")
Back to Journals » Clinical Ophthalmology » Volume 8
Bilateral papillopathy as a presenting sign of pheochromocytoma associated with von Hippel–Lindau disease
Authors Shah V, Zlotcavitch L, Herro A, Dubovy S, Yehoshua Z, Lam BL
Received 15 January 2014
Accepted for publication 12 February 2014
Published 26 March 2014 Volume 2014:8 Pages 623—628
DOI https://doi.org/10.2147/OPTH.S60725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Veeral Shah, Leonid Zlotcavitch, Angela M Herro, Sander R Dubovy, Zohar Yehoshua, Byron L Lam
Bascom Palmer Eye Institute, University of Miami, Miller School of Medicine, Miami, FL, USA
Abstract: A 7-year-old girl presented with decreased vision in both eyes for 1 month. Examination showed visual acuity of 20/50 and 20/60, no afferent pupillary defect, cecocentral scotomas, and bilateral optic disc edema with extensive peripapillary and macular exudates. Magnetic resonance imaging showed multiple cortical and subcortical white matter lesions. Both the laboratory workup and the systemic examination were unrevealing. However, on follow-up, the patient showed episodic elevations of blood pressure as high as 240/160. Further workup revealed elevated urine catecholamines and a right supra-adrenal mass proven to be a pheochromocytoma by histopathologic analysis. The paroxysmal hypertension resolved, and the visual acuity, visual fields, fundus exam, and neuroimaging improved. The patient was lost to follow-up until age 18 when she developed shortness of breath and was found to have multiple pulmonary metastases identified as pheochromocytoma by biopsy. Genetic testing identified a 3p25-26 (c.482 G>A) VHL gene chromosomal mutation consistent with von Hippel–Lindau disease genotype. Multiple peripheral retinal vascular dilations and small retinal capillary hemangioblastomas were also found. This case highlights the importance of recognizing the lability of blood pressure often seen with pheochromocytomas, which may mask the underlying cause of hypertensive papillopathy and retinopathy, a diagnosis of low clinical suspicion in the pediatric population. The case also underscores the importance of thorough systemic workup, including genotyping to detect conditions where pheochromocytoma may be the presenting sign of the disease, such as multiple endocrine neoplasia 2A and 2B, von Hippel–Lindau disease, von Recklinghausen disease, tuberous sclerosis, and Sturge–Weber syndrome.
Keywords: hypertensive encephalopathy, VHL, pheochromocytoma, paroxysmal hypertension
Introduction
von Hippel–Lindau (VHL) disease is an autosomal dominant disorder that is characterized by multisystem predilection to develop tumors and cysts.1–3 The most common tumors manifested include retinal and central nervous system hemangioblastomas, renal cell carcinoma, pheochromocytoma, and pancreatic cystadenoma.1,2 Mean age at diagnosis is the mid-20s, often with retinal angioma as the first manifestation. When pheochromocytoma is the presenting tumor, diagnosis may be more difficult or missed, as often the first symptoms are labile hypertension, anxiety, and palpitations that can be confused for other diagnoses. We report a child with bilateral hypertensive papillopathy as the presenting feature of pheochromocytoma related to VHL disease, which – to our knowledge – has not been previously reported.
Case report
An otherwise healthy 7-year-old girl was evaluated for 1 month of bilateral vision loss. She reported no history of headaches, eye pain, neurological deficits, or other associated symptoms. Her past medical history and family history were negative for eye disease, renal cell carcinoma, or neurologic syndromes. She was on no medications and had no allergies.
The patient was in no distress, and her screening vital signs were normal with a blood pressure of 110/70 mmHg and a heart rate of 95 bpm. Best-corrected visual acuity was 20/50 in the right eye and 20/60 in the left eye, with a normal pupillary examination and intact color vision. Ocular motility and intraocular pressures were normal. Automated visual field testing (Humphrey 30-2 threshold, Swedish Interactive Threshold Algorithm-standard strategy; Carl Zeiss Meditec, Dublin CA, USA) demonstrated bilateral enlarged blind spots and cecocentral scotomas (Figure 1). Fundoscopy showed diffuse bilateral optic nerve head edema with extensive circumferential peripapillary and macular exudation. The optic nerve heads had no spontaneous venous pulsations (Figure 2). The neurologic examination was unremarkable.
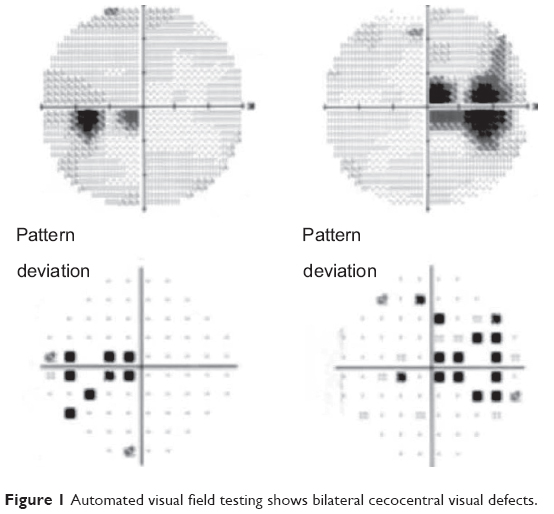 | Figure 1 Automated visual field testing shows bilateral cecocentral visual defects. |
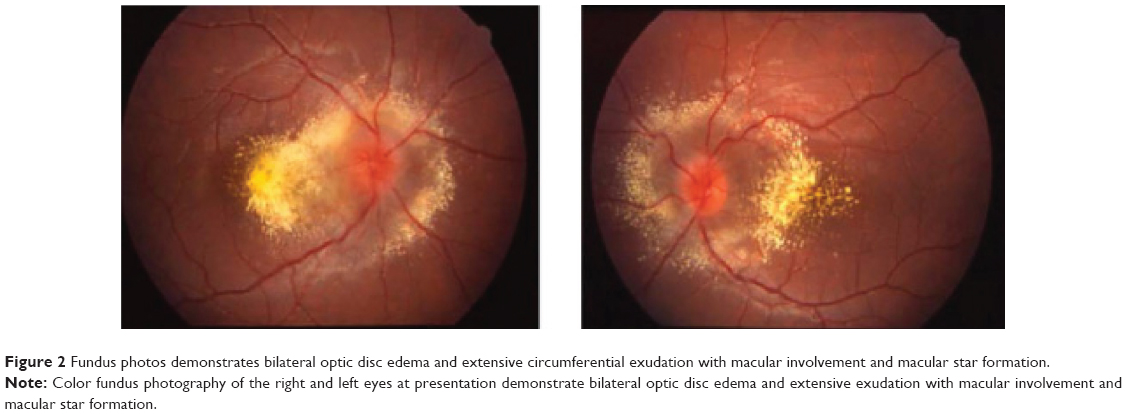 | Figure 2 Fundus photos demonstrates bilateral optic disc edema and extensive circumferential exudation with macular involvement and macular star formation. |
The brain magnetic resonance imaging (MRI) with and without contrast revealed multiple high T2 and fluid-attenuated inversion–recovery (FLAIR) signal lesions found diffusely in the cortical and subcortical white matter region of the frontal and parietal lobes (Figure 3A). One lesion in the left frontal posterior frontal lobe had higher T1 signaling, and a computed tomography (CT) scan confirmed calcifications of this single lesion (Figure 3C and D). A lumbar puncture revealed an opening pressure of 150 mm water with normal cerebrospinal fluid cytology and analysis: red blood cells =1; white blood cell =0; protein =20 mg/dL; glucose =71 mg/dL; immunoglobulin G =1.76 mg/dL; no oligoclonal bands; and no growth in cultures.
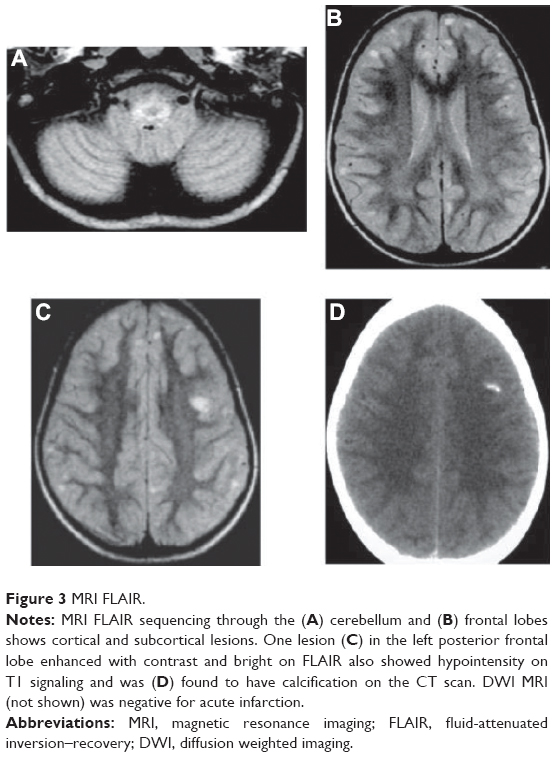 | Figure 3 MRI FLAIR. |
Three days later on follow-up examination, the patient was found to have paroxysmal tachycardia with a heart rate of 180 bpm and blood pressure of 120/75 mmHg. Repeat blood pressure measurements showed episodic elevation in blood pressure reaching as high as 240/160 mmHg. The patient was treated medically for hypertension. In an extensive workup, an abdominal CT scan showed a 3.5×3.0×3.0 cm enhancing noncalcified mass in the right adrenal gland (Figure 4).
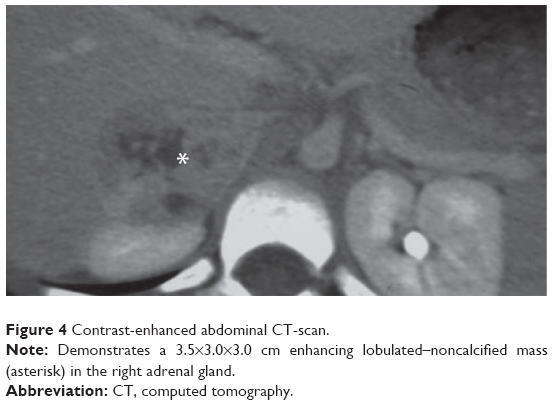 | Figure 4 Contrast-enhanced abdominal CT-scan. |
Laboratory analysis of the patient’s 24-hour urine collection showed elevated vanillylmandelic acid of 15.1 mg (normal <2.3 mg) and random total metanephrines of 22,295 μg/g of creatinine (normal 202–750 ug/g). A scintigraphic distribution of iodine-131-meta-iodobenzylguanidine (I-131 MIBG), an adrenal medullary imaging agent, showed increased radioactive I-MIBG uptake by the right adrenal mass but the absence of pathologic radioactivity elsewhere, and the patient underwent laparoscopic resection of the mass. The pathological specimen showed whorled nests of chromaffin tumor cells (Figure 5) embedded in a fibrovascular stroma. These histopathological findings were consistent with pheochromocytoma.
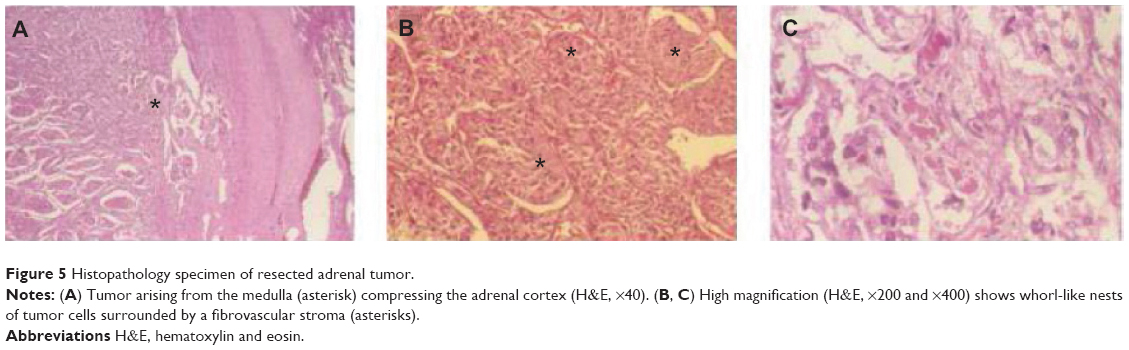 | Figure 5 Histopathology specimen of resected adrenal tumor. |
Postoperatively, the episodic hypertensive episodes resolved and vanillylmandelic acid/metanephrines normalized. After 5 months, the patient’s visual acuity improved to 20/30 in both eyes. Fundoscopically, the optic disc edema and exudates improved, and the postoperative MRI showed a subsequent decrease in white matter lesions (Figure 6).
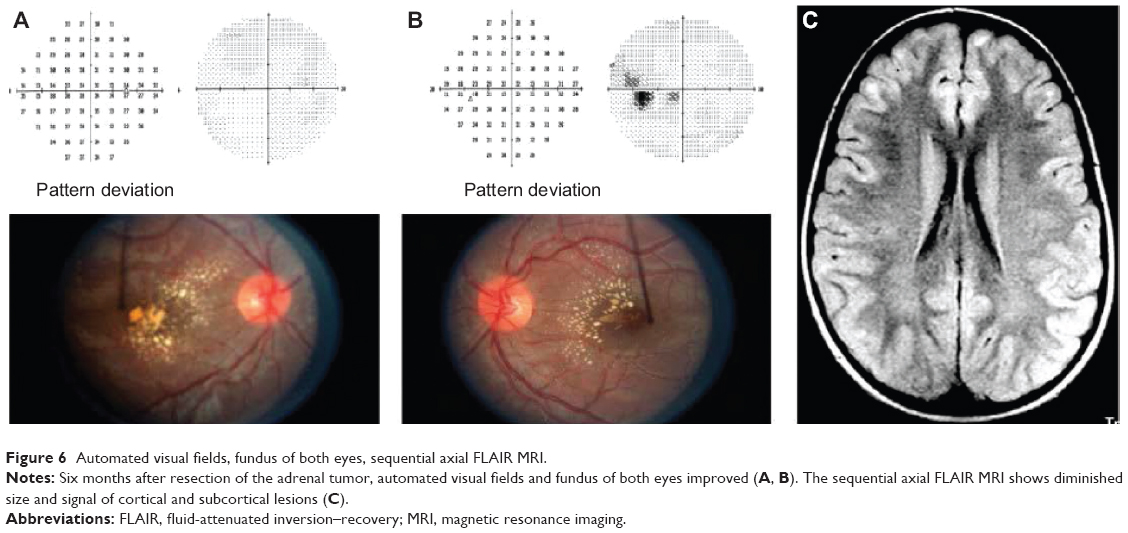 | Figure 6 Automated visual fields, fundus of both eyes, sequential axial FLAIR MRI. |
The patient was lost to follow-up for many years and returned at the age of 18 when she developed shortness of breath over 4 days. She was found to have multiple pulmonary lesions bilaterally; a biopsy showed metastatic pheochromocytoma. Blood pressure at that time was well-controlled on atenolol; however, serum metanephrines were elevated. She underwent serial thoracotomies for resection of the lesions, and she then started on chemotherapy with sunitinib for 12 months. Multiple follow-up metaiodobenzylguanidine (MIBG) scans and urine collections have been negative for metastatic pheochromocytoma. Subsequent genetic testing identified a 3p25-26 (c.482 G>A) chromosomal mutation consistent with a mutation of the VHL gene. RET proto-oncogene sequencing detected no mutation in exons 10, 11, 13, 14, 15, and 16.
The patient’s family history was negative for neuroendocrine disorders, including multiple endocrine neoplasia 2A and 2B, VHL disease, von Recklinghausen disease, tuberous sclerosis, and Sturge–Weber syndrome. Recommendations were made to have family members tested; however, the patient declined as the family lived out of the country. Ophthalmic evaluation at this time showed multiple peripheral vascular dilations and small retinal capillary hemangioblastomas (Figure 7). Repeat imaging and follow-up over a year showed her to be stable without evidence of brain or spinal cord hemangioblastomas, or abdominal pathology, which is concerning for renal cell carcinoma.
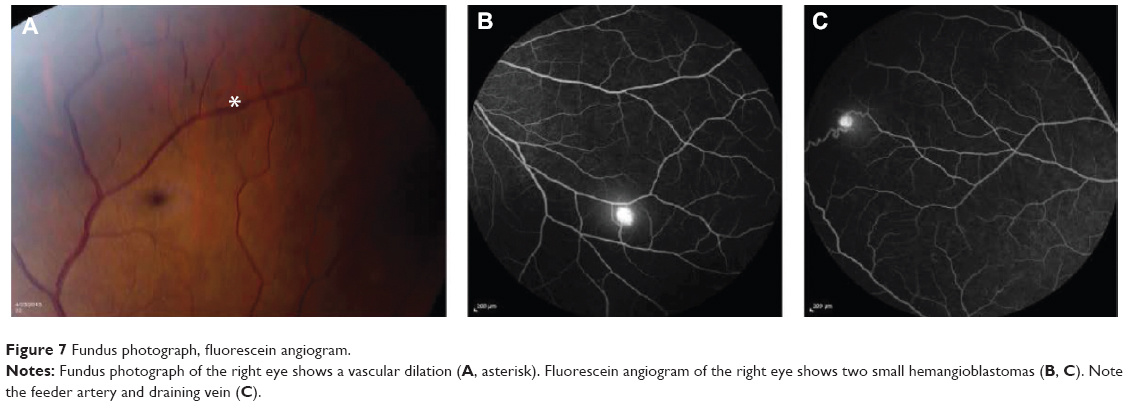 | Figure 7 Fundus photograph, fluorescein angiogram. |
Discussion
Bilateral disc edema in a child may be quite diagnostically challenging, particularly when neuroimaging is negative, and intracranial hypertension is not found. Other potential diagnoses includes: blood dyscrasias; hypertensive papillopathy; neuroretinitis; and anterior ischemic optic neuropathy.4,5 In our case, the appearance of the optic disc edema without the obscuration of the vessels at the disc margin – along with the marked circumferential peripapillary and macular exudations – lowered the clinical suspicion for intracranial hypertension, and the lumbar puncture opening pressure was normal. Our patient also had normal complete blood count, negative inflammatory/infectious laboratory studies, and normal cerebrospinal fluid analysis. However, when the patient presented with tachycardia, serial blood pressure measurements revealed the etiology as malignant hypertension. Blood pressure elevation in the pediatric population to this degree is likely due to renal artery stenosis or pheochromocytoma.6 Given the lability of her vital signs, a pheochromocytoma was favored and eventually found on CT imaging of the abdomen.7
Our case is not the first case reported of pheochromocytoma presenting as bilateral disc edema or macular star formation.8–10 In a few pediatric and adult reported cases, the optic nerve findings were the presenting symptoms, often also linked to a history of headaches, tachycardia, and other signs of elevated catecholamines.8,10 However, to our knowledge, there are no reported cases of bilateral disc edema as the presenting sign of VHL disease, where up to 20% of these patients have pheochromocytoma. The patient was lost to follow-up, and her VHL disease was not diagnosed until many years later with metastatic pheochromocytoma. Had she continued follow-up as suggested, the VHL disease would likely have been discovered earlier. Thorough systemic workup including genotyping to detect conditions where pheochromocytoma may be the presenting sign of the disease, such as multiple endocrine neoplasia 2A and 2B, VHL disease, von Recklinghausen disease, tuberous sclerosis, and Sturge–Weber syndrome, should be considered and undertaken in all patients with pheochromocytoma.
Pheochromocytoma is often not familial, arising from a germline mutation. However, pheochromocytoma is associated with several familial syndromes, including multiple endocrine neoplasia 2A and multiple endocrine neoplasia 2B, VHL disease, von Recklinghausen disease, tuberous sclerosis, and Sturge–Weber syndrome. Further workup should include RET proto-oncogene testing, bone scan, dermatologic examination, and genetic testing and consultation to rule out these disorders.11,12 Although this extensive workup was initially negative, our patient had one calcified cortical lesion seen on CT scan and MRI, which is a minor feature but not a pathognomonic sign of tuberous sclerosis and without any other characteristic signs or symptoms, this diagnosis was not entertained further.13
At the time the patient first presented in 2002, VHL molecular genetic analysis was not yet readily available. The patient was then lost to follow-up for 11 years, and subsequent genetic testing identified the VHL mutation.
Pheochromocytoma is a rare neuroendocrine tumor arising from neoplastic chromaffin cells found in the adrenal medulla and is referred to as paraganglioma when found in extra-adrenal tissues.14 Pheochromocytoma is characterized by the clinical triad of episodic headaches, tachycardia, and sweating; it can lead to fatal hypertensive crisis, myocardial infarction, and arrhythmias.15 Pheochromocytoma presents as sustained hypertension 80%–90% of the time in children, but blood pressure may fluctuate widely.15 Additionally, up to 25% of pediatric pheochromocytoma may be associated with a hereditary component.
The onset of the episodic severe hypertension and disc edema in our case is unknown. Ocular changes may be the initial finding in an asymptomatic patient with hypertension, necessitating a primary care referral. In other cases, such as our own, the visual symptoms are what prompt the patient to seek ophthalmic care.16 Hayreh et al17 through many years of clinical studies and experimental models of hypertension, has described the clinical findings and pathophysiology of the effect of hypertension on the eye.
Because of the vascular differences between the retina, choroid, and optic nerve, each of these anatomic regions responds differently to hypertension.16 The narrowing of the arterioles classically described in hypertensive retinopathy is generally seen after sustained hypertension, particularly with diastolic pressure elevation. The acute changes in the retina with focal intraretinal periarteriolar transudates may be related to the breakdown of autoregulatory mechanisms after an acute rise in blood pressure and are a result of the breakdown of the blood–retinal barrier.17,18
These transudates were also seen in our patient, along with the evidence of a parallel process occurring in the brain, as evidenced by the increased T2 signal in the cortical matter seen on MRI. Hypertensive encephalopathy may produce increased intracranial pressure leading to papilledema, but this did not occur in our case because the lumbar puncture opening pressure was normal.
The blood supply to the optic disc, in contrast, arises via the posterior ciliary arteries and peripapillary choroidal vessels. These vessels are particularly susceptible as they are not governed by the autoregulation principles of retinal arterioles, and the edema is not due to increased blood flow but rather axoplasmic stasis.19 Fluid leakage contributes to axonal swelling, followed by axonal degeneration and gliosis.18 Furthermore, the choroidal vasculature lacks the endothelial tight junctions found in the retina and are open to circulating vasoconstrictors, causing decreased perfusion to the optic nerve head.19
Our case demonstrates that pheochromocytoma may initially present as bilateral papillopathy with a negative initial workup including normal vital signs. This case emphasizes the importance of having a high index of suspicion for paroxysmal hypertension, and repeat blood pressure/heart rate measurements in patients with signs of hypertensive papillopathy.20 This case also highlights the difficulty in diagnosing VHL disease in pediatric patients. VHL disease is an autosomal dominant disease found in approximately 1 in 36,000 live births in the US and is characterized by retinal/central nervous system hemangiomas, renal cell carcinomas, pheochromocytoma, and other neuroendocrine tumors; the manifestations and severity of these tumors are highly variable.1,2,21 VHL disease is only known to be caused by the mutations of the VHL gene on chromosome 3p25-26, and genetic testing can identify 90%–100% of affected individuals. VHL is an autosomal dominant disease; however, an estimated 20% have spontaneous de novo VHL gene mutations.3
Molecular genetic testing is recommended for all individuals with known or suspected VHL disease and all individuals with a single VHL-associated tumor and negative history like our patient. Once the diagnosis is established, surveillance evaluation includes: neurological exam with baseline brain/spine imaging; ophthalmic exam; audiological exam; abdominal ultrasound; blood pressure; and genetic consultation.21 Early identification can significantly reduce morbidity and mortality related to this disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel–Lindau disease. Q J Med. 1990;77(283):1151–1163. | ||
Lonser RR, Glenn GM, Walther M, et al. von Hippel–Lindau disease. Lancet. 2003;361(9374):2059–2067. | ||
Singh AD, Shields CL, Shields JA. von Hippel–Lindau disease. Surv Ophthalmol. 2001;46(2):117–142. | ||
Leavitt JA, Pruthi S, Morgenstern BZ. Hypertensive retinopathy mimicking neuroretinitis in a twelve-year-old girl. Surv Ophthalmol. 1997;41(6):477–480. | ||
Ghauri RR, Lee AG. Optic disk edema with a macular star. Surv Ophthalmol. 1998;43(3):270–274. | ||
Browning AC, Mengher LS, Gregson RM, Amoaku WM. Visual outcome of malignant hypertension in young people. Arch Dis Child. 2001;85(5):401–403. | ||
Francis IR, Korobkin M. Pheochromocytoma. Radiol Clin North Am. 1996;34(6):1101–1112. | ||
I-Linn ZL, Long QB. An unusual case of bilateral optic disk swelling with macular star in a 9-year-old girl. J Pediatr Ophthalmol Strabismus. 2007;44(4):245–247. | ||
Ba-Abbad RA, Nowilaty SR. Bilatreral optic disc swelling as the presenting sign of pheochromocytoma in a child. Medscape J Med. 2008;10(7):176. | ||
Pushpakumari P, Nampoory MR, Vaidyanathan S, Vijayalakshmi A, Visweswaran RK. Bilateral pheochromocytoma with papilloedema. J Assoc Physicians India. 1988;36(12):738–739. | ||
Iliopoulos O, Eng C. Genetic and clinical aspects of familial renal neoplasms. Semin Oncol. 2000;27(2):138–149. | ||
Perel Y, Schlumberger M, Marguerite G. Pheochromocytoma and paraganglioma in children: a report of 24 cases of the French Society of Pediatric Oncology. Pediatr Hematol Oncol. 1997;14(5): 413–422. | ||
Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13(12):624. | ||
Tibbetts MD, Wise R, Forbes B, Hedrick HL, Levin AV. Hypertensive retinopathy in a child caused by pheochromocytoma: identification after a failed school vision screening. J AAPOS. 2012;16(1):97–99. | ||
Dluhy RG. Pheochromocytoma–death of an axiom. N Engl J Med. 2002;346(19):1486–1488. | ||
Oh KT, Moinfar N. Medscape: Ophthalmologic Manifestations of Hypertension. 2012. Available from: http://emedicine.medscape.com/article/1201779-overview. Accessed September 16, 2013. | ||
Hayreh SS, Servais GE, Virdi PS. Fundus lesions in malignant hypertension. V. Hypertensive optic neuropathy. Ophthalmology. 1986;93(1):74–87. | ||
Kishi S, Tso MO, Hayreh SS. Fundus lesions in malignant hypertension. II. A pathologic study of experimental hypertensive optic neuropathy. Arch Ophthalmol. 1985;103(8):1198–1206. | ||
Lee AG, Beaver HA. Acute bilateral optic disk edema with a macular star figure in a 12-year-old girl. Surv Ophthalmol. 2002;47(1):42–49. | ||
Sheps SG, Jiang NS, Klee GG, van Heerden JA. Recent developments in the diagnosis and treatment of pheochromocytoma. Mayo Clin Proc. 1990;65(1):88–95. | ||
Frantzen C, Links TP, Giles RH. Von Hippel–Lindau Disease [webpage on the Internet]. Seattle: University of Washington, Seattle; 2000 [updated June 21, 2012]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1463/. Accessed September 15, 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.