Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 7
Bioavailability of everolimus administered as a single 5 mg tablet versus five 1 mg tablets: a randomized, open-label, two-way crossover study of healthy volunteers
Authors Thudium K, Gallo J, Bouillaud E, Sachs C, Eddy S, Cheung W
Received 30 August 2014
Accepted for publication 13 October 2014
Published 22 January 2015 Volume 2015:7 Pages 11—17
DOI https://doi.org/10.2147/CPAA.S73472
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Arthur E. Frankel
Karen Thudium,1 Jorge Gallo,2 Emmanuel Bouillaud,2 Carolin Sachs,2 Simantini Eddy,1 Wing Cheung1
1Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA; 2Novartis Pharma AG, Basel, Switzerland
Background: The mammalian target of rapamycin (mTOR) inhibitor everolimus has a well-established pharmacokinetics profile. We conducted a randomized, single-center, open-label, two-sequence, two-period crossover study of healthy volunteers to assess the relative bioavailability of everolimus administered as one 5 mg tablet or five 1 mg tablets.
Methods: Subjects were randomized 1:1 to receive everolimus dosed as one 5 mg tablet or as five 1 mg tablets on day 1, followed by a washout period on days 8–14 and then the opposite formulation on day 15. Blood sampling for pharmacokinetic evaluation was performed at prespecified time points, with 17 samples taken for each treatment period. Primary variables for evaluation of relative bioavailability were area under the concentration–time curve from time zero to infinity (AUCinf) and maximum blood concentration (Cmax). Safety was assessed by reporting the incidence of adverse events (AEs).
Results: Twenty-two participants received everolimus as one 5 mg tablet followed by five 1 mg tablets (n=11) or the opposite sequence (n=11). The Cmax of five 1 mg tablets was 48% higher than that of one 5 mg tablet (geometric mean ratio, 1.48; 90% confidence interval [CI], 1.35–1.62). AUCinf was similar (geometric mean ratio, 1.08; 90% CI, 1.02–1.16), as were the extent of absorption and the distribution and elimination kinetics. AEs, all grade 1 or 2, were observed in 54.5% of subjects.
Conclusion: Although the extent of absorption was similar, the Cmax of five 1 mg tablets was higher than that of one 5 mg tablet, suggesting these formulations lead to different peak blood concentrations and are not interchangeable at the dose tested.
Keywords: absorption kinetics, healthy volunteers
Introduction
The mammalian target of rapamycin (mTOR) signaling pathway is important in the regulation of cell growth, proliferation, metabolism, survival, and angiogenesis.1,2 Because activation of mTOR is commonly associated with the pathogenesis of multiple tumor types, inhibition of the pathway provided the rationale for the development of anticancer therapies targeting mTOR.2 Everolimus, an oral mTOR inhibitor, has been studied extensively in patients with multiple tumor types and tuberous sclerosis complex (TSC).3 In addition to its clinical profile, the pharmacokinetics (PK) and pharmacodynamics profiles of everolimus have been well characterized in patients with advanced solid tumors and TSC.4–7 Results of a Phase I study conducted in patients with advanced solid tumors demonstrated sustained activity over 7 days at oral everolimus doses ≥20 mg once weekly.5 Area under the concentration–time curve (AUC) was dose-proportional, but maximum blood concentration (Cmax) increased less than proportionally at doses ≥20 mg once weekly. Everolimus ≥20 mg once weekly or ≥5 mg once daily was recommended as the optimal effective dose. Results of another Phase I study of oral everolimus demonstrated more profound and sustained mTOR pathway-inhibition with everolimus 10 mg once daily than with 50 mg once weekly.4
Everolimus (Afinitor®; Novartis Pharmaceuticals Corp, East Hanover, NJ, USA) is approved in various countries for the treatment of patients with advanced hormone receptor-positive, HER2-negative breast cancer, in combination with exemestane after failure of letrozole or anastrozole; progressive, advanced pancreatic neuroendocrine tumors; renal cell carcinoma after failure of prior sunitinib or sorafenib; renal angiomyolipoma associated with TSC not requiring immediate surgery; and subependymal giant cell astrocytoma associated with TSC not amenable to curative resection.8 As Afinitor, everolimus is available as 2.5 mg, 5 mg, 7.5 mg, and 10 mg tablets, and as 2 mg, 3 mg, and 5 mg dispersible tablets for oral suspension (Afinitor Disperz; Novartis Pharmaceuticals Corp).8 Under the trade names Certican® (Novartis Pharma AG, Basel, Switzerland) and Zortress® (Novartis Pharmaceuticals Corp), everolimus has been approved for the prophylaxis of organ rejection in adult patients who received a renal, hepatic, or cardiac transplant and is available in formulations of 0.25 mg, 0.5 mg, 0.75 mg, and 1 mg tablets, and 0.1 mg and 0.25 mg dispersible tablets.9 Everolimus 1 mg tablets have not been studied in the oncology setting. Although 1 mg and 5 mg everolimus tablets have the same active ingredient and excipients, the proportional composition of the components differs between formulations. It is also not known whether several 1 mg tablets deliver the same peak concentration and bioavailability as a single higher dose tablet.
The purpose of this study was to determine the relative bioavailability of a single 5 mg oral dose of everolimus administered as one 5 mg tablet (reference) or as five 1 mg tablets (test). The secondary objective was to evaluate the safety and tolerability of both formulations.
Subjects and methods
Subjects
Male or female volunteers between 18 and 65 years of age who were in good health, as determined by medical history, current medical condition, physical examination, vital signs, electrocardiogram (ECG), and laboratory tests, were eligible for inclusion in the study. Pregnant or lactating mothers and women of childbearing age and sexually active men (unless they were using contraception during and 8 weeks after dosing) were ineligible. Other exclusion criteria included tobacco or nicotine use within 3 months before screening; use of any prescription drugs within 30 days before dosing or over-the-counter medication or herbal supplement within 14 days of baseline assessments; participation in any clinical investigation within 4 weeks of dosing; known hypersensitivity to rapamycin and its derivatives; and a positive test for HIV antibody, hepatitis B surface antigen, or hepatitis C antibody. Use of CYP3A4 and/or P-glycoprotein inhibitors, inducers, or substrates was prohibited in the 30 days before the first everolimus dose until the end of the study. Consumption of grapefruit (and juice), star fruit (and juice), and cruciferous vegetables within 7 days of baseline assessments and of caffeine within 12 hours of baseline assessments was also prohibited.
The study protocol was approved by Ethics Committee of the Land Berlin, State Office of Health and Social Affairs Berlin, Berlin, Germany. The study was conducted in accordance with the ICH Harmonized Tripartite Guidelines for Good Clinical Practice with applicable local regulations, and with the ethical principles laid down in the Declaration of Helsinki. All participants provided written informed consent before screening.
Study design
This Phase I randomized, single-center, open-label, two-sequence, two-period crossover study was conducted in healthy volunteers (Figure 1). After a 14-day screening period, participants were randomly assigned 1:1 to receive a single 5 mg everolimus tablet on day 1, followed by a washout period on days 8–14 and then five 1 mg everolimus tablets on day 15, or the opposite sequence. All everolimus doses were administered with 240 mL of water under the supervision of study personnel, under fasting conditions. Participants received a standardized, light, low-fat dinner at least 10 hours before study drug administration; after this, no food was allowed until at least 4 hours postdose, when a standard meal was served. Meals were controlled for up to 72 hours postdose. Aside from water for dosing, one fluid was permitted within 1 hour before and after dosing. Water was allowed ad libitum at all other times. End-of-study evaluation was conducted 14 (±2) days after everolimus administration in the second treatment period. Subjects were discontinued from the study in the event of grade 3 or 4 adverse events (AEs), abnormal laboratory values or test procedures, or protocol deviation. Subjects could voluntarily withdraw from the study at any time.
 | Figure 1 Study design. |
Pharmacokinetic assessments
Blood sampling for PK evaluations was performed at prespecified time points, with 17 samples collected during each treatment period: one predose sample collected up to 24 hours before everolimus administration and 16 postdose samples collected 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 24, 48, 72, 96, 120, and 144 hours after everolimus administration. At each time point, 2 mL venous blood was collected into a tube containing dipotassium ethylenediaminetetraacetic acid (K2EDTA) anticoagulant and stored at −20°C or below within 60 minutes of the draw time. Everolimus concentration in whole blood was determined at a central laboratory (WuXi AppTec, Shanghai, China) by a validated method of liquid chromatography mass spectrometry (LC-MS/MS) with solid-phase extraction (lower limit of quantification [LLOQ], 0.300 ng/mL).
Primary variables for evaluation of relative bioavailability were AUC from time zero to infinity (AUCinf) and Cmax. Secondary variables were AUC from time zero to the last observation time point, regardless of whether that concentration is quantifiable (AUCall), AUC from time zero to the 144-hour concentration sampling time (AUC0–144h), time to reach Cmax (Tmax), terminal slope of elimination phase, apparent volume of distribution (Vd/F), total body apparent clearance of drug from the blood (CL/F), elimination half-life (T1/2), and mean resident time (MRT). All PK parameters were analyzed using Phoenix® WinNonlin 6.3 (Pharsight, Mountain View, CA, USA) and noncompartmental methods.
Safety assessments
Safety was assessed throughout the study period and for up to 30 (±2) days following the last everolimus dose by reporting the incidence of AEs and serious AEs (SAEs) and their severity and relationship to study drug. Assessments of hematology and blood chemistry and urinalysis were performed at screening, at each of the two baseline visits, and at the end of study treatment. Vital signs, physical condition, ECG, and body weight were regularly assessed. AEs were graded according to the Common Terminology Criteria for Adverse Events (CTCAE), version 4.03.
Statistical analysis
Because the primary statistical analysis did not include hypothesis testing, no power or power-based sample size calculations were performed. The number of subjects enrolled was driven by the targeted precision of 20% of the two-sided 90% confidence interval (CI) for the ratio of geometric means.
The full analysis set population included all randomly assigned participants. The safety population included all participants who received at least one dose of everolimus. The PK analysis set population included all participants who completed at least one treatment period and had evaluable PK data. Evaluable PK data for any treatment period fulfilled the following criteria: no vomiting within 4 hours of dosing, sufficient sample available for analysis, and no use of CYP3A4 inhibitors, inducers, or substrates for 30 days before the first dose until the end of the study.
Data were analyzed for PK and safety using SAS® version 9.1.3 (SAS Institute, Inc., Cary, NC, USA). Descriptive statistics were used to summarize PK parameters, including geometric and arithmetic means, standard deviation (SD), mean and geometric mean coefficient of variation (CV%), median, minimum, and maximum; only median, minimum, and maximum were used for Tmax. Predose concentrations below the LLOQ were treated as zero, and postdose concentrations below the LLOQ were excluded from summary statistics and from calculation of PK parameters. The primary PK parameters (AUCinf and Cmax) were analyzed separately using a linear mixed model with the log-transformed PK variable as the dependent variable; fitting sequence, period, and treatment as fixed effects; and subject nested within-sequence as a random effect. From the fitted model, the difference in treatment estimates on the log scale and the 90% CI was calculated and back-transformed to provide geometric mean ratios and their two-sided 90% CIs. Missing values were not imputed. Aside from Tmax, similar model-based analyses were conducted for the secondary PK parameters. Tmax was analyzed with point estimate (median difference) and associated ranges provided for comparison.
Results
Subjects
Twenty-two participants were randomly assigned to receive everolimus administered as one 5 mg tablet on day 1 followed by five 1 mg tablets on day 15 (n=11) or as five 1 mg tablets on day 1 followed by one 5 mg tablet on day 15 (n=11). All participants were included in the full analysis set, safety, and PK analysis set populations.
Baseline characteristics were comparable between the sequences (Table 1). In the overall population, median age was 47.5 (range, 25 to 65) years, and most participants (73%) were women.
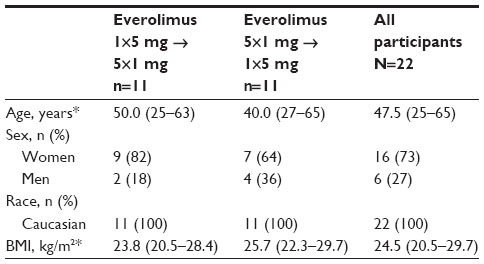 | Table 1 Baseline characteristics |
Pharmacokinetics
The Cmax of five 1 mg tablets was 48% higher than the Cmax of one 5 mg tablet (geometric mean ratio, 1.48; 90% CI, 1.35–1.62) (Table 2 and Figure 2). Because the 90% CI was not within the boundaries of 0.8–1.25, the everolimus formulations of five 1 mg tablets and one 5 mg tablet did not meet bioequivalence criteria in terms of Cmax. The two formulations had similar AUCinf (geometric mean ratio, 1.08; 90% CI, 1.02–1.16) (Table 2).
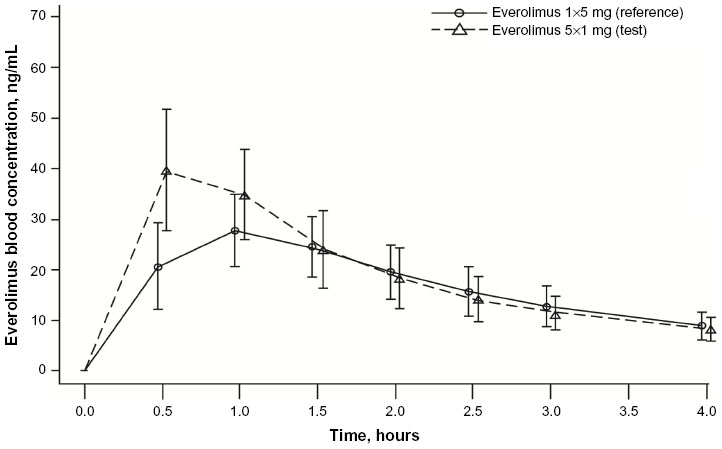 | Figure 2 Arithmetic mean (SD) blood concentration–time profiles from 0 to 4 hours for everolimus administered as five 1 mg tablets and as one 5 mg tablet (PK population, N=22). |
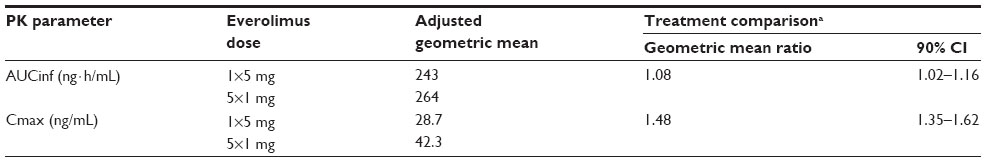 | Table 2 Relative bioavailability of everolimus one 5 mg tablet versus five 1 mg tablets (PK population, N=22) |
Everolimus administered as five 1 mg tablets had a faster absorption rate as demonstrated by a shorter median Tmax than was observed with one 5 mg tablet (0.5 vs 1.0 hour) (Table 3). As assessed by both AUC0–144h and AUCall, total overall exposure was similar for one 5 mg tablet and five 1 mg tablets (Table 3). No differences in T1/2 and MRT were apparent, suggesting similar distribution and elimination kinetics (Table 3). Mean Vd/F of one 5 mg tablet was higher than that of five 1 mg tablets (Table 3), but the geometric mean ratio and 90% CI suggested no statistically significant difference (ratio, 0.92; 90% CI, 0.85–0.99). Everolimus concentration–time profiles at time points beyond 24 hours postdose were comparable between the two treatment formulations (Figure 3). Similar predose trough concentrations (Cmin) are expected after daily administration of the two formulations.
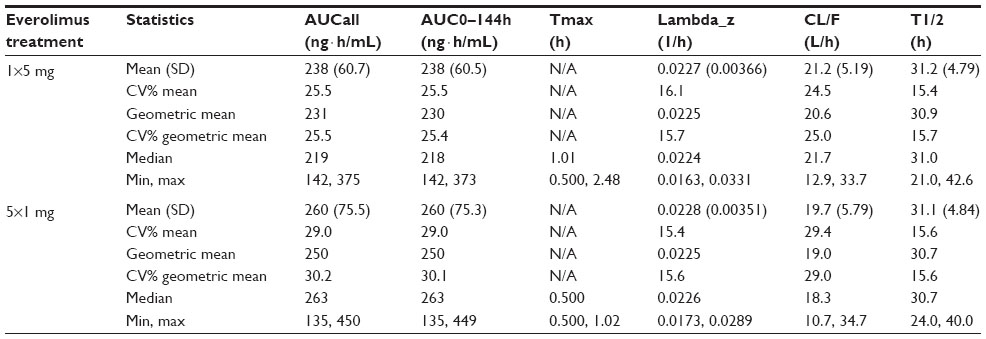 | Table 3 Summary of secondary PK parameters of everolimus by treatment (PK population, N=22) |
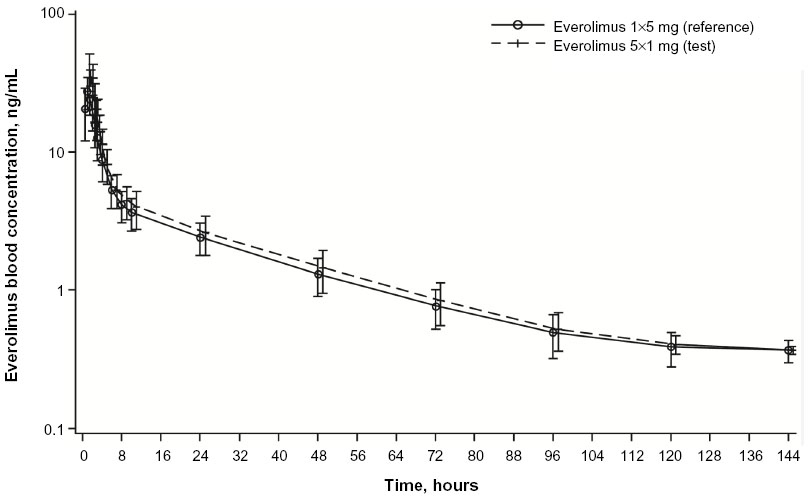 | Figure 3 Arithmetic mean (SD) blood concentration–time profiles for everolimus administered as five 1 mg tablets and as one 5 mg tablet (PK population, N=22). |
Safety
Overall, 12 (54.5%) participants experienced at least one AE during the study (Table 4). The most frequently reported AEs were headache (27.3%) and nasopharyngitis, nausea, dry skin, and myalgia (9.1% each). Seven (31.8%) participants experienced AEs determined to be related to everolimus, with headache (18.2%) being the most common. Other AEs suspected to be related to everolimus were nausea, diarrhea, vomiting, oral herpes, myalgia, dizziness, and dry skin. All AEs were grade 1 or 2, and no AE led to study discontinuation. No clinically significant changes in laboratory parameters, vital signs, or ECG intervals were observed during the study.
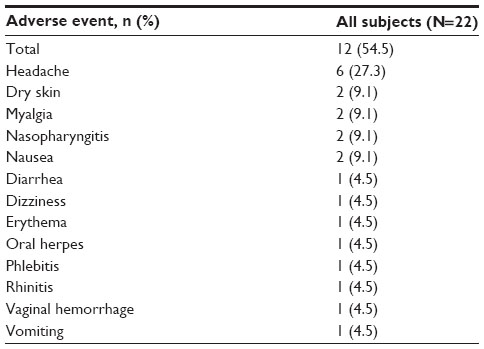 | Table 4 Number of subjects who experienced adverse events, regardless of study drug relationship (safety population, N=22) |
Discussion
Everolimus efficacy and safety in the oncology setting are well established, and everolimus is currently approved in multiple countries for treatment of patients with various types of cancer.8 The relationship between dose and the pharmacodynamic response of everolimus in patients with cancer has been established.4,5 Following oral administration, everolimus is rapidly absorbed, with a Tmax of 1–2 hours.5 Steady state is reached by week 2 after daily administration.5 The steady state AUC is dose-proportional for dosages of 5 mg to 70 mg weekly and 5 mg to 10 mg daily.5 In addition, Cmin was highly predictive of AUC, with a coefficient of determination of 0.96.5 A meta-analysis of everolimus in clinical oncology trials demonstrated that a twofold increase in Cmin increased the likelihood of tumor size reduction (odds ratio, 1.40; 95% CI, 1.23–1.60), was associated with a trend toward reduced risk of progression-free survival events (risk ratio [RR], 0.90; 95% CI, 0.69–1.18), and increased the risk of grade ≥3 stomatitis (RR, 1.49; 95% CI, 1.05–2.10) and pulmonary (RR, 1.93; 95% CI, 1.12–3.34) and metabolic (RR, 1.30; 95% CI, 1.02–1.65) events.10
Although everolimus is available as a 1 mg tablet, this formulation has not been evaluated in the oncology setting, and it is not known whether several 1 mg tablets deliver the same Cmax and bioavailability as a single tablet of a higher dose. Results of this healthy volunteer study, which used a well-established, standard crossover design, suggest that everolimus dosed as five 1 mg tablets does not have the same bioavailability as one 5 mg tablet. As assessed by Cmax, the bioequivalence criteria were not met, despite similar AUCinf. The Cmax was 48% higher with five 1 mg tablets than with one 5 mg tablet. In addition, Tmax was 0.5 hour with five 1 mg tablets and 1.01 hours with one 5 mg tablet. The distribution and elimination kinetics (CL/F, T1/2, and terminal slope of elimination phase parameters) were similar with both everolimus formulations.
The safety profile of everolimus administered as 5 mg tablets to patients with advanced solid tumors has been well established. Common class effect AEs include mucositis, stomatitis, skin toxicities, pulmonary toxicities, hyperlipidemia, and hyperglycemia.11 Results of a Phase I dose-escalation study of oral everolimus conducted in patients with advanced solid tumors found dose-limiting toxicities to be stomatitis and fatigue at the 50 mg once-weekly dosage and hyperglycemia at the 10 mg daily dosage.5 Conversely, the safety profile of everolimus administered as several 1 mg tablets in patients with cancer has not been evaluated. In this single-dose study of healthy volunteers, both formulations were well tolerated, with 12.5% of subjects experiencing at least one AE during the study period, all of which were grade 1 or 2. The most common AE was headache. No serious AEs were reported, no AE led to treatment discontinuation, and no new safety concerns were identified.
In conclusion, everolimus formulations of one 5 mg tablet and five 1 mg tablets did not meet bioequivalence criteria in terms of Cmax despite similar AUCinf in this Phase I study of healthy volunteers. In addition to the consideration that the safety profile of several 1 mg tablets in patients with advanced solid tumors is unknown, this finding suggests that these formulations are not interchangeable.
Acknowledgments
This study was funded by Novartis Pharmaceuticals Corporation, Inc. (East Hanover, NJ, USA). Editorial assistance was provided by Cathy Winter, PhD, and Melanie Leiby, PhD (both from ApotheCom, Yardley, PA, USA) and funded by Novartis Pharmaceuticals Corporation.
Disclosure
Drs Thudium, Eddy, and Cheung are employees of Novartis Pharmaceuticals Corporation. Drs Gallo, Bouillaud, and Sachs are employees of Novartis Pharma AG.
References
Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7(4):209–219. | |
Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27(13):2278–2287. | |
Lebwohl D, Anak O, Sahmoud T, et al. Development of everolimus, a novel oral mTOR inhibitor, across a spectrum of diseases. Ann N Y Acad Sci. 2013;1291:14–32. | |
Tabernero J, Rojo F, Calvo E, et al. Dose- and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008;26(10):1603–1610. | |
O’Donnell A, Faivre S, Burris HA, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26(10):1588–1595. | |
Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010; 363(19):1801–1811. | |
Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2):a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824. | |
AFINITOR® (everolimus) tablets for oral administration. AFINITOR DISPERZ (everolimus tablets for oral suspension) [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014. | |
ZORTRESS® (everolimus) tablets for oral use [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2013. | |
Ravaud A, Urva SR, Grosch K, Cheung WK, Anak O, Sellami DB. Relationship between everolimus exposure and safety and efficacy: meta-analysis of clinical trials in oncology. Eur J Cancer. 2014;50(3):486–495. | |
Sankhala K, Mita A, Kelly K, Mahalingam D, Giles F, Mita M. The emerging safety profile of mTOR inhibitors, a novel class of anticancer agents. Target Oncol. 2009;4(2):135–142. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.