Back to Journals » International Journal of Nanomedicine » Volume 11
Biocompatible nanocarriers that respond to oxidative environments via interactions between chitosan and multiple metal ions
Authors Zhang S, Xia L, Ding C, Wen L, Wan W, Chen G
Received 29 January 2016
Accepted for publication 13 April 2016
Published 13 June 2016 Volume 2016:11 Pages 2769—2784
DOI https://doi.org/10.2147/IJN.S105339
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Shichang Zhang, Liye Xia, Chenchen Ding, Lu Wen, Weihua Wan, Gang Chen
Department of Pharmaceutics, Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China
Abstract: Hydrogen peroxide (H2O2) functions as an early damage signal contributing to the oxidative stress response and can act as a trigger in smart oxidation-responsive drug delivery systems that are currently in development. Current H2O2-triggered oxidation-responsive polymeric systems are usually derived from chemical synthesis and rarely include natural polymers. Herein, we report two series of nanoparticle (NP) complexes prepared with the biopolymer chitosan (CS) and four different metal ions (Cu2+, Ca2+, Zn2+, and Fe3+), defined as CSNPs-metal complexes (Series 1) and CS-metal complexes NPs (Series 2), which responded to oxidation by dissolving upon H2O2 exposure. Experiments examining Nile red release and H2O2-triggered degradation confirmed that both series of complexes showed better sensitivity to oxidation than the CSNPs alone. Furthermore, preliminary cytotoxicity and histological observations indicated that the two series exhibited little or no cytotoxicity and generated a mild inflammatory response. Our work provides a novel and promising strategy for developing NPs for use as intelligent oxidation-responsive systems.
Keywords: oxidation-responsive system, chitosan, nanoparticles, hydrogen peroxide, metal complexes
Introduction
Reactive oxygen species (ROS) such as hydrogen peroxide (H2O2), superoxide, and hydroxide radicals play dual roles as both beneficial and harmful species that can alter the cellular redox balance under oxidative stress conditions. This phenomenon has driven research focused on creating ROS-specific detection systems and ROS-responsive micro- or nanocarriers.1–3 Recently, numerous polymers designed to sense specific cellular oxidants in vivo, including H2O2 and hypochlorous acid, have been proposed for oxidation-responsive applications.2 However, these chemosynthetic polymers contain groups that are usually time-consuming and/or environmentally unfriendly to produce, such as metallocenes, (poly) pyridyl metal complexes, polyconjugated sequences, thiols, thioethers (sulfides), and stannanes.4
Chitosan (CS) is a biodegradable, biocompatible, and natural polymer with low immunogenicity. When in nanoparticle (NP) form, CS has been reported to attenuate H2O2-induced stress injury.5 Moreover, the metal complexes (CS–Cu, CS–Zn) display high scavenging on the superoxide anion radical and hydroxyl radical produced by the EDTANa2·Fe (II)-H2O2 system.6 In addition, it has been shown that H2O2 in the presence of metal ions such as Cu2+ leads to Fenton-based reactions that lead to the oxidative degradation of NPs containing sp2-hybridized bonds and can also cause the degradation of NPs.7,8 Based on these background studies, we theorized that NPs containing CS and metal ions would be capable of serving as nanocarriers in response to H2O2. Here, we systematically synthesized two series of NPs prepared with CS and various metal ions using two methods (Figure S1). Method 1 generated the CSNPs-metal complexes (Series 1). In method 1, CSNPs were first prepared via ionic gelation of sodium tripolyphosphate (TPP), a nontoxic cross-linking agent widely used because of its multivalent properties,9 and then four different types of metal ions (Cu2+, Ca2+, Zn2+, and Fe3+) were chelated with the CSNPs. Method 2 produced CS-metal complex NPs (Series 2). In method 2, CS-metal complexes were created first, using CS and the four different metal ions. This reaction was followed by the ionic gelation of TPP, which resulted in the formation of Series 2. To our knowledge, no studies have systematically investigated the oxidation responsiveness of NPs fabricated with CS and metal ions until now.
Materials and methods
Materials
CS (deacetylation: 80%–85%, ~100 kDa) was purchased from Yuhuan Ocean Biochemical Co. (Zhejiang, People’s Republic of China). TPP, zinc sulfate, H2O2, and acetic acid were purchased from Guangzhou Chemical Reagent Factory (Guangzhou, People’s Republic of China). Sodium hydroxide was purchased from Guangdong Guanghua Chemical Reagent Factory (Shantou, People’s Republic of China). Calcium chloride was purchased from Tianjin Guangcheng Chemical Co. Ltd. (Tianjin, People’s Republic of China). Copper sulfate, Tween 80, and ethanol were purchased from Tianjin Yongda Chemical Co. Ltd. (Tianjin, People’s Republic of China). Ferric sulfate was purchased from Tianjin Fuchen Chemical Co. Ltd. (Tianjin, People’s Republic of China). Distilled water was supplied by Guangzhou Watson’s Food & Drinks Co. Ltd. (Guangzhou, People’s Republic of China). Nile red was purchased from Guangzhou Qiyun Biotechnology Co. Ltd. (Guangzhou, People’s Republic of China). All the other reagents were of analytical grade.
Preparation of CS-metal complexes
CS (50 mg) was dissolved in 1% (v/v) diluted acetic acid to form a 2.5 mg/mL CS solution. Desired quantities of metal ions (5 mL, 4 mg/mL CuCl2, 6 mg/mL CaCl2 solution, 6 mg/mL ZnSO4, and 8 mg/mL Fe2(SO4)3, respectively) were added to the CS solutions (20 mL) and mixed. The pH value was increased to 6.0 by the addition of NaOH solution. After stirring for 3 hours, each mixture was poured into an excess of acetone. The precipitates were collected by filtering, repeatedly washed with ethanol, and dried under vacuum until a constant weight was observed.9
Preparation of NPs
Preparation of CSNPs
CSNPs were prepared based on the ionic gelation of positively charged amino groups on CS and negatively charged phosphate groups on TPP. Briefly, CS (50 mg) was dissolved in 1% (v/v) acetic acid to form a 2.5 mg/mL CS solution and then adjusted to pH 5.3–5.5 with 1 mol/L NaOH. TPP solution (8 mL, 1 mg/mL) was slowly added to the CS solution (20 mL) and stirred magnetically at 500 rpm, and this reaction was performed for 30 minutes.10 The products were purified by centrifugation at 3,000 rpm for 10 minutes, and the supernatants were decanted and analyzed after freeze-drying and dissolution.
Preparation of Series 1
The desired quantities of metal ions (5 mL, 4 mg/mL CuCl2, 6 mg/mL CaCl2, 6 mg/mL ZnSO4, and 8 mg/mL Fe2(SO4)3, respectively) were added to the CSNPs solution (20 mL, 2.5 mg/mL) and stirred magnetically at 200 rpm for 3 hours. The purification processes of the products were the same as the purification of CSNPs.
Preparation of Series 2
CS-metal complexes (50 mg) were dissolved in 1% (v/v) acetic acid to form 2.5 mg/mL CS-metal solutions and were then adjusted to pH 5.3–5.5 with 1 mol/L NaOH. TPP solution (8 mL, 1 mg/mL) was slowly added to the CS-metal solutions (20 mL) and stirred magnetically at 500 rpm, and this reaction was performed for 30 minutes. The purification processes of the products were the same as the purification of CSNPs.
Preparation of NPs loaded with Nile red
Nile red (1 mg) was dissolved in Tween 80 (1 g) and was then mixed with TPP solution (8 mL, 1 mg/mL) under magnetic stirring at room temperature. Then the follow-up processes were the same as the purification of blank CSNPs, Series 1 and Series 2.
Characterization methods
Transmission electron microscopy analysis
The morphologies of the NPs were examined with a transmission electron microscope (HITACHI H-7650, Hitachi Ltd., Tokyo, Japan) operating with a voltage of 80 kV.
Dynamic light scattering analysis
The particle size and the polydispersity index of CSNPs, Series 1 and Series 2 were determined using a dynamic light scattering (DLS) instrument (Delsa™ Nano C, Beckman Coulter, Brea, CA, USA).
X-ray diffractometry analysis
The physical form (crystalline or amorphous) of the CS, CSNPs, Series 1, and CS-metal complexes of Series 2 were obtained using an X-ray diffractometer (Bruker Smart 1000 CCD, Bruker Corporation, Billerica, MA, USA). The scanning speed was 2°/min, and scanning angles ranged from 5° to 80°.
Fourier transform infrared spectroscopy analysis
The structural characteristics of the CS, CSNPs, Series 1, and CS-metal complexes of Series 2 were detected using Fourier transform infrared spectroscopy (FTIR) (Spectrum 100, PerkinElmer Inc., Waltham, MA, USA) and recorded in KBr pellets with four scans at a resolution of 4 cm−1 over a wave number range of 4,000-450 cm−1.
Thermogravimetric and differential thermal analysis (TGA-DTA)
TGA-DTA measurements were obtained by using a STA 449 F3 Jupiter® (NETZSCH-Gerätebau GmbH, Selb, Germany) instrument. TGA-DTA of CS, CSNPs, Series 1, and CS-metal complexes of Series 2 were performed under a nitrogen atmosphere at a heating rate of 10°C/min, and the weight loss was analyzed.
Nile red release
An amount of 2 mL of H2O2 (2,000, 1,000, 300, or 100 mM) was added to 2 mL of CSNPs, Series 1 or Series 2 loaded with Nile red, and the solutions were incubated at 37°C. At various time points, the solutions were collected, and the Nile red level was determined by a fluorospectrophotometer (RF-5301PC, Shimadzu Corp., Kyoto, Japan). Each point was measured in triplicate.11
Oxidation-turbidity measurements
The oxidation-responsive reactions in the presence of H2O2 (1,000, 500, 150, or 50 mM) proceeded at 37°C with the time-dependent changes in turbidity (optical density at a wavelength of 400 nm) monitored by ultraviolet–visible spectroscopy (Shimadzu UV-2500) and photographed using a digital camera (three replicates).
Cytotoxicity tests
RAW264.7 macrophages were incubated for 24 hours at a density of 1×106 cells/well in a 24-well plate until reaching approximately 80% confluency. After treatment with the NPs (25–500 μg/mL) and incubation for 48 hours, the cells were treated with Annexin-V Alexa Fluor 647/propidium iodide (PI) for 15 minutes, and flow cytometry (BD FACScalibur flow cytometer, BD Biosciences, San Jose, CA, USA) was performed.
Biocompatibility study
Mice (18–22 g) were randomly divided into ten groups (n=7) and CSNPs and Series 1 and Series 2 were administered at doses of 1,000 mg/kg by intraperitoneal injection. Then, the mortality rate of mice was observed. All of the mice were sacrificed 2 weeks after injection, and their organs (heart, liver, lung, kidney, and brain) were dissected and embedded in paraffin. Paraffin sections were stained with hematoxylin and eosin and examined under a microscope (OPTEC BDS300, Chongqing, People’s Republic of China). All mice were obtained from the Laboratory Animal Center of Southern Medical University (Guangzhou, People’s Republic of China). Animal protocols were approved by the Animal Ethics Committee (Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China) for the use of experimental animals and all animal studies were carried out according to the Guide for Care and Use of Laboratory Animals. These animals had free access to food and water and were allowed a 1-week acclimation period before starting the experiment.
Results and discussion
In our work, both NPs series were prepared simply and under mild reaction conditions. The prepared NP solutions all exhibited the Tyndall effect and were transparent and clear, except for the CSNPs-Cu and CSNPs-Fe, which were light blue and brown, respectively. Transmission electron microscopy was performed to directly observe the NPs and showed that both Series 1 and Series 2 were nearly spherical (Figure 1). DLS was performed to provide evidence of NPs’ formation and indicated that both Series 1 and Series 2 had a narrow size distribution in the range of 200–400 nm (Figure 2A).
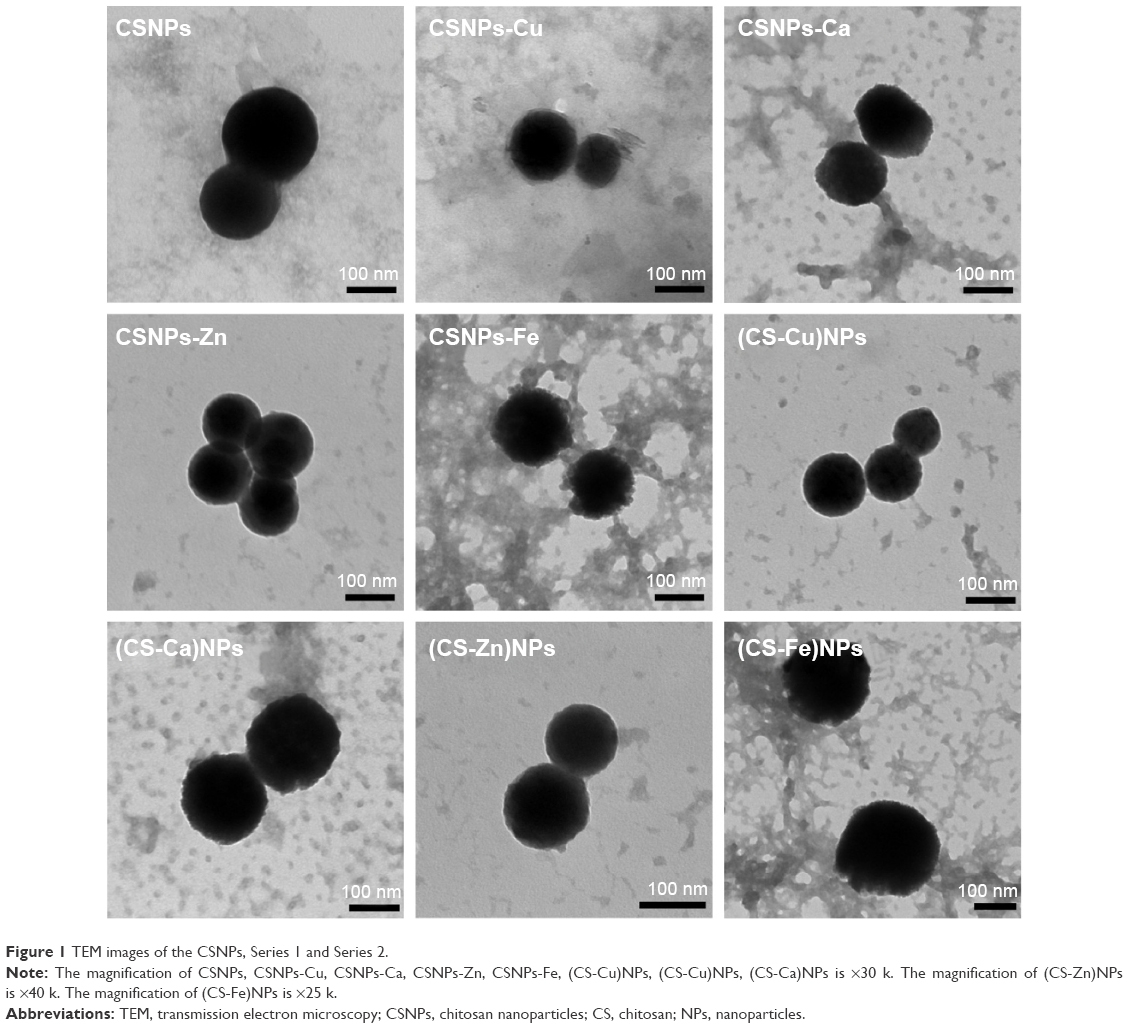 | Figure 1 TEM images of the CSNPs, Series 1 and Series 2. |
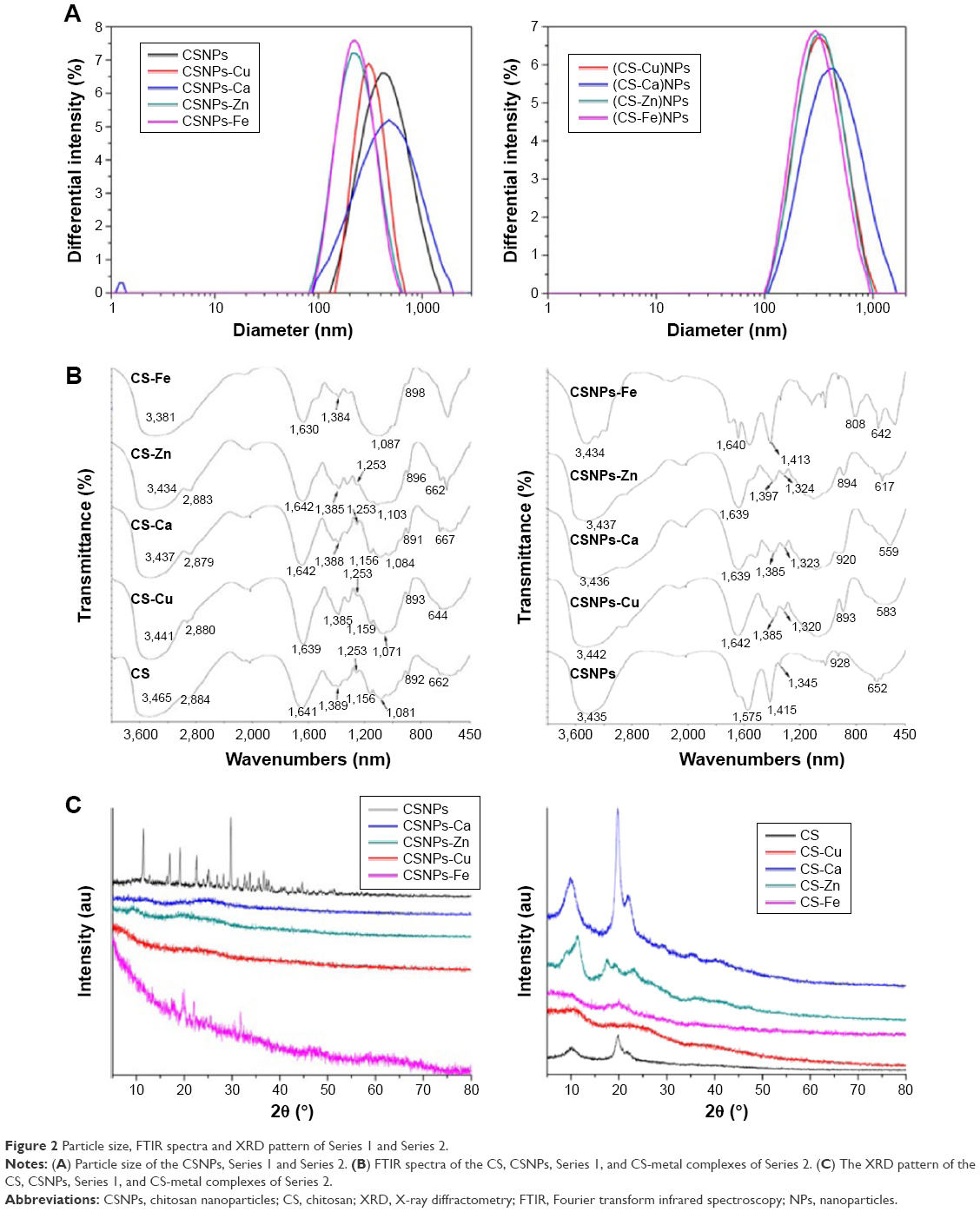 | Figure 2 Particle size, FTIR spectra and XRD pattern of Series 1 and Series 2. |
The FTIR spectra of CS, Series 1, and CS-metal complexes of Series 2 were acquired to provide a direct readout of complex formation. The peak around 3,435 cm−1 in Figure 2B was assigned to the -NH2 and -OH groups of CS, and the absorption bands of CS-metal complexes were much lower than that of CS. Moreover, the absorption bands at 1,641 cm−1 (δNH), 1,389 cm−1 (νCN), 1,253 cm−1 (νCN), and 662 cm−1 (ωNH) shifted to lower frequencies or decreased (Table S1), further demonstrating that the metal ions interacted with the -NH2 groups of CS. Similarly, the absorption bands at 1,081 cm−1 (νC-OH) and 1,156 cm−1 (ωC-O-C) decreased or disappeared, indicating that the metal ions also reacted with -OH groups in spite of the weak interactions. Additionally, the red shift or disappearance of the band at 2,884 cm−1 (νC-H) and the decrease of the band at 892 cm−1 (corresponding to the ring stretching vibration of CS skeleton) suggested that the coordination of metal ions weaken the skeleton vibration of CS, thus leading to the change in crystal structure of CS. In brief, the interaction of CS (-NH2 and -OH) and the metal ions caused a decrease in electron density of -NH2 and -OH and reduced the bond force constant of CS chemical bonds, subsequently related band absorptions all shifted to lower frequencies. Comparison of the spectra of CS and CSNPs showed red shifts from 3,465 to 3,435 cm−1, 1,641 to 1,575 cm−1, and 662 to 652 cm−1 (Figure 2B, Table S2), and the band at 1,389 cm−1 (νCN) was divided into two peaks at 1,415 and 1,345 cm−1, suggesting that TPP interacted with the -NH2 groups of CS. In addition, the disappearance of the band at 1,156 cm−1 (ωC-O-C) indicated that inter- and intra-molecular hydrogen bonding within CS was enhanced by the cross-linked effect of TPP. In the spectra of CSNPs and Series 1, the peaks at 3,435 cm−1 (CSNPs νNH and νOH), 1,575 cm−1 (CSNPs δNH), 1,415 cm−1, and 1,435 cm−1 (CSNPs νCN) all underwent shifts, especially the red shifts of the bands at 1,415 and 1,435 cm−1, indicating that there is competition between the metal ions and TPP to bind to the –NH2 of CS during the formation of Series 1. For this reason, in the spectrum of CSNPs, the band at 2,884 cm−1 disappeared and the band at 892 cm−1 decreased, but those peaks could be weakly observed when the complexes formed. These results, which were similar to those of previous studies,12,13 confirmed that the metal ions took part in the coordination.
X-ray diffractometry (XRD) analysis was performed to compare the crystallinities of CS, CSNPs, and Series 1. CS had three peaks at 2θ=10.2°, 19.8°, and 21.8° (corresponding to the reflections of the (020)h, (200)h, and (220)h planes of CS, respectively), which was in agreement with the literature14 (Figure 2C). In the XRD patterns of CSNPs, the characteristic CS peaks diminished and disappeared and many new diffraction peaks appeared, which resulted from the formation of new regular crystalline phases. This result was in accord with the result of FTIR, which confirmed that the cross-linked effect of TPP enhanced inter- and intra-molecular hydrogen bonding of CS. Except for a few new diffraction peaks in CSNPs-Fe spectrum, it was interesting that no peaks or amorphous humps were observed in the diffraction patterns of Series 1, indicating that the metal ions chelated with the -OH and -NH2 groups destroyed the new regular crystalline phase of CS with TPP. And this result further demonstrated that TPP and the metal ions had a competitive relationship, which was discussed in the FTIR section. Meanwhile, to validate formation of CS-metal complexes of Series 2, we examined the crystallinity of CS-metal complexes. In the XRD patterns of CS-Cu and CS-Fe, the characteristic CS peaks were not observed, suggesting that the Cu2+ and Fe3+ had chelated with CS. New diffraction peaks shown by CS-Zn XRD pattern and a slight change in CS-Ca spectrum also indicated that complexes had been formed. Compared to the CS-metal complex of Series 2 diffraction peaks, the metal ions within Series 1 destroyed the intramolecular CS hydrogen bonds more effectively because more metal ions were on the surface of CSNPs than CS, in agreement with previously published results.9 In addition, the XRD patterns of the prepared samples were compared with the Powder Diffraction Files found in the International Centre for Diffraction Data database, which showed that there were no CuCl2, CaCl2, ZnSO4·7H2O, and Fe2(SO4)3 crystals within CSNPs.
TGA-DTA was carried out to evaluate the thermostability of two NPs series. The differential thermogravimetric (DTG) curves of CSNPs (Figure 3 and Table S3) showed three significant losses of weight. The weight loss at 50°C–150°C was due to moisture evaporation, the weight loss at 200°C–300°C corresponded to CS decomposition, and the weight loss at 400°C–500°C was due to the elimination of cross-linking between the -NH3+ and the TPP phosphate groups. Series 1 showed two losses of weight due to strong coordination between the CSNPs and the metal ions, and the major peak shifted to 200°C–300°C. Two peaks on the DTG curves of CS and the CS-metal complexes of Series 2 are shown in Figure 3 and Table S4. The first peak occurred at approximately 88°C, whereas the second peak occurred at 220°C–320°C. The characteristic temperatures for the thermal degradation of CS and CS-metal complexes are provided in Table S4. At the first stage, the characteristic temperatures of CS-metal complexes were higher than that of CS, because the reaction of CS with the metal ions generated some pores, allowing water molecules to enter these holes and cause crystallization. At the second stage, compared to CS, the characteristic temperature of CS-metal complexes significantly changed, which was attributed to the deformation of the strong hydrogen bonds formed by CS and the metal ions.
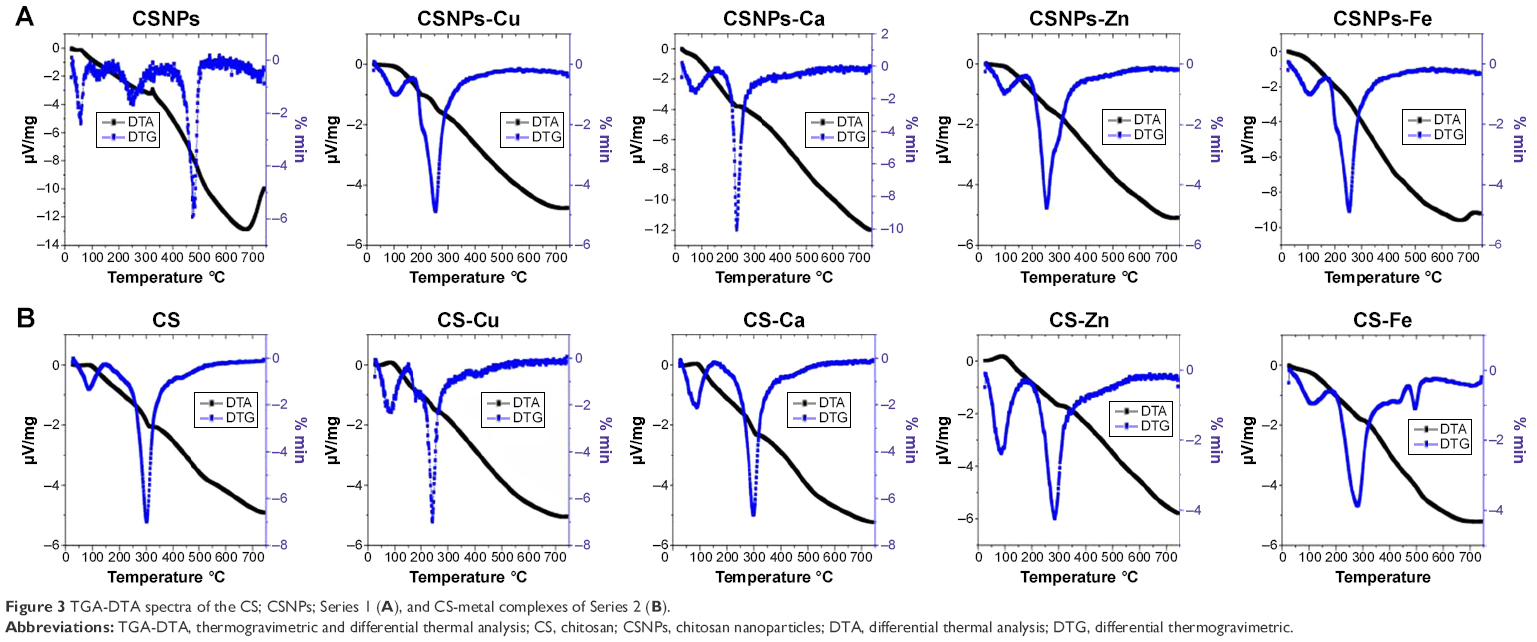 | Figure 3 TGA-DTA spectra of the CS; CSNPs; Series 1 (A), and CS-metal complexes of Series 2 (B). |
To evaluate the H2O2-triggered release response of Series 1 and Series 2 in vitro, NPs encapsulating solvatochromic Nile red dye were generated. The reactions were allowed to proceed at 37°C, and the time-dependent changes in Nile red fluorescence at 637 nm were monitored. The fluorescence of Nile red is quenched in aqueous solvents, thus spectral parameters were chosen to provide the highest readings in polar environments with low water content and negligible fluorescence in environments with higher water content. The Nile red encapsulation efficiency of nanocarriers was above 80% (Figure S2 and Table S5). Although Nile red was approximately stable in H2O2 even in a high concentration (see Figure S3), the fluorescence intensities of Series 1 and Series 2 decreased dramatically compared to that of the CSNPs when exposed to various concentration gradients of H2O2, as shown in Figure 4. The observed release rates of Nile red were as follows: Series 1> Series 2> CSNPs. The XRD results suggested that the molecular structures of the two series were looser than that of CSNPs due to lower crystallinity. The relatively loose structures led to H2O2 easily penetrating Series 1 and Series 2. Furthermore, as degradation of the amorphous regions occurs before degradation of the crystalline regions,15 the release of Nile red from Series 1 and Series 2 were faster than that of Nile red from CSNPs. Based on these analyses, the destruction of crystallinity resulting from the metal ions might play an important role in the increase of NPs’ oxidation responsiveness. Additionally, the CSNPs’ surfaces could offer more binding sites to interact with metal ions, which could lead to more metal ions destroying the crystallinity of CSNPs and explain why Series 1 reacted more easily with H2O2.
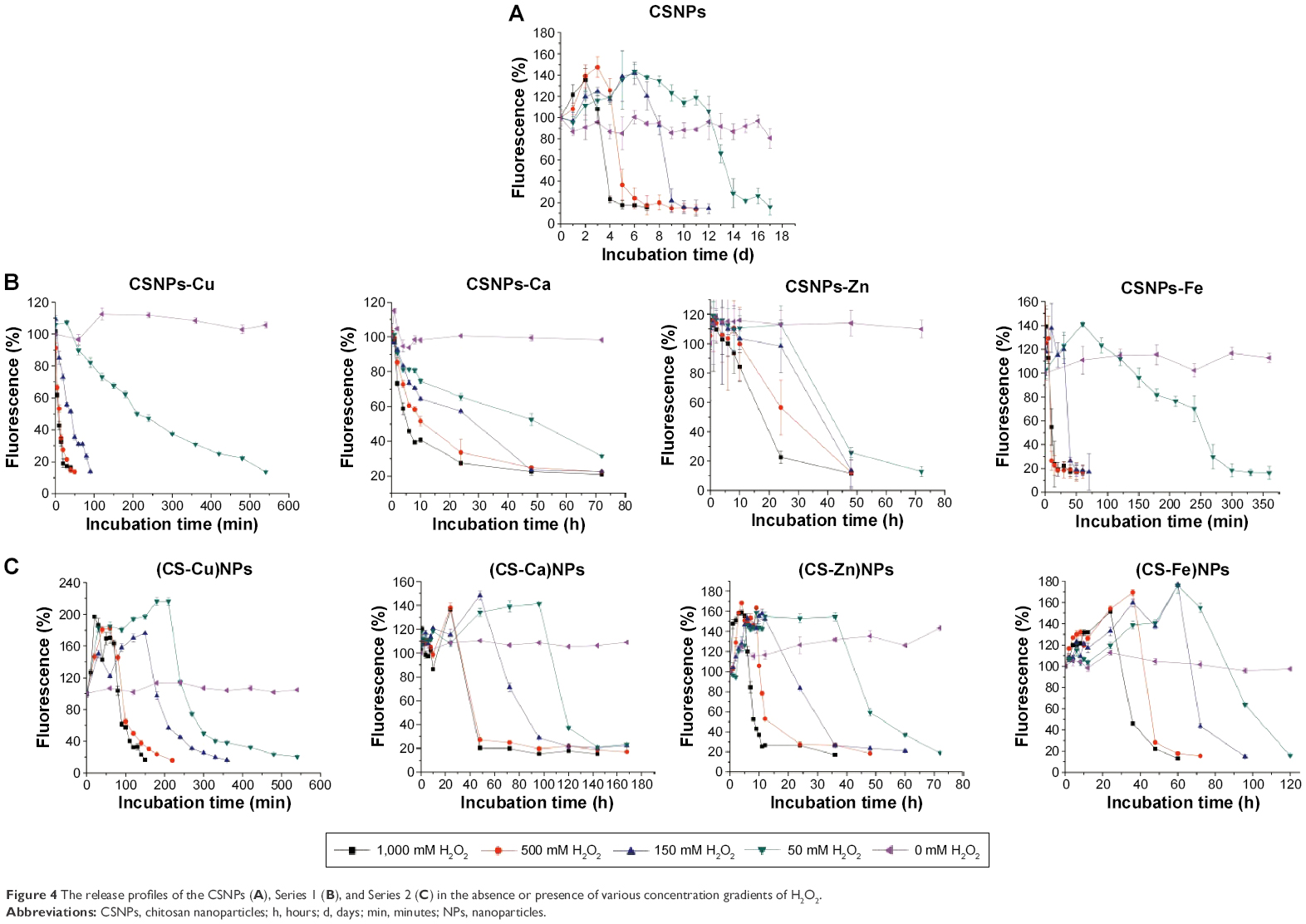 | Figure 4 The release profiles of the CSNPs (A), Series 1 (B), and Series 2 (C) in the absence or presence of various concentration gradients of H2O2. |
The oxidation responsiveness of Series 1 and Series 2 was investigated further using turbidity measurements. Changes in the optical density of polymer NP suspensions are indicative of nanosized polymers changing into small soluble molecules upon exposure to H2O2.16 As shown in Figure 5, in the presence of various concentration gradients of H2O2, the tendency toward turbidity of Series 1 and Series 2 increased at the beginning and then decreased, which was similar to the CSNPs results that had been further supported by DLS analysis (Figure S4 and Table S6). Overall, the increase of NPs’ suspension turbidity was attributed to the swelling of the NPs; oxidation reactions increased the hydrophilic character of the polymer chains, which therefore caused swelling without solubilization. As time passed, oxidation reactions decreased the number of polymer chains, which therefore caused solubilization. Notably, the swelling stage was not observed for the CSNPs-Cu because Cu2+ was sensitive enough to the oxidative environment due to its Fenton-like reaction,17 the CSNPs-Cu reaction occurred too quickly to be observed. Furthermore, both Nile red release and turbidity measurements confirmed that the two series exhibited more effective oxidation responsiveness than CSNPs alone.
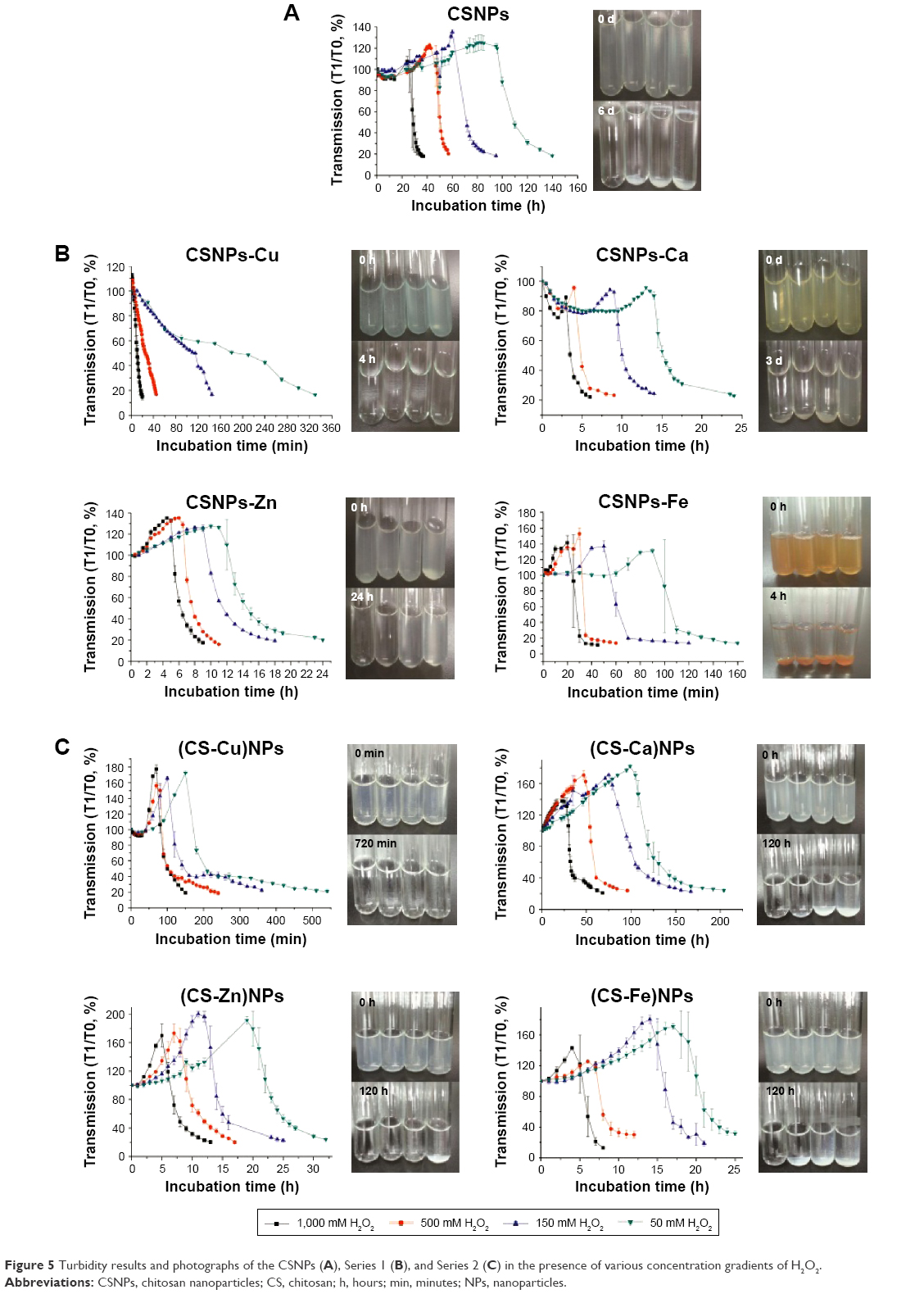 | Figure 5 Turbidity results and photographs of the CSNPs (A), Series 1 (B), and Series 2 (C) in the presence of various concentration gradients of H2O2. |
The cytotoxicity of Series 1 and Series 2 was evaluated at different concentrations using RAW264.7 macrophages and flow cytometry to assess the biocompatibility of the NPs (Figure 6 and Figure S5). The cytotoxicity observed in cells treated with various concentrations of Series 1 and Series 2 suggested that no significant cytotoxicity differences were observed up to concentrations of 200 (CSNPs-Zn) and 500 μg/mL (CSNPs-Cu). Note that with increasing concentrations of NPs, more -NH3+ groups become present, and therefore higher cytotoxicity is observed.18
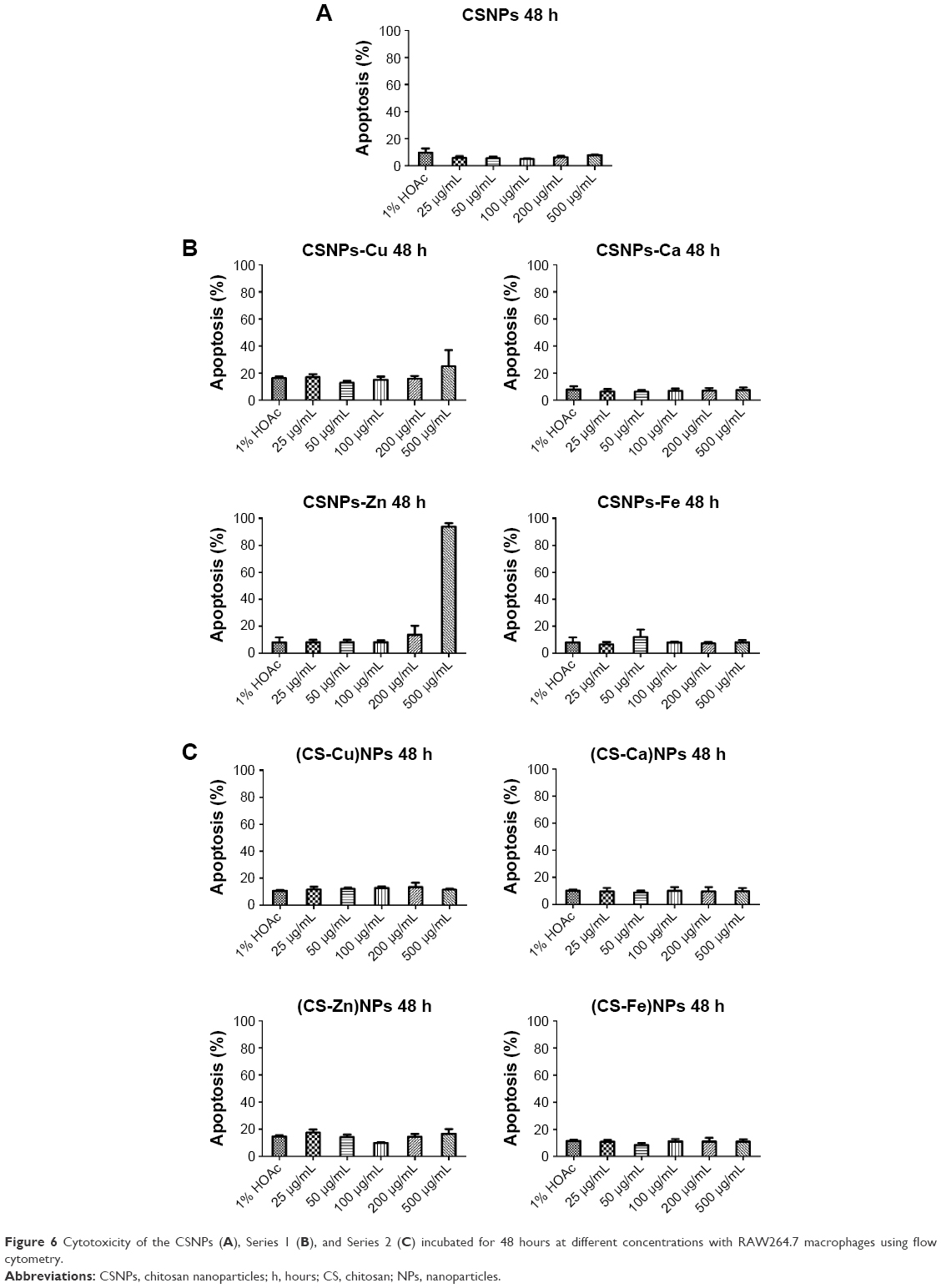 | Figure 6 Cytotoxicity of the CSNPs (A), Series 1 (B), and Series 2 (C) incubated for 48 hours at different concentrations with RAW264.7 macrophages using flow cytometry. |
Series 1 and Series 2 were then administered by intraperitoneal injection to evaluate the potential toxic effects of the NPs, and sections stained with hematoxylin and eosin are shown in Figure S6. There was no significant histological evidence of toxicity in the heart, lung, kidney, or brain tissues. The livers of mice treated with CSNPs-Zn and CSNPs-Cu showed different degrees of inflammatory reactions, which corresponded to the degree of cytotoxicity. These preliminary observations, together with the cytotoxicity results, suggest that both Series 1 and Series 2 exhibited little toxicity in vivo, and that Series 2 showed a better overall biocompatibility compared with Series 1.
Conclusion
In conclusion, we have developed two series of biocompatible and oxidation-responsive NPs using the biopolymer CS and four different metal ions. The two series were produced with two different methods to combine the metal ions and the -OH and -NH2 groups on the CS chains. The oxidation-responsiveness, cytotoxicity, and biocompatibility of the resulting complexes were ascertained. In this paper, we report that two different preparative methods can generate NPs with different oxidation-responsive and biocompatible properties. Compared to Series 2, Series 1 exhibited better sensitivity according to Nile red release and turbidity test results. However, cytotoxicity testing and histological observations indicated that Series 2 demonstrated very low cytotoxicity and inflammation, suggesting good biocompatibility. We believe these two series of biocompatible NPs show potential for use as nanocarriers for therapeutic agents, for in vivo imaging, and for applications in the treatment of oxidative stress-associated diseases.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (grant number 81573618), the Pearl River S&T Nova Program of Guangzhou (grant number 2013J2200059), and the Cultivation Foundation for Distinguished Young Teachers in Higher Education of Guangdong (grant number Yq2013099).
Disclosure
The authors report no conflicts of interest in this work.
References
Martin RE, Cao Z, Bulleid NJ. Regulating the level of intracellular hydrogen peroxide: the role of peroxiredoxin IV. Biochem Soc Trans. 2014;42(1):42–46. | ||
Broaders KE, Grandhe S, Fréchet JM. A biocompatible oxidation-triggered carrier polymer with potential in therapeutics. J Am Chem Soc. 2010;133(4):756–758. | ||
de Gracia Lux C, Joshi-Barr S, Nguyen T, et al. Biocompatible polymeric nanoparticles degrade and release cargo in response to biologically relevant levels of hydrogen peroxide. J Am Chem Soc. 2012;134(38):15758–15764. | ||
Lallana E, Tirelli N. Oxidation-responsive polymers: which groups to use, how to make them, what to expect from them (biomedical applications). Macromol Chem Phys. 2013;214(2):143–158. | ||
Wen ZS, Liu LJ, Qu YL, OuYang XK, Yang LY, Xu ZR. Chitosan nanoparticles attenuate hydrogen peroxide-induced stress injury in mouse macrophage RAW264.7 cells. Mar Drugs. 2013;11(10):3582–3600. | ||
Liu S, Xing R, Y HH, Li PC. Scavenging Effects of the Metal Complexes of Chitosan and Carboxymethyl Chitosan on Oxygen Free Radical. J Funct Polym. 2004;4(17):645–648. | ||
Lalwani G, Xing W, Sitharaman B. Enzymatic degradation of oxidized and reduced graphene nanoribbons by lignin peroxidase. J Mater Chem B Mater Biol Med. 2014;2(37):6354–6362. | ||
Xing W, Lalwani G, Rusakova I, Sitharaman B. Degradation of graphene by hydrogen peroxide. Part Part Syst Charact. 2014;31(7):745–750. | ||
Wang X, Du Y, Fan L, Liu H, Hu Y. Chitosan-metal complexes as antimicrobialagent: synthesis, characterization and structure-activity study. Polym Bullet. 2005;55(1):105–113. | ||
Fan W, Yan W, Xu Z, Ni H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf B Biointerfaces. 2012;90:21–27. | ||
Lee D, Bae S, Ke Q, et al. Hydrogen peroxide-responsive copolyoxalate nanoparticles for detection and therapy of ischemia–reperfusion injury. J Control Release. 2013;172(3):1102–1110. | ||
Qi L, Xu Z, Jiang X, Hu C, Zou X. Preparation and antibacterial activity of chitosan nanoparticles. Carbohydr Res. 2004;339(16):2693–2700. | ||
Sun Z, Lv F, Cao L, Liu L, Zhang Y, Lu Z. Multistimuli-responsive, moldable supramolecular hydrogels cross-linked by ultrafast complexation of metal ions and biopolymers. Angew Chem Int Ed Engl. 2015;54(27):7944–7948. | ||
Osorio-Madrazo A, David L, Trombotto S, Lucas JM, Peniche-Covas C, Domard A. Kinetics study of the solid-state acid hydrolysis of chitosan: evolution of the crystallinity and macromolecular structure. Biomacromolecules. 2010;11(5):1376–1386. | ||
Qin CQ, Du YM, Xiao L. Effect of hydrogen peroxide treatment on the molecular weight and structure of chitosan. Polym Degrad Stab. 2002;76(2):211–218. | ||
Napoli A, Valentini M, Tirelli N, Müller M, Hubbell JA. Oxidation-responsive polymeric vesicles. Nat Mater. 2004;3(3):183–189. | ||
Wang S. A comparative study of Fenton and Fenton-like reaction kinetics in decolourisation of wastewater. Dyes Pigments. 2008;76(3):714–720. | ||
Fischer D, Li Y, Ahlemeyer B, Krieglstein J, Kissel T. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24(7):1121–1131. |
Supplementary materials
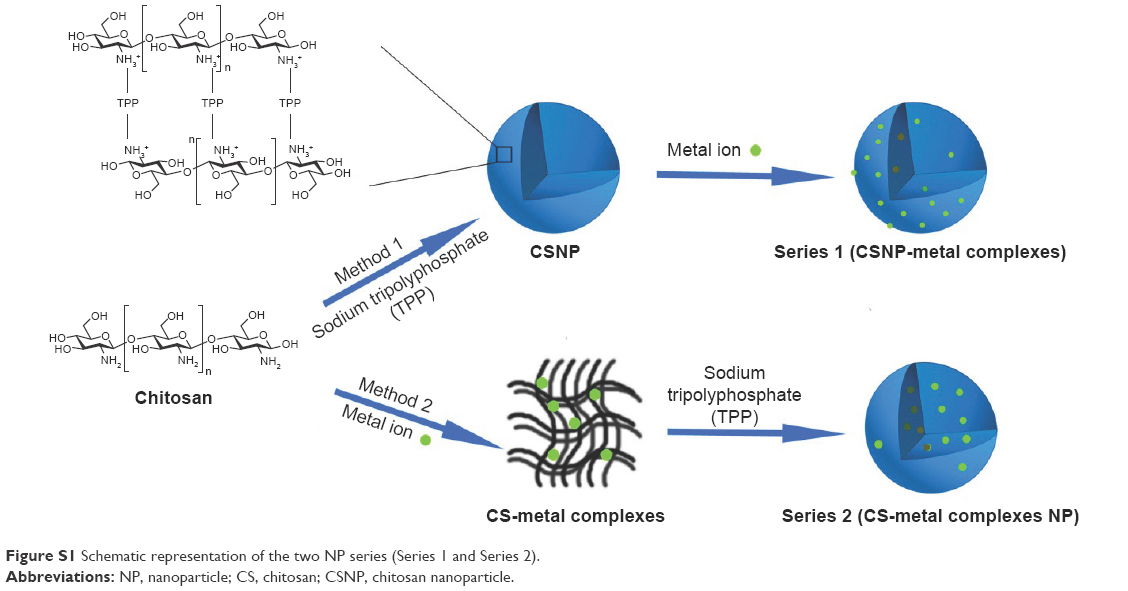 | Figure S1 Schematic representation of the two NP series (Series 1 and Series 2). |
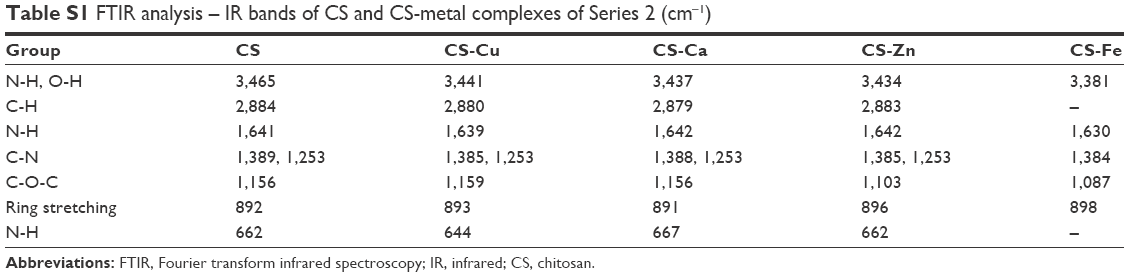 | Table S1 FTIR analysis – IR bands of CS and CS-metal complexes of Series 2 (cm−1) |
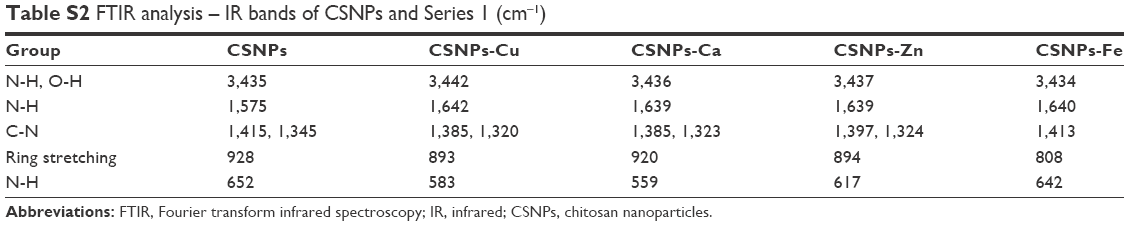 | Table S2 FTIR analysis – IR bands of CSNPs and Series 1 (cm−1) |
 | Table S3 TGA-DTA – Tmax for the thermal degradation of CSNPs and Series 1 (oC) |
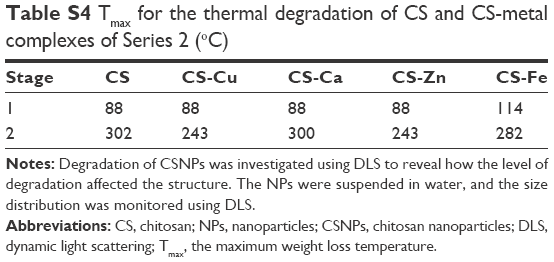 | Table S4 Tmax for the thermal degradation of CS and CS-metal complexes of Series 2 (oC) |
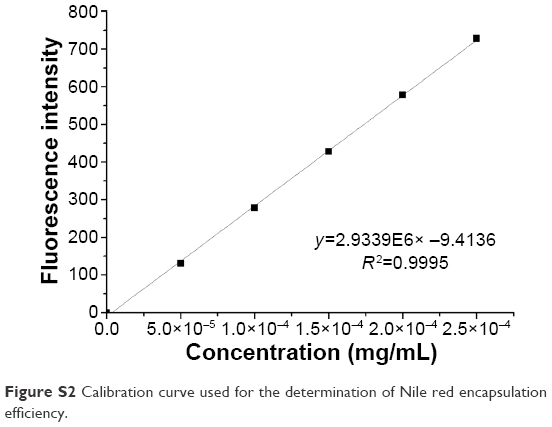 | Figure S2 Calibration curve used for the determination of Nile red encapsulation efficiency. |
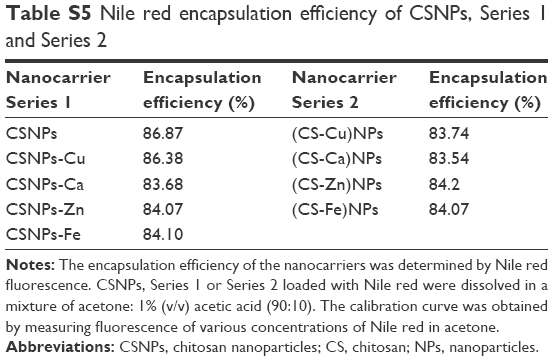 | Table S5 Nile red encapsulation efficiency of CSNPs, Series 1 and Series 2 |
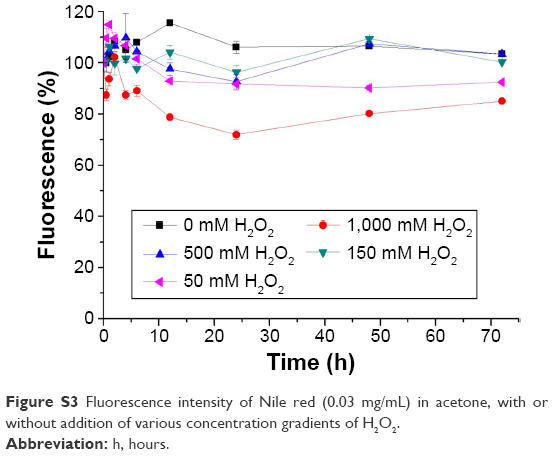 | Figure S3 Fluorescence intensity of Nile red (0.03 mg/mL) in acetone, with or without addition of various concentration gradients of H2O2. |
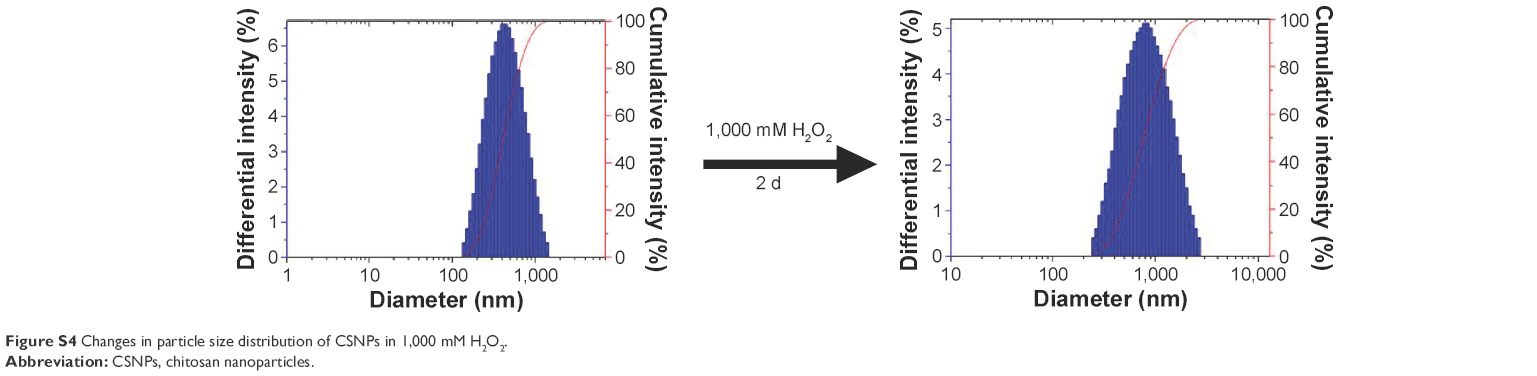 | Figure S4 Changes in particle size distribution of CSNPs in 1,000 mM H2O2. |
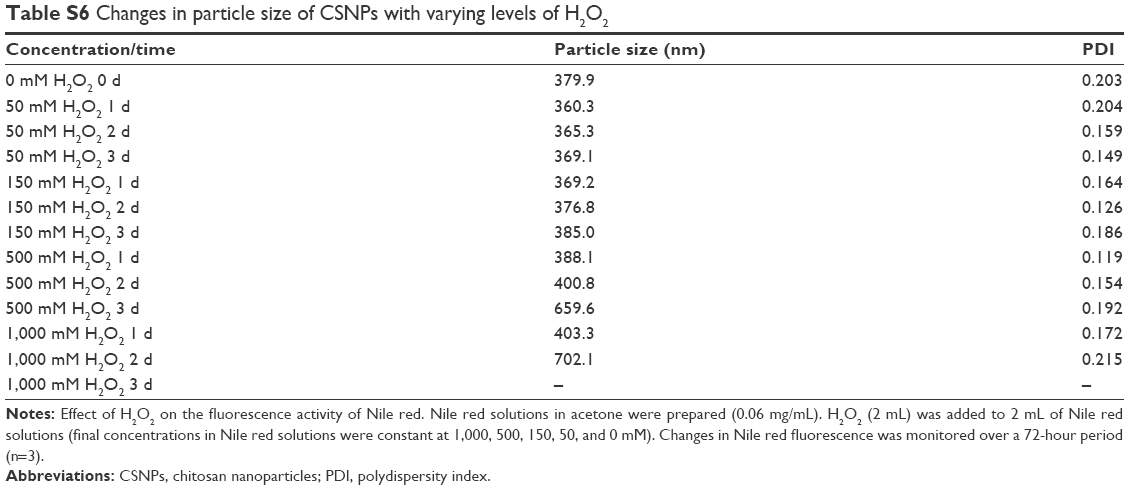 | Table S6 Changes in particle size of CSNPs with varying levels of H2O2 |
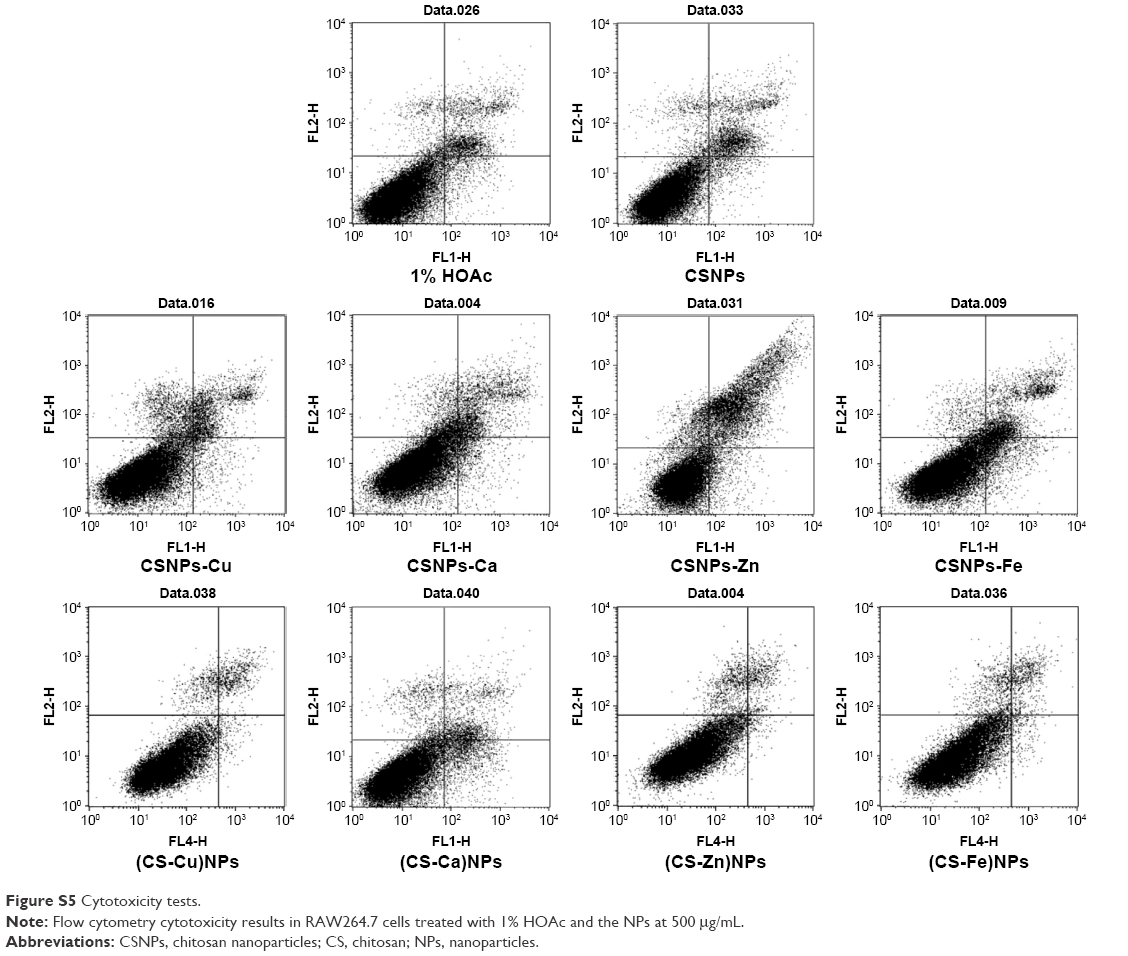 | Figure S5 Cytotoxicity tests. |
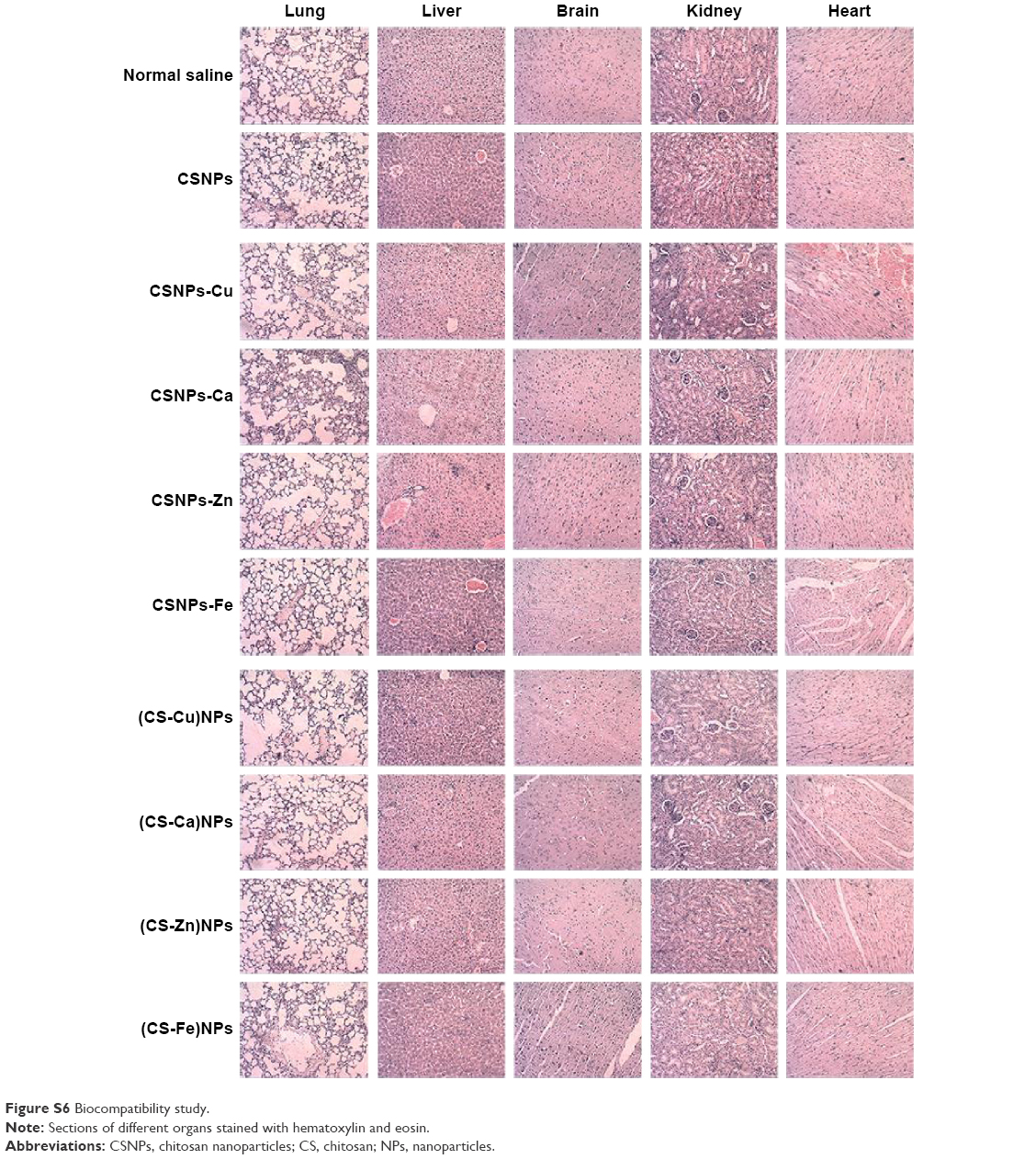 | Figure S6 Biocompatibility study. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.