medium for 1 week, the cells were lysed with 0.2% Triton X-100 for 20 min. The cell lysate was mixed with assay buffer. Absorbance of p-nitrophenol at 405 nm was recorded every minute for a maximum of 16 min using a microplate reader (Synergy HT; BioTek Instruments, Winooski, VT, USA). The level of ALP activity of the cell lysate in IU/L (μmol/L/min) was calculated from the equation: (ODSample t − ODSample 0) × Reaction Vol ×35.3/(ODCalibrator − ODH2O) × Sample Vol × t, where OD is the optical density at 405 nm, t is the time in min, and ODSample 0 is the optical density at 0 min. The peak value for each material was used for comparison among groups (N=6).
Statistical analyses
Data sets acquired from each assay were evaluated for their normality (Shapiro–Wilk test) and equal variance assumptions (modified Levene test). If those assumptions appeared to be violated, the data sets were nonlinearly transformed to satisfy the assumptions prior to the use of parametric statistical methods. For MTT assay, the data were analyzed by one-factor analysis of variance (ANOVA), and Tukey pairwise comparison procedures at α =0.05.
For qRT-PCR, because the chance of finding one or more significant differences in testing six hypotheses (ie, six genes) with the same specimens (type I error) is 26.49%, the Dunn–Sidak correction was used for controlling the familywise error rate in multiple comparison procedures. Accordingly, the critical value (α =0.05) was reduced to 0.0085 for each osteogenic marker, so test results with P<0.0085 were only considered significantly different from the GADPH endogenous control.
For analysis of ALP enzymatic activity, data from the three groups were analyzed with one-factor ANOVA and Tukey multiple comparison procedures, with α =0.05.
Results
 | Table S1 Forward and reverse primers employed for qRT-PCR |
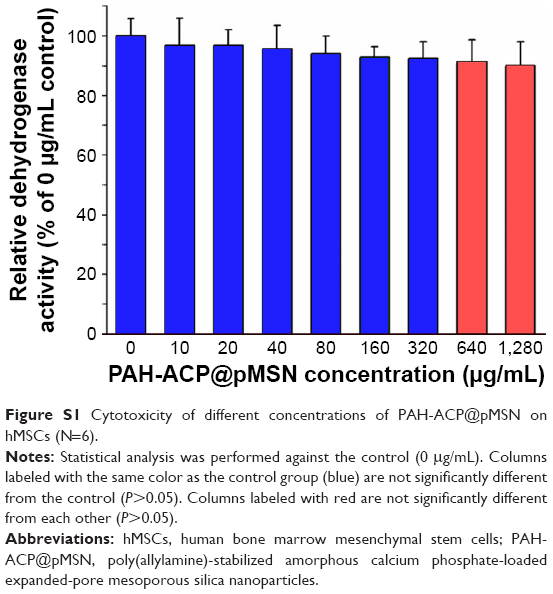 | Figure S1 Cytotoxicity of different concentrations of PAH-ACP@pMSN on hMSCs (N=6). |
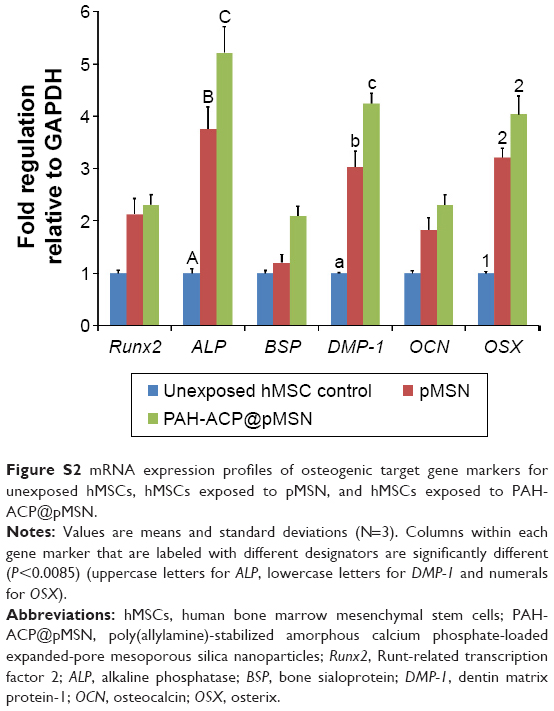 | Figure S2 mRNA expression profiles of osteogenic target gene markers for unexposed hMSCs, hMSCs exposed to pMSN, and hMSCs exposed to PAH-ACP@pMSN. |
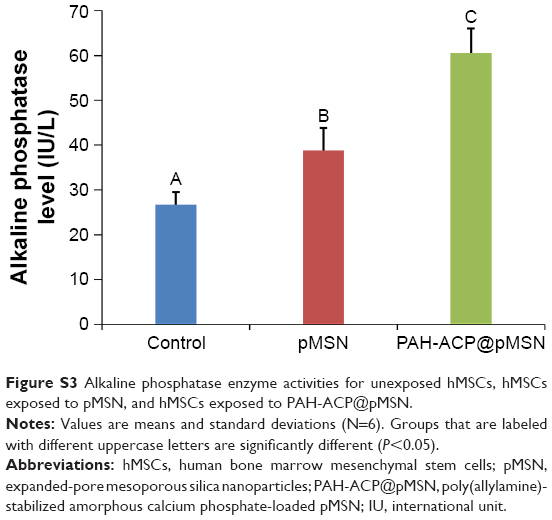 | Figure S3 Alkaline phosphatase enzyme activities for unexposed hMSCs, hMSCs exposed to pMSN, and hMSCs exposed to PAH-ACP@pMSN. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.