Back to Journals » Journal of Inflammation Research » Volume 19
Bidirectional Crosstalk Between Intestinal Epithelium and Immune Microenvironment in Inflammatory Bowel Disease: Mechanisms and Therapeutic Implications
Authors Shen J, Du S, Zhang Y, Li H ![]() , Liu X
, Liu X ![]() , Jing J
, Jing J
Received 8 May 2025
Accepted for publication 18 November 2025
Published 8 January 2026 Volume 2026:19 538988
DOI https://doi.org/10.2147/JIR.S538988
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Jinghan Shen,* Simin Du,* Yuyue Zhang, HongKun Li, XingYan Liu, Jie Jing
School and Hospital of Stomatology, Zunyi Medical University, Zunyi, Guizhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jie Jing, School and Hospital of Stomatology, Zunyi Medical University, Zunyi, Guizhou, People’s Republic of China, Email [email protected]
Abstract: Inflammatory bowel disease (IBD), encompassing Crohn’s disease and ulcerative colitis, is characterized by chronic mucosal inflammation driven by dysregulated interactions between intestinal epithelial cells (IECs) and immune components. This review systematically explores the dynamic interplay between epithelial barrier integrity and immune-microenvironmental regulation in IBD pathogenesis. We highlight the dual roles of innate immunity (neutrophils, macrophages, dendritic cells, and innate lymphoid cells) and adaptive immunity (Th1, Th17, and Treg cells) in orchestrating inflammatory cascades and mucosal repair. It also describes the interaction between microbial metabolites and the intestinal microenvironment.Key mechanisms include neutrophil extracellular trap (NET)-mediated epithelial damage, macrophage polarization modulated by ROS/NOX4 signaling, and IL-22/STAT3-driven epithelial regeneration. Additionally, we dissect the Wnt/β-catenin and bile acid-TGR5 (Takeda G-protein-coupled receptor 5) pathways in intestinal stem cell renewal. Emerging therapeutic strategies targeting epithelial-immune axes, such as anti-IL-23/IL-17 biologics and MSC-derived exosomes, are critically evaluated. By integrating recent advances in single-cell omics and preclinical models, this review underscores the necessity of precision medicine approaches to restore immune-epithelial homeostasis. This paper also introduces the current application of organoids—a novel emerging technology—in experimental research.Future research should prioritize spatial-temporal mapping of cellular interactions and leverage organoids to advance translational validation of dual-target therapies to bridge mechanistic insights into clinical practice.
Keywords: intestinal epithelial cells, inflammatory, innate immunity, adoptive immunity, pathogenesis
Introduction
IBD, encompassing CD and UC, represents a spectrum of chronic inflammatory intestinal disorders with distinct pathological features.1 CD predominantly affects the terminal ileum and colon, demonstrating transmural inflammation that may involve the entire gastrointestinal tract. The etiopathogenesis involves complex interactions between genetic susceptibility and dysregulated immune responses. In contrast, UC is characterized by continuous superficial inflammation limited to the colonic mucosa and rectum, with autoimmune mechanisms targeting intestinal epithelial components.2 Clinical observations reveal significant comorbidity between IBD and extraintestinal immune-mediated conditions including psoriasis, ankylosing spondylitis, primary sclerosing cholangitis, and oral mucosal lesions,3,4 underscoring the systemic nature of immune dysregulation in IBD pathophysiology.
Contemporary investigations highlight the pivotal role of both innate and adaptive immunity in perpetuating IBD-related inflammation. While historical research focused on aberrant T-cell responses as disease drivers, current paradigms emphasize synergistic contributions from innate immune mechanisms. CD pathogenesis typically involves Th1-polarized responses, whereas UC demonstrates atypical Th2-mediated pathology. Emerging evidence substantiates the critical involvement of Th17 cell-derived inflammatory mediators across IBD subtypes.5
The IBD microenvironment demonstrates profound imbalance between pro-inflammatory Th1/Th17 cell populations and regulatory T-cell (Treg) subsets, facilitating macrophage activation, plasma cell differentiation, and neutrophil infiltration into intestinal mucosa - pathophysiological hallmarks contributing to tissue destruction.6
Intestinal epithelial architecture comprises four principal cell lineages: nutrient-absorbing enterocytes, mucin-secreting goblet cells, antimicrobial peptide-producing Paneth cells, and hormone-releasing enteroendocrine cells. This cellular consortium collaborates with the stratified mucus layer to maintain physicochemical barriers against luminal pathogens. Post-injury epithelial regeneration relies on intestinal stem cells (ISCs) localized at crypt bases, which orchestrate mucosal homeostasis through controlled proliferative activity.
The ISC niche constitutes a complex microenvironment containing epithelial derivatives, stromal components, and smooth muscle elements, enriched with paracrine growth factors and extracellular matrix proteins. Core regulatory mechanisms involve Wnt3 ligand signaling, epidermal growth factor (EGF) receptor activation, and bone morphogenetic protein (BMP) inhibition, which collectively modulate stem cell quiescence-proliferation dynamics and tissue repair processes.7
IBD pathogenesis fundamentally involves compromised mucosal barrier integrity that disrupts intestinal immune homeostasis. This comprehensive review systematically analyzes the dynamic interplay between epithelial compartment constituents and immune microenvironment modulators during innate and adaptive immune activation cascades.
Intestinal Physiology
The gastrointestinal mucosa comprises diverse cellular constituents: absorptive enterocytes, mucin-secreting goblet cells, antimicrobial Paneth cells, stromal mesenchymal cells, and immunocompetent cell populations. A continuous epithelial barrier connected via intercellular junction complexes integrates resident immunological components, with secretory goblet cells and Paneth cells strategically positioned within this structural framework. The subepithelial lamina propria contains mesenchymal derivatives encompassing fibroblasts, contractile myofibroblasts, and perivascular pericytes. Functionally, this polarized epithelium mediates selective nutrient absorption while maintaining strict compartmentalization against luminal contents.8,9 Mucosal defense mechanisms employ synergistic interactions between the glycocalyx-enriched mucus layer, epithelial tight junction complexes, and commensal microbiota, all modulated through intricate immune system signaling.10 Dynamic bidirectional communication occurs between epithelial components and both microbial consortia and immune mediators.
Maintenance of intestinal barrier integrity relies on perpetual epithelial renewal driven by crypt-residing ISCs. These multipotent progenitor cells give rise to distinct epithelial lineages that undergo coordinated migration along crypt-villus axes in the small intestine or colonic mucosal surfaces.11 Regulatory control of ISC proliferation and differentiation involves paracrine signaling from adjacent stromal niche components, including epithelial progeny and mesenchymal cells. Contemporary research highlights immune cell populations as pivotal regulators of mucosal homeostasis and tissue regeneration.6,12
Environmental challenges activate niche cellular communication networks that induce cytokine and growth factor secretion, initiating ISC-dependent repair mechanisms. Multiple stromal niche-derived signaling pathways have been characterized as essential modulators of stem cell kinetics and mucosal restoration processes.13
The gut-associated lymphoid tissue (GALT) and lamina propria (LP) maintain organized immunological networks. The LP compartment contains both innate and adaptive immune effectors, with T lymphocytes accounting for approximately 30% of resident cells. CD4+ TCR-positive cells constitute the predominant mucosal T cell population, reflecting systemic circulation patterns. Homeostatic balance among effector T cell subsets preserves intestinal immune tolerance, with functional dysregulation demonstrating strong correlation with pathological progression.14 Dendritic cells and tissue-resident macrophages populate the healthy LP, orchestrating bacterial antigen presentation to lymphocytes and IgA synthesis. Phagocytic macrophages execute microbial clearance while maintaining immunoregulatory capacity. Under physiological conditions, T cell activation is restrained by Treg populations secreting IL-10, which suppresses pro-inflammatory cytokine production (IL-12/IL-23) and pathogenic Th17 responses.15 IL-10 deficiency preferentially activates Th17 pathways, thereby increasing colitis susceptibility.
While essential for antimicrobial defense and neoplastic surveillance, intestinal immunity requires precise regulation to prevent aberrant responses against self-antigens and commensal microorganisms. Non-immune epithelial components engage in crosstalk with immunological elements to mediate mucosal homeostasis and inflammatory bowel disease pathogenesis.8
Collectively, perturbations in commensal microbiota composition disrupt immune homeostasis, fostering pro-inflammatory microenvironments that initiate systemic inflammatory cascades.
Functional Significance of Intestinal Epithelial Cells in IBD Pathogenesis
ISCs represent the principal regenerative mechanism for mucosal injury repair.16 The mucosal regenerative process requires coordinated cellular interactions among epithelial populations, growth mediators, stem cell niches, and immune effectors, with microbiota-derived modulatory signals exerting regulatory control. This reparative mechanism necessitates biphasic regulation: primary attenuation of inflammatory burden followed by epithelial-specific tissue reconstitution.
During the initial inflammatory phase (24-hour post-injury), neutrophil infiltration constitutes the predominant cellular response within damaged epithelial regions. The early mucosal microenvironment becomes characterized by microbial-derived pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) from cellular debris. These molecular signatures activate pattern recognition receptors, initiating leukocyte recruitment and pro-inflammatory cytokine polarization. Macrophage populations demonstrate significant expansion at lesion sites through PAMP/DAMP-mediated activation, differentiating into M1 phenotypes that predominantly secrete TNF-α, IL-1β, IL-6, and IL-23. This acute inflammatory phase typically resolves within 48–72 hours, coinciding with DAMP/PAMP clearance and apoptotic neutrophil accumulation. Subsequent macrophage phenotype switching to M2 anti-inflammatory states enhances TGF-β and IL-10 production, thereby promoting inflammatory resolution.
Post-acute phase neutrophil reduction (days 1–3) contrasts with persistent recruitment mechanisms in chronic inflammation, mediated through sustained bacterial chemotactic signals. While inflammatory activation initiates essential repair cascades, dysregulated or excessive inflammatory responses exhibit inhibitory effects on epithelial regenerative processes.17
Immune Regulation of Intestinal Stem Cells in Microbiota–Immune–Epithelial
ISCs are essential for maintaining gut homeostasis, as they control the renewal of the intestinal epithelium through carefully balanced processes of proliferation and differentiation. The gut microbiota engages in close crosstalk with the epithelial layer via diverse mechanisms, including immune and metabolic signaling, establishing a strong link between microbial processes and ISC activity. Key bacterial metabolites—including short-chain fatty acids, bile acids, and tryptophan derivatives—modulate both intestinal stem cells and the surrounding immune microenvironment. Consequently, elucidating the regenerative mechanisms of ISCs within the context of IBD, along with the interplay among bacterial metabolites, ISCs, and immune components, may offer novel insights into IBD pathogenesis and inform future treatment strategies.
Regenerative Mechanisms of Intestinal Stem Cells in IBD Pathophysiology
Persistent mucosal inflammation represents the central pathomechanism underlying IBD progression, characterized by sustained inflammatory mediator influx from chronic mucosal injury. Prolonged exposure to proinflammatory cytokines including TGF-β, TNF-α, and IL-23 within the intestinal microenvironment activates critical signaling pathways such as NF-κB and RIPK-mediated NOD2 stimulation, perpetuating inflammatory infiltration while concurrently inhibiting epithelial barrier restoration—key pathophysiological determinants of IBD chronicity.17
Compromised intestinal barrier integrity constitutes a fundamental etiopathogenic component. Clinical investigations demonstrate significant downregulation of mercaptopyruvate sulfurtransferase (MPST) expression in colonic specimens from UC patients, CD cases, and dextran sodium sulfate (DSS)-treated murine models. This sulfotransferase exerts cytoprotective effects through modulation of AKT-mediated apoptotic cascades in IECs.18 AKR1B8 deficiency exacerbates epithelial barrier dysfunction, evidenced by elevated TLR4 expression in IEC-specific knockout murine models during DSS-induced colitis, with TLR4-mediated IL-1β and IL-6 overproduction demonstrating significant pathogenic contributions.19
Pathological epithelial cell migration patterns facilitate mucosal transmigration events that exacerbate IBD progression. TGF-β synergizes with regenerating islet-derived (Reg) family proteins and intestinal trefoil factor (ITF) genes to coordinate critical mucosal repair processes.20,21 Mucosal microenvironmental cytokines including matrix metalloproteinases (MMPs), keratinocyte growth factor (KGF), and trefoil factor family (TFF) proteins regulate these migratory dynamics, with human ITF demonstrating capacity to orchestrate coordinated cell movement through ERK-JAK/STAT3 pathway crosstalk.22
Effective epithelial restitution requires dynamic reorganization of cellular adhesion complexes. Migratory front epithelial cells develop actin-rich protrusions for extracellular matrix anchorage, facilitating collective cell migration. Phosphorylated calmodulin (P-calmodulin), minimally expressed in healthy IECs, shows significant upregulation in inflamed mucosa. Its targeted depletion accelerates IEC migration through Src kinase/Rac1 GTPase activation coupled with myosin II-dependent transcriptional reprogramming.23 Differentiated IECs employ polyamine-dependent molecular mechanisms to enhance myosin II-mediated motility during epithelial repair processes.24
ISC proliferation and differentiation dynamics form the cornerstone of mucosal regeneration, with regulatory dysregulation directly contributing to IBD pathogenesis. The Wnt/β-catenin signaling axis emerges as a pivotal regulator of IEC proliferation, differentiation, and mucosal repair. Canonical Wnt/β-catenin signaling interacts with β-hydroxybutyrate (BHB) metabolites and pro-inflammatory cascades (NF-κB, JAK/STAT3, MAPK), modulating inflammatory mediator production through cyclooxygenase-2 (COX-2) and interleukin-8 (IL-8) transcriptional regulation.24
Metabolic Interaction
Short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate, are organic acids generated through bacterial fermentation of predominantly undigested carbohydrates within the intestinal lumen. These SCFAs supply approximately 60–70% of the energy needed by colonic epithelial cells and contribute to 5–15% of the overall caloric intake in humans. Among them, butyrate exhibits immunomodulatory properties by influencing chemotaxis and adhesion of immune cells.25 It also regulates neutrophil migration to sites of inflammation mediated by intestinal epithelial cells.26 Moreover, butyrate is instrumental in suppressing proinflammatory responses in lamina propria macrophages and modulating cytokine production in T cells.Acetate and propionate have been demonstrated to enhance ISC stemness in organoid models. For instance, Akkermansia muciniphila promotes the expression of Wnt-related genes and elevates synthesis of SCFAs like acetate, thereby supporting ISC stemness.27 In a separate study, microbial fucose fermentation by Akkermansia was shown to boost propionate production, which subsequently enhances intestinal stem cell function via a mechanism dependent on Wnt signaling.28
Tryptophan, an essential amino acid for mammals, is crucial for maintaining intestinal homeostasis by strengthening the intestinal barrier and regulating the composition and metabolic activity of the gut microbiota. These effects are largely mediated through two key pathways: the promotion of immune tolerance and the reinforcement of intestinal barrier function, both of which are modulated by the gut microbial community. Studies indicate that tryptophan notably enhances the synthesis of short-chain fatty acids (SCFAs) by specific bacterial groups such as Clostridium, unclassified bacteria, and Vibrio acetobacter spp and contribute to intestinal barrier integrity through the upregulation of tight junction (TJ) proteins and by influencing immune cell–microbiota crosstalk. Study find tryptophan supplementation was found to counteract the adverse gut microbiota metabolism, especially changing the production of SCFAs.29
Bile acids (BAs), metabolites in the gut.Endogenous luminal bile acid release demonstrates sufficiency to stimulate ISC renewal and post-injury tissue reconstitution.30 BA-TGR5 signals promote epithelial regeneration by activating the transmembrane receptor TGR5 in ISC compartments and restoring ISC function and promoting intestinal regeneration. During intestinal regeneration, TGR5 activation downstream triggers the activation of YAP; simultaneously, the activation of SRC kinase is also essential for the TGR5-signal-induced YAP-dependent regenerative program. But,chronic activation of the BA signaling could be dangerous which could contribute to IBD pathogenesis through the same pathway.31 In another research finds Chronic consumption of a high-fat diet (HFD) elevates intestinal levels of deoxycholic acid (DCA), which in turn activates the ABL1–YAP1 signaling pathway. This activation disrupts the differentiation of ISCs into goblet and Paneth cells, leading to impaired intestinal barrier function.32
Crosstalk Between Innate Immunity and Intestinal Epithelium in IBD Pathogenesis
Innate immune activation in IBD is initiated through damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) recognition by pattern recognition receptors (PRRs). Receptor-interacting protein kinases (RIPKs) serve as pivotal modulators of both inflammatory cascades and programmed cell death pathways activated via innate immune detection mechanisms. Notably, RIPK2 orchestrates intricate signaling cascades downstream of bacterial-sensing nucleotide-binding oligomerization domain (NOD) 1/2 receptors – fundamental regulators of intestinal immune homeostasis and inflammatory responses.
The intestinal innate defense network encompasses dendritic cells, heterogeneous macrophage populations, epithelial components, and contractile myofibroblasts that collectively respond to antigenic stimuli. These cellular sentinel systems coordinate antigen processing and T lymphocyte activation processes, thereby bridging innate and adaptive immunity. Mucosal innate immunity operates through synergistic interactions among neutrophil granulocytes, mononuclear phagocytes, dendritic cell subsets, innate lymphoid cells (ILCs), and natural killer (NK) cell effector populations.33,34
Neutrophilic Granulocytes
Neutrophilic granulocytes constitute the predominant leukocyte population in systemic circulation, functioning as primary immunological sentinels through rapid chemotactic mobilization to sites of microbial invasion or tissue injury. Following intestinal barrier compromise,33 granulocytic infiltration initiates via cytokine-mediated chemotactic gradients orchestrated by IL-1β, IL-6, and TNF-α signaling pathways. These recruited effector cells mediate pathogenic clearance through coordinated effector mechanisms including: (1) phagocytic engulfment of microorganisms, (2) generation of reactive oxygen species (ROS), (3) exocytosis of cytotoxic granular enzymes, and (4) formation of NETs - chromatin-based structures that immobilize and neutralize microbial pathogens through histone-modulated antimicrobial activity.
Within gastrointestinal pathophysiology, myeloid-derived cells exhibit dualistic biological functionalities. Controlled neutrophilic responses promote mucosal regeneration via matrix remodeling and phagocytic clearance mechanisms. Conversely, excessive cellular infiltration coupled with defective apoptotic clearance pathways sustains pro-inflammatory signaling cascades. This dysregulation initiates self-perpetuating inflammatory cycles that exacerbate chronic mucosal injury, a pathognomonic feature of IBD progression.35,36 Emerging evidence underscores the pivotal role of epithelial interface dynamics in regulating granulocyte activity, revealing reciprocal communication mechanisms that potentiate both barrier dysfunction and inflammatory mediator secretion.
Peptidylarginine deiminase 4 (PAD4), a critical molecular regulator of neutrophilic effector functions, mediates chromatin decondensation during NETosis while concurrently releasing enzyme-laden extracellular vesicles into epithelial microenvironments. Complementary research demonstrates that CKMT1-guanosine molecular complexes disrupt intercellular junctional integrity through Rho GTPase pathway modulation, establishing synergistic pro-inflammatory niches. Experimental colitis models elucidate three distinct mechanisms of NET-mediated epithelial compromise: 1) Structural disruption of apical junctional complexes (ZO-1, E-cadherin); 2) Caspase-dependent apoptotic induction; and 3) Pattern recognition receptor activation on epithelial surfaces.37,38
The intestinal epithelium orchestrates granulocyte recruitment through multifaceted chemoattractant systems:
- Pro-inflammatory cytokines: IL-8 (CXCL8), IL-6, and alarmin IL-33
- Chemokine networks: Epithelial-derived neutrophil-activating peptide (ENA-78/CXCL5), CXCL7, IFN-γ-inducible protein 10 (CXCL10), and macrophage inflammatory protein-3α (CCL20)
- Lipid mediators: Leukotriene B4 (LTB4) and hepoxilin-derived chemotactic factors
- Matrix remodeling enzymes: Stromelysin-1 (MMP-3) and matrilysin (MMP-7).39
Transmural neutrophil migration involves coordinated β2 integrin (CD11b/CD18) interactions with specialized surface receptors: immunoglobulin superfamily members (CD47/SIRP-α complexes), junctional adhesion molecules (JAM-L/CAR heterotypic interactions), complement regulatory proteins (CD55-CD97 pairs), and ectonucleotidase systems (CD39/CD73 adenosine signaling regulators).
Beyond classical antimicrobial functions, granulocytes modulate epithelial dynamics through non-cell-autonomous mechanisms. Current research identifies neutrophil-derived microvesicles containing regulatory miRNAs that influence IEC proliferation and apoptosis. Nevertheless, the molecular mechanisms governing these processes remain poorly understood, representing significant knowledge gaps requiring systematic exploration. Clinically pertinent observations demonstrate enhanced NETotic potential in IBD epithelia through exosome-mediated transfer of pro-inflammatory cargo, establishing pathophysiological connections between mucosal inflammation and thromboembolic sequelae.40
Advanced imaging methodologies (intravital microscopy, organoid co-culture systems) elucidate spatiotemporal regulation of neutrophil-epithelial interactions during mucosal repair. Emerging paradigms position granulocytes as dynamic modulators of epithelial stem cell niches through Wnt/β-catenin pathway regulation and Notch signaling activation. Transcriptomic profiling reveals novel epithelial-derived danger signals (S100 proteins, HMGB1) that prime neutrophil effector functions in IBD microenvironments. These collective findings establish granulocyte-epithelial crosstalk as a promising therapeutic target for mucosal healing in inflammatory bowel diseases.
Monocytes/Macrophages
Intestinal macrophage populations derive from bone marrow-derived monocytes that differentiate into three functionally distinct subsets defined by their specialized physiological roles: microbial defense, tissue restoration, and immune regulation. Antimicrobial macrophages mediate pathogen clearance through cytokine production (IFN-γ, TNF-α) and antigen-presenting cell (APC) functions. Tissue-reparative macrophages differentiate under IL-4 stimulation from granulocytic or lymphocytic precursors, facilitating structural regeneration processes. Immunoregulatory macrophages develop through IL-10 signaling, corticosteroid exposure, or apoptotic cell contact, executing anti-inflammatory modulation through specialized secretory profiles.41,42
In IBD pathogenesis, mucosal injury sites release pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) that trigger inflammatory cascades. CCR2-mediated recruitment of Ly6C++ monocytes to intestinal tissues occurs through CCL2/MCP-1 chemotactic gradients. Epithelial-derived IL-8 and TGF-β further orchestrate monocyte trafficking into lamina propria compartments.43 IL-10RA-deficient macrophages demonstrate dysregulated IL-23 overproduction, which stimulates Th17/ILC3-mediated IL-22 secretion. This cytokine cascade amplifies intestinal epithelial cell (IEC) antimicrobial peptide expression and neutrophil infiltration, thereby accelerating IBD progression.44 NOX4-derived ROS drive macrophage polarization toward pro-inflammatory M1 phenotypes, perpetuating inflammatory cascades and compromising mucosal barrier integrity.45
Counterintuitively, macrophage-epithelial crosstalk maintains critical homeostatic functions. α-TREM-1-mediated CD177 induction in neutrophils facilitates macrophage-IEC coordination that promotes mucosal repair mechanisms. Macrophage-secreted cytokines (IL-6, IL-8, IFN-γ, TGF-β1) enhance epithelial maturation and barrier reinforcement through tight junction protein modulation.45 PTPN2 emerges as a critical regulator of macrophage-IEC synergy, maintaining junctional adhesion molecule-A (JAM-A) expression and epithelial junction stability. Genetic PTPN2 deficiency disrupts these cytoprotective interactions.46
Experimental colitis models demonstrate macrophage-mediated epithelial cytotoxicity. GPR84-deficient M1 macrophages exhibit attenuated capacity to compromise Caco-2 monolayer integrity compared to wild-type (WT) counterparts, indicating GPR84’s mechanistic involvement in macrophage-driven epithelial damage.47
Dendritic Cells (DCs)
Dendritic cell populations, comprising conventional (cDCs) and plasmacytoid (pDCs) subsets, serve as primary immunological sentinels orchestrating antigenic surveillance. In IBD pathophysiology, DC-epithelial crosstalk exerts pivotal regulatory control over disease progression and inflammatory cascades. The CCL25 chemokine derived from intestinal epithelium interacts with CCR9 receptors expressed on DCs, facilitating DC chemotaxis toward mucosal compartments.48,49
This bidirectional signaling axis dynamically regulates inflammatory homeostasis. Activated DCs induce epithelial secretion of pro-inflammatory cytokines (IL-6, TNF-α), thereby perpetuating inflammatory initiation and maintenance mechanisms. TNFR1/2 receptor signaling exacerbates ileitis through dual targeting of TLR5-expressing Paneth cells and dendritic cell populations.50 Experimental colitis models demonstrate TNF-α-mediated DC regulation of IL-22 binding protein (IL-22BP), which antagonizes IL-22-dependent epithelial regeneration.51 Conversely, counter-regulatory pathways exist wherein DCs ameliorate colitis through IFN-γ-mediated induction of epithelial anti-inflammatory programming.52,53
Fibrinogen-like protein 2 (FGL2) emerges as a critical regulatory node, exerting biphasic modulation of IKK/NF-κB activation in both epithelial cells and lamina propria DCs, thereby precisely calibrating intestinal inflammatory states.54
Within the intestinal mucosa, DCs acquire luminal antigens through trans-epithelial sampling, subsequently priming T lymphocytes via MHC-II molecular complex presentation. This immunogenic priming process establishes adaptive immunity in T lymphocytes, thereby propagating pro-inflammatory cascades within gastrointestinal tissues. Notably, mucosal epithelial components exhibit functional MHC-like molecule expression, suggesting complementary antigen-presenting capabilities that may synergize with DC functions.55
DC-epithelial intercellular communication plays a central role in mucosal barrier restitution during inflammatory episodes. DC activation promotes epithelial proliferative responses while attenuating pathological inflammation induced by barrier disruption. Paradoxically, this cellular crosstalk concurrently amplifies pro-inflammatory mediator production (IL-1β, IL-6, IL-23) through reciprocal signaling mechanisms, thereby accelerating IBD progression. Such synergistic interactions can instigate focal inflammatory cascades, ultimately leading to mucosal erosion and ulcerative pathogenesis.56
The DC-epithelial axis manifests bidirectional regulatory complexity in IBD pathogenesis, concurrently governing immunopathological processes, inflammatory modulation, and tissue repair mechanisms. Systematic elucidation of molecular determinants underlying these cellular interactions will enhance mechanistic understanding of IBD pathophysiology and facilitate development of targeted therapeutic strategies.
Innate Lymphocytes
Innate lymphoid cells (ILCs) constitute critical immunomodulatory components in the gastrointestinal ecosystem, orchestrating host-pathogen interface regulation and maintaining mucosal immune homeostasis. These evolutionarily conserved effector populations demonstrate a tripartite classification system based on distinct developmental pathways and effector functions: Group 1 encompasses cytotoxic ILC1s and natural killer (NK) cells; Group 2 contains type 2 cytokine-producing ILC2s; Group 3 comprises IL-17/IL-22-secreting ILC3s alongside lymphoid tissue-organizing cells.57 Emerging research frontiers have significantly advanced our understanding of dynamic bidirectional communication pathways between ILC subsets and gastrointestinal epithelial compartments, particularly during critical phases of mucosal wound resolution and tissue regenerative processes.
Intestinal mucosal restitution processes involve complex immunoregulatory mechanisms.58 During inflammatory bowel disease pathogenesis, ILC-mediated pathological mechanisms are predominantly mediated through tripartite pathogenic axes (Figure 1), including: 1) Microbiota composition modulation, 2) Tight junction protein homeostasis disruption, and 3) Proinflammatory cytokine cascade activation (notably IL-17/IL-22 overproduction), synergistically promoting disease exacerbation.
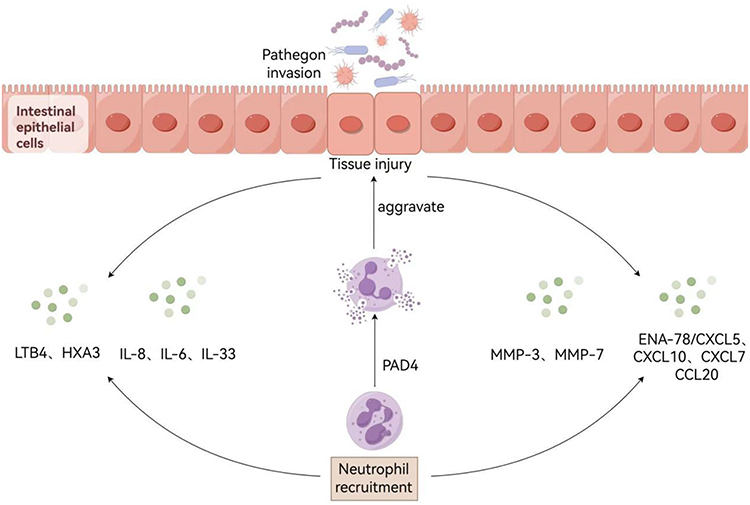 |
Figure 1 Illustrates the molecular mechanisms mediating interactions between neutrophils and intestinal epithelium during epithelial injury. Following breach of epithelial integrity, a cascade of inflammatory mediators is released, including: (i) cytokines (IL-8, IL-6, IL-33); (ii) chemokines (ENA-78/CXCL5, CXCL10, CXCL7, CCL20); (iii) lipid-derived mediators (LTB4, HXA3); and (iv) matrix metalloproteinases (MMP-3, MMP-7), which collectively mediate neutrophil recruitment to inflammatory foci. Concomitantly, protein-arginine deiminase 4 (PAD4) facilitates NET formation through extracellular vesicles (EVs) secreted into intestinal epithelial cells. Notably, mitochondrial creatine kinase 1 (CKMT1) demonstrates epithelial barrier-disrupting activity, thereby exacerbating the inflammatory response through compromised mucosal integrity. |
Group 1 innate lymphoid cells (ILC1s) demonstrate dual regulatory functions in intestinal barrier homeostasis. Although essential for antimicrobial defense, genetic ablation of cellular FLICE-inhibitory protein (c-FLIP) in NKp46+ ILC populations induces chronic colitis via IL-7/IL-15 signaling axis destabilization. These survival-critical cytokines, derived from stromal fibroblasts and mucosal epithelium, position ILC1 subsets as key mediators in inflammatory amplification cascades and epithelial repair pathway dysregulation.59
The ILC2 subpopulation exhibits significant mucosal protective properties against epithelial damage. IL-33-mediated activation of colonic ILC2s initiates type 2 immune-mediated repair mechanisms that counteract toxin-induced epithelial injury through amphiregulin (AREG)-dependent pathways.60 Genetic ablation of transcription factor T-bet in ILC precursors enhances IL-7 receptor alpha (IL-7Rα)-dependent ILC2 expansion, potentially ameliorating immune hyperactivation and colitis progression via type 2 response potentiation.59,61 Mechanistic investigations demonstrate that ILC2-derived AREG critically promotes goblet cell hyperplasia and mucin glycoprotein biosynthesis (particularly MUC2) - fundamental processes for barrier preservation during inflammatory insults.59 Single-cell transcriptomic profiling has uncovered novel ILC2-epithelial communication networks involving tuft cell-derived IL-25 and ILC2-secreted IL-13, suggesting previously unrecognized regulatory pathways in mucosal regeneration.Tuft cells release IL-25 and other inflammatory mediators. IL-25 stimulates ILC2s to secrete IL-13, which acts on epithelial crypt progenitors to induce their differentiation into tuft and goblet cells. This process further activates a subset of IL-25R+ST2-ILC2s, termed inflammatory ILC2s, thereby establishing a positive feedback loop that amplifies type 2 inflammation.59
Group 3 ILCs display paradoxical regulatory roles in mucosal homeostasis. Maladaptive activation induces epithelial damage through IL-22 overproduction, triggering pathogenic neutrophil infiltration and extracellular matrix degradation.62 Conversely, controlled ILC3-epithelial crosstalk facilitates mucosal regeneration via growth factor-mediated restitution mechanisms. Microbial metabolite sensing through free fatty acid receptor 2 (FFAR2) on colonic ILC3s upregulates retinoic acid receptor-related orphan receptor gamma t (RORγt) expression, potentiating IL-22-driven epithelial repair and antimicrobial peptide production via PI3K/Akt/mTOR signaling pathway activation.63,64
The ILC3 secretome comprises pleiotropic mediators including leukemia inhibitory factor (LIF) and IL-26 that bidirectionally regulate epithelial dynamics. While LIF stimulates crypt progenitor proliferation through JAK/STAT3 pathway activation, IL-26 inhibits epithelial growth while inducing TNF/IL-8 production in ILC3s, establishing autocrine proinflammatory feedback loops.65 These cells further coordinate goblet cell functional specialization via IL-22-dependent Notch signaling regulation, constituting essential mucosal defense systems. Current research priorities include elucidating spatiotemporal regulation of ILC-epithelial interactions during mucosal damage responses. Emerging data implicate Wnt/β-catenin signaling as a pivotal pathway governing ILC3-dependent epithelial stem cell activation (Figure 2).
 |
Figure 2 Crosstalk between intestinal epithelial cells and innate immune populations. The dynamic interplay between innate immune components and intestinal epithelial cells (IECs) serves as a critical regulatory mechanism in maintaining intestinal homeostasis while potentially driving pathological tissue remodeling. Epithelium-derived interleukin-8 (IL-8) and transforming growth factor-beta (TGF-β) facilitate monocyte recruitment to the lamina propria microenvironment. NADPH oxidase 4 (NOX4)-mediated ROS generation promotes M1 macrophage polarization, exacerbating IBD pathogenesis through enhanced mucosal barrier disruption and pro-inflammatory signaling. DCs secrete pro-inflammatory mediators including interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), which induce IEC production of inflammatory effectors, thereby perpetuating chronic inflammatory cascades. Dysregulated activation of group 3 innate lymphoid cells (ILC3s) mediates epithelial damage via IL-22 overproduction, triggering neutrophil chemoattractant synthesis in IECs and subsequent neutrophilic infiltration-induced tissue destruction. Conversely, this cellular crosstalk facilitates mucosal repair through multiple mechanisms: Macrophage-derived cytokines (IL-6, IL-8, interferon-gamma [IFN-γ], and TGF-β1) enhance epithelial cytoprotection, while IL-33-activated ILC2s produce amphiregulin (AREG) to promote tissue integrity. Furthermore, ILC3-derived IL-22 exhibits protective properties by stimulating IEC secretion of antimicrobial peptides, thereby reinforcing the epithelial barrier against pathogenic bacterial invasion and maintaining intestinal mucosal homeostasis. |
The sophisticated interplay between ILC subsets and epithelial constituents encompasses multi-tiered signaling networks and cytokine feedback loops, particularly during mucosal injury responses. Deciphering these molecular interactions advances our comprehension of intestinal immunopathology while revealing novel therapeutic targets including:
- IL-22/IL-22 binding protein (IL-22BP) axis regulation
- FFAR2/RORγt signaling pathway inhibition
- AREG-mediated epithelial repair potentiation
- These mechanistic insights possess significant translational potential for developing targeted immunotherapies in inflammatory bowel diseases and colitis-associated carcinogenesis.
The Interplay Between Adaptive Immunity and Gastrointestinal Epithelium in Inflammatory Bowel Disease Pathogenesis
Ecological perturbations within the intestinal microbiota, concomitant with structural deterioration of mucosal barriers in IBD, substantially amplify microbial antigen exposure. This process initiates hyperactivation cascades impacting both innate and adaptive immune systems. The primary immune activation cascade involves myeloid-derived cells responding to microbial-associated molecular patterns (MAMPs) and damage-associated molecular patterns (DAMPs) through inflammatory mediator secretion, subsequently priming lymphocyte populations via antigen presentation mechanisms. A defining characteristic of IBD pathophysiology manifests as pathological infiltration of activated T lymphocytes and their associated pro-inflammatory cytokine milieu within intestinal tissues.66,67
Adaptive immune activation fundamentally relies on cytokine microenvironments modulated by antigen-presenting cell (APC) functionality. Despite exhibiting delayed initiation kinetics compared to innate immune responses, this system demonstrates antigen-specific precision. Following luminal antigen penetration through compromised mucosal barriers, dendritic cells and related APCs coordinate intricate signaling networks that polarize circulating lymphocytes toward distinct pro-inflammatory or anti-inflammatory phenotypes. Subsequent leukocyte homing to inflammatory foci occurs through sequential molecular interactions: 1) Integrin-mediated endothelial tethering, 2) Chemokine receptor-dependent activation, 3) Selectin-mediated firm adhesion, and 4) Transendothelial migration into inflamed tissues - processes critically governed by endothelial adhesion molecule expression patterns.68,69 The principal cellular mediators of adaptive immunity comprise three key lymphocyte populations:CD4+ helper T lymphocytes, Cytotoxic CD8+ T cells, Antibody-secreting B lymphocytes, Antigen-driven differentiation of CD4+ T cells generates multiple effector subsets: Th1 (IFN-γ predominant), Th2 (IL-4/IL-13 producing), Th17 (IL-17 secreting), Treg (immunosuppressive).
Notably, unconventional lymphocyte populations including natural killer T (NKT) cells and γδ T cells serve as immunological interfaces bridging innate and adaptive immunity. B lymphocyte contributions extend beyond immunoglobulin production to encompass cytokine secretion and antigen presentation capabilities - all mechanistically implicated in IBD pathogenesis.70,71 Current pathophysiological models emphasize adaptive immune dysregulation, particularly Th17/Treg imbalance and pathogenic B cell activation, as central drivers of IBD progression through perpetuation of mucosal inflammation.72
TH1 Lymphocytes
T helper 1 (Th1) cells serve as critical cellular effectors in antimicrobial immunity, specializing in the eradication of intracellular pathogens including parasitic protozoa, viral particles, and intracellular bacterial species. This CD4+ T lymphocyte subset orchestrates cell-mediated immune responses through two principal mechanisms: direct cytotoxic activity and cytokine-mediated immunoregulation. The characteristic cytokine profile of Th1 cells, particularly IFN-γ and TNF-αfacilitates cross-talk between innate and adaptive immune systems through paracrine activation of myeloid cells (macrophages, neutrophils) and structural components (epithelial cells, stromal fibroblasts). In IBD pathogenesis, Th1-mediated responses drive mucosal inflammation through dysregulated immune activation.73
Mechanistically, IFN-γ exerts multifaceted cytotoxic effects on intestinal epithelial lineages: mucus-producing goblet cells, antimicrobial peptide-secreting Paneth cells, and ISCs. While IFN-γ alone induces epithelial damage, its synergistic interaction with TNF-α potentiates enterocyte cytotoxicity through activation of the caspase-8/JAK1/2/STAT1 signaling axis. Molecular analyses demonstrate that IFN-γ receptor engagement initiates sequential phosphorylation events: JAK1/JAK2 kinase activation followed by STAT1 transcriptional activation. This signaling cascade mediates both cellular responses to IFN-γ exposure and TNF-α/IFN-γ co-induced apoptotic pathways in intestinal epithelium.74,75
Experimental models elucidate IFN-γ’s pivotal role in lymphocyte-mediated stem cell injury. The destruction of intestinal stem cell niches represents a pathological hallmark in graft-versus-host disease and autoimmune disorders, where T lymphocytes eliminate both allogeneic and autologous intestinal organoids via IFN-γ-dependent mechanisms. Transcriptomic analyses reveal that IFN-γ directly reprograms ISCs through apoptotic gene activation, a process attenuated by pharmacological inhibition of JAK/STAT signaling pathways.76
The IFN-γ/TNF-α synergy extends to macrophage polarization dynamics in IBD pathophysiology. Tissue macrophages in IBD patients predominantly exhibit pro-inflammatory M1 polarization, enhanced through Th1 cytokine-driven classical activation. This polarization state amplifies antigen presentation capacity and inflammatory mediator secretion, exacerbating epithelial barrier dysfunction. Pathogenic feedback loops emerge through Th1-derived IL-2/IFN-γ stimulation of macrophage activation, establishing self-perpetuating Th1/STAT3-M1 macrophage circuits that drive intestinal tissue destruction.77
Notably, type I interferons (IFN-α/β) contribute to intestinal immunopathology through innate immune activation. Clinical studies implicate IFN-α in mucosal homeostasis disruption via caspase-1/IL-18/IFN-γ axis activation, inducing epithelial barrier failure through pyroptotic cell death. This interferon-mediated apoptotic cascade demonstrates conserved functionality across celiac disease and IBD progression, underscoring the therapeutic potential of interferon signaling modulation in gastrointestinal inflammation.
TH17 Lymphocytes
TH17 lymphocyte populations exhibit distinct anatomical localization within the gastrointestinal lamina propria, functioning as critical sentinels against invasive microbial pathogens. The lineage specification of these cells requires coordinated activation of STAT3 signaling and RORγt-mediated transcriptional regulation, processes dynamically modulated by commensal microbiota and local cytokine networks. Through specialized secretory activity, TH17 cells establish immunoregulatory microenvironments via production of effector molecules including IL-17, IL-21, IL-22, and IL-26, which collectively maintain intestinal homeostasis through pleiotropic mechanisms.78–80
IL-17-mediated paracrine signaling induces proliferative responses in gastrointestinal epithelial cells while enhancing mucosal defense through three principal mechanisms: (1) upregulation of polymeric immunoglobulin receptor (pIgR) expression, (2) augmentation of secretory IgA production, and (3) induction of antimicrobial peptide synthesis - biological processes essential for mucosal restitution. Contrastingly, during active IBD phases, IL-17 synergizes with TNF-α to initiate pathological cascades in epithelial cells, promoting concurrent secretion of inflammatory mediators (IL-12/IL-23), chemotactic factors (CXCL8), and tissue-remodeling proteases (MMPs). This cytokine crosstalk facilitates TH17 transdifferentiation toward TH1-like phenotypes,81,82 While inducing pro-inflammatory mediators (IL-6, TNF-α, GM-CSF) that coordinate neutrophilic infiltration and activation, ultimately driving mucosal injury progression.83
The maintenance of gastrointestinal immune homeostasis requires precise bidirectional communication between mucosal lymphocytes and epithelial components. Epithelial-derived IL-17C emerges as a pivotal inflammatory mediator during microbial challenges, mediating reciprocal activation of TH17 lymphocytes. Mechanistic investigations demonstrate that TH17-secreted TNF-α, IL-17A, and IL-22 transcriptionally upregulate IL-17C in colonic epithelia through NF-κB/AP-1-dependent pathways, establishing IL-17C as a dynamic biomarker reflecting TH17-mediated inflammatory processes.84
IL-22, another TH17-associated cytokine, demonstrates context-dependent pleiotropy. While undetectable in healthy colonic mucosa, its expression becomes significantly elevated during intestinal inflammation, particularly in Crohn’s disease pathogenesis. Experimental models reveal IL-22 enhances mucosal barrier integrity through dual mechanisms: (1) stimulating epithelial migratory capacity and (2) inducing antimicrobial peptide synthesis. Paradoxically, IL-22 simultaneously activates NF-κB/AP-1/MAPK pathways in subepithelial myofibroblasts, promoting secretion of pro-inflammatory mediators (IL-6, IL-8, IL-11) that exacerbate IBD progression.84,85
Completing the TH17 effector triad, IL-26 - structurally homologous to IL-10 family cytokines - engages enterocyte receptor complexes to mediate proliferative regulation and inflammatory gene induction. Clinical analyses demonstrate elevated IL-26 concentrations in active Crohn’s disease lesions, reflecting shared pro-inflammatory signaling pathways with IL-22 through Akt/STAT activation. This cytokine potentiates inflammatory cascades in IBD epithelia while modulating intestinal epithelial cell survival pathways, confirming its pathogenic significance in disease progression.86,87
Regulatory T Cells and TH17 Lymphocytes
Treg serve as critical regulators of immune tolerance, maintaining peripheral immune homeostasis through suppression of excessive immune activation. This specialized T cell population comprises two distinct subsets: thymus-derived natural Treg (nTreg) and peripherally induced adaptive Treg (pTreg). The immunosuppressive function of Tregs primarily involves secretion of anti-inflammatory cytokines, particularly TGF-β and IL-10, which mediate suppression of effector lymphocyte activity. Notably, intestinal tissues from IBD patients exhibit paradoxical increases in Treg infiltration, potentially indicating compensatory regulatory mechanisms to counteract heightened pro-inflammatory responses.
The differentiation pathways of Th17 effector cells and regulatory T lymphocytes demonstrate reciprocal regulatory relationships, both dependent on TGF-β signaling cascades. Experimental studies reveal that IL-6/IL-21 synergism with TGF-β promotes CD4+ T cell differentiation toward Th17 lineage, while TGF-β predominance in anti-inflammatory environments drives Treg development. This delicate balance critically influences autoimmune disease pathogenesis, with IBD patients demonstrating substantial Th17 cell infiltration in intestinal mucosa alongside elevated IL-17 concentrations - a characteristic cytokine of Th17-mediated inflammatory responses.88,89
IL-10, an immunoregulatory cytokine produced by Tregs, inhibits epithelial-derived CCL20 chemokine expression, which becomes significantly upregulated during IBD progression. As the primary ligand for CCR6 receptors, CCL20 mediates tissue-specific recruitment of CCR6-expressing leukocyte populations including B lymphocytes, Th17 cells, and regulatory T cell subsets. While pro-inflammatory cytokines (IL-17, TNF-α, IFN-γ) stimulate CCL20 production across various tissue compartments, IL-10 exerts counter-regulatory effects through transcriptional inhibition. Co-expression of CCR6 on both Th17 and Treg populations facilitates their coordinated migration to CCL20-enriched inflammatory sites. Maintenance of mucosal immune equilibrium requires strict regulation of the CCL20/CCR6 axis, with IBD patients exhibiting elevated colonic CCL20 levels that correlate with Th17/Treg dysregulation and mucosal inflammation (Figure 3).90,91
 |
Figure 3 Illustrates the complex interplay between adaptive immunity and intestinal epithelium. Th1 lymphocytes secrete interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α), cytokines capable of inducing structural damage to intestinal epithelial cells (IECs). This pathological mechanism involves M1 macrophage activation that mediates mucosal tissue injury. IFN-γ demonstrates direct cytotoxicity toward both mature IECs and ISCs, with its synergistic interaction with TNF-α potentiating IEC destruction. In contrast, Th17 cells exhibit dual regulatory functions: IL-17 secretion stimulates IEC proliferation, while IL-22 enhances epithelial migration and upregulates antimicrobial defensin production, collectively reinforcing intestinal barrier integrity. Furthermore, Th17-derived IL-26 demonstrates additional regulatory capacity in modulating IEC proliferation dynamics. Treg cells maintain mucosal homeostasis primarily through immunosuppressive cytokines, including transforming growth factor-β (TGF-β) and interleukin-10 (IL-10), which counterbalance pro-inflammatory Th1 and Th17 responses. Notably, CCL20 chemokine expression exhibits significant elevation in IBD, with epithelial-derived CCL20 demonstrating multifold increases. This chemokine’s expression is regulated through opposing mechanisms: IL-17, TNF-α, and IFN-γ serve as potent inducers, while IL-10 functions as a transcriptional repressor. |
Intestinal epithelial cells (IECs) constitute the principal source of IL-18 during CD4+ T cell-mediated intestinal inflammation. Murine models demonstrate IL-18’s dual regulatory functions: suppression of physiological Th17 differentiation while enhancing Foxp3+ Treg immunosuppressive capacity to mitigate colitis. Mechanistic studies indicate that IL-18 receptor signaling in Tregs is essential for colitis prevention, whereas epithelial-derived IL-18 regulates colonic T cell homeostasis through IL-18R1-dependent mechanisms.92,93 However, clinical data reveal a positive correlation between epithelial IL-18 overproduction and IBD severity. Experimental evidence from IEC-specific IL-18R knockout models demonstrates protection against dextran sulfate sodium (DSS)-induced colitis, suggesting excessive IL-18 signaling may impair epithelial barrier function through modulation of monolayer permeability.94,95
B Lymphocyte Biology
B lymphocytes orchestrate humoral immune responses through three principal mechanisms: antibody synthesis, antigen presentation to T lymphocytes, and cytokine-mediated immunomodulation (IL-2, IL-4, IFN-γ, TGF-β, GM-CSF). Cellular activation commences within mesenteric lymphoid tissues, with activated B cells subsequently undergoing CXCR5-dependent chemotaxis toward intestinal mucosa where terminal differentiation into immunoglobulin-secreting plasma cells occurs. This coordinated process establishes localized antibody-mediated immune protection while simultaneously modulating inflammatory cascades through IL-10 secretion and regulatory T cell interactions. Histologically, plasma cells - representing terminal B cell differentiation products - demonstrate preferential localization in bone marrow and lamina propria compartments. Immunoglobulin subclass analysis reveals IgA-secreting plasma cells as the predominant intestinal population, whereas IgG-secreting counterparts exhibit strong association with inflammatory milieus. Pathological observations in ulcerative colitis (UC) patients demonstrate both quantitative expansion of mucosal plasma cell infiltrates and qualitative shifts in antibody-secreting cell ratios, with immunohistochemical analyses confirming predominant IgG secretion within inflamed intestinal mucosa. These findings collectively implicate plasma cell dysregulation in UC pathogenesis.96,97
IL-33, a dual-function cytokine within the IL-1 superfamily, serves as a critical molecular interface coordinating innate and adaptive immunity. This nuclear-associated alarmin is constitutively expressed by barrier epithelial cells lining pulmonary, integumentary, gastrointestinal, and reproductive mucosal surfaces. Murine colitis models elucidate IL-33’s capacity to drive differentiation of immunosuppressive regulatory B cell subsets (CD25+ B220+ and IL-10+ CD19+ phenotypes), effectively attenuating intestinal inflammation through multiple mechanisms.98 Complementary genetic studies demonstrate exacerbated colitis susceptibility in IL-33-deficient mice, mechanistically linked to defective B lymphocyte-dependent IgA production - a vital process for maintaining mucosal immune homeostasis. These experimental insights establish IL-33-mediated IgA potentiation as a promising therapeutic strategy for inflammatory bowel disease through targeted suppression of pathological inflammation.99
Immune and Metabolic Dysregulation in IBD
Current research has summarized the effects of interactions between intestinal epithelium and immune cells on the intestinal microenvironment in IBD. Recent studies indicate that these interactions influence gut microbiota metabolism, including impaired glycolysis, disrupted lipid metabolism, and mitochondrial dysfunction, thereby perpetuating intestinal inflammation.
Inflammation is characterized by a metabolic shift toward promoting glycolysis and lactate production. Hexokinase (HK) catalyzes the first step of glycolysis, and inhibiting HK2 in epithelial cells provides protective effects against mouse colitis. Intestinal epithelial cells are the primary source of HK2 expression. Studies reveal that HK2 expression is upregulated in inflammatory tissues relative to non-inflammatory tissues from the same patient, regardless of the type of intestinal inflammation. Furthermore, clearing HK2 from intestinal epithelial cells (IECs) alleviates acute intestinal inflammation, suppresses cell death, and alters mitochondrial function. RNA sequencing data indicate that both epithelial erosion and immune cell infiltration into the submucosa coexist during intestinal inflammation. Although infiltrating macrophages appear most relevant, other cell types (eg, B cells) also show increased proportions. Macrophage expansion may be particularly significant, potentially disrupting tight junction complexes between IECs by producing IL-1β, thereby increasing intestinal permeability, promoting epithelial cell loss, and inducing subsequent alterations in HK2 expression.100,101
Obesity is commonly associated with metabolic abnormalities, and elevated IL-1β levels have been detected in certain patients with active IBD and obesity. GLP-1 receptor agonists (GLP-1 RAs), which influence both intestinal metabolism and immune function, significantly lower pro-inflammatory cytokines like IL-1β. These agents may therefore provide therapeutic advantages in obese individuals with IBD by targeting overlapping metabolic and inflammatory pathways. Enteric endocrine cells (EECs) are crucial in resolving inflammation. In EECs, TNF-α activates the NF-κB pathway, leading to the production of IL-17C, which worsens inflammation in IBD. EECs are also targeted by GLP-1/GLP-1R signaling. Preclinical studies using DSS-induced colitis models demonstrate that GLP-1 RAs alleviate intestinal inflammation by enhancing IL-22 production from beneficial gut bacteria such as Firmicutes, Proteobacteria, and Lactobacillus reuteri. Furthermore, treatment with GLP-1 RAs inhibits the differentiation of T helper cells into Th1 and Th17 subtypes, reducing secretion of IFN-γ, TNF-α, and IL-17.102 Conversely, GLP-1 promotes Th2 and Treg cell polarization, thereby increasing anti-inflammatory cytokines including IL-10 and IL-5.103,104
Among dietary factors, high-fat diets (HFD) are strongly linked to the development of IBD. Obesity driven by HFD is associated with immune dysregulation, promoting chronic inflammation and elevating the risk of autoimmune and inflammatory disorders. Treg cells, identified by stable expression of FOXP3 and CD25, play a key immunosuppressive role and help maintain immune tolerance in IBD. Studies show that HFD triggers immune cell activation in the colon and small intestine, markedly decreasing CD4+Foxp3+ Treg cells.105 This loss of Treg cells weakens control over pro-inflammatory responses, accelerating IBD progression. Recent findings indicate that Treg cells rely on lipid metabolism and preferentially use polyunsaturated fatty acids (PUFAs) for phospholipid synthesis. Diets high in PUFAs, such as arachidonic acid (AA) and its precursors, disrupt Treg cell function. HFD also causes gut microbiota imbalance and increases intestinal barrier permeability, activating resident immune cells. Elevated redox activity then generates excessive ROS, inducing lipid peroxidation and ferroptosis in Treg cells. This breakdown in immune tolerance raises susceptibility to colitis. Moreover, HFD lowers levels of glutathione peroxidase 4 (GPX4) in colonic Treg cells, and GPX4 deficiency further promotes ferroptosis, worsening HFD-induced intestinal inflammation.106 These insights advance the understanding of IBD mechanisms and highlight potential therapeutic targets for restoring Treg-mediated immune balance.107
Additionally,Inflammatory bowel diseases are characterized by a reduction in Paneth cell (PC) numbers, and impaired PC function is known to contribute to ileal pathogenesis in Crohn’s disease (CD). It has been observed that mitochondrial dysfunction in the intestinal epithelium during inflammation induces a metabolic imbalance, which leads to diminished stemness and the adoption of a dysfunctional PC phenotype.108 This may be related to the downregulation of Prohibitin 1 (PHB1), a major component protein of the mitochondrial inner membrane. Experiments revealed that mitochondrial dysfunction leads to reduced levels of PHB1 and inhibits the growth of intestinal organoids and epithelial repair. This inhibition can be mitigated by exogenous polyamine putamine.109
Immune-Fibrotic Interactions in IBD
In the progression of IBD, persistent intestinal inflammation combined with recurrent tissue injury often results in secondary intestinal fibrosis (IF). This is especially prevalent in CD patients, where it commonly presents as fibrosis and obstruction. The following section discusses the interplay of immune factors with elements such as TGF-β and fibroblasts in IBD-associated intestinal fibrosis.110 Although extracellular matrix (ECM) deposition is a physiological requirement for tissue repair, excessive ECM accumulation causes tissue stiffening and functional deficits, largely driven by fibroblasts and myofibroblasts. Currently, thickening of the lamina propria is viewed as a principal factor in luminal obstruction.111
Regarded as a central pro-fibrotic growth factor, TGF-β1 is up-regulated in nearly all fibrotic disorders, including CD.IL-6, largely produced by monocytes, epithelial cells, and mesenchymal cells, is established to contribute to IBD pathogenesis. Its expression is increased within the muscular layers of CD strictures, can be induced by TGF-β1, and participates in aggravating fibrosis in CD.112 Furthermore, several interleukins from both immune and non-immune sources modulate fibrotic processes. Members of the IL-1 family, for example, exhibit pro-fibrotic traits: IL-1α, derived from epithelial cells, activates fibroblasts and contributes to intestinal inflammation, while IL-1β, mainly from mononuclear phagocytes, acts pro-inflammatorily in the gut. Stimulation with IL-1β leads myofibroblasts to upregulate production of type I and IV collagen, IL-8, MCP-1, and MMP-1, all of which are key ECM constituents. IL-33 can be secreted by immune cells or non-immune cells like fibroblasts and SMCs, and via its receptor ST2, it promotes pro-inflammatory and Th2-type immune activity. Th2 responses are linked to fibrosis in most organs, and IL-33/ST2 signaling encourages epithelial-mesenchymal transition (EMT) and fibroblast proliferation, thus advancing fibrosis.113
TNF and TL1A (TNF-like ligand 1A) are recognized drivers of intestinal inflammation. They promote IF indirectly by amplifying inflammation or through direct actions on mesenchymal cells. The interaction between TL1A and its functional receptor DR3 fosters both inflammatory and fibrotic responses.114 Research indicates their potential role in epithelial-mesenchymal crosstalk and inflammation-mediated intestinal fibrosis in CD. It is noteworthy that expression of TL1A in myofibroblasts is influenced by proinflammatory signals such as TNF-α, IL-1α, and IFN-γ. These signals may act directly on myofibroblasts or indirectly by triggering epithelial cells to release soluble factors that subsequently induce TL1A expression in myofibroblasts.115
Research on the Treatment of IBD Using Intestinal Organoids
Human intestinal organoids represent an ideal model system widely used in studying gastrointestinal physiology and immunopathology. By leveraging patient-derived intestinal organoids, researchers can gain deeper insights into key physiological and immunopathological aspects of intestinal diseases, offering a robust platform for uncovering disease mechanisms. Based on this model, organoid research has furthered the understanding of IBD and contributed to the development of new therapeutic approaches.116 It has been established that under defined culture conditions, purified intestinal stem cells can be maintained long-term as intestinal epithelial cells and subsequently self-organize into three-dimensional organoid structures, with morphological variations depending on the culture conditions.117 In this system, scientists have successfully modeled IBD pathology and achieved conventional in vitro IL-4/IL-13-induced M2 macrophage differentiation within organoids. Moreover, conditioned medium from organoid cultures induces immunomodulatory effects mediated by CD5 antigen-like molecules, further clarifying the interactions between intestinal epithelial cells and mucosal immune cells.
IL-22 activates its downstream target, activating transcription factor 3 (ATF3), which is involved in the IL-22/pSTAT3 signaling pathway to support tissue repair and intestinal homeostasis. Stimulation of IECs by IL-22 initiates a signaling cascade that results in STAT3 phosphorylation and promotes IEC proliferation through ATF3 activation. Studies using mouse colon organoids to mimic IBD have shown that induction of the IL-22 pathway enhances host defense and wound repair, thereby slowing disease progression. Similarly, organoid-based experiments indicate that IL-28 controls intestinal crypt proliferation in wild-type organoids via the IL-28RA–STAT1 pathway and upregulates several genes associated with critical functions such as cytokine production, immune response, and positive regulation of wound healing. These findings suggest that IL-28–mediated STAT1 phosphorylation in epithelial cells helps balance intestinal homeostasis.
Drawing on many advances, a emerging strategy involves harvesting ISCs from patient lesions via endoscopic biopsy, expanding them ex vivo using established organoid culture methods, and then transplanting the cells to target sites via endoscopic delivery. Organoid-based experiments have further shown that postbiotics generated by co-fermenting Lactobacillus fermentum and Lactobacillus delbrueckii can protect and restore intestinal barrier function following acute injury in a dose-dependent manner. This approach supports both intestinal therapy and mechanistic studies using organoid models.118 However, several challenges remain in translating organoid transplantation into clinical practice, including refinement of endoscopic cell delivery systems, quality assurance of cultured ISCs, and identification of optimal efficacy evaluation metrics. Thus, the therapeutic potential of intestinal organoid transplantation for IBD warrants further validation through extensive clinical trials.119
Conclusions and Future Perspectives
The primary therapeutic objective in IBD management centers on achieving complete mucosal restoration to ameliorate clinical symptoms, reduce disease recurrence, and prolong surgical remission. Key bacterial metabolites such as SCFAs, tryptophan metabolites, and bile acids serve as crucial messengers through which gut microbiota regulate host physiology. By directly or indirectly modulating intestinal-skin communication ISC function, immune responses, and epithelial barrier integrity, they play a significant role in the pathogenesis of intestinal diseases like IBD.Additionally, the complex bidirectional interplay between epithelial components and immunocompetent cells poses substantial challenges for functional epithelial barrier reconstitution. Notably, the successful mucosal healing observed in specific IBD patient subgroups underscores the therapeutic efficacy of next-generation biologics (eg, anti-IL-23p19, JAK inhibitors) targeting immunomodulatory pathways. Organoid model studies further elucidate that specific gut microbiota (such as Akkermansia muciniphila) or their metabolites exert dose-dependent protective effects under acute injury conditions, promoting epithelial repair and barrier function recovery. This discovery provides robust preclinical evidence for developing targeted therapies based on the microbiome.
The development of innovative molecular biomarkers and precision diagnostic platforms remains crucial for optimizing treatment stratification and monitoring therapeutic efficacy. Emerging therapeutic domains encompass three key therapeutic avenues: immunomodulators with enhanced tissue specificity, stem cell-derived regenerative therapies, and targeted microbiome modulation technologies—all requiring systematic preclinical and clinical evaluation. Future research should prioritize deciphering the molecular characteristics of epithelial-immune signaling networks and translating these mechanistic discoveries into personalized treatment strategies, enabling precision and customized medicine.
Abbreviations
IBD, Inflammatory bowel disease; UC, Ulcerative colitis; CD, Crohn’s disease; IECs, Intestinal epithelial cells; ISCs, Intestinal stem cells; APC, Antigen-Presenting Cell; Treg, Regulatory T cells; NKT, Natural Killer T cells; IFN-γ, Interferon-gamma; TNF-α, Tumor Necrosis Factor-alpha; IL, Interleukin; MMPs, Matrix Metalloproteinases; GM-CSF, Granulocyte-Macrophage Colony-Stimulating Factor; RORγt, Retinoic Acid-Related Orphan Nuclear Receptor gamma; NF-κB, Nuclear Factor kappa-light-chain-enhancer of activated B cells; TGF-β, Transforming Growth Factor-beta; GALT, Gut-Associated Lymphoid Tissue; PAMPs, Pathogen-Associated Molecular Patterns; DAMPs, Damage-Associated Molecular Patterns; M1, Classically Activated Macrophages; M2, Alternatively Activated Macrophages; cDCs, Conventional Dendritic Cells; EGF, epidermal growth factor; BMP, bone morphogenetic protein; LP, lamina propria; MPST, significant downregulation of mercaptopyruvate sulfurtransferase; ITF, intestinal trefoil factor; KGF, keratinocyte growth factor; P-calmodulin, Phosphorylated calmodulin; β-hydroxybutyratecy; COX-2, clooxygenase-2; GSDMB, mesenchymal stem cellMSC Gasdermin B; FPR1, formyl peptide receptor 1; PRRS, pattern recognition receptors; RIPKs, Receptor-interacting protein kinases; NOD, oligomerization domain, NK, natural killer; ROS, reactive oxygen species; NETs, neutrophil extracellular traps,; PAD4, Peptidylarginine deiminase 4; LTB4, Leukotriene B4; JAM-A, junctional adhesion molecule-A; WT, wild-type; FGL2, Fibrinogen-like protein 2; c-FLIP, cellular FLICE-inhibitory protein; FFAR2, free fatty acid receptor 2; RORγt, receptor-related orphan receptor gamma; LIF, leukemia inhibitory factor; IL-22BP, IL-22 binding protein; NKT, natural killer T; IFN-γ, interferon-gamma; TNF-α, tumor necrosis factor-alpha; pIgR, polymeric immunoglobulin receptor; nTreg, natural Treg; TGF-β, transforming growth factor-beta.
Acknowledgments
We thank Figdraw for providing drawing services.
Funding
This work was supported by Guizhou Provincial Basic Research Program (Natural Science) (NO. ZK [2021]440), the PhD Fund of Scientific Research Foundation of School and Hospital of Stomatology, Zunyi Medical University (NO. KY2020-14).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28(1):573–20. doi:10.1146/annurev-immunol-030409-101225
2. Huldani H, Margiana R, Ahmad F, et al. Immunotherapy of inflammatory bowel disease (IBD) through mesenchymal stem cells. Int Immunopharmacol. 2022;107:108698. doi:10.1016/j.intimp.2022.108698
3. Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014;13:3–10. doi:10.1016/j.autrev.2013.06.004
4. Lauritano D, Boccalari E, Di Stasio D, et al. Prevalence of oral lesions and correlation with intestinal symptoms of inflammatory bowel disease: a systematic review. Diagnostics. 2019;9. doi:10.3390/diagnostics9030077
5. Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and treg cells. Curr Opin Immunol. 2009;21:274–280. doi:10.1016/j.coi.2009.05.021
6. Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–418. doi:10.1038/nature09637
7. Sailaja BS, He XC, Li L. The regulatory niche of intestinal stem cells. J Physiol. 2016;594:4827–4836. doi:10.1113/jp271931
8. Saez A, Herrero-Fernandez B, Gomez-Bris R, Sánchez-Martinez H, Gonzalez-Granado JM. Pathophysiology of inflammatory bowel disease: innate immune system. Int J Mol Sci. 2023;24. doi:10.3390/ijms24021526
9. Chang JT. Pathophysiology of inflammatory bowel diseases. N Engl J Med. 2020;383:2652–2664. doi:10.1056/NEJMra2002697
10. Chen Y, Cui W, Li X, Yang H. Interaction between commensal bacteria, immune response and the intestinal barrier in inflammatory bowel disease. Front Immunol. 2021;12:761981. doi:10.3389/fimmu.2021.761981
11. Blander JM. On cell death in the intestinal epithelium and its impact on gut homeostasis. Curr Opin Gastroenterol. 2018;34:413–419. doi:10.1097/mog.0000000000000481
12. Biton M, Haber AL, Rogel N, et al. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell. 2018;175:1307–1320.e1322. doi:10.1016/j.cell.2018.10.008
13. Hu D, Yan H, He XC, Li L. Recent advances in understanding intestinal stem cell regulation. F1000Res. 2019;8. doi:10.12688/f1000research.16793.1
14. Goto Y, Ivanov II. Intestinal epithelial cells as mediators of the commensal-host immune crosstalk. Immunol Cell Biol. 2013;91:204–214. doi:10.1038/icb.2012.80
15. Zhou L, Sonnenberg GF. Essential immunologic orchestrators of intestinal homeostasis. Sci Immunol. 2018. doi:10.1126/sciimmunol.aao1605
16. Xie J, Li L, Deng S, et al. Slit2/Robo1 mitigates DSS-induced ulcerative colitis by activating autophagy in intestinal stem cell. Int J Biol Sci. 2020;16:1876–1887. doi:10.7150/ijbs.42331
17. Deng F, Wu Z, Zou F, Wang S, Wang X. The Hippo-YAP/TAZ signaling pathway in intestinal self-renewal and regeneration after injury. Front Cell Dev Biol. 2022;10:894737. doi:10.3389/fcell.2022.894737
18. Zhang J, Cen L, Zhang X, et al. MPST deficiency promotes intestinal epithelial cell apoptosis and aggravates inflammatory bowel disease via AKT. Redox Biol. 2022;56:102469. doi:10.1016/j.redox.2022.102469
19. Deng Q, Yao Y, Yang J, et al. AKR1B8 deficiency drives severe DSS-induced acute colitis through invasion of luminal bacteria and activation of innate immunity. Front Immunol. 2022;13:1042549. doi:10.3389/fimmu.2022.1042549
20. Anzai S, Kawamoto A, Nagata S, et al. TGF-β promotes fetal gene expression and cell migration velocity in a wound repair model of untransformed intestinal epithelial cells. Biochem Biophys Res Commun. 2020;524:533–541. doi:10.1016/j.bbrc.2020.01.108
21. Le J, Zhang DY, Zhao Y, Qiu W, Wang P, Sun Y. ITF promotes migration of intestinal epithelial cells through crosstalk between the ERK and JAK/STAT3 pathways. Sci Rep. 2016;6:33014. doi:10.1038/srep33014
22. Naydenov NG, Lechuga S, Zalavadia A, et al. P-Cadherin regulates intestinal epithelial cell migration and mucosal repair, but is dispensable for colitis associated colon cancer. Cells. 2022:11. doi:10.3390/cells11091467
23. Rao JN, Li J, Li L, Bass BL, Wang JY. Differentiated intestinal epithelial cells exhibit increased migration through polyamines and myosin II. Am J Physiol. 1999;277:G1149–1158. doi:10.1152/ajpgi.1999.277.6.G1149
24. Liang X, Li C, Song J, et al. HucMSC-exo promote mucosal healing in experimental colitis by accelerating intestinal stem cells and epithelium regeneration via wnt signaling pathway. Int J Nanomed. 2023;18:2799–2818. doi:10.2147/ijn.S402179
25. Liu H, Wang J, He T, et al. Butyrate: a double-edged sword for health? Adv Nutr. 2018;9:21–29. doi:10.1093/advances/nmx009
26. Vinolo MA, Rodrigues HG, Hatanaka E, Hebeda CB, Farsky SH, Curi R. Short-chain fatty acids stimulate the migration of neutrophils to inflammatory sites. Clin Sci. 2009;117:331–338. doi:10.1042/cs20080642
27. Kim S, Shin YC, Kim TY, et al. Mucin degrader Akkermansia muciniphila accelerates intestinal stem cell-mediated epithelial development. Gut Microbes. 2021;13:1–20. doi:10.1080/19490976.2021.1892441
28. Duan C, Wu J, Wang Z, et al. Fucose promotes intestinal stem cell-mediated intestinal epithelial development through promoting Akkermansia-related propanoate metabolism. Gut Microbes. 2023;15:2233149. doi:10.1080/19490976.2023.2233149
29. Zheng J, Sun T, Qin T, et al. Tryptophan attenuates chronic restraint stress-induced intestinal injury through modulation of intestinal barrier integrity and gut microbiota homeostasis. Nutrients. 2025;17. doi:10.3390/nu17060975
30. Zhao X, Ma L, Dai L, et al. TNF‑α promotes the malignant transformation of intestinal stem cells through the NF‑κB and Wnt/β‑catenin signaling pathways. Oncol Rep. 2020;44:577–588. doi:10.3892/or.2020.7631
31. Sorrentino G, Perino A, Yildiz E, et al. Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology. 2020;159:956–968.e958. doi:10.1053/j.gastro.2020.05.067
32. Mao T, Xu X, Liu L, et al. ABL1‒YAP1 axis in intestinal stem cell activated by deoxycholic acid contributes to hepatic steatosis. J Transl Med. 2024;22:1119. doi:10.1186/s12967-024-05865-6
33. Gomez-Bris R, Saez A, Herrero-Fernandez B, Rius C, Sanchez-Martinez H, Gonzalez-Granado JM. CD4 T-Cell subsets and the pathophysiology of inflammatory bowel disease. Int J Mol Sci. 2023;24. doi:10.3390/ijms24032696
34. Giambra V, Pagliari D, Rio P, et al. Gut microbiota, inflammatory bowel disease, and cancer: the role of guardians of innate immunity. Cells. 2023:12. doi:10.3390/cells12222654
35. Drury B, Hardisty G, Gray RD, Ho GT. Neutrophil extracellular traps in inflammatory bowel disease: pathogenic mechanisms and clinical translation. Cell Mol Gastroenterol Hepatol. 2021;12:321–333. doi:10.1016/j.jcmgh.2021.03.002
36. Danne C, Skerniskyte J, Marteyn B, Sokol H. Neutrophils: from IBD to the gut microbiota. Nat Rev Gastroenterol Hepatol. 2024;21:184–197. doi:10.1038/s41575-023-00871-3
37. Wang S, Song Y, Wang Z, et al. Neutrophil-derived PAD4 induces citrullination of CKMT1 exacerbates mucosal inflammation in inflammatory bowel disease. Cell Mol Immunol. 2024;21:620–633. doi:10.1038/s41423-024-01158-6
38. Lin EY, Lai HJ, Cheng YK, et al. Neutrophil extracellular traps impair intestinal barrier function during experimental colitis. Biomedicines. 2020;8. doi:10.3390/biomedicines8080275
39. Kang L, Fang X, Song YH, et al. Neutrophil-epithelial crosstalk during intestinal inflammation. Cell Mol Gastroenterol Hepatol. 2022;14:1257–1267. doi:10.1016/j.jcmgh.2022.09.002
40. Zhang L, Zheng B, Bai Y, et al. Exosomes-transferred LINC00668 contributes to thrombosis by promoting NETs formation in inflammatory bowel disease. Adv Sci. 2023;10(e2300560). doi:10.1002/advs.202300560
41. Delfini M, Stakenborg N, Viola MF, Boeckxstaens G. Macrophages in the gut: masters in multitasking. Immunity. 2022;55:1530–1548. doi:10.1016/j.immuni.2022.08.005
42. Zhang K, Guo J, Yan W, Xu L. Macrophage polarization in inflammatory bowel disease. Cell Commun Signal. 2023;21:367. doi:10.1186/s12964-023-01386-9
43. Yu D, Zhao Y, Wang H, et al. IL-1β pre-stimulation enhances the therapeutic effects of endometrial regenerative cells on experimental colitis. Stem Cell Res Ther. 2021;12:324. doi:10.1186/s13287-021-02392-9
44. Bernshtein B, Curato C, Ioannou M, et al. IL-23-producing IL-10Rα-deficient gut macrophages elicit an IL-22-driven proinflammatory epithelial cell response. Sci Immunol. 2019;4. doi:10.1126/sciimmunol.aau6571
45. Han C, Sheng Y, Wang J, et al. NOX4 promotes mucosal barrier injury in inflammatory bowel disease by mediating macrophages M1 polarization through ROS. Int Immunopharmacol. 2022;104:108361. doi:10.1016/j.intimp.2021.108361
46. Spalinger MR, Sayoc-Becerra A, Santos AN, et al. PTPN2 regulates interactions between macrophages and intestinal epithelial cells to promote intestinal barrier function. Gastroenterology. 2020;159:1763–1777.e1714. doi:10.1053/j.gastro.2020.07.004
47. Zhang Q, Chen LH, Yang H, et al. GPR84 signaling promotes intestinal mucosal inflammation via enhancing NLRP3 inflammasome activation in macrophages. Acta Pharmacol Sin. 2022;43:2042–2054. doi:10.1038/s41401-021-00825-y
48. Yang ZJ, Wang BY, Wang TT, et al. Functions of dendritic cells and its association with intestinal diseases. Cells. 2021;10. doi:10.3390/cells10030583
49. Kuttke M, Hromadová D, Yildirim C, et al. PI3K signaling in dendritic cells aggravates DSS-Induced colitis. Front Immunol. 2022;13:695576. doi:10.3389/fimmu.2022.695576
50. Jiménez-Cortegana C, Palomares F, Alba G, et al. Dendritic cells: the yin and yang in disease progression. Front Immunol. 2023;14:1321051. doi:10.3389/fimmu.2023.1321051
51. Ninnemann J, Winsauer C, Bondareva M, et al. TNF hampers intestinal tissue repair in colitis by restricting IL-22 bioavailability. Mucosal Immunol. 2022;15:698–716. doi:10.1038/s41385-022-00506-x
52. Muzaki AR, Tetlak P, Sheng J, et al. Intestinal CD103(+)CD11b(-) dendritic cells restrain colitis via IFN-γ-induced anti-inflammatory response in epithelial cells. Mucosal Immunol. 2016;9:336–351. doi:10.1038/mi.2015.64
53. Thelemann C, Eren RO, Coutaz M, et al. Interferon-γ induces expression of MHC class II on intestinal epithelial cells and protects mice from colitis. PLoS One. 2014;9(e86844). doi:10.1371/journal.pone.0086844
54. Li T, Chen RR, Gong HP, et al. FGL2 regulates IKK/NF-κB signaling in intestinal epithelial cells and lamina propria dendritic cells to attenuate dextran sulfate sodium-induced colitis. Mol Immunol. 2020;117:84–93. doi:10.1016/j.molimm.2019.11.001
55. Wang Q, Chen F, Peng Y, Yi X, He Y, Shi Y. Research progress of interleukin-27 in inflammatory bowel disease. Inflamm Bowel Dis. 2024;30:303–310. doi:10.1093/ibd/izad153
56. Rees WD, Sly LM, Steiner TS. How do immune and mesenchymal cells influence the intestinal epithelial cell compartment in inflammatory bowel disease? Let’s crosstalk about it! J Leukoc Biol. 2020;108:309–321. doi:10.1002/jlb.3mir0120-567r
57. Lockhart A, Mucida D, Bilate AM. Intraepithelial lymphocytes of the intestine. Annu Rev Immunol. 2024;42:289–316. doi:10.1146/annurev-immunol-090222-100246
58. Binienda A, Ziolkowska S, Hauge IH, Salaga M. The role of immune and epithelial stem cells in inflammatory bowel disease therapy. Curr Drug Targets. 2020;21:1405–1416. doi:10.2174/1389450121666200504074922
59. Schulz-Kuhnt A, Neurath MF, Wirtz S, Atreya I. Innate lymphoid cells as regulators of epithelial integrity: therapeutic implications for inflammatory bowel diseases. Front Med. 2021;8:656745. doi:10.3389/fmed.2021.656745
60. Frisbee AL, Saleh MM, Young MK, et al. IL-33 drives group 2 innate lymphoid cell-mediated protection during clostridium difficile infection. Nat Commun. 2019;10:2712. doi:10.1038/s41467-019-10733-9
61. Garrido-Mesa N, Schroeder JH, Stolarczyk E, et al. T-bet controls intestinal mucosa immune responses via repression of type 2 innate lymphoid cell function. Mucosal Immunol. 2019;12:51–63. doi:10.1038/s41385-018-0092-6
62. Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. 2019;10:315. doi:10.1038/s41419-019-1540-2
63. Cochez PM, Michiels C, Hendrickx E, et al. AhR modulates the IL-22-producing cell proliferation/recruitment in imiquimod-induced psoriasis mouse model. Eur J Immunol. 2016;46:1449–1459. doi:10.1002/eji.201546070
64. Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol. 2018;53:465–474. doi:10.1007/s00535-017-1401-7
65. Guo Y, Liu Y, Rui B, et al. Crosstalk between the gut microbiota and innate lymphoid cells in intestinal mucosal immunity. Front Immunol. 2023;14:1171680. doi:10.3389/fimmu.2023.1171680
66. Ramos GP, Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. 2019;94:155–165. doi:10.1016/j.mayocp.2018.09.013
67. Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi:10.1038/ni1511
68. Arseneau KO, Cominelli F. Targeting leukocyte trafficking for the treatment of inflammatory bowel disease. Clin Pharmacol Ther. 2015;97:22–28. doi:10.1002/cpt.6
69. Veny M, Fernández-Clotet A, Panés J. Controlling leukocyte trafficking in IBD. Pharmacol Res. 2020;159:105050. doi:10.1016/j.phrs.2020.105050
70. Raouia F, Mariem BJ, Nesrine E, et al. CD4(+) T-cell subsets and cytokine signature in pemphigus foliaceus clinical stratification beyond the th1/Th2 paradigm. Curr Mol Med. 2025;25:485–495. doi:10.2174/0115665240305096240611064617
71. Kałużna A, Olczyk P, Komosińska-Vassev K. The role of innate and adaptive immune cells in the pathogenesis and development of the inflammatory response in ulcerative colitis. J Clin Med. 2022;11. doi:10.3390/jcm11020400
72. Caza T, Landas S. Functional and phenotypic plasticity of CD4(+) T cell subsets. Biomed Res Int. 2015;2015:521957. doi:10.1155/2015/521957
73. Monteleone G, Trapasso F, Parrello T, et al. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol. 1999;163:143–147.
74. Hartman ML, Czyz M. BCL-G: 20 years of research on a non-typical protein from the BCL-2 family. Cell Death Differ. 2023;30:1437–1446. doi:10.1038/s41418-023-01158-5
75. Zeissig S, Bojarski C, Buergel N, et al. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut. 2004;53:1295–1302. doi:10.1136/gut.2003.036632
76. Takashima S, Martin ML, Jansen SA, et al. T cell-derived interferon-γ programs stem cell death in immune-mediated intestinal damage. Sci Immunol. 2019;4. doi:10.1126/sciimmunol.aay8556
77. Ruan S, Xu L, Sheng Y, et al. Th1 promotes M1 polarization of intestinal macrophages to regulate colitis-related mucosal barrier damage. Aging. 2023;15:6721–6735. doi:10.18632/aging.204629
78. Łukasik Z, Gracey E, Venken K, Ritchlin C, Elewaut D. Crossing the boundaries: IL-23 and its role in linking inflammation of the skin, gut and joints. Rheumatology. 2021;
79. Akiyama M, Alshehri W, Suzuki K, Shimanuki K, Saito K, Kaneko Y. Clinical features, pathogenesis, and treatment of inflammatory bowel disease-associated spondyloarthritis. Autoimmun Rev. 2025;24:103853. doi:10.1016/j.autrev.2025.103853
80. Schmitt H, Neurath MF, Atreya R. Role of the IL23/IL17 pathway in crohn’s disease. Front Immunol. 2021;12:622934. doi:10.3389/fimmu.2021.622934
81. Swedik SM, Madola A, Cruz MA, Llorens-Bonilla BJ, Levine AD. Th17-Derived cytokines synergistically enhance IL-17C production by the colonic epithelium. J Immunol. 2022;209:1768–1777. doi:10.4049/jimmunol.2200125
82. Verstockt B, Salas A, Sands BE, et al. IL-12 and IL-23 pathway inhibition in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2023;20:433–446. doi:10.1038/s41575-023-00768-1
83. Bunte K, Beikler T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int J Mol Sci. 2019. doi:10.3390/ijms20143394
84. Hundorfean G, Neurath MF, Mudter J. Functional relevance of T helper 17 (Th17) cells and the IL-17 cytokine family in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:180–186. doi:10.1002/ibd.21677
85. Zenewicz LA. IL-22 binding protein (IL-22BP) in the regulation of IL-22 biology. Front Immunol. 2021;12:766586. doi:10.3389/fimmu.2021.766586
86. Brand S, Beigel F, Olszak T, et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol. 2006;290:G827–838. doi:10.1152/ajpgi.00513.2005
87. Dambacher J, Beigel F, Zitzmann K, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. 2009;58:1207–1217. doi:10.1136/gut.2007.130112
88. Yan JB, Luo MM, Chen ZY, He BH. The function and role of the Th17/Treg cell balance in inflammatory bowel disease. J Immunol Res. 2020;2020:8813558. doi:10.1155/2020/8813558
89. Yang Y, Deng Y, Zhang G, et al. α-mangostin derivatives ameliorated mouse DSS-induced chronic colitis via regulating Th17/Treg balance. Mol Immunol. 2024;166:110–118. doi:10.1016/j.molimm.2023.11.013
90. Meitei HT, Jadhav N, Lal G. CCR6-CCL20 axis as a therapeutic target for autoimmune diseases. Autoimmun Rev. 2021;20:102846. doi:10.1016/j.autrev.2021.102846
91. Kulkarni N, Meitei HT, Sonar SA, et al. CCR6 signaling inhibits suppressor function of induced-Treg during gut inflammation. J Autoimmun. 2018;88:121–130. doi:10.1016/j.jaut.2017.10.013
92. Harrison OJ, Srinivasan N, Pott J, et al. Epithelial-derived IL-18 regulates Th17 cell differentiation and Foxp3⁺ Treg cell function in the intestine. Mucosal Immunol. 2015;8:1226–1236. doi:10.1038/mi.2015.13
93. Mu J, Maeda K, Ohashi A, et al. Monoclonal antibodies against mature interleukin-18 ameliorate colitis and repair goblet cell function. Dig Dis Sci. 2024;69:2573–2585. doi:10.1007/s10620-024-08453-2
94. Wu J, Zhang X, Wu D, Jin O, Gu J. Evaluation of causal associations between interleukin-18 levels and immune-mediated inflammatory diseases: a Mendelian randomization study. BMC Med Genomics. 2023;16:306. doi:10.1186/s12920-023-01744-z
95. Pizarro TT, Michie MH, Bentz M, et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J Immunol. 1999;162:6829–6835.
96. Wang X, Zhu Y, Zhang M, Wang H, Jiang Y, Gao P. Ulcerative colitis is characterized by a decrease in regulatory B cells. J Crohns Colitis. 2016;10:1212–1223. doi:10.1093/ecco-jcc/jjw074
97. Paulos CM, Wrzesinski C, Kaiser A, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117:2197–2204. doi:10.1172/jci32205
98. Zhu J, Xu Y, Zhu C, et al. IL-33 induces both regulatory B cells and regulatory T cells in dextran sulfate sodium-induced colitis. Int Immunopharmacol. 2017;46:38–47. doi:10.1016/j.intimp.2017.02.006
99. Williams MA, O’Callaghan A, Corr SC. IL-33 and IL-18 in inflammatory bowel disease etiology and microbial interactions. Front Immunol. 2019;10:1091. doi:10.3389/fimmu.2019.01091
100. Jin T, Lu H, Zhou Q, et al. H 2 S-Releasing versatile montmorillonite nanoformulation trilogically renovates the gut microenvironment for inflammatory bowel disease modulation. Adv Sci. 2024;11(14):e2308092. doi:10.1002/advs.202308092
101. Coccia M, Harrison OJ, Schiering C, et al. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med. 2012;209:1595–1609. doi:10.1084/jem.20111453
102. Pang J, Feng JN, Ling W, Jin T. The anti-inflammatory feature of glucagon-like peptide-1 and its based diabetes drugs-therapeutic potential exploration in lung injury. Acta Pharm Sin B. 2022;12:4040–4055. doi:10.1016/j.apsb.2022.06.003
103. Migliorisi G, Gabbiadini R, Dal Buono A, et al. GLP-1 receptor agonists in IBD: exploring the crossroads of metabolism and inflammation. Front Immunol. 2025;16:1610368. doi:10.3389/fimmu.2025.1610368
104. Neurath MF, Artis D, Becker C. The intestinal barrier: a pivotal role in health, inflammation, and cancer. Lancet Gastroenterol Hepatol. 2025;10:573–592. doi:10.1016/S2468-1253(24)00390-X
105. Luck H, Tsai S, Chung J, et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. 2015;21:527–542. doi:10.1016/j.cmet.2015.03.001
106. Seiler A, Schneider M, Förster H, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-Mediated cell death. Cell Metab. 2008;8:237–248. doi:10.1016/j.cmet.2008.07.005
107. Yan J, Zeng Y, Guan Z, et al. Inherent preference for polyunsaturated fatty acids instigates ferroptosis of Treg cells that aggravates high-fat-diet-related colitis. Cell Rep. 2024;43. doi:10.1016/j.celrep.2024.114636
108. Khaloian S, Rath E, Hammoudi N, et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut. 2020;69:1939–1951. doi:10.1136/gutjnl-2019-319514
109. Cairns CA, Chen T, Han N, et al. Polyamines regulate mitochondrial metabolism essential for intestinal epithelial renewal and wound healing. Am J Physiol Gastrointest Liver Physiol. 2025:
110. Di Mola FF, Friess H, Scheuren A, et al. Transforming growth factor-βs and their signaling receptors are coexpressed in crohn’s disease. Ann Surg. 1999;229(1):67–75.
111. Rieder F, Mukherjee PK, Massey WJ, Wang Y, Fiocchi C. Fibrosis in IBD: from pathogenesis to therapeutic targets. Gut. 2024;73:854–866. doi:10.1136/gutjnl-2023-329963
112. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587:555–566. doi:10.1038/s41586-020-2938-9
113. Imai J, Kitamoto S, Sugihara K, et al. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019;12:632–643. doi:10.1038/s41385-019-0138-4
114. Bamias G, Filidou E, Goukos D, et al. Crohn’s disease-associated mucosal factors regulate the expression of TNF-like cytokine 1A and its receptors in primary subepithelial intestinal myofibroblasts and intestinal epithelial cells. Transl Res. 2017;180:118–130.e112. doi:10.1016/j.trsl.2016.08.007
115. Solitano V, Jairath V, Ungaro F, Peyrin-Biroulet L, Danese S. TL1A inhibition for inflammatory bowel disease treatment: from inflammation to fibrosis. Med. 2024;5:386–400. doi:10.1016/j.medj.2024.03.010
116. Zhang MJ, Chan SX, Jia ZG, Lv C, Chen JJ, Hong SC. Roles of intestinal stem cells in inflammatory bowel disease pathogenesis. World J Stem Cells. 2025;17:107639. doi:10.4252/wjsc.v17.i8.107639
117. Liu W, Wang Q, Bai Y, et al. Potential application of intestinal organoids in intestinal diseases. Stem Cell Rev Rep. 2024;20:124–137. doi:10.1007/s12015-023-10651-w
118. Cercamondi CI, Bendik I, Eckhardt E, et al. A postbiotic derived from lactobacillaceae protects intestinal barrier function in a challenge model using colon organoid tubules. Foods. 2025;14. doi:10.3390/foods14071173
119. Kakar S, Derouet MF, Zhang L, et al. Development of a tissue-specific bioscaffold for intestinal stem cell culture. PLoS One. 2025;20:e0328898. doi:10.1371/journal.pone.0328898
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.