")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Biochemical characterization of the interactions between doxorubicin and lipidic GM1 micelles with or without paclitaxel loading
Authors Leonhard V, Alasino R, Bianco I, Garro A, Heredia V, Beltramo D
Received 7 November 2014
Accepted for publication 13 December 2014
Published 6 May 2015 Volume 2015:10(1) Pages 3377—3388
DOI https://doi.org/10.2147/IJN.S77153
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Victoria Leonhard,1,2 Roxana V Alasino,1,2 Ismael D Bianco,1–3 Ariel G Garro,1 Valeria Heredia,1 Dante M Beltramo1,2,4
1Centro de Excelencia en Productos y Procesos de Córdoba (CEPROCOR), Córdoba, Argentina; 2Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina; 3Departamento de Ciencias Exactas, Físicas y Naturales, Universidad Nacional de La Rioja, La Rioja, Argentina; 4Laboratorio de Biotecnología, Facultad de Ciencias Químicas, Universidad Católica de Córdoba, Córdoba, Argentina
Abstract: Doxorubicin (Dox) is an anthracycline anticancer drug with high water solubility, whose use is limited primarily due to significant side effects. In this study it is shown that Dox interacts with monosialoglycosphingolipid (GM1) ganglioside micelles primarily through hydrophobic interactions independent of pH and ionic strength. In addition, Dox can be incorporated even into GM1 micelles already containing highly hydrophobic paclitaxel (Ptx). However, it was not possible to incorporate Ptx into Dox-containing GM1 micelles, suggesting that Dox could be occupying a more external position in the micelles. This result is in agreement with a higher hydrolysis of Dox than of Ptx when micelles were incubated at alkaline pH. The loading of Dox into GM1 micelles was observed over a broad range of temperature (4°C–55°C). Furthermore, Dox-loaded micelles were stable in aqueous solutions exhibiting no aggregation or precipitation for up to 2 months when kept at 4°C–25°C and even after freeze–thawing cycles. Upon exposure to blood components, Dox-containing micelles were observed to interact with human serum albumin. However, the amount of human serum albumin that ended up being associated to the micelles was inversely related to the amount of Dox, suggesting that both could share their binding sites. In vitro studies on Hep2 cells showed that the cellular uptake and cytotoxic activity of Dox and Ptx from the micellar complexes were similar to those of the free form of these drugs, even when the micelle was covered with albumin. These results support the idea of the existence of different nano-domains in a single micelle and the fact that this micellar model could be used as a platform for loading and delivering hydrophobic and hydrophilic active pharmaceutical ingredients.
Keywords: cancer drugs, nano-domains, drug delivery, hydrophobic interactions
Introduction
Despite the wide range of currently available strategies for chemotherapeutic treatment, prognosis of many types of cancer remains poor for many patients; therefore, there is a clear need for new therapies that improve outcomes.
Some of the most common drawbacks of cancer drugs are related to their low solubility, poor bioavailability, and undesirable toxic side effects, the last being a concern for doxorubicin (Dox).
Dox is an anthracycline used primarily in the treatment of childhood solid tumors, soft tissue sarcomas, lymphomas, and breast cancer.1–4 The use of Dox is limited by significant side effects like cardiotoxicity, nephrotoxicity, hepatotoxicity, and also by its short circulation lifetime. Dox is a tetracyclic quinoid aglycone linked to an amino sugar that has a pKa of 8.3; thus, it is a hydrophilic weak amphipathic base usually incorporated in the aqueous interior of preformed vesicles such as liposomes, using ionic or pH gradients.5 However, these gradients are unstable and the incorporated drugs tend to leak out at a rate which strongly depends on the composition of the bilayer. Physical entrapment of Dox, mostly by hydrophobic effect, in self-assembled systems based on block copolymers, has been also reported,6–8 as well as its incorporation in the cross-linked ionic core of some polymeric micelles through electrostatic interactions.8
More recently, nanomaterials have arisen as a promising alternative to cancer therapeutics; they can enhance the delivery and treatment efficiency of anticancer drugs. In this context, to be used for drug delivery, nanomaterials must be biocompatible, have suitable biodegradation kinetics, be easy to process, possess adequate mechanical properties, and be compatible with the drug to be transported. In the last 20 years, many strategies based on nanoscale (10–200 nm) vehicles, such as liposomes,9 polymeric micelles,10,11 and polymer particles,12 have been increasingly used in a wide variety of approaches for therapeutic drug delivery devices.
In previous studies, we demonstrated that lipidic micelles composed of monosialoglycosphingolipid (GM1) spontaneously load high amounts of a hydrophobic cancer drug such as paclitaxel (Ptx).13 This micellar structure has already been proposed for loading small hydrophobic molecules that localize in the more internal region of the micelle, with the fatty acid chains of gangliosides.14 Moreover, GM1 micelles bind human serum albumin (HSA) via hydrophobic interactions.15 This offers additional advantages because many cancer cells overexpress gp60 receptors specific for albumin. This mechanism could facilitate the transport of the complexes with albumin to the tumor tissue where its interaction with the SPARC protein may result in intratumoral accumulation of albumin-bound paclitaxel.16,17
In order to gain greater insights into the particular behavior of this micellar structure, we evaluated the interactions between a water-soluble cytotoxic drug such as Dox and GM1 ganglioside micelles and characterized the structures resulting from this interaction.
Furthermore, considering the special features of micelle polarity described earlier, we also evaluated whether this structure could simultaneously load hydrophobic and hydrophilic drugs into a single GM1 micelle and how the incorporation of these drugs regulates the subsequent interaction of the micelle with HSA. This would provide a transportation system which, in addition to being able to individually load drugs of very different polarities, it might be able to load drugs simultaneously, becoming a multidrug delivery system.
Materials and methods
Materials
Purified monosialoganglioside GM1 from pig brain was obtained from TRB Pharma S.A. (Buenos Aires, Argentina), Dox–HCl (Dox) was from ELEA (Buenos Aires, Argentina), and Ptx was from Yunnan Smandbet Co. Ltd (Kunming, People’s Republic of China). Flutax-1 (7-O-[N-(4′-fluoresceincarbonyl)-l-alanyl]taxol) was purchased from Calbiochem (San Diego, CA, USA). Purified HSA 20% (w/v) was obtained from Laboratorio de Hemoderivados, Universidad Nacional de Córdoba (Córdoba, Argentina). All other chemicals used were of analytical grade.
Methods
Standard procedure for studying the interactions between Dox and Ptx with GM1 micelles
Stock solutions of GM1 with a concentration of 250 mg mL−1 were prepared in bidistilled water 24 hours prior to use as described in Leonhard et al.13 Briefly, the solutions were maintained at 4°C for 24 hours. They were then centrifuged at 50,000× g for 15 minutes and the supernatant was filtered through 0.22 μm.
Stock solutions of Dox (24 mg mL−1) were prepared in 145 mM of NaCl. Solutions of Dox were slowly added to the solution of GM1 micelles in order to attain different Dox–GM1 molar ratios, ie, 1/15, 1/10, 1/5, 1/2.5, 1/1, 1/0.7. These mixtures were incubated at 4°C for 24 hours. Unbound Dox was removed by dialysis against 100 volumes of bidistilled water for 24 hours at 4°C.
Stock solutions of Ptx (50 mg mL−1) were prepared in dimethylsulfoxide (DMSO). The solutions were added to GM1 or Dox–GM1 micellar solutions and then stirred for 2 minutes before incubation at 4°C for 24 hours and dialyzation for 24 hours at 4°C against 100 volumes of bidistilled water to remove all DMSO. The removal of DMSO in the solution was determined spectrophotometrically by measuring absorbance at 227 nm at different times until this value remained constant.
When studying the interactions of Dox with Ptx–GM1, stock solutions of Dox described earlier were slowly added to Ptx–GM1 micelles in order to attain the desired Dox–GM1 molar ratios. These mixtures were incubated at 4°C for 24 hours. Unbound Dox was removed by dialysis against 100 volumes of bidistilled water for 24 hours at 4°C.
Determination of GM1 concentration
Ganglioside concentrations were measured by the modified colorimetric resorcinol assay, described by Miettinen and Takki-Luukkainen.18 Briefly, 1 mL of resorcinol reagent was added to 1 mL of the samples and heated to 100°C for 15 minutes. (Resorcinol reagent: 2 mg of resorcinol powder dissolved in 0.1 mL of bidistilled water +0.8 mL of 37.9% [w/v] HCl +2.5 μL of 0.1 M CuSO4 + amount of bidistilled water so as to reach 1 mL). The samples were then allowed to cool and the chromophore developed was extracted with 2.5 mL of n-butyl acetate: n-butanol (85/15 by vol). After centrifugation at 2,500× g for 5 min, the supernatants were removed and measured spectrophotometrically at 580 nm.
Determination of Dox concentration
Dox concentration was determined by absorbance at 490 nm using a calibration curve performed with a standard solution of Dox in 145 mM NaCl, as described by Abraham et al.19
Determination of Ptx concentration
Ptx was determined as described in Leonhard et al.13 Briefly, Ptx was extracted from micelles with 10 volumes of ethyl acetate. Samples were then centrifuged at 2,500× g for 5 minutes; the organic layer was transferred to a clean tube and evaporated to dryness at 40°C. The dried residue was solubilized in 1 volume of ethanol.
Ptx concentration was measured using a Curosil B C18 column (250×3.20 mm inner diameter, particle size 5 μm) and a Curosil B C18 guard column (30×4.60 mm inner diameter, particle size 5 μm) supplied by Phenomenex. The mobile phase was 60% (v/v) acetonitrile and 40% (v/v) bidistilled water. Flow rate was 0.7 mL min−1 and the eluent was monitored at 227 nm. Chromatography was performed at ambient temperature (20°C) and the calibration curve of Ptx was linear in a range from 5 to 150 μg.
Structural characterization of Dox–GM1 and Dox–Ptx–GM1 mixed micelles
Chromatographic analysis
Samples were run on an Åkta Explorer 100 system (GE Healthcare Life Science, Buenos Aires, Argentina) as described in Leonhard et al.13 Briefly, samples containing GM1 (10 mg mL−1) were loaded into a Superdex 200® column, equlibrated with 50 mM phosphate buffer (pH 7.0) and 150 mM NaCl at a rate of 0.4 mL min−1. The elution pattern of GM1 and Dox were monitored using an UV detector at 227 and 490 nm, respectively. GM1 and Dox levels were quantified as described. The molecular weights (MWs), corresponding to the various elution peaks, were extrapolated from a calibration curve made up of globular proteins of known MW as albumin (67,000 Da), lactoglobulin (35,000 Da), and human immunoglobulin G (160,000 Da) using the formula: log MW = a. eslution volume + b.
Determination of particle size by dynamic light scattering
Average particle size of the aqueous solutions of micelles with GM1 (10 mg mL−1) were measured by dynamic light scattering (DLS), which was performed on a Delsa™ Nano Submicron Particle Size and Zeta Potential Particle Analyzer at a fixed scattering angle of 165°. Data were analyzed by Delsa Nano Beckman Coulter software (version 2.2) with CONTIN analysis method.
Physical stability of Dox–GM1 and Dox–Ptx–GM1 mixed micelles
Stability of micelles to high-speed centrifugation
Dox–GM1 and Dox–Ptx–GM1 micelles were centrifuged at 25,000, 50,000, or 100,000× g for 1 hour at 20°C in an XL-90 ultracentrifuge (Beckman Coulter Argentina S.A., Buenos Aires, Argentina). Afterward, the supernatants were dialyzed for 24 hours to remove unbound molecules before determining GM1, Ptx, and Dox concentrations as previously described.
Stability of micelles in solution
Dox–GM1 and Dox–Ptx–GM1 micelles were stored for 40 days at 4°C. Aliquots of samples were taken at various time periods and dialyzed for 24 hours prior to quantification of soluble Dox and Ptx as described. Dialysis conditions were as follows: membrane tubing was from SpectraPor with a MW cut-off of 10,000 Da, 1 mL of sample was dialyzed against 1,000 mL of bidistilled water. Dox and Ptx were quantified in the sample and dialyzed.
Effect of freeze–thawing cycles and lyophilization
Dox–GM1 and Dox–Ptx–GM1 micelles were frozen at −80°C, and after 24 hours samples were kept at room temperature for about 2 hours until complete thawing, centrifuged at 15,000× g for 10 minutes and dialyzed for 24 hours before quantifying the concentrations of Dox, Ptx, and GM1 that remained soluble.
Moreover, lyophilized micelles were dissolved in their initial volume, filtered through a 0.22-μm pore, and dialyzed for 24 hours before measuring soluble Dox, Ptx, and GM1 as described.
Chemical stability of Dox in Dox–GM1 and Dox–Ptx–GM1 micelles
We evaluated the chemical stability of Dox loaded into the micelles when subjected to a denaturing condition such as the alkaline environment where ester groups are hydrolyzed. For this, we incubated Dox–GM1 and Dox–Ptx–GM1 micelles at pH 10 and at room temperature (25°C±1°C). At various time periods, aliquots of the samples were taken and the amount of soluble Dox and Ptx was quantified as described. Stock solution of free Dox and Ptx was used as a control for the kinetics of chemical hydrolysis at alkaline pH.
Characterization of the interaction of Dox–GM1 and Dox–PtxGM1 micelles with HSA
Dox–GM1 micelles of different molar ratios (1/20, 1/5, and 1/1), with or without preloaded Ptx (Ptx–GM1: 1/20 molar ratio) and with a GM1 concentration of 10 mg mL−1, were incubated for 24 hours with purified HSA (1/1 HSA–GM1 w/w ratio) at pH 3 and 37°C. Samples were then returned to pH 7 and the interaction was studied by chromatographic analysis and DLS as described.
Determination of HSA concentration
The amount of albumin associated with the micelles was determined using a Coomassie Brilliant blue assay or by direct absorbance at 280 nm.20
In vitro biological effect of Dox in Dox–GM1 and Dox–GM1–HSA micelles on cell cultures
Hep2 (human epithelial carcinoma of larynx) cell lines were grown as described in Leonhard et al.13 Briefly, cells were grown in minimum essential medium supplemented with 10% of fetal bovine serum (NATOCOR, Córdoba, Argentina) at 37°C with 5% CO2. Then, the cell monolayers were incubated for 15, 30, 60, 120, and 240 minutes and for 24 hours at 4°C and 37°C with increasing concentration of Dox, Dox–GM1, and Dox–GM1–HSA micelles. After this incubation, the medium was removed and fresh medium was added.
After 24 hours at 4°C or 37°C, viable cells were measured by an 3-4,5 dimethylthiazol-2,5 diphenyl tetrazolium bromide staining assay. The half maximal inhibitory concentration (IC50) values were graphically estimated.
Characterization of the cellular uptake of Dox and Ptx from Dox–GM1 and Dox–Ptx–GM1 micelles with and without HSA
Hep2 cell lines were grown as described. Dox, Ptx, Dox–GM1, and Dox–Ptx–GM1 micelles with and without HSA, with 1% of Flutax-1 (a fluorescently labeled derivative of Ptx) were diluted in minimum essential medium to a final concentration of 30 μg mL−1 of Dox and 10 μg mL−1 of Ptx.
Each sample was incubated with Hep2 cell monolayers at 70% confluence for 15, 30, and 60 minutes at 37°C. Cells were then washed out with phosphate-buffered saline, and the incorporated drugs were observed by fluorescent microscopy.
Results
Interactions between Dox and GM1 micelles under different physicochemical conditions
Gangliosides are double-tailed anionic glycosphingolipids with a complex polar head group of several sugar units. In particular, GM1 is a monosialoganglioside with four sugar units in its polar head group and due to thermodynamic and geometric constrains, it forms spherical micelles in aqueous media at a very low concentration (critical micellar concentration of 10−8–10−10 M).13,21–23
In order to start studying how Dox, a highly water-soluble drug, interacts with GM1 micelles, solutions of Dox were slowly added to a solution of GM1 micelles in order to attain different Dox–GM1 molar ratios, ie, 1/15, 1/10, 1/5, 1/2.5, 1/1, 1/0.7. These samples were incubated at 4°C for 24 hours and unbound Dox was removed by dialysis against bidistilled water as detailed in the Materials and Methods section. Figure 1 shows that there is a spontaneous incorporation of Dox into GM1 micelles, reaching saturation around 10 mg mL−1 per 50 mg mL−1 of GM1.
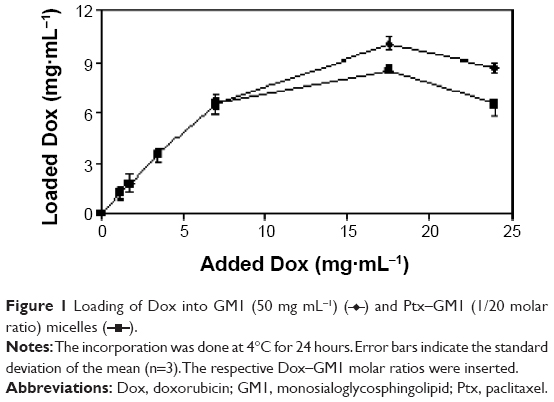 | Figure 1 Loading of Dox into GM1 (50 mg mL−1) ( |
 ) and Ptx–GM1 (1/20 molar ratio) micelles (
) and Ptx–GM1 (1/20 molar ratio) micelles ( ).
).The results demonstrate that these micelles can incorporate up to five times more Dox compared to current commercial products based on liposomal formulations containing 2 mg mL−1.24
The encapsulation efficiency calculated from these varies according to the different Dox–GM1 molar ratios. Up to 1/2.5 molar ratio, the incorporation efficiency is greater than 95%, whereas at 1/1 and 1/0.7 ratios, it drops to 55% and 35%, respectively.
Effect of medium pH on Dox–GM1 interaction
It was observed that the reduction of pH from 7 to 2 did not affect the ability of GM1 to incorporate Dox, demonstrating that Dox–GM1 interaction does not involve the sialic acid molecules of gangliosides (pKa ~2.6). In addition, it was observed that the presence of up to 0.5 M NaCl did not prevent Dox incorporation into GM1 micelles, again suggesting that the association involves mostly hydrophobic rather than electrostatic interactions (data not shown).
Effect of temperature on Dox–GM1 interaction
Previous studies showed that the incorporation of Ptx in GM1 micelles is favored with increasing temperature to 55°C.13 Such condition produces a release of the water bound to the oligosaccharide chain resulting in a significant reduction of the hydrophilic portion which is associated with a small expansion of the hydrophobic portion of the micelle.25–28
We found that, unlike what was observed with Ptx, changes in temperature do not significantly affect the incorporation of Dox in GM1 micelles (data not shown). This result is in disagreement with what was obtained with Ptx, a highly hydrophobic drug, probably due to the fact that Dox is a water-soluble molecule whose entry into the micelle is insensitive to its changes in hydrophobicity.
Ability of GM1 micelles to load hydrophobic and hydrophilic drugs into the same structure
Having demonstrated the ability of GM1 micelles to incorporate drugs with such different physicochemical characteristics, a highly hydrophobic drug like Ptx and a hydrophilic one like Dox, we wondered if it would be possible to incorporate both molecules simultaneously into the same micellar nanostructure. Figure 1 shows that Ptx–GM1 micelles are able to incorporate large amounts of Dox, similarly to GM1 micelles, reaching a saturation level around 8 mg mL−1. In this case, the encapsulation efficiency is also higher than 95% up to the molar ratio Dox–GM1 1/2.5, while at 1/1 and 1/0.7 molar ratios it drops to 45% and 25%, respectively.
However, it was also noted that Ptx incorporation into GM1 is impaired by the presence of Dox in the micelles. Thus, when a low amount of Ptx is added (up to 0.5 mg mL−1), its incorporation into Dox–GM1 micelles is 63% lower than the incorporation into pure GM1 micelles. Curiously, the addition of larger amounts of Ptx (>0.5 mg mL−1) caused precipitation of most of the Ptx, rendering mixed micelles with only around 10% of the Ptx than the amount that could be solubilized when mixed with Dox-free GM1 micelles (Figure 2).
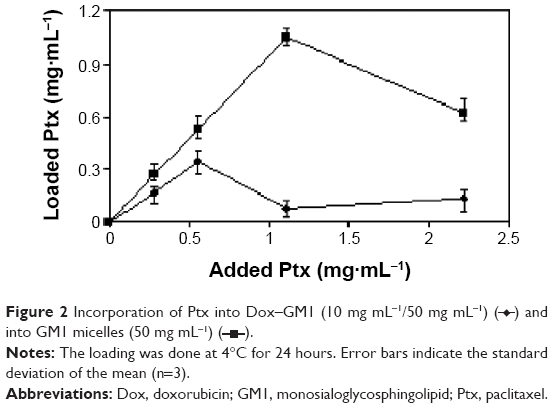 | Figure 2 Incorporation of Ptx into Dox–GM1 (10 mg mL−1/50 mg mL−1) ( |
These results clearly suggest that Dox induced a change in the micellar structure that hinders the subsequent entry of Ptx into the hydrophobic region of the micelles.
Structural characterization of Dox–GM1 and Dox–Ptx–GM1 micelles
In order to gain further insight into the structure and thermodynamic equilibrium of the micelles, we evaluated the elution profile of Dox–GM1 and Dox–Ptx–GM1 micelles at different molar ratios, using a size exclusion chromatography column, and their average size by DLS.
The chromatographic patterns obtained show that GM1 presents two elution peaks, one equivalent to a globular protein of 365 kDa and the other corresponding to the population of monomers that are in equilibrium with the micelles (Figure 3A, B).
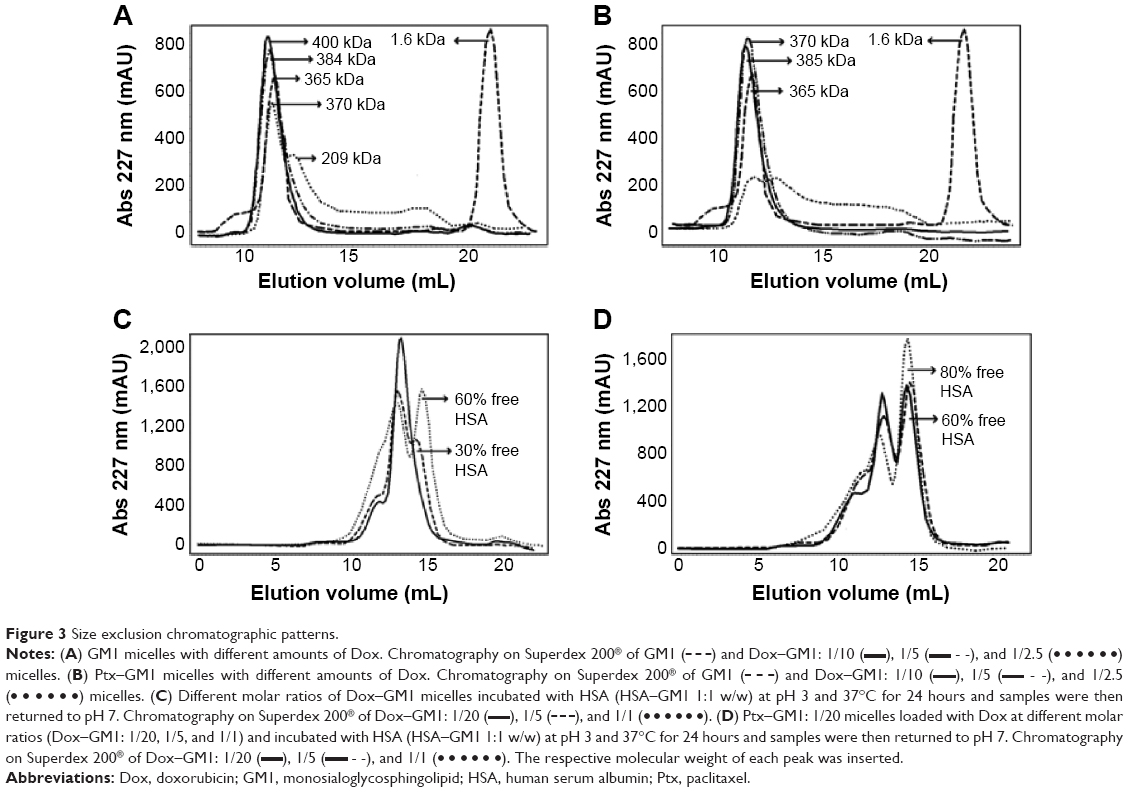 | Figure 3 Size exclusion chromatographic patterns. |
 ), 1/5 (
), 1/5 (The loading of Dox into GM1 micelles led to changes in the hydrodynamic radius that correlated with the amount of drug incorporated into the micelles. At molar ratios from 1/10 up to 1/5 (Dox–GM1), the incorporation of the drug produced a slight increase in the hydrodynamic radius of the micelles from 365 kDa to 390 kDa. However, when the amount of Dox incorporated into the micelles was increased up to 1/2.5 Dox–GM1 molar ratios, two populations of micelles with hydrodynamic radius of 370 and 210 kDa were found (Figure 3A).
These changes in the hydrodynamic radius as a function of the amount of Dox loaded into the micelles were also observed with its incorporation into Ptx–GM1 (1/20 molar ratio) micelles. Again, at 1/2.5 Dox–GM1 molar ratio, micelles seemed to be unstabilized, leading to two principal peaks with hydrodynamic radius of 370 and 210 kDa and to a gradient of new micellar populations of widely differing sizes (Figure 3B).
Interestingly, it was noticed that the elution peak corresponding to the monomers of GM1 disappeared completely after incorporating Ptx, Dox, or both drugs simultaneously, suggesting that the presence of either drug produces a displacement of the equilibrium toward the micellar state indicating that mixed micelles are thermodynamically more stable than pure GM1 micelles.
At this point, knowing the high loading capacity and the homogeneity of the Dox–GM1 1/5 molar ratio micellar population, we selected this proportion to continue our studies.
In agreement with the results obtained by chromatographic analysis of these mixed micelles, their average size as measured by DLS was not modified by the incorporation of Dox into GM1 or Ptx–GM1 micelles at 1/51 molar ratio (Table 1).
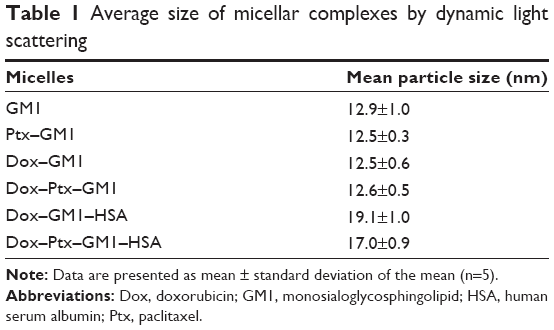 | Table 1 Average size of micellar complexes by dynamic light scattering |
Physical stability of Dox–GM1 and Dox–Ptx–GM1 micelles
As shown in Table 2, the amounts of Dox and Ptx that remained soluble after centrifugation of Dox–GM1 and Dox–Ptx–GM1 micelles at high speeds, was above 90% in all cases.
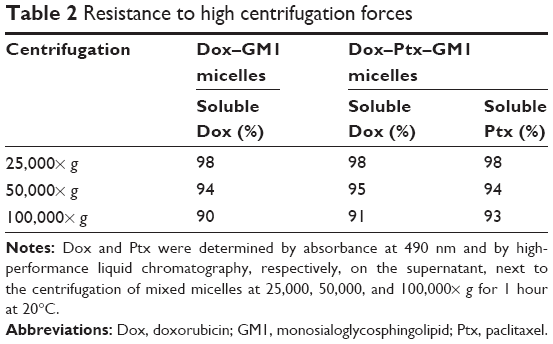 | Table 2 Resistance to high centrifugation forces |
Furthermore, both micelles remained stable in solution for at least 2 months at 4°C and 25°C and neither Dox nor Ptx was released from the micelles after freeze–thawing cycles or lyophilization procedures (data not shown).
We also evaluated the stability of micelles loaded with Dox and Dox–Ptx dialyzed for 72 hours. It was observed that the entire initial drug loaded remained soluble within the dialysis bag. This result agrees with those observed when passing the mixed micelles through a size exclusion column indicating that the incorporation of either Dox or Ptx shifts the equilibrium toward the micellar form and that the mixed drug–GM1 complex structure formed is thermodynamically and kinetically stable in solution (data not shown).
Chemical stability of Dox in Dox–GM1 and Dox–Ptx–GM1 micelles
The incubation of Dox–GM1 and Dox–Ptx–GM1 micelles under pH conditions where the drugs are unstable (pH 10) resulted in about 17% and 25% of Dox hydrolyzed, respectively. This result should be compared with the percentage of free drug hydrolyzed that exceeds 90% after 6 hours (Figure 4).
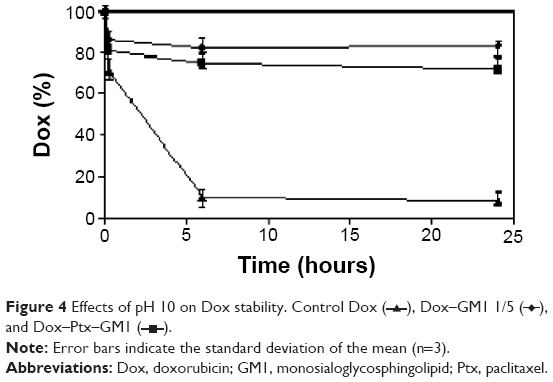 | Figure 4 Effects of pH 10 on Dox stability. Control Dox ( |
 ), Dox–GM1 1/5 (
), Dox–GM1 1/5 (These results differ from what happens with Ptx, in which case no hydrolysis of the drug was observed when incorporated into GM1 micelles.13 This different susceptibility to alkaline hydrolysis suggests that Dox could be located in a more external domain of the micelle, thus rendering the drug more exposed to the external aqueous environment.
Interaction of Dox–GM1 and Dox–Ptx–GM1 micelles with HSA
In previous reports we demonstrated that Ptx–GM1 micelles spontaneously associate with HSA.15 Further studies revealed that the amount of albumin that ends up being associated with micelles reached saturation at 1/1 HSA–GM1 w/w ratio and also that at 55°C and at pH 3, where HSA is reversibly denatured and exposes part of its hydrophobic residues, the interaction is favored.29–31 Considering these results together and the fact that Dox might end up located in more external domains of GM1 micelles, we wondered whether the incorporation of Dox into GM1 or Ptx–GM1 micelles could have any effect on their ability to interact with HSA.
Although Dox–GM1 complexes also show the ability to spontaneously interact with HSA, the amount of protein that binds to the micelle in this case depends largely on the amount of incorporated drug. At 1/20 Dox–GM1 molar ratio, the amount of albumin that binds to the micelles reaches a 1/1 (w/w) HSA–GM1 saturation level. However, as the amount of Dox loaded in the micelle increases to 1/5 and 1/2.5 Dox–GM1 molar ratios, the percentage of bound albumin falls to 70% and 40%, respectively (Figure 3C). Similar results were observed for Dox–Ptx–GM1 micelles, as the amount of Dox increases, the quantity of albumin that binds to the micelle decreased (Figure 3D). In this case, the effect is even more pronounced and the amount of albumin that binds to these micelles is much lower than that which binds to the micelles loaded only with Dox falling to 40% for 1/20 and 1/5 Dox–GM1 molar ratios and to 20% for 1/1 molar ratio.
Effect of albumin on the size of Dox–GM1 or Dox–Ptx–GM1 complexes
Another significant change observed in drug–GM1 mixed micelles after HSA binding was that the interaction induced a significant increase in the size of the structures, reaching an average size of 19.1±1.0 and 17.0±0.9 nm for Dox–GM1–HSA and Dox–Ptx–GM1–HSA complexes, respectively (Table 1). This result was expected because HSA is a molecule that has a considerable size and therefore its association with the micelle produces an increase in the size of the micellar structure. In both cases, the binding of albumin to Dox–GM1 or Dox–Ptx–GM1 micelles seems to be driven by hydrophobic interactions, because it was not prevented or removed in the presence of 0.5 M NaCl (data not shown).
Characterization of the in vitro biological effect of Dox–GM1 and Dox–GM1–HSA micelles
Hep2 cells were used to assess the biological effect of Dox–GM1 and Dox–GM1–HSA micelles compared to the effect of the free drug in 150 mM NaCl as a control. The results of Figure 5, performed at 37°C for 24 hours, show similar IC50 values of Dox whether the drug is as free drug in saline, in Dox–GM1, or in Dox–GM1–HSA micelles. It should be noted that studies conducted at shorter incubation periods (15, 30, 45, 60, and 240 minutes) gave similar results, without significant differences observed between the free drug and the micellar formulations. In addition, in studies performed at 4°C versus 37°C and after 60 minutes of cell incubation with 10 μg mL−1 of Dox or Dox–GM1, the cytotoxic effect of Dox was only observed in those cultures incubated at 37°C, whereas no effect was observed in those incubated at 4°C (data not shown), suggesting the involvement of active transport mechanisms in the uptake of both free drug and Dox–GM1 micelles.
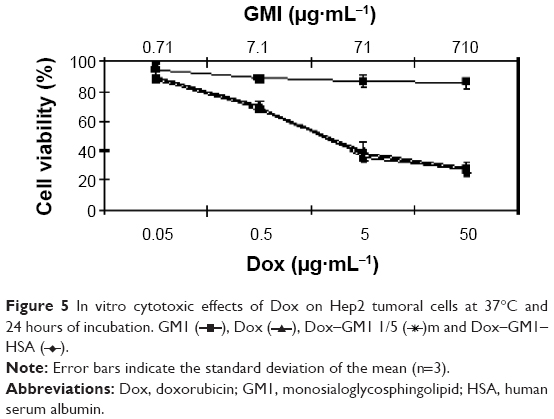 | Figure 5 In vitro cytotoxic effects of Dox on Hep2 tumoral cells at 37°C and 24 hours of incubation. GM1 ( |
 )m and Dox–GM1–HSA (
)m and Dox–GM1–HSA (Characterization of the cellular uptake of Dox and Ptx loaded into Dox–GM1 and Dox–Ptx–GM1 micelles with and without HSA
The natural fluorescence of Dox was used to evaluate the kinetics of the cellular uptake of Dox from GM1 micelles compared with the uptake of the free form of the drug. We also assessed whether the interaction of Dox–GM1 micelle with HSA affects the cellular uptake of Dox. The kinetic profile of naturally fluorescent Dox delivery from Dox–GM1 and Dox–GM1–HSA micelles to Hep2 cell line is shown in Figure 6. The images show that there are subtle differences in the cellular uptake of Dox from the different formulations, being slightly higher when the drug is free or associated to GM1 micelles without bound albumin. Regarding Dox location inside the cells, it is observed that in the case of the free drug and that loaded in GM1 micelles, the drug takes a little longer to reach its site of action, the cell nucleus, than in the case of the formulation containing HSA.
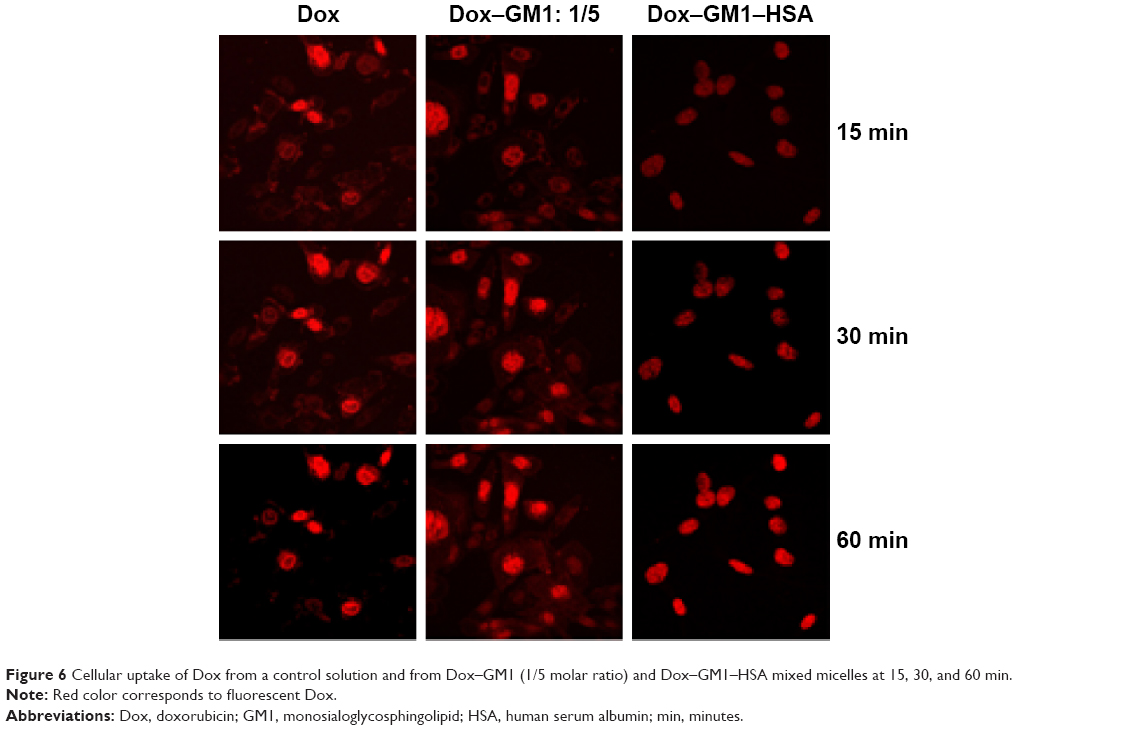 | Figure 6 Cellular uptake of Dox from a control solution and from Dox–GM1 (1/5 molar ratio) and Dox–GM1–HSA mixed micelles at 15, 30, and 60 min. |
We also used a fluorescently labeled derivative of Ptx to assess its cellular uptake. As shown in Figure 7, the kinetic profile of Dox and Ptx delivery from Dox–Ptx–GM1 and Dox–Ptx–GM1–HSA micelles was similar to that from control solutions.
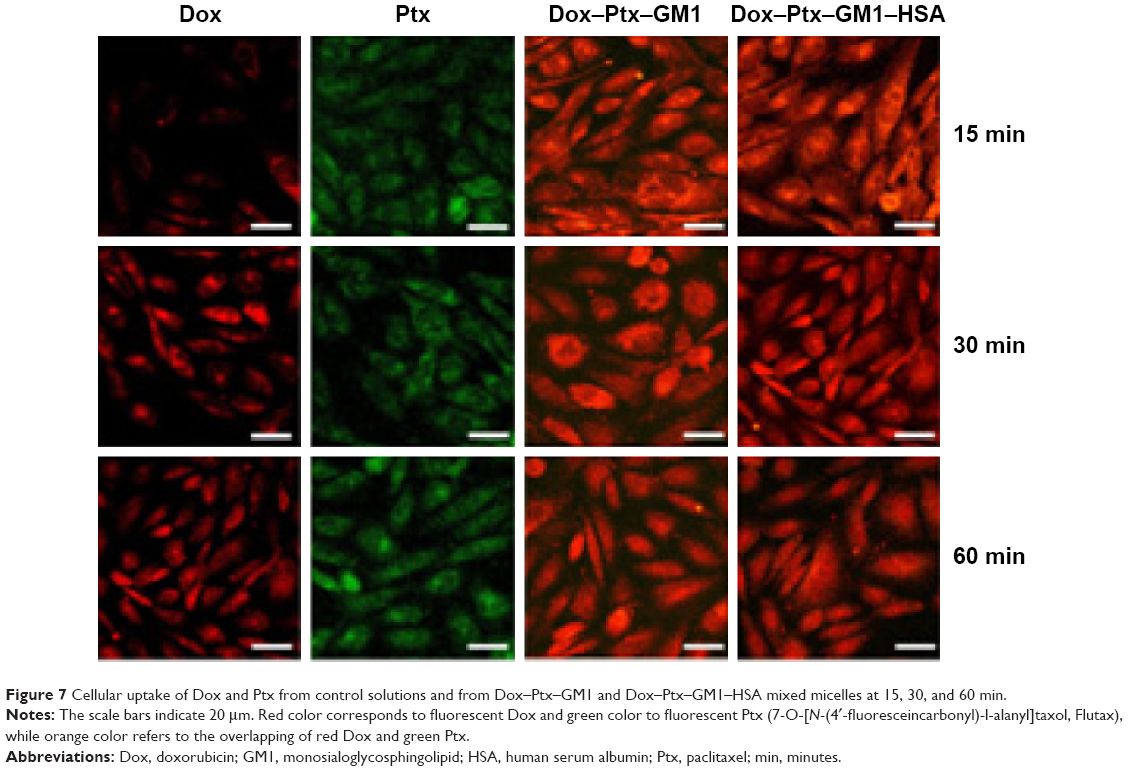 | Figure 7 Cellular uptake of Dox and Ptx from control solutions and from Dox–Ptx–GM1 and Dox–Ptx–GM1–HSA mixed micelles at 15, 30, and 60 min. |
Discussion
As previously mentioned, the use of Dox is limited by its serious side effects. Liposomal formulations of Dox have shown a strong reduction of the toxicity and an increase in blood circulation time. However, these formulations also generate a high cardiotoxic effect which strongly limits their use.32–34 More recently, Dox was loaded into synthetic multicomponent structures such as pH-sensitive micelles or amphiphilic block copolymers that self-assemble into nanoscopic micelles and accommodate Dox in the hydrophobic core,35–41 or under enzymatically activated pro-drug of Dox covalently bound to copolymers micelles.42
In this work, we evaluated the interactions between GM1 nano-micelles and Dox and characterize the behavior and physicochemical properties of the structures resulting from this interaction. Our results show that GM1 micelles load spontaneously higher amount of Dox (10 mg mL−1) per unit volume than, for example, classical liposomal formulations of Dox (2 mg mL−1).24 This loading capacity of GM1 is not altered by changes in pH or in the ionic strength of the medium, implying that the hydrophobic effect is the main driving force involved in the interaction. Moreover, the ability of GM1 micelles to load Dox was not affected by temperature, which may be related to the fact that, as the Dox is a hydrophilic drug, the temperature rise in the aqueous medium does not produce a change in the repulsion between molecules, as seen in the case of Ptx.13
We demonstrated that this Dox–GM1 complex is stable at different physical treatments such as freeze–thawing, high-speed centrifugation, and lyophilization–resuspension. Moreover, as in the case of Ptx,13 drug loading produces a shift in the monomer–micelle equilibrium to the aggregated form preventing disassembly of micelles due to dilution, providing greater stability.
DLS analysis of Dox–GM1 complexes in a molar ratio of 1/5 showed that these have an average size of about 12 nm, which is similar to that of polymeric micelles,39 but very different from the size of commercial liposomes, which have an average size around 100 nm. This confers a benefit over the latter, allowing micelles to evade elimination by the reticuloendothelial system, a common problem with liposomes. In this respect, many reports describe that the addition of a minimum amount of GM1 in the lipid mixture is sufficient to cause an increase in the average life of the liposome and also a differential tissue distribution,43,44 which could represent a potential advantage in the use of GM1 micelles.
In addition, the chromatographic profile of Dox–GM1 of different molar ratios shows that the size of micelles changes with increasing proportions of Dox in the structure. This result suggests differences in the stability of Dox–GM1 complexes at different molar ratios. Initially, the micelle enlarges with Dox loading; however, when a critical point is reached, the additional quantities of drug destabilize the structure, showing a blasting of structures of different molecular size. This phenomenon is observed with Dox but not with Ptx,13 suggesting that these modifications arise from changes in the areas occupied by Dox in the micelle. This is probably due to the fact that Dox is located in areas of the micelle described as critical for the stability of the aggregated structure formed by gangliosides.45
Moreover, many reports16,46 describe that albumin increases the antitumor capacity through its binding to gp160, promoting endothelial transcytosis of the formulations. In this work we confirm that Dox–GM1 micelles retain the ability to spontaneously interact with albumin forming a ternary complex of Dox–GM1–HSA with a size of around 19 nm. However, in this case there is a competitive relationship between the amount of Dox incorporated into the micelle and the amount of albumin that binds, which suggests that Dox and albumin partially share common sites in the micelle.
On the other hand, considering published results regarding the ability of GM1 micelles to load Ptx added to those described here in relation to Dox–GM1 interaction, we confirm that GM1 micelle can load hydrophobic (Ptx) and hydrophilic (Dox) drugs together into the same micellar unit with a differential localization of Ptx and Dox in view of each polarity. The fact that supports the presence of this gradient of polarity is the existence of a specific loading sequence, first Ptx in the most inner part, and then Dox in a more superficial area of the structure. The initial loading of Dox determines a barrier precluding the subsequent entry of Ptx to the innermost of the micelle.
The in vitro studies to evaluate the cytotoxic effect of Dox showed that Dox–GM1 and Dox–GM1–HSA micelles have IC50 values similar to those of the free drug and, in turn, these IC50 values are similar to those obtained with other delivery systems of Dox.47,48
Finally, the cellular uptake of Ptx and Dox from the micellar complexes was similar to that of the free form of these drugs, only slightly affected when the micelles are associated to albumin, proving that the superficial presence of the protein does not impair the interaction of the micelles with the cells and the subsequent drug release.
Conclusion
The present study described a self-assembled GM1 micellar system able to load and release hydrophobic and hydrophilic drugs like Ptx, Dox, or both that, in addition, interact spontaneously with HSA. These results suggest that GM1 micelles could be used as a system that would allow multiple drug combinations with actives of very different physical and chemical characteristics in a single formulation.
In addition, GM1 micelles have several advantages as a drug delivery system such as homogeneous composition, self-assembly with very low critical micellar concentration (10−8–10−10 M), small size, spontaneous loading of hydrophobic and hydrophilic drugs as well as interaction with albumin.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Acknowledgments
The present work was supported by grants from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) to DMB and IDB (PIP 11220100100502 and PIP 11220090100732).
Disclosure
RVA, IDB, and DMB are researchers and VL is a research fellow of CONICET. All authors confirm that there are no other conflicts of interest associated with this work.
References
Rivera E, Valero V, Arun B, et al. Phase II study of pegylated liposomal doxorubicin in combination with gemcitabine in patients with metastatic breast cancer. J Clin Oncol. 2003;21:3249–3254. | ||
Shah JJ, Orlowski RZ, Thomas SK. Role of combination bortezomib and pegylated liposomal doxorubicin in the management of relapsed and/or refractory multiple myeloma. Ther Clin Risk Manag. 2009;5:151–159. | ||
Swenson CE, Bolcsak LE, Batist G, et al. Pharmacokinetics of doxorubicin administered i.v. as myocet (TLC D-99; liposome-encapsulated doxorubicin citrate) compared with conventional doxorubicin when given in combination with cyclophosphamide in patients with metastatic breast cancer. Anticancer Drugs. 2003;14(3):239–246. | ||
Cuong NV, Hsieh MF. Recent advances in pharmacokinetics of polymeric excipients used in nanosized anti-cancer drugs. Curr Drug Metab. 2009;10:842–850. | ||
Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993; 1151(2):201–215. | ||
Kwon G, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Block copolymer micelles for drug delivery: loading and release of doxorubicin. J Control Release. 1997;48(2–3):195–201. | ||
Chen W, Meng F, Cheng R, Zhong Z. pH-sensitive degradable polymersomes for triggered release of anticancer drugs: a comparative study with micelles. J Control Release. 1997;142(1):40–46. | ||
Kim JO, Kabanov AV, Bronich TK. Polymer micelles with cross-linked polyanion core for delivery of a cationic drug doxorubicin. J Control Release. 2009;138(3):197–204. | ||
Torchilin V. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. | ||
Kakizawa Y, Kataoka K. Block copolymer micelles for delivery of gene and related compounds. Adv Drug Deliv Rev. 2002;54(2):203–222. | ||
Yokoyama M. Polymeric micelles as a new drug carrier system and their required considerations for clinical trials. Expert Opin Drug Deliv. 2010;7(2):145–158. | ||
Amin C, Mackman N, Key NS. Microparticles and cancer. Pathophysiol Haemost Thromb. 2008;36(3–4):177–183. | ||
Leonhard V, Alasino RV, Bianco ID, Garro AG, Heredia V, Beltramo DM. Self-assembled micelles of gangliosides as nanodelivery vehicles of taxanes. J Control Release. 2012;162(3):619–627. | ||
Leonhard V, Beltramo DM, Alasino RV, Bianco ID, inventor; Conicet-Consejo Nac. De Investigaciones Científicas Y Técnicas, Ceprocor – Centro De Excelencia En Prod. Y Procesos De Córdoba, assignee. Water-soluble pharmaceutical composition comprising at least one therapeutically active substance having hydrophobic properties and at least one compound selected from among sialoglycosphingolipids, glycosphingolipids or a mixture of sialoglycosphingolipids and glycosphingolipids. Patent WO/2011/113981. 2011 Sep 22. | ||
Leonhard V, Alasino RV, Bianco ID, Garro AG, Heredia V, Beltramo DM. Selective binding of albumin to gm1 ganglioside micelles containing paclitaxel. J Nanomed Nanotechnol. 2012;4:159. | ||
Gradishar WJ. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin Pharmacother. 2006;7:1041–1053. | ||
Fukunaga-Kalabis M, Herlyn M. Unraveling mysteries of the multifunctional protein SPARC. J Invest Dermatol. 2007;127:2497–2498. | ||
Miettinen T, Takki-Luukkainen IT. Use of butyl acetate in determination of sialic acid. Acta Chem Scand. 1959;13:856–858. | ||
Abraham SA, Edwards K, Karlsson G, et al. Formation of transition metal-doxorubicin complexes inside liposomes. Biochim Biophys Acta. 2002;1565(1):41–54. | ||
Garro AG, Beltramo DM, Alasino RV, Leonhard V, Heredia V, Bianco ID. Reversible exposure of hydrophobic residues on albumin as a novel strategy for the formulation of nanodelivery vehicles of taxanes. Int J Nanomedicine. 2011;6:1193–1200. | ||
Ulrich-Bott B, Wiegandt H. Micellar properties of glycosphingolipids in aqueous media. J Lipid Res. 1984;25:1233–1245. | ||
Sonnino S, Cantú L, Corti M, Acquotti D, Venerando B. Aggregative properties of gangliosides in solution. Chem Phys Lipids. 1994;71:21–45. | ||
Maggio B, Albert J, Yu RK. Thermodynamic-geometric correlations for the morphology of self-assembled structures of glycosphingolipids and their mixtures with dipalmitoylphosphatidylcholine. Biochim Biophys Acta. 1988;945(2):145–160. | ||
Tardi PG, Boman NI, Cullis PR. Review Liposomal Doxorubicin. J Drug Target. 1996;4(3):129–140. | ||
Cantú L, Corti M, Del Favero E, Muller E, Raudino A, Sonnino S. Thermal hysteresis in ganglioside micelles investigated by differential scanning calorimetry and light-scattering. Langmuir. 1999;15: 4975–4980. | ||
Orthaber D, Glatter O. Time and temperature dependent aggregation behaviour of the ganglioside GM1 in aqueous solution. Chem Phys Lipids. 1998;92(1):53–62. | ||
Hirai M, Takizawa T. Intensive extrusion and occlusion of water in ganglioside micelles with thermal reversibility. Biophys J. 1998;74:3010–3014. | ||
Hayakawa T, Hirai M. Hydration and thermal reversibility of glycolipids depending on sugar chains. Eur Biophys J. 2002;31(1):62–72. | ||
Wallevik K, Hvidt A. Conformational changes in human serum albumin as revealed by Hydrogen-Deuterium Exchange Studies. J Biol Chem. 1972;247:1530–1535. | ||
El Kadi N, Taulier N, Le Huérou JY, et al. Unfolding and refolding of bovine serum albumin at acid pH: ultrasound and structural studies. Biophys J. 2006;91(9):3397–3404. | ||
Muzammil S, Kumar Y, Tayyab S. Molten globule-like state of human serum albumin at low pH. Eur J Biochem. 1999;266:26–32. | ||
Uziely B, Jeffers S, Isacson R, et al. Liposomal doxorubicin: antitumor activity and unique toxicities during two complementary phase I studies. J Clin Oncol. 1995;13:1777–1785. | ||
Lotem M, Hubert A, Lyass O, et al. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch Dermatol. 2000;136:1475–1480. | ||
Elbayoumi TA, Torchilin VP. Tumor-specific antibody-mediated targeted delivery of DoxilR reduces the manifestation of auricular erythema side effect in mice. Int J Pharm. 2008;357:272–279. | ||
Kim D, Gao ZG, Lee ES, Bae YH. In vivo evaluation of doxorubicin-loaded polymeric micelles targeting folate receptors and early endosomal pH in drug-resistant ovarian cancer. Mol Pharm. 2009;6(5): 1353–1362. | ||
Kim D, Lee ES, Oh KT, Gao ZG, Bae YH. Doxorubicin-loaded polymeric micelle overcomes multidrug resistance of cancer by double-targeting folate receptor and early endosomal pH. Small. 2008;4(11):2043–2050. | ||
Lee ES. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103(2):405–418. | ||
Cuong NV, Jiang JL, Li YL, Chen JR, Jwo SC, Hsieh MF. Doxorubicin-loaded PEG-PCL-PEG micelle using xenograft model of nude mice: effect of multiple administration of micelle on the suppression of human breast cancer. Cancers. 2011;3:61–78. | ||
Dube N, Shu JY, Dong H, et al. Evaluation of doxorubicin-loaded 3-helix micelles as nanocarriers. Biomacromolecules. 2013;14(10): 3697–3700. | ||
Duan X, Xiao J, Yin Q, et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7(7):5858–5869. | ||
Guo X, Shi C, Yang G, Wang J, Cai Z, Zhou S. Dual-responsive polymer micelles for target-cell-specific anticancer drug delivery. Chem Mater. 2014;26(15):4405–4418. | ||
Ruiz-Hernández E, Hess M, Melen GJ, et al. PEG-pHPMAm-based polymeric micelles loaded with doxorubicin-prodrugs in combination antitumor therapy with oncolytic vaccinia viruses. Polym Chem. 2014; 5:1674–1681. | ||
Allen TM, Chonn A. Large unilamellar liposomes with low uptake into the reticuloendothelial system. FEBS Lett. 1987;223:42–46. | ||
Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85:6949–6953. | ||
Cantu L, Corti M, Acquotti D, Sonnino S. Aggregation properties of gangliosides: influence of the primary and secondary structure of the headgroup. J Phys IV. 1993;3(C1):57–64. | ||
Ibrahim NK, Desai N, Legha S, et al. Phase I and pharmacokinetic study of ABI-007, a Cremophor-free, protein-stabilized, nanoparticle formulation of paclitaxel. Clin Cancer Res. 2002;8:1038–1044. | ||
Hayashi K, Tatsui T, Shimanouchi T, Umakoshi H. Enhanced cytotoxicity for colon 26 cells using doxorubicin-loaded sorbitan monooleate (Span 80) vesicles. Int J Biol Sci. 2013;9(2):142–148. | ||
Nie Y, Ji L, Ding H, et al. Cholesterol derivatives based charged liposomes for doxorubicin delivery: preparation, in vitro and in vivo characterization. Theranostics. 2012;2(11):1092–1103. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.