Back to Journals » Clinical Ophthalmology » Volume 8
Bimatoprost 0.01% or 0.03% in patients with glaucoma or ocular hypertension previously treated with latanoprost: two randomized 12-week trials
Authors Myers J, Vold SD, Zaman F, Williams J, Hollander D
Received 14 December 2013
Accepted for publication 28 January 2014
Published 27 March 2014 Volume 2014:8 Pages 643—652
DOI https://doi.org/10.2147/OPTH.S59197
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Jonathan S Myers,1 Steven Vold,2 Fiaz Zaman,3 Julia M Williams,4 David A Hollander4
1Wills Eye Hospital, Philadelphia, PA, USA; 2Vold Vision, PLLC, Bentonville, AR, USA; 3Houston Eye Associates, Houston, TX, USA; 4Allergan, Inc., Irvine, CA, USA
Background: The purpose of this study was to evaluate the intraocular pressure (IOP)-lowering efficacy and safety of bimatoprost 0.01% or 0.03% as monotherapy in patients treated with latanoprost 0.005% monotherapy who require additional IOP lowering for their ocular hypertension or open-angle glaucoma.
Methods: Two prospective, investigator-masked, randomized, parallel-group, multicenter studies enrolled patients with baseline IOP ≥20 mmHg after ≥30 days of latanoprost 0.005% monotherapy. Patients were randomized to 12 weeks of study treatment (study 1, bimatoprost 0.01% once daily or bimatoprost 0.01% once daily plus brimonidine 0.1% three times daily; study 2, bimatoprost 0.03% once daily or bimatoprost 0.03% once daily plus fixed-combination brimonidine 0.2%/timolol 0.5% twice daily). Patient evaluations at weeks 4 and 12 included IOP at 8 am, 10 am, and 4 pm and safety assessments. Results in the monotherapy study arms (bimatoprost 0.01% or 0.03%) are presented.
Results: Latanoprost-treated baseline mean diurnal IOP (± standard error of the mean) was 22.2±0.3 mmHg and 22.1±0.4 mmHg in the bimatoprost 0.01% and bimatoprost 0.03% treatment arms, respectively (P=0.957). In both treatment arms, mean (± standard error of the mean) reduction in IOP from latanoprost-treated baseline was statistically significant at each time point at both follow-up visits (P<0.001), ranging from 3.7±0.4 (17.0%) mmHg to 4.4±0.4 (19.9%) mmHg with bimatoprost 0.01% and from 2.8±0.5 (12.8%) mmHg to 3.9±0.5 (16.7%) mmHg with bimatoprost 0.03%. Mean percentage IOP reduction from latanoprost-treated baseline was numerically greater with bimatoprost 0.01% than with bimatoprost 0.03% throughout follow-up. The incidence of conjunctival hyperemia of mild or greater severity increased from latanoprost baseline after 12 weeks of treatment only in the bimatoprost 0.03% treatment arm.
Conclusion: Many patients who do not reach their target IOP on latanoprost can achieve additional IOP lowering and maintain monotherapy by replacing latanoprost with bimatoprost. Reductions in IOP from latanoprost baseline were larger with bimatoprost 0.01% than with bimatoprost 0.03%, and bimatoprost 0.01% had a more favorable tolerability profile.
Keywords: bimatoprost, intraocular pressure, latanoprost, monotherapy, prostaglandin, prostamide
Introduction
Treatment for ocular hypertension and glaucoma aims to reduce intraocular pressure (IOP) to protect against damage to the optic nerve and progressive loss of visual field. Once-daily prostaglandins and prostamides have become first-line therapy for many patients with glaucoma or ocular hypertension.1 The prostamide bimatoprost and the prostaglandins latanoprost and travoprost reduce IOP effectively2 and are systemically safe and generally well tolerated.3
Monotherapy is usually preferred over use of multiple IOP-lowering medications to achieve IOP control.4 Treatment with a single medication minimizes drug exposure and the possibility of ocular and systemic adverse effects and avoids the inconvenience of using multiple eye drops, which can be a barrier to adherence, as well as the additional cost of add-on therapy. When patients treated with a prostaglandin or prostamide (PG/PM) need additional IOP lowering, switching within class to a different PG/PM may provide further IOP reduction5 and is recommended before adding another medication.4,6
Among the PG/PM medications, the prostamide bimatoprost has excellent IOP-lowering efficacy.7,8 Bimatoprost 0.03% has been seen in meta-analyses to achieve greater IOP lowering compared with travoprost or latanoprost.8 The most frequent side effect of bimatoprost 0.03% treatment has been conjunctival hyperemia.7 To improve its tolerability profile while maintaining its efficacy in reducing IOP, the original bimatoprost 0.03% ophthalmic solution was reformulated,9 and bimatoprost 0.01% was introduced in the United States in 2010. In a Phase III clinical study, bimatoprost 0.01% demonstrated equivalent IOP lowering to bimatoprost 0.03% and was associated with less frequent and less severe conjunctival hyperemia.9
Two randomized clinical trials with similar design were conducted to evaluate therapies for patients with glaucoma or ocular hypertension using latanoprost who need additional IOP lowering. One of the studies tested bimatoprost 0.01% alone or in combination with brimonidine 0.1%, and the other study tested bimatoprost 0.03% alone or in combination with a fixed combination of brimonidine 0.2% and timolol 0.5%. Because maintaining monotherapy is preferred, we present here the results of the study arms that evaluated the efficacy and safety of bimatoprost 0.01% or bimatoprost 0.03% monotherapy in patients previously treated with latanoprost who needed additional IOP lowering.
Materials and methods
Two multicenter, masked, randomized, controlled clinical studies evaluated the IOP-lowering efficacy and safety of prostamide regimens in patients with glaucoma or ocular hypertension. The studies were conducted at 17 sites in the USA from February 2012 to October 2012 (study 1) and eleven sites in the USA from December 2009 to September 2010 (study 2) in compliance with Good Clinical Practice regulations and guidelines. An institutional review board approved the study protocol at each site, and all patients provided written informed consent. The studies are registered at ClinicalTrials.gov with the identifiers NCT0152517310 (study 1) and NCT0117088411 (study 2).
Patient selection for study participation was similar in the two studies. Entry criteria for both studies included diagnosis of open-angle glaucoma, chronic angle-closure glaucoma with patent iridotomy, or ocular hypertension, best-corrected visual acuity of 20/100 or better in both eyes, and currently using no more than two (study 1) or three (study 2) IOP-lowering medications at screening. IOP in the study eye was required to be ≥20 mmHg and <34 mmHg at 8 am and 10 am (study 1) or at 8 am (study 2) at baseline after at least a 30-day run-in on monotherapy with latanoprost 0.005% (Falcon Pharmaceuticals, Ltd, Fort Worth, TX, USA [study 1] or Pfizer Inc., New York, NY, USA [study 2]). Key exclusion criteria for both studies included active ocular disease other than glaucoma or ocular hypertension that would interfere with the study interpretation, corneal abnormality that would prevent accurate applanation tonometry, history of or active ocular infection or inflammation, history of any intraocular surgery or glaucoma laser surgery within 3 months prior to enrollment, uncontrolled systemic disease, end-stage glaucomatous visual field loss, and known sensitivity or allergy to the study medications or their components.
A schematic of the study designs is shown in Figure 1. Each study had four study visits, ie, screening, baseline, week 4, and week 12. At the screening visit (2 to 30 days prior to the baseline visit), patients who were using any IOP-lowering medication other than latanoprost discontinued the medication and began a 30-day washout period. Patients who were using latanoprost at screening continued on latanoprost monotherapy; all other patients began latanoprost monotherapy. Open-label latanoprost 0.005% was dispensed to patients at the screening visit with instructions to administer the drop once daily in the evening. The baseline visit was scheduled after at least 30 days of latanoprost monotherapy.
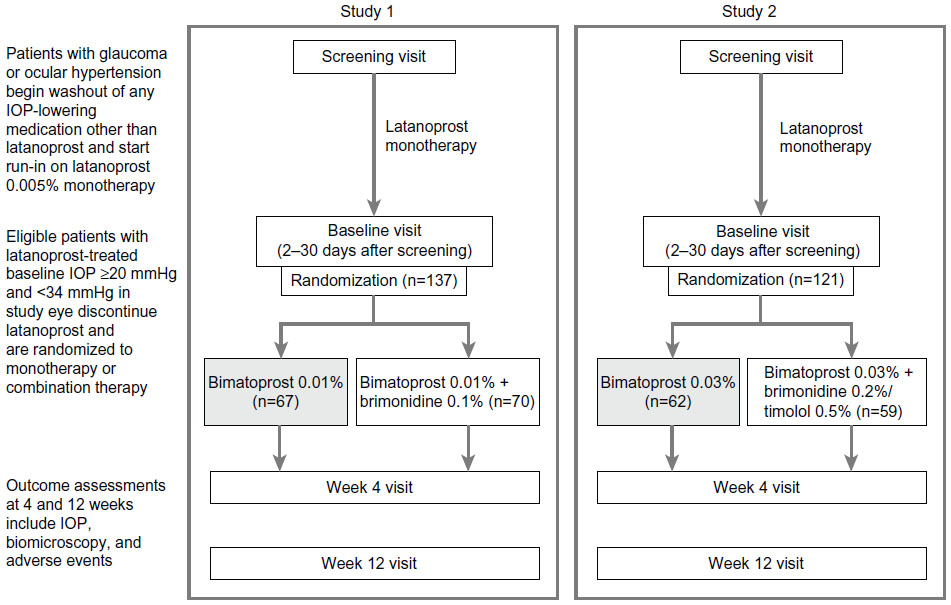 | Figure 1 Study design. |
Following evaluations at the baseline visit, patients who met all eligibility criteria, including baseline latanoprost-treated IOP ≥20 mmHg in the study eye, were enrolled and randomized in a 1:1 ratio to one of two treatment arms. In study 1, the treatment arms were bimatoprost 0.01% (Lumigan® 0.01%) monotherapy and bimatoprost 0.01% plus brimonidine 0.1% (Alphagan® P 0.1%) combination therapy. In study 2, the treatment arms were bimatoprost 0.03% (Lumigan 0.03%) monotherapy and bimatoprost 0.03% plus fixed-combination brimonidine 0.2%/timolol 0.5% combination therapy. All study medications other than latanoprost were manufactured by Allergan, Inc. (Irvine, CA, USA) and dispensed in over-labeled bottles supplied in identical packaging. Patients in the monotherapy treatment arms (bimatoprost 0.01% or 0.03%) also received an artificial tear (three times daily in study 1, twice daily in study 2) for masking monotherapy versus combination therapy. In each treatment arm, bimatoprost 0.01% or 0.03% was dosed once daily in the evening at 8 pm.
Patient assessments were identical in the two studies. IOP was measured in each eye by Goldmann applanation at 8 am, 10 am, and 4 pm at baseline, week 4, and week 12. Two measurements of IOP were taken, and if they differed by ≤2 mmHg, the mean of those measurements was recorded on the case report form and used for analysis. If the measurements differed by >2 mmHg, an additional measurement was taken, and the median of the three measurements was recorded on the case report form and used for analysis. The primary efficacy outcome measures were IOP and change from baseline IOP at 8 am, 10 am, and 4 pm at weeks 4 and 12, diurnal IOP averaged over the 8 am, 10 am, and 4 pm time points, change from baseline diurnal IOP, achievement of diurnal IOP <14, 15, 16, 17, or 18 mmHg, and achievement of a ≥15% reduction in diurnal IOP from baseline. The primary efficacy endpoint was mean change in diurnal IOP from baseline at week 12 in study 1 and mean diurnal IOP at week 12 in study 2. Safety measures included adverse events and slit-lamp biomicroscopy. Biomicroscopy findings were graded on a scale of 0 (none), +0.5 (trace), +1 (mild), +2 (moderate), and +3 (severe). Corneal erosion was evaluated with fluorescein staining on biomicroscopic examination.
This report focuses on the study arms evaluating monotherapy (bimatoprost 0.01% or 0.03%). Efficacy results were evaluated in the per protocol population of all patients who completed 12 weeks of treatment without significant protocol violations. If both eyes were eligible for the study, results were analyzed for the eye with the worse IOP at baseline, or if the IOP was the same in both eyes, the right eye. Observed values were used for the analyses with no imputation of missing values. Safety results were evaluated in all patients who received study treatment. JMP 7.0 software (SAS Institute Inc., Cary, NC, USA) and a two-sided alpha level of 0.05 were used for statistical analysis. Analysis of change in IOP and percentage change in IOP from baseline used analysis of covariance models with treatment group as a fixed effect and baseline IOP as the covariate. Within-group changes in IOP from baseline were analyzed with paired t-tests. Baseline mean IOP was compared between groups with t-tests. Comparisons of categorical variables used Fisher’s exact tests.
Results
Patient characteristics and disposition
A total of 67 patients were randomized to bimatoprost 0.01% monotherapy and 62 patients were randomized to bimatoprost 0.03% monotherapy following at least 30 days of latanoprost monotherapy. Baseline characteristics of these patients are listed in Table 1. Most patients in each treatment arm were diagnosed with glaucoma and were using IOP-lowering medication at screening (Table 1).
 | Table 1 Baseline patient characteristics |
Study completion rates were 92.5% with bimatoprost 0.01% and 96.8% with bimatoprost 0.03%. Four (6.0%) patients treated with bimatoprost 0.01% discontinued due to adverse events (eye pain and dizziness, anxiety, cardiac ischemia, exacerbation of depression) and one (1.5%) patient was lost to follow-up. In the bimatoprost 0.03% treatment arm, two (3.2%) patients discontinued due to adverse events (allergic conjunctivitis, uncontrolled IOP). Fifty-nine (88.1%) patients treated with bimatoprost 0.01% and 58 (93.5%) treated with bimatoprost 0.03% completed the study without significant protocol violations and were included in the efficacy analyses.
Efficacy outcomes
At baseline on latanoprost, mean diurnal IOP (± standard error of the mean) was 22.2±0.3 mmHg (22.6±0.3 mmHg at 8 am, 22.1±0.3 mmHg at 10 am, and 21.8±0.4 mmHg at 4 pm) in the bimatoprost 0.01% treatment arm and 22.1±0.4 mmHg (23.3±0.3 mmHg at 8 am, 21.8±0.4 mmHg at 10 am, and 21.3±0.5 mmHg at 4 pm) in the bimatoprost 0.03% treatment arm. There was no statistically significant difference between treatment arms in latanoprost-treated baseline mean diurnal IOP (P=0.957) or latanoprost-treated baseline mean IOP at 8 am, 10 am, or 4 pm. After replacement of latanoprost, mean IOP at follow-up (8 am, 10 am, and 4 pm at weeks 4 and 12) ranged from 17.7 mmHg to 18.8 mmHg with bimatoprost 0.01% and from 18.1 mmHg to 20.1 mmHg with bimatoprost 0.03%. Mean IOP was numerically lower in the bimatoprost 0.01% treatment arm at each follow-up time point (Figure 2).
 | Figure 2 Mean IOP at each time point. Error bars, standard error of the mean. |
In both treatment arms, the reduction in IOP from latanoprost-treated baseline was statistically significant at each time point at both follow-up visits (P<0.001). The mean IOP reduction from latanoprost-treated baseline ranged from 3.7±0.4 mmHg to 4.4±0.4 mmHg with bimatoprost 0.01% and from 2.8±0.5 mmHg to 3.9±0.5 mmHg with bimatoprost 0.03% and was statistically significantly larger with bimatoprost 0.01% at two of six time points (P≤0.035) and numerically larger with bimatoprost 0.01% at five of six follow-up time points (Figure 3A). The mean percentage IOP reduction from latanoprost-treated baseline ranged from 16.6% to 19.6% with bimatoprost 0.01% and from 11.6% to 16.2% with bimatoprost 0.03% and was statistically significantly larger with bimatoprost 0.01% at two of six time points (P≤0.019) and numerically larger with bimatoprost 0.01% at all six follow-up time points (Figure 3B).
 | Figure 3 Mean change (A) and mean percentage change (B) in intraocular pressure from latanoprost baseline at each time point during follow-up. Error bars, standard error of the mean. *P≤0.035 versus bimatoprost 0.03%. |
Latanoprost-treated baseline mean diurnal IOP was within 0.1 mmHg in the two studies (22.2 mmHg and 22.1 mmHg in the bimatoprost 0.01% and 0.03% treatment arms, respectively). At 4 and 12 weeks after replacement of latanoprost with bimatoprost, mean diurnal IOP was 0.7–0.9 mmHg lower in the bimatoprost 0.01% treatment arm than in the bimatoprost 0.03% treatment arm (Table 2). In accord, the mean reduction in diurnal IOP from latanoprost-treated baseline was 0.8–1.0 mmHg larger in the bimatoprost 0.01% treatment arm than in the bimatoprost 0.03% treatment arm (Table 2).
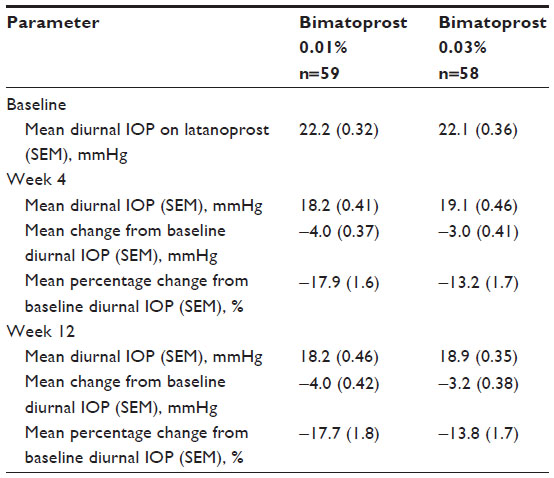 | Table 2 Diurnal intraocular pressure |
As the study eligibility criteria required IOP ≥20 mmHg after run-in on latanoprost monotherapy, no patients had diurnal IOP <18 mmHg at the latanoprost-treated baseline. After 12 weeks of bimatoprost treatment, 52.5% of patients treated with bimatoprost 0.01% and 32.1% of patients treated with bimatoprost 0.03% had diurnal IOP <18 mmHg (P=0.038, Figure 4). The percentage of patients achieving a 15% or greater decrease in diurnal IOP from latanoprost-treated baseline diurnal IOP at week 12 was 61.0% in the bimatoprost 0.01% treatment arm and 48.2% in the bimatoprost 0.03% treatment arm (P=0.193).
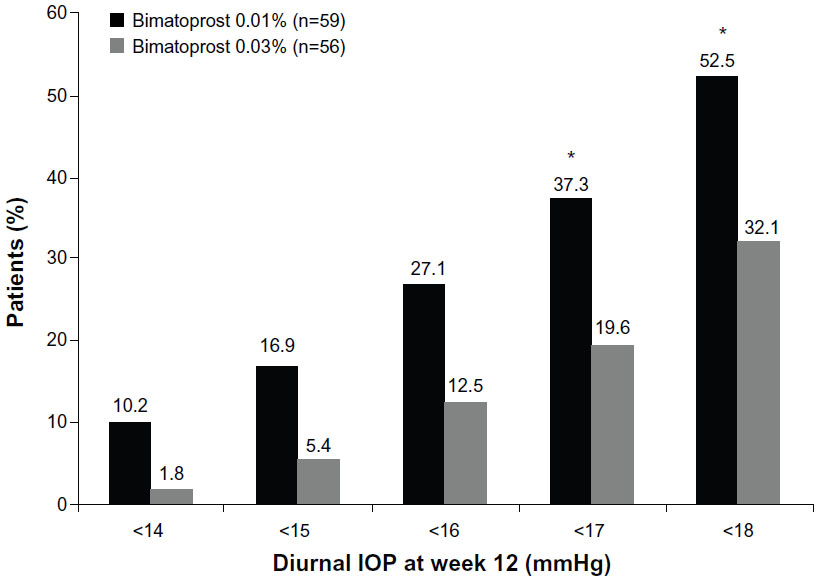 | Figure 4 Percentage of patients achieving specific diurnal intraocular pressures at week 12. *P≤0.041 versus bimatoprost 0.03%. |
Safety outcomes
All randomized patients received the assigned study treatment and were included in the safety analyses. Adverse events were reported in nine (13.4%) patients treated with bimatoprost 0.01% and eight (12.9%) patients treated with bimatoprost 0.03% (Table 3). Ocular adverse events were reported in six (9.0%) patients treated with bimatoprost 0.01% and seven (11.3%) patients treated with bimatoprost 0.03%. There were two serious adverse events (cardiac ischemia that required prolonged hospitalization and exacerbation of depression disorder) in the bimatoprost 0.01% group; both were considered to be unrelated to treatment. Because no adverse event was reported in more than one patient in either bimatoprost group (Table 3), the incidence of each adverse event in the bimatoprost 0.01% group was 1.5%, and the incidence of each adverse event in the bimatoprost 0.03% group was 1.6%.
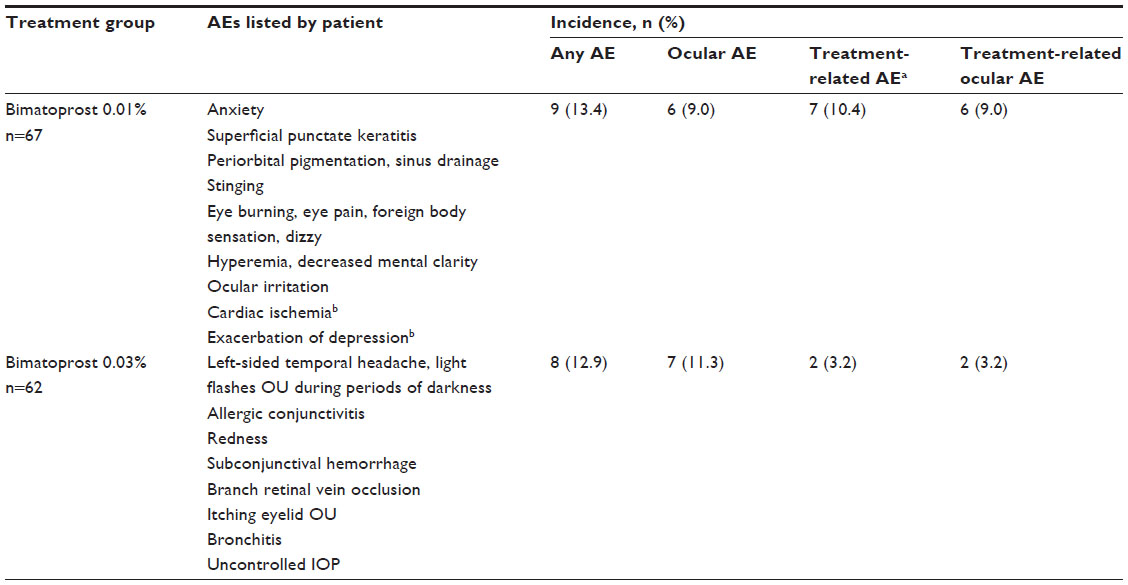 | Table 3 Adverse events |
Conjunctival hyperemia was evaluated in the study eye of patients on biomicroscopic examination. Mean scores of conjunctival hyperemia remained in the none-to-trace range with both bimatoprost 0.01% and bimatoprost 0.03% (Figure 5A). In the bimatoprost 0.01% treatment arm, the percentage of patients with mild or greater severity of conjunctival hyperemia was 9.0% at latanoprost-treated baseline, 10.9% after 4 weeks of bimatoprost 0.01%, and 8.1% after 12 weeks of bimatoprost 0.01%. In the bimatoprost 0.03% treatment arm, the percentage was 1.6% at latanoprost-treated baseline, 14.5% after 4 weeks of bimatoprost 0.03%, and 10.0% after 12 weeks of bimatoprost 0.03%. At week 12, the change from latanoprost-treated baseline in the percentage of patients with conjunctival hyperemia of mild or greater severity was −0.9% in the bimatoprost 0.01% treatment arm and +8.4% in the bimatoprost 0.03% treatment arm (Figure 5B).
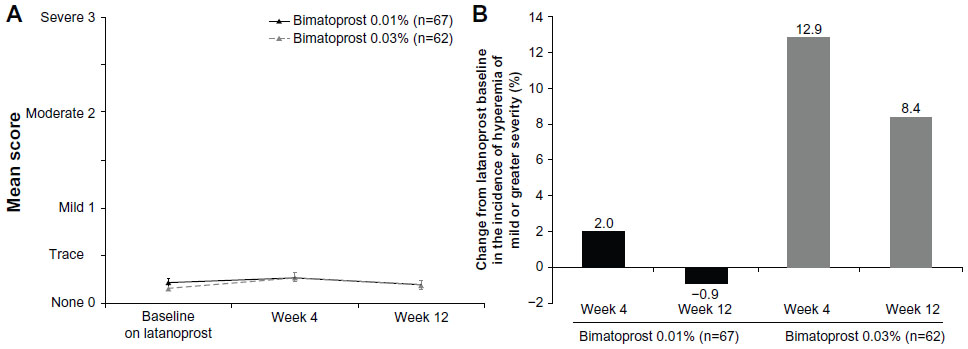 | Figure 5 Severity of conjunctival hyperemia on biomicroscopic examination. (A) Mean severity scores. (B) Change from latanoprost-treated baseline in the percentage of patients with mild or greater severity of conjunctival hyperemia. Error bars, standard error of the mean. |
Corneal staining with fluorescein in the study eye was also evaluated on biomicroscopy. In the bimatoprost 0.01% treatment arm, the percentage of patients with trace or greater severity in corneal staining was 6.0% at latanoprost-treated baseline, 6.7% after 4 weeks of bimatoprost 0.01%, and 4.8% after 12 weeks of bimatoprost 0.01%. In the bimatoprost 0.03% treatment arm, the percentage was 8.1% at latanoprost-treated baseline, 11.3% after 4 weeks of bimatoprost 0.03%, and 10.0% after 12 weeks of bimatoprost 0.03%. There were no cases of severe corneal staining in either treatment arm.
Discussion
These studies evaluated the safety and efficacy of bimatoprost 0.01% and bimatoprost 0.03% in patients on latanoprost who required additional IOP lowering. The results showed significant additional mean IOP lowering after replacement of latanoprost with either bimatoprost formulation. Both the efficacy and the safety results, however, favored the bimatoprost 0.01% formulation. Bimatoprost 0.01% provided a greater percentage reduction in IOP from latanoprost-treated baseline at all six follow-up time points and was associated with less conjunctival hyperemia and ocular staining compared with bimatoprost 0.03%. The 2010 Physician Quality Reporting Initiative from the Centers for Medicare and Medicaid Services considers intervention to be successful if it produces at least a 15% reduction in IOP from the preintervention level (AAO Preferred Practice Pattern).12 At week 12, a ≥15% reduction in IOP was achieved by 61.0% of patients who replaced latanoprost with bimatoprost 0.01% relative to 48.2% with bimatoprost 0.03%.
It is recommended that patients using a PG/PM who have inadequate IOP lowering switch within class to another PG/PM before adding other therapy.4,6 Previous studies have shown that many patients with either no response or an inadequate response to latanoprost demonstrate additional IOP lowering upon a switch to bimatoprost 0.03%.13–16 The present results are confirmatory and further suggest that patients on latanoprost who need additional IOP lowering may obtain even greater benefit from a switch to bimatoprost 0.01%. Bimatoprost 0.01% demonstrated a numerical advantage in lower mean IOP at each follow-up time point in the studies, and a majority of patients achieved diurnal IOP <18 mmHg after 12 weeks of bimatoprost 0.01% treatment. In addition, the reduced drug concentration in bimatoprost 0.01% results in improved tolerability compared with bimatoprost 0.03%.
Conjunctival hyperemia associated with PG/PM treatment typically peaks within 4 weeks of starting medication.17,18 After 4 weeks of treatment, bimatoprost 0.01% was associated with an increase in the incidence of hyperemia from the latanoprost baseline of only 2.0% compared with 12.9% for bimatoprost 0.03%. By week 12, the rate of hyperemia had improved to be below the latanoprost baseline for bimatoprost 0.01% but was maintained at an elevated level with bimatoprost 0.03%. These results suggest a more favorable tolerability profile of bimatoprost 0.01% compared with bimatoprost 0.03%, consistent with the results of head-to-head clinical comparisons of the formulations.9 Corneal staining scores also suggested a more favorable tolerability profile of bimatoprost 0.01%, as the percentage of patients with corneal staining had decreased from latanoprost-treated baseline by week 12 only in the bimatoprost 0.01% treatment arm.
In a Phase III clinical trial, bimatoprost 0.01% was associated with a lower incidence and less severe hyperemia compared with bimatoprost 0.03%.9 In contrast with the Phase III trial, all patients in the present studies were run in on latanoprost, and there was no washout prior to beginning bimatoprost treatment. Previous studies have shown that bimatoprost-related hyperemia is reduced in patients switched directly to bimatoprost from latanoprost.19 The present analysis demonstrates that there appears to be a discernible difference in the hyperemia profiles of bimatoprost 0.01% and bimatoprost 0.03% even in patients run in on latanoprost. The results are consistent with a recent observational study (the Canadian Lumigan RC Early Analysis Review [CLEAR] study) showing a significant decrease in conjunctival hyperemia in patients switched from bimatoprost 0.03% to bimatoprost 0.01%.20 Moreover, bimatoprost 0.01% was associated with a low incidence of conjunctival hyperemia even in treatment-naïve patients.21
When a patient using a single IOP-lowering medication needs additional IOP lowering, switching within PG/PM class rather than adding a second medication may allow the patient to retain the many benefits of monotherapy. These benefits include reduced risk of side effects, lower costs, and an uncomplicated treatment regimen that enhances compliance.22 The studies presented demonstrate that patients treated with latanoprost monotherapy who need additional IOP lowering can benefit from a switch to bimatoprost monotherapy, and further suggest that outcomes of a switch to bimatoprost 0.01% are most favorable.
A limitation of the present analysis is that the two bimatoprost formulations were tested in separate studies. Although the studies were almost identical in study design, and the study populations were similar in demographic characteristics and latanoprost-treated baseline diurnal IOP, differences in the study populations could have affected the results. The study population consists of patients with IOP of 20 mmHg or higher on latanoprost who may benefit from additional IOP lowering, though the historical untreated baseline IOP of patients was not taken into account. Patients who may not achieve significant IOP lowering with latanoprost, described in the literature as 4%–19% of the general population,23,24 were not specifically excluded. Also, although deemed equivalent by regulatory authorities, the latanoprost used for run-in was from two different manufacturers. There was a difference between studies in the incidence of hyperemia on latanoprost at baseline (9.0% in the bimatoprost 0.01% treatment arm compared with 1.6% in the bimatoprost 0.03% treatment arm), yet the change from baseline hyperemia, as well as the incidence of hyperemia at 12 weeks, was reduced in the bimatoprost 0.01% treatment arm compared with the bimatoprost 0.03% treatment arm. A direct head-to-head comparison study would allow further comparison of the effects of bimatoprost 0.01% and bimatoprost 0.03% in patients using latanoprost who need additional IOP lowering.
The inclusion criteria of baseline IOP ≥20 mmHg could have led to a regression to the mean in subsequent measurements. However, diurnal IOP at baseline was >22 mmHg in each study, suggesting that the IOP on latanoprost was sustained above 20 mmHg and not a chance observation. Further, the reduction from baseline IOP with bimatoprost 0.01% (mean diurnal 4.0 mmHg) and bimatoprost 0.03% (mean diurnal 3.2 mmHg) is greater than the expected fluctuation in IOP between visits in treated patients. Inadequate time on latanoprost could be argued to have undermined the measured efficacy of latanoprost. However, a 30-day run-in has been used in multiple prior studies.18,25
In summary, these studies have demonstrated that many patients treated with latanoprost who need lower IOP may achieve additional IOP lowering and maintain monotherapy by switching to bimatoprost. Of the available bimatoprost formulations, bimatoprost 0.01% had the more favorable efficacy and safety profile.
Acknowledgments
This study was sponsored by Allergan, Inc., Irvine, CA, USA. Writing and editorial assistance was provided to the authors by Kate Ivins of Evidence Scientific Solutions, Philadelphia, PA, USA, and was funded by Allergan Inc. All authors met the International Committee of Medical Journal Editors authorship criteria. Neither honoraria nor payments were made for authorship. Principal investigators and sites for study 1 were: Louis Alpern, El Paso, TX; Jason Bacharach, Petaluma, CA; James Boyce, Garden Grove, CA; Douglas Day, Atlanta, GA; El-Roy Dixon, Albany, GA; Russell Hayhurst, Austin, TX; David Hillman, Chicago, IL; Barry Katzman, San Diego, CA; Joshua Kim, Sarasota, FL; Jonathan Myers, Philadelphia, PA; Arvind Neelakantan, Dallas, TX; William Rand, Pompano Beach, FL; Ehsan Sadri, Huntington Beach, CA; Gail Schwartz, Baltimore, MD; Greg Sulkowski, Louisville, KY; Mark Weiss, Tulsa, OK; and Fiaz Zaman, Houston, TX. Principal investigators and sites for study 2 were: Louis Alpern, El Paso, TX; Douglas Day, Atlanta, GA; Barry Katzman, San Diego, CA; Blythe Monheit, Austin, TX; Parag Parekh, Boston, MA (currently Brookville, PA); Steven Rauchman, Mission Hills, CA; Benjamin Rubin, Potomac, MD; Daniel Stegman, Clifton, NJ; Steven Vold, Rogers, AR; Ruth Williams, Wheaton, IL; and Fiaz Zaman, Houston, TX.
Disclosure
JSM is a consultant for Allergan, Inotek, and Sucampo, is on the speaker’s bureaus for Alcon, Allergan, Haag Streit, New World Medical, and Sucampo, and has received research funds from Alcon, Allergan, Diopsys, Glaukos, and Inotek. SV is a consultant for and has received research funds from Aeon, Alcon, AqueSys, ForSight Labs, Glaukos Corporation, InnFocus, Iridex, iScience Interventional, Ivantis, and Transcend Medical, is a consultant for Carl Zeiss Meditec and NeoMedix, is on the speaker’s bureau for and has received research funds from Allergan, is on the speaker’s bureau for Merck and Co, has received research funds from Calhoun Vision, Solx, and Bausch and Lomb, is an investor and consultant for Truevision Systems, and is an investor and has received product royalties for Ocunetics. FZ is a consultant for Allergan and has received research funds from Alcon, Allergan, Bausch and Lomb, and Merck. JMW and DAH are employees of Allergan.
References
McKee HD, Gupta MS, Ahad MA, Saldana M, Innes JR. First-choice treatment preferences for primary open-angle glaucoma in the United Kingdom. Eye (Lond). 2005;19(8):923–924. | |
van der Valk R, Webers CA, Lumley T, Hendrikse F, Prins MH, Schouten JS. A network meta-analysis combined direct and indirect comparisons between glaucoma drugs to rank effectiveness in lowering intraocular pressure. J Clin Epidemiol. 2009;62(12):1279–1283. | |
Hommer A. A review of preserved and preservative-free prostaglandin analogues for the treatment of open-angle glaucoma and ocular hypertension. Drugs Today (Barc). 2010;46(6):409–416. | |
European Glaucoma Society. Terminology and Guidelines for Glaucoma. 2008. Available from: http://www.eugs.org/eng/EGS_guidelines.asp. Accessed February 7, 2014. | |
Law SK, Song BJ, Fang E, Caprioli J. Feasibility and efficacy of a mass switch from latanoprost to bimatoprost in glaucoma patients in a prepaid Health Maintenance Organization. Ophthalmology. 2005;112(12):2123–2130. | |
Coleman AL. Evidence-based management of glaucoma: recommendations of an expert panel. In: Glaucoma Disease Management Guide. PDR. 1st ed. Montvale, NJ, USA: Thomson PDR; 2004. | |
Aptel F, Cucherat M, Denis P. Efficacy and tolerability of prostaglandin analogs: a meta-analysis of randomized controlled clinical trials. J Glaucoma. 2008;17(8):667–673. | |
Kymes SM, Burk C, Feinman T, Williams JM, Hollander DA. Demonstration of an online tool to assist managed care formulary evidence-based decision making: meta-analysis of topical prostaglandin analog efficacy. Ther Clin Risk Manag. 2011;7:283–290. | |
Katz LJ, Cohen JS, Batoosingh AL, Felix C, Shu V, Schiffman RM. Twelve-month, randomized, controlled trial of bimatoprost 0.01%, 0.0125%, and 0.03% in patients with glaucoma or ocular hypertension. Am J Ophthalmol. 2010;149(4):661–671. e661. | |
Allergan. A Safety and Efficacy Study of ALPHAGAN® P and LUMIGAN® in Subjects Previously Treated With Latanoprost for Glaucoma and Ocular Hypertension. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2012 [updated October 7, 2013]. Available from http://clinicaltrials.gov/ct2/show/NCT01525173. NLM identifier: NCT01525173. Accessed March 3, 2014. | |
Allergan. Comparing Safety and Efficacy of Combigan® and Lumigan® With Lumigan® Alone in Glaucoma or Ocular Hypertension Subjects Treated With Xalatan®. In: ClinicalTrials.gov [website on the Internet]. Bethesda, MD: US National Library of Medicine; 2010 [updated December 7, 2011]. Available from http://clinicaltrials.gov/ct2/show/NCT01170884?term=NCT01170884&rank=1. NLM identifier: NCT01170884. Accessed March 3, 2014. | |
American Academy of Ophthalmology. Primary Open-Angle Glaucoma. Preferred Practice Guidelines. San Francisco, CA: American Academy of Ophthalmology; 2010. | |
Williams RD. Efficacy of bimatoprost in glaucoma and ocular hypertension unresponsive to latanoprost. Adv Ther. 2002;19(6):275–281. | |
Gandolfi SA, Cimino L. Effect of bimatoprost on patients with primary open-angle glaucoma or ocular hypertension who are nonresponders to latanoprost. Ophthalmology. 2003;110(3):609–614. | |
Bournias TE, Lee D, Gross R, Mattox C. Ocular hypotensive efficacy of bimatoprost when used as a replacement for latanoprost in the treatment of glaucoma and ocular hypertension. J Ocul Pharmacol Ther. 2003;19(3):193–203. | |
Kammer JA, Katzman B, Ackerman SL, Hollander DA. Efficacy and tolerability of bimatoprost versus travoprost in patients previously on latanoprost: a 3-month, randomised, masked-evaluator, multicentre study. Br J Ophthalmol. 2010;94(1):74–79. | |
Higginbotham EJ, Schuman JS, Goldberg I, et al. One-year, randomized study comparing bimatoprost and timolol in glaucoma and ocular hypertension. Arch Ophthalmol. 2002;120(10):1286–1293. | |
Crichton AC, Vold S, Williams JM, Hollander DA. Ocular surface tolerability of prostaglandin analogs and prostamides in patients with glaucoma or ocular hypertension. Adv Ther. 2013;30(3):260–270. | |
Kurtz S, Mann O. Incidence of hyperemia associated with bimatoprost treatment in naive subjects and in subjects previously treated with latanoprost. Eur J Ophthalmol. 2009;19(3):400–403. | |
Crichton AC, Nixon DR, Simonyi S, et al. An observational study of bimatoprost 0.01% in patients on prior intraocular pressure-lowering therapy: the Canadian Lumigan RC Early Analysis Review (CLEAR) trial. Clin Ophthalmol. 2013;7:1–8. | |
Nixon DR, Simonyi S, Bhogal M, et al. An observational study of bimatoprost 0.01% in treatment-naive patients with primary open angle glaucoma or ocular hypertension: the CLEAR trial. Clin Ophthalmol. 2012;6:2097–2103. | |
Whitson JT. Glaucoma: a review of adjunctive therapy and new management strategies. Expert Opin Pharmacother. 2007;8(18):3237–3249. | |
Rossetti L, Gandolfi S, Traverso C, et al. An evaluation of the rate of nonresponders to latanoprost therapy. J Glaucoma. 2006;15(3):238–243. | |
Sakurai M, Higashide T, Takahashi M, Sugiyama K. Association between genetic polymorphisms of the prostaglandin F2alpha receptor gene and response to latanoprost. Ophthalmology. 2007;114(6):1039–1045. | |
Stewart WC, Day DG, Sharpe ED, Dubiner HB, Holmes KT, Stewart JA. Efficacy and safety of timolol solution once daily vs timolol gel added to latanoprost. Am J Ophthalmol. 1999;128(6):692–696. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.