Back to Journals » International Journal of Nanomedicine » Volume 10 » Special Issue on diverse applications in Nano-Theranostics
Biocompatible and biodegradable fibrinogen microspheres for tumor-targeted doxorubicin delivery
Authors Joo JY, Park GY ![]() , An SSA
, An SSA ![]()
Received 11 May 2015
Accepted for publication 20 July 2015
Published 1 September 2015 Volume 2015:10(Special Issue on diverse applications in Nano-Theranostics) Pages 101—111
DOI https://doi.org/10.2147/IJN.S88381
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Supplementary video presented by Joo et al.
Views: 523
Jae Yeon Joo, GilYong Park, SeongSoo A An
Department of Bionano Technology, Gachon Medical Research Institute, Gachon University, Seongnam-si, Republic of Korea
Abstract: In the development of effective drug delivery carriers, many researchers have focused on the usage of nontoxic and biocompatible materials and surface modification with targeting molecules for tumor-specific drug delivery. Fibrinogen (Fbg), an abundant glycoprotein in plasma, could be a potential candidate for developing drug carriers because of its biocompatibility and tumor-targeting property via arginine–glycine–aspartate (RGD) peptide sequences. Doxorubicin (DOX), a chemotherapeutic agent, was covalently conjugated to Fbg, and the microspheres were prepared. Acid-labile and non-cleavable linkers were used for the conjugation of DOX to Fbg, resulting in an acid-triggered drug release under a mild acidic condition and a slow-controlled drug release, respectively. In vitro cytotoxicity tests confirmed low cytotoxicity in normal cells and high antitumor effect toward cancer cells. In addition, it was discovered that a longer linker could make the binding of cells to Fbg drug carriers easier. Therefore, DOX–linker–Fbg microspheres could be a suitable drug carrier for safer and effective drug delivery.
Keywords: biocompatibility, anti-cancer drug, micro-structure, cytotoxicity, bio-conjugation, tumor targeting
Introduction
In recent years, various drug delivery techniques have emerged with the development of novel drug carriers, such as nanoparticles1 and microspheres.2 The drug carriers can improve drug delivery efficacy by preventing degradation of drugs from environmental factors,3 by increasing their solubility,4 by improving the pharmacokinetic properties of drugs that are difficult to deliver, and by controlling drug release. Even though some of them showed remarkable efficiency, suggesting their possible usage in clinical practice, their applications are still limited because of their toxicity or that of their metabolites, causing severe damage in tissues and organs.5 Therefore, many researches have focused on the usage of nontoxic and biocompatible materials for safer drug delivery. In particular, protein-based drug carriers have received considerable interest, since proteins are naturally degraded into amino acids and excreted from human body with lower immunogenicity.6 For example, biocompatible and biodegradable fibrinogen (Fbg) microspheres showed favorable cytocompatibility in the C2C12 cell line.7
On the other hand, chemotherapy is a prime choice for cancer treatment, and many antitumor drugs have been discovered and introduced in clinical practice. Unfortunately, chemotherapeutic agents are coupled with unintended adverse effects, such as tissue and organ damage,8 because the administered drugs circulate through the bloodstream and attack the rapidly dividing cells, which results in the destruction of normal cells as well as abnormal tumor cells. To minimize the side effect of the drugs, target-specific drug delivery techniques were introduced with various surface modifications of drug carriers, based on interactions between targeting moieties and receptors that are specifically over-expressed on the surface of tumor cells.9 In other words, tumor specificity of anticancer drugs could be significantly enhanced by combining targeting molecules with either the drugs or drug carriers. For example, an arginine–glycine–aspartate (RGD) peptide as a tumor-targeting moiety was conjugated onto the albumin nanoparticles with gemcitabine, inhibiting pancreatic tumor cell growth.10 Furthermore, various targeting molecules, such as folic acid11 and biotin,12 have been extensively investigated, demonstrating improved tumor specificity and reduced toxicity to normal cells.
Fbg, a 340-kDa glycoprotein in plasma, is closely involved in hemostasis. During hemostasis, Fbg is converted into fibrin by activated thrombin in the presence of calcium ions, causing a cross-link between fibrin molecules, thereby forming a fibrin clot to prevent blood loss. After completing the wound healing process, fibrin is naturally degraded by fibrinolysis.13 Since Fbg exists naturally in human body and goes through the degradation process without any immunogenicity, Fbg can be used as a nontoxic and biocompatible material for the development of drug carriers. In addition to its biocompatibility, Fbg possesses tumor targeting property through its arginine-glycine-aspartate (RGD) peptide sequence. RGD peptide, well-known as a tumor targeting peptide, has high affinity for αvβ3 integrin, which is over-expressed on the surface of various cancer cells.14,15 Based on the specific interaction between RGD peptide and αvβ3 integrin, RGD peptide can be extensively conjugated to various drug carriers, showing enhanced cellular uptake by tumor cells.10,16,17 Therefore, the biocompatibility of Fbg and the RGD sequence present in Fbg may offer a great potential for use in developing biocompatible drug delivery carriers as well as for targeting cancer cells.
The major aim of this study was to investigate the suitability of Fbg as a targeted delivery carrier for antitumor drugs. Doxorubicin (DOX), a commonly used anticancer drug, was conjugated to Fbg, and the microspheres were developed. Two different types of linkers were used for conjugating DOX to Fbg with the intent of creating a pH-responsive and slow drug release based on biodegradability. The method used for the fabrication of Fbg microspheres, characterizations of the particles, and different in vitro studies are described in this report.
Materials and methods
Materials
Fbg from bovine plasma and 2-iminothiolane (2-IT) were purchased from Sigma-Aldrich (Republic of Korea). Doxorubicin hydrochloride was obtained from LC Laboratories (Woburn, MA, USA) and 3-maleimidopropionic acid hydrazonium trifluoroacetic acid (3-MAH) was acquired from Toronto Research Chemicals Inc. (Ontario, Canada). Succinimidyl-[(N-maleimidopropionamido)-tetraethylene glycol] ester (SM(PEG)4) and succinimidyl-[(N-maleimidopropionamid)- dodecaethylene glycol] ester (SM(PEG)12) were purchased from Thermo Scientific (Rockford, IL, USA). Tissue plasminogen activator (t-PA) was acquired from Calbiochem and plasminogen was acquired from EMD Millipore (Darmstadt, Germany). The Celltiter-glo® luminescent cell viability assay kit was obtained from Promega Ltd (Seoul, Republic of Korea). NIH-3T3 and SH-SY5Y cell lines were kindly provided by NanoEntek Inc. (Seoul, Republic of Korea). Gibco® Dulbecco’s Modified Eagle’s Medium (DMEM), Gibco® Roswell Park Memorial Institute (RPMI) 1640, Gibco® penicillin–streptomycin (pen–strep), and Hyclone™ fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Seoul, Republic of Korea). All chemicals used in this experiment were of analytical grade. Institutional Review Board approval was not sought for the following research. All principles outlined in the Declaration of Helsinki were followed.
Synthesis of DOX–linker–Fbg conjugates using acid-labile linkers
DOX was conjugated to Fbg using acid-labile cross-linkers;18 3-MAH (L1, n=1) for acid-triggered drug release, and each conjugation concept was described in Figure 1A. DOX (1.5 mg, 2.76 μmol) was mixed with 3-MAH (0.88 mg, 2.97 μmol) in 1 mL of phosphate-buffered saline (PBS) (pH 7.2), and stirred with an orbital shaker (250 rpm) at 25°C in the dark for 24 hours. The resultant reaction produced DOX–linker [DOX–(3-MAH)] complexes through a hydrazone bond. To introduce thiol (–SH) groups on the Fbg molecules, Fbg (100 mg, 0.294 μmol) was dissolved in 2 mL of PBS (pH 7.2) containing 5 mM ethylenediaminetetraacetic acid, mixed with 2-IT (0.4 mg, 2.91 μmol), and stirred at 25°C for 1 hour. The DOX–linker solution was added to the thiolated-Fbg solution, and the reaction mixture was shaken at 25°C in the dark for 48 hours. After that, the DOX–linker–Fbg solution was purified using a size-exclusive Zeba™ Spin Desalting Column with 7 kDa molecular weight cut-off (MWCO) Thermo Fisher Scientific (Seoul, Republic of Korea) at 2,400 rpm for 2 minutes to remove the unconjugated DOX and linkers. Absorbance of the collected DOX–linker–Fbg solution was measured with micro-plate reader (Victor 3 Multilabel Counter, PerkinElmer Inc., Waltham, MA, USA) at 490 nm, and the conjugation efficacy was calculated from a standard curve of DOX concentration gradient.
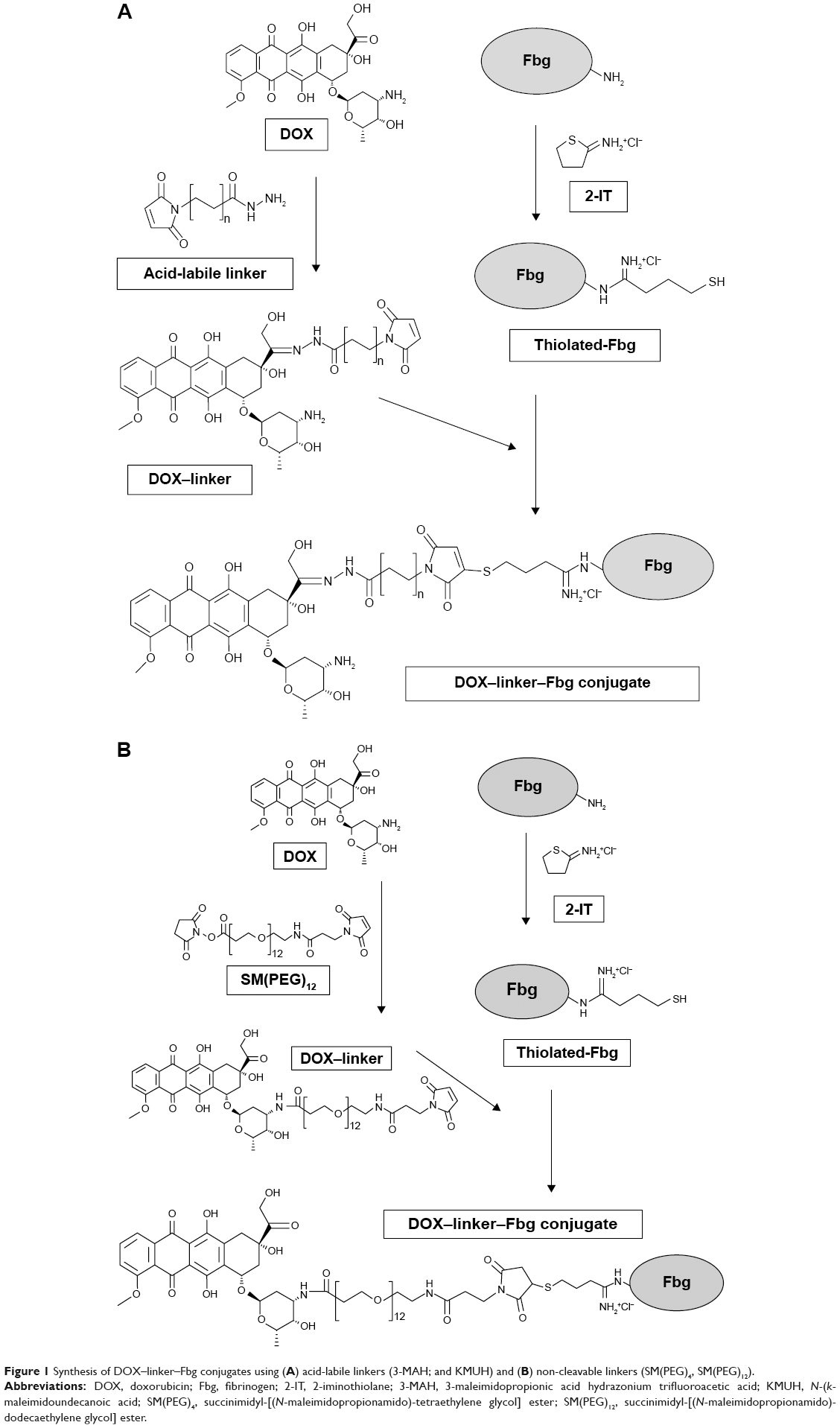 | Figure 1 Synthesis of DOX–linker–Fbg conjugates using (A) acid-labile linkers (3-MAH; and KMUH) and (B) non-cleavable linkers (SM(PEG)4, SM(PEG)12). |
Synthesis of DOX–linker–Fbg conjugates using non-cleavable linkers
To investigate the length effect of linkers, DOX was covalently attached to Fbg by non-cleavable linkers: SM(PEG)4 (L3, n=4) and SM(PEG)12 (L5, n=12), as outlined in Figure 1B. DOX (1.5 mg, 2.76 μmol) was mixed with SM(PEG)4 (1.6 mg, 3.12 μmol) or SM(PEG)12 (2.6 mg, 3.00 μmol) in 1 mL of PBS (pH 7.2) and stirred using an orbital shaker (250 rpm) at 25°C for 24 hours, forming stable DOX–linker (DOX–L3, DOX–L5) complexes through an amide bond. To introduce thiol (–SH) groups on the Fbg molecules, Fbg (100 mg, 0.294 μmol) was dissolved in 2 mL of PBS (pH 7.2) containing 5 mM ethylenediaminetetraacetic acid, mixed with 2-IT (0.4 mg, 2.91 μmol), and stirred at 25°C for 1 hour. The DOX–linker solution was added to the thiolated-Fbg solution, and the reaction mixture was shaken at 25°C for 12 hours. After that, the DOX–linker–Fbg solution was purified using a size-exclusive Zeba spin desalting column with 7 K MWCO (Thermo) at 1,000× g for 2 minutes to remove the unconjugated DOX and linkers. Absorbance of the collected DOX–linker–Fbg solution was measured using Victor 3 at 490 nm, and the conjugation efficacy was calculated from the DOX standard curve.
Preparation of Fbg microspheres
Microspheres were prepared with each DOX–linker–Fbg conjugate solution, using a modified water-in-oil (W/O) emulsification/solvent extraction method. The conjugate solution was constantly injected in soy oil (100 mL) using a syringe pump (1 mL/min), and stirred with an overhead stirrer (600 g) for 30 minutes to form stable a W/O emulsion. Then, the emulsified solution was gradually heated up to 60°C for 1 hour using a hot plate. Acetone was slowly added to extract the solvent and evaporate the water. Once the aqueous phase was evaporated, solid microspheres were formed after continuous addition of acetone. The solution was stirred for another 10 minutes to remove any residual solvent. The microspheres were collected by centrifugation at 1,000 rpm for 3 minutes, washed thrice with acetone, and dried in a vacuum chamber for 1 day.
Characterization of Fbg microspheres
Loading of DOX in Fbg microspheres was confirmed by a confocal microscope (D-eclipse; Nikon Corporation, Tokyo, Japan). The light source approximately 480 nm was used for excitation of DOX, and the emission light was collected by opening the 580 nm filter. A field emission scanning electron microscope (FE-SEM, HITACHI S-4,700) was used to capture images of the Fbg microspheres for morphological analysis. After the microspheres were sputter-coated with platinum for 120 seconds, the images are acquired.
Degradation test of Fbg microspheres
To evaluate the biodegradable characteristics of Fbg microspheres, fibrinolysis of microspheres was observed by the addition of plasmin and t-PA. The Fbg microspheres (20 mg/mL) were suspended by Tris buffered saline (45 μL), and treated with CaCl2 (10 μL of 50 mM), t-PA (20 μL of 20 μg/mL), and plasminogen (25 μL of 0.1 mM). The mixture was incubated at 37°C with rotation. After 4 hours, the microspheres were collected for SEM analysis.
Release profiles of DOX from the DOX–linker–Fbg microspheres with different pHs
A Slide-A-Lyzer® MINI dialysis device with a 20 K MWCO cellulose membrane (Thermo) was used for DOX release study, and PBS (pH 7.3 and 5.0) was used as a releasing media. The DOX–(3-MAH)–Fbg (30 mg), DOX–SM(PEG)4–Fbg (20 mg), or DOX–SM(PEG)12–Fbg (15 mg) microspheres, equivalent to 100 μg of DOX, which was calculated from drug loading efficiencies, was put in the device. Free DOX (100 μg) and bare-Fbg microspheres were incorporated into two more devices containing PBS (pH 7.3), which were used as a positive control and a negative control, respectively. Each device was kept on an orbital shaker (120 g) at 37°C for continuous shaking. Aliquots, containing released DOX, were taken at the predetermined time intervals, refilled with the same amount of fresh PBS, and the fluorescence intensity was measured at 480 nm for excitation and 560 nm for emission using Victor 3 for 7 days.
In vitro cytotoxic effect of DOX–linker–Fbg microspheres
SH-SY5Y (human neuroblastoma) and NIH-3T3 (mouse fibroblast) cell lines were cultured in DMEM medium containing 10% FBS and 1% pen–strep at 37°C with 5% CO2 in an incubator. The cytotoxicity of the DOX-loaded Fbg microspheres was evaluated by performing a celltiter-glo® luminescent cell viability assay. The cells were seeded into a 96-well opaque plate (~1×104 cells/well) with 100 μL of the culture medium and incubated for 1 day, enabling sufficient attachment of the cells onto the plate. The culture medium was replaced with culture media containing the DOX–linker–Fbg microspheres, bare-Fbg microspheres, and free-DOX solution. Prior to treatment, all the microspheres were sterilized with alcohol and dispersed in the culture medium. The amount of Fbg microspheres was determined to be equivalent to 10 μg of DOX. After adding the Fbg microspheres to the plate, the cells were incubated at 37°C with 5% CO2 for 24 hours. The celltiter-glo® reagent was prepared by complete mixing of celltiter-glo® substrate and the buffer. Reagent (100 μL) was added to each well, followed by mixing on the plate for 2 minutes using an orbital shaker (350 rpm) for cell lysis. Then, the plate was incubated at 25°C for stabilization of the luminescent signal. The luminescence was recorded using Victor 3 with an integration time of 1 second per well.
Statistical analysis
All the experiments were conducted in triplicate or more, and the results were presented as mean ± standard error. Significant differences were compared by Student’s unpaired t-test with P<0.05 as a significant level.
Results
DOX loading efficacy in DOX–linker–Fbg conjugates
For the acid-specific drug release, a hydrazine part of 3-MAH was conjugated to a 13-C carbonyl group of DOX, forming a hydrazone bond, which is hydrolyzed under a mild acidic condition (~pH 5),18 and a maleimide part of 3-MAH was conjugated with a thiol group of Fbg, forming a thioester bond. Consequently, 22.44%±2.13% of DOX was loaded onto Fbg, and approximately three DOX molecules seemed to be attached to each Fbg molecule. For the non-cleavable drug conjugation, the N-hydroxysuccinimide ester part of SM(PEG)4 and SM(PEG)12 was conjugated to a 3′-C amino group of DOX, forming a relatively stable amide bond, and a maleimide group of SM(PEG)4 and SM(PEG)12 was cross-linked to Fbg by a thioester bond. DOX (33.10%±2.41%) was loaded onto Fbg with SM(PEG)4, having a DOX/Fbg ratio of 4.11, and 45.25%±2.45% of DOX was carried to Fbg with SM(PEG)12, showing 5.95 molecules on a Fbg molecule. The overall results of DOX loading are summarized in Table 1.
 | Table 1 Summary of DOX loading with different linkers |
Characterization of the Fbg microspheres
Using a confocal microscope, it was revealed that DOX molecules were evenly distributed in the DOX–linker–Fbg microspheres. The bare-Fbg microspheres (without DOX loading) could not be observed by confocal microscope image, whereas the other three kinds of microspheres clearly showed DOX loading in the form of red-colored microspheres with similar shape as DOX–3-MAH–Fbg microspheres, as shown in Figure 2.
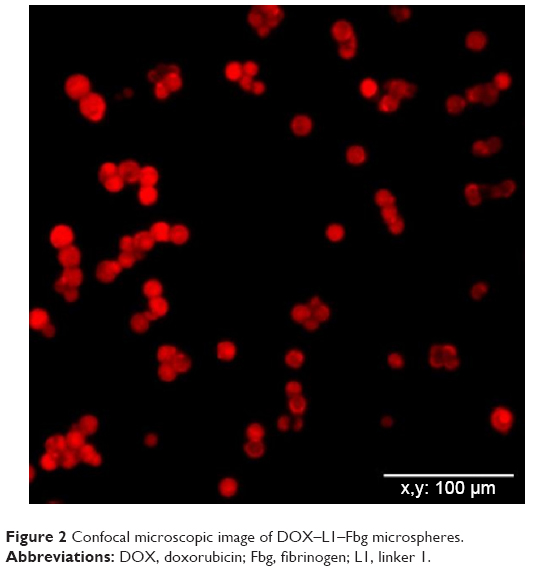 | Figure 2 Confocal microscopic image of DOX–L1–Fbg microspheres. |
Outer morphology, surface fine structure, degradation form were observed using a SEM. Spherical and porous Fbg microspheres with approximately 20 μm size were prepared, with a nano-structured surface (Figure 3A–C). Likewise, DOX–linker 1–Fbg microspheres were fabricated with approximately 10 μm diameter and highly porous surface (Figure 3D–F). Degradation of bare-Fbg microspheres was analyzed (Figure 4A and B), degradation of DOX–(3-MAH)–Fbg microspheres was analyzed (Figure 4C and D), and Degradation of DOX–SM(PEG)12–Fbg was analyzed (Figure 4E and F).
 | Figure 3 SEM images of Fbg microspheres. |
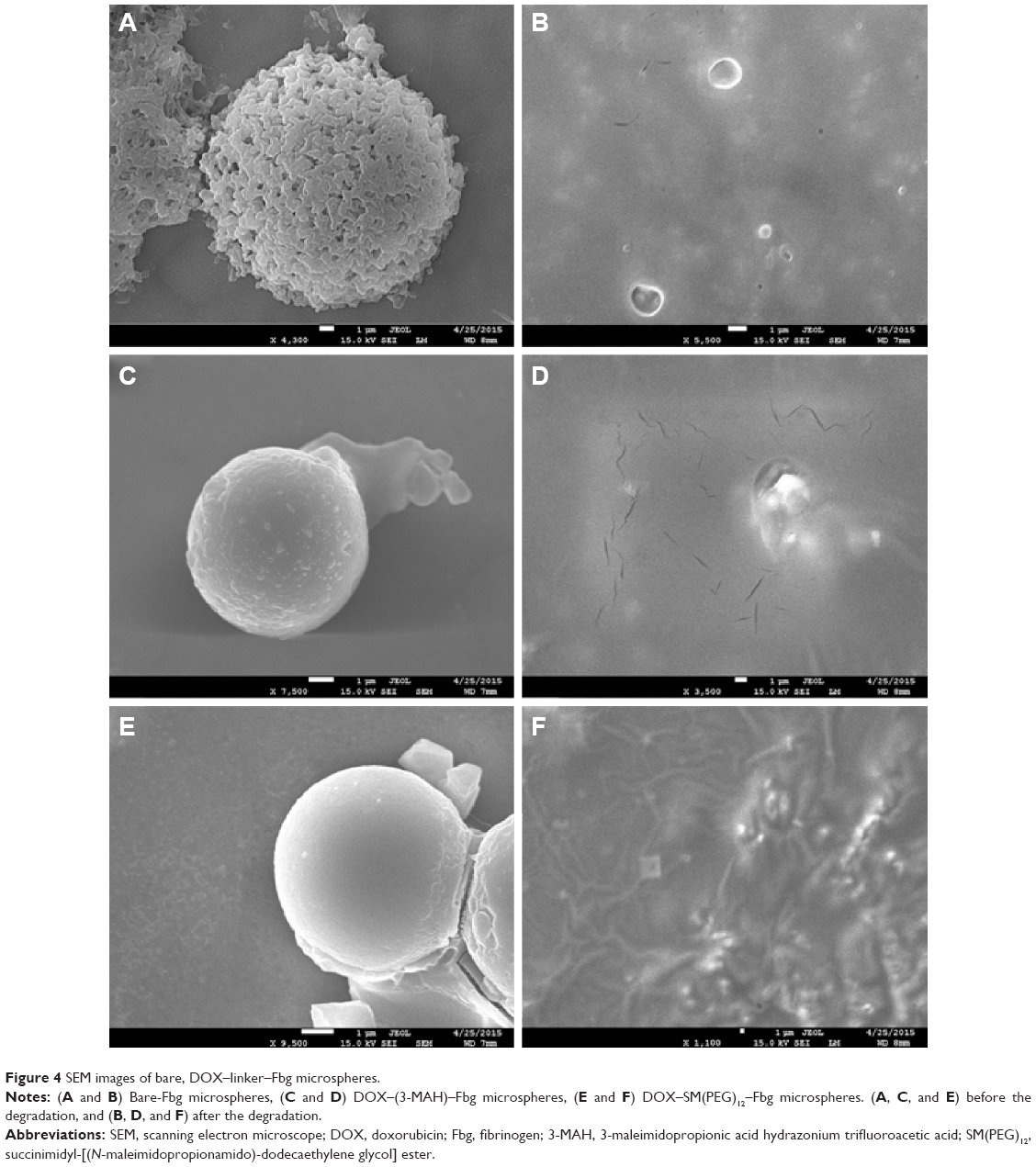 | Figure 4 SEM images of bare, DOX–linker–Fbg microspheres. |
In vitro DOX releasing profile
A significant difference was identified in the release of DOX–(3-MAH)–Fbg microspheres. At pH 5.0, a higher relative fluorescence unit (RFU), which correlated with DOX release, was detected than a relative fluorescence unit at pH 7.3, definitely showing the acid-specific cleavage of 3-MAH. Its release profile represented the initial burst release within 48 hours, reaching a threshold of DOX release. After 48 hours, DOX release occurred in a slightly increasing manner. For a more detailed comparison, DOX release from all the microspheres was compared at 48 hours, indicating remarkable DOX release of DOX–3-MAH–Fbg microspheres at pH 5.0, as illustrated in Figure 5.
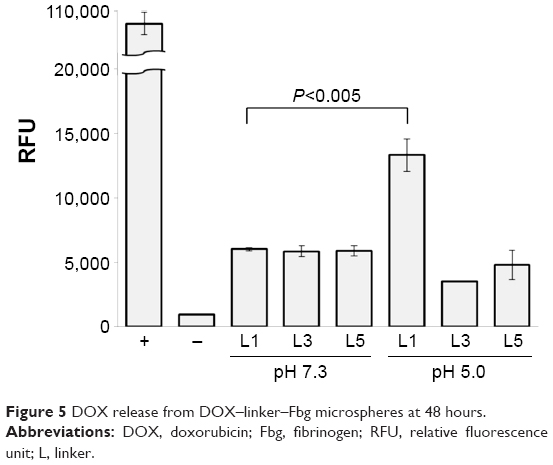 | Figure 5 DOX release from DOX–linker–Fbg microspheres at 48 hours. |
Meanwhile, a gradual increasing release was observed from both DOX–SM(PEG)4–Fbg and DOX–SM(PEG)12–Fbg microspheres at pH 7.3 in a controlled manner, while DOX release was stagnant at pH 5.0. An amide bond is stable, and the hydrolysis of amide bonds occurs at a slow rate19,20 under an un-catalyzed condition. Hence, cleavage of linkers by proteolysis might be promoted by some activated enzymes, which become active at neutral pH. The overall DOX releasing profile in PBS with different pHs is summarized in Figure 6.
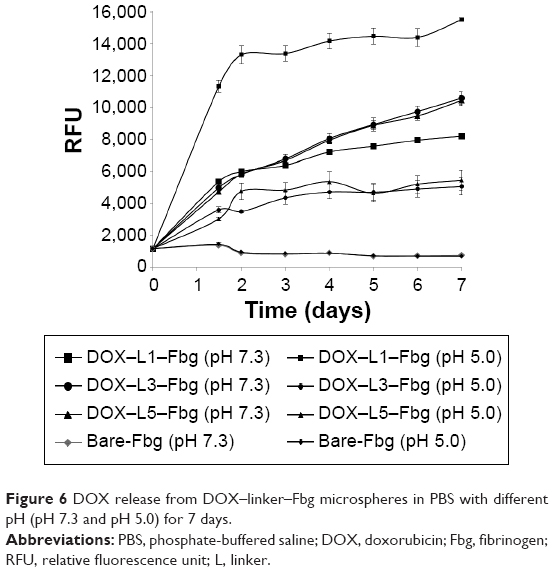 | Figure 6 DOX release from DOX–linker–Fbg microspheres in PBS with different pH (pH 7.3 and pH 5.0) for 7 days. |
Cytotoxic effect of DOX–linker–Fbg microspheres on the cells
The cytotoxicity of the DOX–linker–Fbg microspheres was evaluated with NIH-3T3 as a normal fibroblast and SH-SY5Y as an abnormal tumor cell line by performing the celltiter-glo® luminescence assay (Figure 7). Free-DOX solution (0.39 μg/mL) was added to the cells, revealing 43.90% of NIH-3T3 cell viability and 21.23% of SH-SY5Y cell viability. Higher toxicity to normal cells and abnormal tumor cells was observed, without low specificity to the targeting of tumor cells, which demonstrates a potential side effect of DOX. However, when bare-Fbg microspheres were treated, slightly increased cell viability was measured in both cell lines, due to the cytocompatiblity of Fbg.
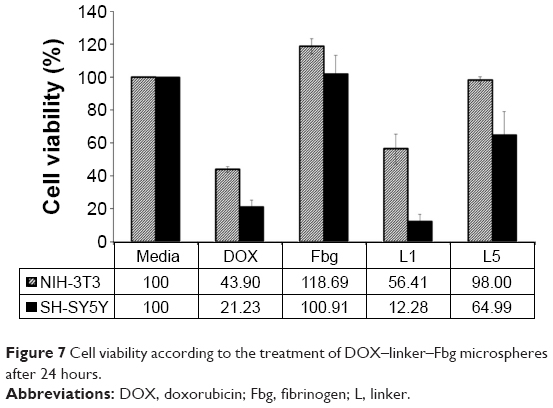 | Figure 7 Cell viability according to the treatment of DOX–linker–Fbg microspheres after 24 hours. |
The treatment with DOX–(3-MAH)–Fbg microspheres on the cells decreased both cell viabilities exceedingly, especially in the SH-SY5Y cell line (12.28%). Interestingly, in comparison with free-DOX solution treatment, DOX–(3-MAH)–Fbg microspheres indicated lower toxicity in the normal cells and higher toxicity in the abnormal tumor cells, implying the increased tumor specificity than that of the free-DOX treatment.
For the DOX–linker–Fbg microspheres with the non-cleavable linkers [SM(PEG)4 and SM(PEG)12], mild toxicity was confirmed in the tumor cells, but there was increased cell viability with SM(PEG)4, and maintained cell viability with SM(PEG)12. Because SM(PEG)4 and SM(PEG)12 cleavage may occur by proteolysis of the amide bond, their release rate appeared slower than 3-MAH cleavage, but they could potentially release the DOX continuously over a long time, enabling the controlled and sustained release of DOX.
Discussion
Even though DOX is one of the highly effective anticancer drugs in chemotherapy, its clinical usage was still limited because of their adverse effects. For safer DOX delivery, biocompatible drug carriers with various drug conjugation techniques have been designed, reducing damage to the normal tissues and organs. As it possesses two conjugatable sites on 13-C carbonyl and 3′-C amino positions (Figure 8), DOX can be conjugated with several cross-linkers by forming a hydrazone bond and an amide bond, respectively. A hydrazone bond, cleavable under a mild acidic condition and stable in neutral pH, enables acid-triggered DOX release in a lysosomal or cancerous environment. Using this bond, DOX molecules were conjugated to diverse novel drug carriers with moderate antitumor efficacy.21,22 On the other hand, DOX could be conjugated with an amide bond at a 3′-C position for the preparation of enzymatically cleavable prodrugs.23,24 In this article, we prepared the DOX–linker–Fbg microspheres with different linkers to evaluate their feasibility as a novel cancer drug delivery system.
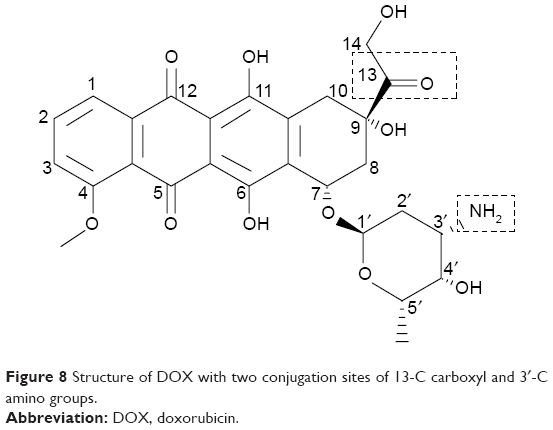 | Figure 8 Structure of DOX with two conjugation sites of 13-C carboxyl and 3′-C amino groups. |
DOX loading was confirmed by a confocal microscope, and the loading efficacy after conjugation steps was comparable to the published results.24,25 By adding tenfold thiolating agent (2-IT) to Fbg, enough conjugation sites were generated on the Fbg, resulting in higher loading efficacy. Moreover, unconjugated DOX and linkers, which could induce unintended reactions, were completely removed by purification with a desalting column. Therefore, the prepared DOX–linker–Fbg microspheres might be nontoxic and safe, yet might still contain a relatively high amount of DOX.
The SEM images showed morphology of the prepared microspheres and their surface fine structure. In the fabrication step, an overhead stirrer was used to form a W/O emulsion, and heat (65°C–75°C) was applied to extract the solvent from Fbg microspheres. Little denaturation by heat occurs during a fabrication step, but prepared microspheres still maintain its major functions.26 By controlling stirring speed and heat, we could prepare the spherical microspheres with highly porous surface structure, offering cells sufficient space for attachment.27 However, the differences between bare-Fbg microspheres and DOX–linker–Fbg microspheres were found on their surface density. This difference might be due to some changes in Fbg properties during the conjugation steps, such as increased thiol groups and slight degradation. In this study, we determined a degradability of bare and Dox–linker–Fbg microspheres. All the Fbg microspheres were fully degraded within 4 hours by plasminogen.
The releasing profiles of DOX from DOX–linker–Fbg microspheres were obtained for 7 days in PBS with two different pHs, representing the extracellular matrix environment of tumor and normal cells. Designed to be cleaved under a mild acidic condition, the DOX–(3-MAH)–Fbg microspheres indicated a significant initial burst release profile for 48 hours at pH 5.0, while the same microspheres showed slow release at pH 7.3. Many DOX delivery carriers, conjugated by a hydrazone bond, demonstrated similar release profiles to our result.25,28 Interestingly, unexpected DOX releases were observed with three kinds of linkers at pH 7.3. The hydrazone bond by 3-MAH is stable at neutral pH, and the amide bond by SM(PEG)4 and SM(PEG)12 is a more stable linkage regardless of pH. After 14 days, the amount of released DOX at pH 7.3 from the three different DOX–linker–Fbg microspheres reached levels similar to those from DOX–(3-MAH)–Fbg microspheres at pH 5.0, and DOX was continuously released (data is not shown). Continuous protein degradation might result in a continuous increase of DOX release, supporting the conclusion regarding the unexpected DOX release.
The in vitro antitumor effect of DOX–linker–Fbg microspheres was evaluated with NIH-3T3 and SH-SY5Y. The biocompatibility of Fbg was achieved with the addition of bare-Fbg microspheres, exhibiting increased cell growth. Meanwhile, treatment with free-DOX seemed highly toxic to both normal and abnormal cancer cells; as a small molecule, DOX can easily pass through the cell membrane without any specificity, leading to cell death. Compared to free-DOX treatment, DOX–(3-MAH)–Fbg microspheres revealed lesser cytotoxicity to the normal cells and higher cytotoxicity to neuroblastoma, which may increase its specificity to tumor cells. P-glycoprotein, a membrane transporter inhibiting the uptake of toxic substances, is known to be over-expressed in cancer cells with resistance against several drugs.29 Therefore, a drug-loaded carrier system, such as DOX–linker–Fbg microspheres, might be a solution for inhibiting action of P-glycoprotein, resulting in enhanced antitumor efficacy. In the case of the microspheres with non-cleavable linkers, DOX–SM(PEG)12–Fbg microspheres showed slightly higher toxicity than DOX–SM(PEG)4–Fbg microspheres, suggesting that a longer linker could mediate increased interaction between the microspheres and the cells. Similarly, the published result of Toita et al explained that more flexible longer linkers could make the binding of drug carriers to cells easier.30 Therefore, the feasibility of the DOX–linker–Fbg microsphere-based drug carrier system was confirmed by the cell experiments.
According to our hypothesis, the DOX–linker–Fbg microspheres can be injected into the muscle or the tumor tissue, following which they may go through the enzymatic degradation process and get converted into smaller movable DOX–linker–Fbg conjugates. The smaller fragments can specifically interact with cancer cells through RGD-αvβ3 integrin binding, thereby entering into the cells. Finally, DOX may be released into the cytosol or extracellular matrix of cancer cells due to the acidic condition and more enzymatic cleavage, leading to specific cancer cell death. To investigate the pathway of DOX delivery from the DOX–linker–Fbg microspheres, in vivo animal studies will be conducted.
Conclusion
The DOX–linker–Fbg microspheres were prepared with two kinds of linkers: acid-labile linkers and non-cleavable linkers. Their porous nano-structured surfaces can provide sufficient space for cell attachment, thereby enhancing the interactions with the cells. The release study demonstrated acid-specific DOX release from DOX–(3-MAH)–Fbg microspheres by the cleavage of hydrazone bond. In the cell experiments, enhanced antitumor effect was observed in DOX–(3-MAH)–Fbg microspheres compared to free-DOX treatment. Meanwhile, investigating the influence of linker length indicated that a longer linker was able to make binding of the drug carriers with the cells easier, resulting in higher antitumor efficacy. In addition, the biocompatibility of Fbg was confirmed. Since Fbg possesses biocompatibility and tumor-targeting property with RGD peptide sequences, we expect that Fbg microspheres drug carrier systems can be effectively used for chemotherapy. Supplementary materials http://www.dovepress.com/get_supplementary_file.php?f=88381.pdf and https://www.youtube.com/watch?v=UqonwTB7Ms4.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (NRF-2012R1A2A2A03046819).
Disclosure
The authors report no conflicts of interest in this work.
References
Mishra D, Hubenak JR, Mathur AB. Nanoparticle systems as tools to improve drug delivery and therapeutic efficacy. J Biomed Mater Res Part A. 2013;101(12):3646–3660. | ||
Thompson CJ, Hansford D, Higgins S, Rostron C, Hutcheon GA, Munday DL. Evaluation of ibuprofen-loaded microspheres prepared from novel copolyesters. Int J Pharm. 2007;329(1–2):53–61. | ||
Schmidts T, Dobler D, von den Hoff S, Schlupp P, Garn H, Runkel F. Protective effect of drug delivery systems against the enzymatic degradation of dermally applied DNAzyme. Int J Pharm. 2011; 410(1–2): 75–82. | ||
Bailly N, Thomas M, Klumperman B. Poly(N-vinylpyrrolidone)-block-poly(vinyl acetate) as a drug delivery vehicle for hydrophobic drugs. Biomacromolecules. 2012;13(12):4109–4117. | ||
Poland CA, Duffin R, Kinloch I, et al. Carbon nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat Nanotechnol. 2008;3(7):423–428. | ||
Bessa PC, Machado R, Nurnberger S, et al. Thermoresponsive self-assembled elastin-based nanoparticles for delivery of BMPs. J Controlled Rel. 2010;142(3):312–318. | ||
Rajangam T, Paik H-J, An SSA. Development of fibrinogen microspheres as a biodegradable carrier for tissue engineering. BioChip J. 2011;5(2):175–183. | ||
Shaikh AY, Shih JA. Chemotherapy-induced cardiotoxicity. Current Heart Failure Rep. 2012;9(2):117–127. | ||
Zhong Y, Meng F, Deng C, Zhong Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules. 2014;15(6):1955–1969. | ||
Ji S, Xu J, Zhang B, et al. RGD-conjugated albumin nanoparticles as a novel delivery vehicle in pancreatic cancer therapy. Cancer Biol Ther. 2012;13(4):206–215. | ||
Mansoori GA, Brandenburg KS, Shakeri-Zadeh A. A comparative study of two folate-conjugated gold nanoparticles for cancer nanotechnology applications. Cancers. 2010;2(4):1911–1928. | ||
Lin GY, Lv HF, Lu CT, et al. Construction and application of biotin–poloxamer conjugate micelles for chemotherapeutics. J Microencapsul. 2013;30(6):538–545. | ||
Hawiger J. Formation and regulation of platelet and fibrin hemostatic plug. Hum Pathol. 1987;18(2):111–122. | ||
Pidgeon GP, Tang K, Cai YL, Piasentin E, Honn KV. Overexpression of platelet-type 12-lipoxygenase promotes tumor cell survival by enhancing αvβ3 and αvβ5 integrin expression. Cancer Res. 2003;63(14): 4258–4267. | ||
Trikha M, Zhou Z, Timar J, et al. Multiple roles for platelet GPIIb/IIIa and αvβ3 integrins in tumor growth, angiogenesis, and metastasis. Cancer Res. 2002;62(10):2824–2833. | ||
Nasongkla N, Shuai X, Ai H, et al. cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew Chem (Engl). 2004; 43(46):6323–6327. | ||
Xu W, Luo T, Li P, et al. RGD-conjugated gold nanorods induce radiosensitization in melanoma cancer cells by downregulating alpha(v)beta(3) expression. Int J Nanomed. 2012;7:915–924. | ||
Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. | ||
Radzicka A, Wolfenden R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. J Am Chem Soc. 1996;118(26):6105–6109. | ||
Smith RM, Hansen DE. The pH-rate profile for the hydrolysis of a peptide bond. J Am Chem Soc. 1998;120(35):8910–8913. | ||
Dong DW, Xiang B, Gao W, Yang ZZ, Li JQ, Qi XR. pH-responsive complexes using prefunctionalized polymers for synchronous delivery of doxorubicin and siRNA to cancer cells. Biomaterials. 2013;34(20): 4849–4859. | ||
Yuan H, Luo K, Lai Y, et al. A novel poly(l-glutamic acid) dendrimer based drug delivery system with both pH-sensitive and targeting functions. Mol Pharm. 2010;7(4):953–962. | ||
Calderon M, Graeser R, Kratz F, Haag R. Development of enzymatically cleavable prodrugs derived from dendritic polyglycerol. Bioorg Med Chem Lett. 2009;19(14):3725–3728. | ||
Zhong YJ, Shao LH, Li Y. Cathepsin B-cleavable doxorubicin prodrugs for targeted cancer therapy (Review). Int J Oncol. 2013;42(2): 373–383. | ||
Dong DW, Tong SW, Qi XR. Comparative studies of polyethylenimine-doxorubicin conjugates with pH-sensitive and pH-insensitive linkers. J Biomed Mater Res Part A. 2013;101(5):1336–1344. | ||
Marx G, Mou X, Hotovely-Salomon A, et al. Heat denaturation of fibrinogen to develop a biomedical matrix. J Biomed Mater Res B Appl Biomater. 2008;84(1):49–57. | ||
Alamdari OG, Seyedjafari E, Soleimani M, Ghaemi N. Micropatterning of ECM proteins on glass substrates to regulate cell attachment and proliferation. Avicenna J Med Biotechnol. 2013;5(4):234–240. | ||
Wu DC, Cammarata CR, Park HJ, Rhodes BT, Ofner CM 3rd. Preparation, drug release, and cell growth inhibition of a gelatin: doxorubicin conjugate. Pharm Res. 2013;30(8):2087–2096. | ||
Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights. 2013;7:27–34. | ||
Toita R, Murata M, Tabata S, et al. Development of human hepatocellular carcinoma cell-targeted protein cages. Bioconj Chem. 2012;23(7): 1494–1501. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.