Back to Journals » Infection and Drug Resistance » Volume 19
Beyond Thresholds: CMV Replication Kinetics as Early Predictors of Clinical Risk in Transplant Recipients
Authors Kleiboeker HL ![]() , Descourouez JL, Saddler C, Jorgenson MR
, Descourouez JL, Saddler C, Jorgenson MR
Received 1 October 2025
Accepted for publication 19 December 2025
Published 7 January 2026 Volume 2026:19 547565
DOI https://doi.org/10.2147/IDR.S547565
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chi H. Lee
Hanna L Kleiboeker,1 Jillian L Descourouez,1 Chris Saddler,2 Margaret R Jorgenson1
1Department of Pharmacy, UW Health, Madison, WI, USA; 2Department of Medicine, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA
Correspondence: Margaret R Jorgenson, UW Health, 600 Highland Ave, Madison, WI, 53792, USA, Email [email protected]
Abstract: Cytomegalovirus (CMV) is a pervasive beta herpes virus which establishes latency after primary infection. CMV infection after solid organ transplant is common and associated with negative outcomes for both the allograft and patient. The medications that are available can be associated with significant toxicity and do not provide a cure. Reliable protection from recurrence and freedom from antiviral therapy requires development of CMV-specific cell-mediated immunity (CMI). This can be difficult to achieve after transplant in the setting of immunosuppressive medications due to the complexity of the immune response required to control the virus and often leads to prolonged antiviral courses and lengthy monitoring periods. While assays exist to measure CMI, they are not widely available and can pose challenges related to cost and collection restrictions. Conversely, viral replication trends utilizing widely available molecular diagnostics in tandem with risk factor assessment can be used as surrogate markers for the presence and development of CMI which can guide need for antiviral prophylaxis or treatment. The purpose of this review is to establish a foundation for the immunology of CMV and widely used molecular diagnostics, highlight CMV-specific CMI testing as an emerging molecular diagnostic tool, justify how the currently available antiviral drugs have limitations in their utility and outline the opportunity to use viral replication and replication kinetics, rather than absolute viral load thresholds, as a tool to understand the complex interplay of the host’s immune response and the virus’ capacity for replication while guiding clinical decision making.
Keywords: transplantation, immunosuppression, infection, outcomes
Introduction
Cytomegalovirus (CMV) is a ubiquitous beta herpes virus. Seropositivity rates in the United States are 40–80% and worldwide can be >90%.1 Exposure and primary infection typically occur in childhood, followed by lifelong latency.2 Infection in immunocompetent individuals is typically asymptomatic; however, primary infection in the immunocompromised host, such as after solid organ transplant (SOT), can be severe, particularly in those patients who were not exposed prior to transplant (R-) and received a graft from a donor who was exposed (D+).3 Activation has been associated with negative outcomes after transplant, so prevention strategies are warranted. That said, there is no treatment currently available to “cure” this latent herpes viral infection; currently available treatments are virostatic. These medications can inactivate the virus but rely on the host immune system to eliminate it, which can be problematic in immunosuppressed patient populations such as SOT recipients and result in prolonged treatment courses and recurrent infections. Treatment strategies ultimately rely on immune priming and subsequent development or reconstitution of host immunity to provide sustained remission and long-term freedom from antiviral drugs, associated toxicities and resistance. This development of CMV-specific cell-mediated immunity (CMI) is the goal of CMV prevention and treatment in SOT.4 Although assays have been developed to detect CMV-specific CMI, their clinical use and reliability have not been completely established and access can be limited by pragmatic factors such as collection, processing and shipping restrictions. Currently, the best strategy for detection of CMI is clinical demonstration of the ability to maintain in latency via serial CMV viral load monitoring via molecular diagnostics, which are widely available. When used optimally, monitoring viral load trends and replication kinetics not only predicts the presence of CMI early after activation but can also be used at end of treatment to predict recurrence and during prophylaxis utilizing the preemptive monitoring approach to dictate preemptive treatment. Replication kinetics can also be used to predict development of resistance and indicate when adjustment of therapy or immunosuppression medications is needed. This is particularly important given lack of well-established viral load thresholds that are associated with clinical outcomes. This review aims to provide background on why replication kinetics are important, summarize how they are measured, describe shortcomings of current immunoassays and antiviral therapeutics and finally review how we have used replication kinetics as a readily accessible tool to guide clinical decision making in the management of CMV in immunocompromised patient populations at our center.
Immunology of Cytomegalovirus and Detection
After transmission, CMV infections are considered to have three distinct and dynamic phases characterized by different immune responses: acute infection (phase I), persistence (Phase II) and latency/reactivation (Phase III).5 Phase I is characterized by the innate immune system’s response to the initial CMV infection. This phase is primarily moderated by natural killer (NK) cells and type I interferon (IFN-I).6,7 IFN-I is triggered in two systemic phases: an initial phase that peaks 10–12 hours after infection and a delayed phase that occurs 36–48 hours after infection directed by dendritic cells.8,9 These dendritic cells produce IL-12 and IL-18, which further contributes to NK cell activation and priming the adaptive immune response. Other components of the innate immune system come into play during phase I including macrophages localized to lymphoid tissues, which can prevent infection into select cells; mast cells, which can produce chemokines that draw CD8 T cells to the site of infection; and neutrophils, which can activate TNF-related apoptosis inducing ligand (TRAIL).10–12 During Phase I, serology will show a positive IgM response.
Phase II is characterized by the adaptive immune system’s response. During the first week after infection, CMV-specific CD4 and CD8 T cell responses are primed and go on to launch a population of stable memory T cells.13,14 A specific type of T cells also expands during phase II (and later in phase III): inflationary memory cells, which are maintained by CD8 T cells and do not demonstrate expected signs of T cell exhaustion that can be characteristic of chronic viral infections.15,16 Inflationary memory cells typically display an effector-memory phenotype (Tem).17 Observed CD4 and CD8 T cell response is quite broad, involving up to 20% of all circulating T cells.18 CMV-specific CD4 T cells with a cytolytic phenotype (CD4-cytotoxic T lymphocytes, CD4-CTL) are produced and induced during this time.19 Humoral immune responses are also present during Phase II. The CMV IgM response of phase I tapers off, and CMV IgG response steadily increases through phase II (and later in phase III).19 Memory B cells are developed though the timing of production in phase II is not yet understood. During Phase II, serology will show positive IgM and IgG responses.
Phase III is characterized by viral latency in a number of different cell types including myeloid cells, hematopoietic stem cells, endothelial cells, and epithelial cells.20 The viral genome is stored as extra-chromosomal genetic material in these cells and tissues for the life of the host. Gene expression is restricted and does not perpetuate viral replication in latency. Importantly,
CMV can be reactivated to a fully replicative virion under select conditions.20 These conditions include critical illness, clinical stressors such as other infections or select underlying conditions, and introduction of immunosuppression such as that introduced after solid organ transplant to protect the allograft from rejection. During Phase III, serology will consistently show a positive IgG response; the durability of IgG response explains why CMV IgG (as opposed to CMV IgM or the combination of CMV IgG and IgM) is recommended by consensus guidelines for pre-transplant testing of both donors and recipients in solid organ transplant.21,22
Further complicating the complex interplay between the host’s immune system and CMV is the immunomodulatory nature of CMV. CMV is able to curb NK cell activating ligands and the expression of their receptors on the surface of infected cells, as well as inhibiting the expression of TRAIL receptors which inhibits NK-mediated cell death.23,24 CMV has a number of mechanisms which can be used to avoid detection by CD4 and CD8 T cells. CMV can cause downregulation of major histocompatibility complex (MHC) expression on cell membranes and peptide presentation to T cells.25 CMV may also reduce the expression of costimulatory molecules and promote inhibitory ligands, as well as promoting cell death of T cells that face infected dendritic cells.25 CMV can express IL-10, which may contribute to the virus’ ability to replicate during latency by suppressing T cell function.26 Lastly, CMV has the ability to impact T cell function by affecting both chemokines and their receptors to downregulate the inflammatory environment triggered by the host’s immune responses.27
Molecular Diagnostics
Molecular diagnostics play an important role in the detection and treatment of CMV. The first such iteration, the pp65 antigenemia assay, detected a CMV-specific antigen (pp65) in peripheral blood leukocytes; however, this assay had several key flaws including sample processing restrictions, limited utility in patients with leukopenia and lack of standardization between centers.28,29 Given these limitations, use of the pp65 antigenemia assay has fallen out of favor and consensus guidelines no longer recommend its use.21,22
Subsequently, molecular assays that detect CMV nucleic acids (NAT) have become the preferred method for detection of viral replication. While CMV RNA is a very specific for viral replication, there is currently not a commercially available assay at this time. Currently, available assays detect and amplify CMV DNA; however, clinicians must be aware that these highly sensitive assays can amplify latent viral DNA in addition to active replication. For this reason, quantitative NAT assays are preferred over qualitative assays, as absolute viral load can be used to differentiate between latent virus (low-level CMV DNAemia) and active replication (high-level CMV DNAemia).30,31 Samples from plasma and whole blood can be used for the CMV quantitative NAT (QNAT) assay though samples from whole blood tend to have a higher viral load compared to samples from plasma.32–34 A number of commercial assays, which are approved by the US Food and Drug Administration, are available, though use of laboratory-developed assays still occurs.
In an effort to improve commutability of the available assays, the World Health Organization developed an international standard for CMV NAT technology.35 While this improved agreement between various assays, significant differences remain leading consensus guidelines to recommend using a single testing platform and specimen type for a given patient.21,22,36 If this is not feasible, clinicians must take into account interlaboratory variability due to factors such as extraction method and platform, sample volume, amplicon size and variation of primer-binding site.21,22
CMV QNAT assays typically have a lower and upper limit of quantification. Lower limits of quantification are typically 100–500 IU/mL (2–2.7 log10 IU/mL) for commercially available and laboratory-developed assays; newer automated assays may have lower limits of quantification at 34.5 IU/mL (1.5 log10 IU/mL). Given the increasing sensitivity of CMV QNAT assays, clinicians must take into account the logarithmic nature of the assays. In general, changes of <0.5 log10 IU/mL likely lack clinical significance; when viral loads fall below 1000 IU/mL (3 log10 IU/mL), the threshold for significant change increases to 0.7 log10 IU/mL.21 Therefore, it is important to note CMV QNAT assay results using log10 IU/mL to prevent overinterpretation of changes in CMV viral load that lack clinical significance.
Cell-Mediated Immunity Testing
Cellular immunity is crucial to controlling CMV replication and maintaining the virus in latency. Given the importance of CMV-specific cell-mediated immunity (CMI), several immune function assays have been developed to quantify cellular immunity responses against CMV. The first type of immune function assay uses Enzyme-Linked Immunosorbent Assay (ELISA) testing on a whole blood sample to detect IFN-gamma release by CD8 T cells after stimulation with a peptide cocktail designed to mimic CMV proteins.37 There is a commercially available ELISA-based immune function assay, but it is only available in Europe. The second type of immune function assay uses Enzyme-Linked Immunosorbent Spot (ELISpot) testing on peripheral blood mononuclear cells from a whole blood sample to detect IFN-gamma release by CD4 and CD8 T cells after stimulation with CMV-specific antigens pp65 and IE-1; of note, CD4 and CD8 T cell responses cannot be distinguished with this assay.38 The third type of immune function assay uses intracellular cytokine staining (ICS) on a whole blood sample to detect INF-gamma release by CD4 and CD8 T cells after stimulation with CMV-specific antigens pp65 and IE-1; of note, CD4 and CD8 T cell responses can be differentiated with this assay.39 Only one immune function assay is commercially available in the United States at present: Eurofins ViraCor CMV inSIGHT T-Cell Immunity Panel (ICS assay). T-SPOT. CMV (ELISpot assay) is available as a laboratory-developed test.
Early evidence examining the utility of CMV immune function assays in various clinical scenarios is available. Studies using testing in the pre-transplant or early post-transplant period have been completed in seropositive transplant recipients.40–42 Key takeaways from these studies indicated that a positive immune function assay obtained pre-transplant may not accurately predict post-transplant CMV-specific CMI as induction immunosuppression may disrupt the immune response; a positive immune function assay obtained 14 days post-transplant may predict post-transplant CMV-specific CMI and can be used to stop prophylaxis or reduce the intensity of monitoring; and immune function assays can be used post-transplant in seropositive recipients with potential passive antibody transfer to determine true CMV status. Studies using immune function assay testing in the post-transplant period have been completed in conjunction with antiviral prophylaxis43,44 or preemptive therapy.45,46 Key takeaways from the studies completed in conjunction with antiviral prophylaxis included that negative immune function assay results at the end of prophylaxis can be used to continue prophylaxis or transition to post-prophylaxis surveillance, including in patients who received lymphocyte depleting induction. Key takeaways from the studies completed while on preemptive therapy included selecting ongoing monitoring (rather than antiviral treatment) for asymptomatic seropositive patients with viremia and CMV-specific CMI, as well as transitioning patients who lack CMV-specific CMI to secondary prophylaxis after completion of treatment given the high risk of relapse/recurrence.
In summation, at present the currently available literature supports several potential timepoints in the peri- and post-transplant period where clinicians could reasonably consider using an immune function assay. The first timepoint is during or at the end of the primary prophylaxis period in seropositive patients to assess for post-transplant reconstitution of immunity with the intent of stopping prophylaxis or forgoing post-prophylaxis surveillance monitoring if the patient demonstrates CMV-specific CMI. The second timepoint, in addition to an assessment of patient-specific risk factors, is after completion of treatment to assess if secondary prophylaxis should be considered. Clinicians must also recognize that the clinical utility of immune function assays is limited by the fact that an immune function assay only represents a patient’s immune status at a snapshot in time; if the patient’s net burden of immunosuppression is increased by events such as intensification of or change in maintenance immunosuppression, treatment of rejection, re-transplantation or acute infection, the result of the prior immune function assay no longer has the same predictive potential. Furthermore, clinicians must also recognize logistical barriers to the implementation of immune function assays, including collection, processing and shipping restrictions. Future studies utilizing the immune function assay commercially available in the United States will need to be conducted to better understand the use of these assays in the pre-, peri-, and post-transplant settings.
Absolute Lymphocyte Count
In scenarios where immune function assays are unable to be obtained for any reason, non-specific metrics such as CD4+ T-cell count, nonspecific T-cell immune responses and absolute lymphocyte count (ALC) can be obtained to stratify the risk of CMV disease.47–49 Of these metrics, ALC is tested routinely as part of a complete blood count with differential post-solid organ transplant and results are obtained more rapidly than other available metrics, making it the most advantageous in this patient population. ALC thresholds have been associated with post-transplant CMV outcomes. In heart transplant, an ALC < 500 cells/µL on post-operative day 7 was found to be an independent risk factor for early CMV infection.50 An ALC below 610 cells/µL has been shown to be associated with the development of CMV infection in both a general cohort of kidney transplant recipients and a subgroup of high-risk (D+/R-) kidney transplant recipients.51 A study of heart, liver and kidney transplant recipients found that an ALC > 1080 cells/µL may be protective against relapse of CMV disease.52 In the absence of data from immune function assays, ALC may serve as an appropriate surrogate marker for CMV-specific CMI in solid organ transplant recipients at discrete timepoints post-transplant.
Antivirals for Treatment and Prophylaxis
Antivirals aimed at preventing and treating CMV are limited. These available agents are virostatic and include DNA polymerase inhibitors (ganciclovir, foscarnet, cidofovir), kinase inhibitors (maribavir) and terminase inhibitors (letermovir). These antiviral agents work by different mechanisms to inactivate virus but rely on the host immune system to work concomitantly for clearance. (Table 1) This can lead to prolonged treatment courses in immunocompromised patients, which is problematic given the associated toxicity that increases with ongoing exposure. Resistance is also a major issue, given the many evasive tactics employed by the virus, and can develop quickly particularly in the setting of suboptimal or prolonged exposure.53
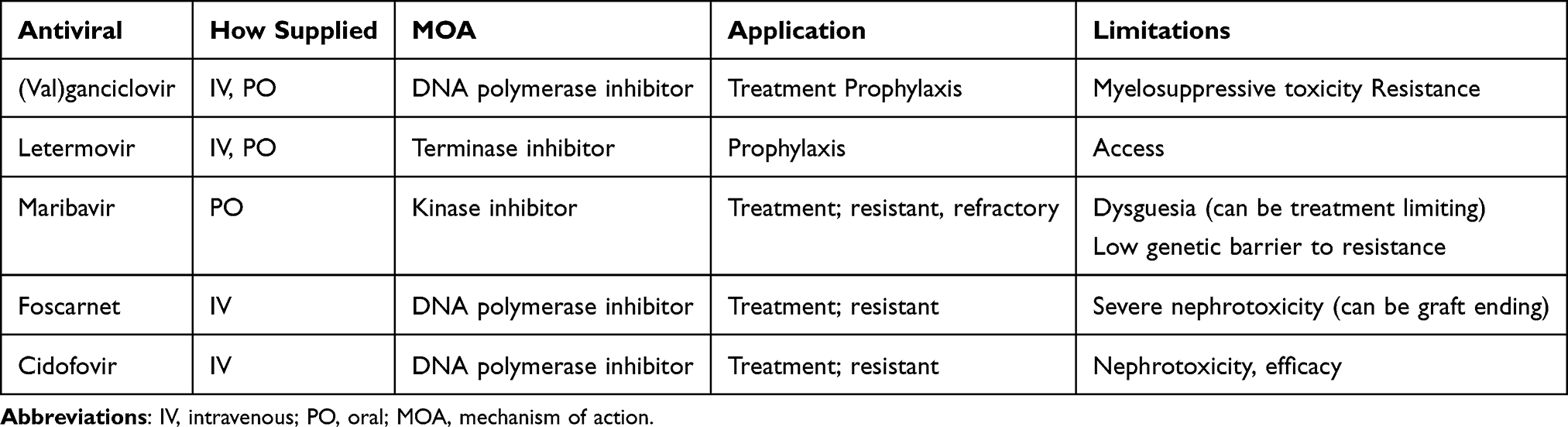 |
Table 1 Anti-Cytomegaloviral Antivirals |
(Val)ganciclovir
Ganciclovir and its oral prodrug, valganciclovir, are the gold standard antiviral agents for treatment and prophylaxis of CMV.21,22 Ganciclovir interferes with viral replication via phosphorylation and subsequent creation of ganciclovir triphosphate which competitively inhibits binding of deoxyguanosine triphosphate to DNA polymerase. This inhibition prevents viral synthesis and replication. Ganciclovir is currently only available intravenously; valganciclovir is the oral equivalent. Use is limited by leukopenia due to bone marrow suppression via inhibition of DNA synthesis in hematopoietic progenitor cells. Bone marrow suppression can be more pronounced in SOT recipients as they are already at risk for leukopenia and neutropenia due to use of lymphocyte-depleting induction agents, maintenance immunosuppression such as mycophenolic acid and other prophylaxis agents such as trimethoprim-sulfamethoxazole. Toxicity is more pronounced with higher doses and ongoing exposure.54,55 Leukopenia can be a rate limiting factor in the ability to use (val)ganciclovir for CMV prophylaxis. Patients may need to be converted to alternative prophylaxis agents or strategies to relieve the bone marrow suppression caused by valganciclovir. Additionally, viral resistance to (val)ganciclovir can occur, which develops due to mutations in CMV UL97 viral kinase or UL54 DNA polymerase genes.56 Risk factors for development include prolonged exposure and suboptimal dosing of (val)ganciclovir.57 When resistance develops, it is difficult to manage given significant tolerability issues with currently available treatment options. Additionally, resistance in associated with morbidity, mortality and negative graft outcomes.58 Guidelines currently recommend maribavir or foscarnet for patients with proven ganciclovir resistance, with choice of antiviral agent dependent on clinical scenario.21,22
Foscarnet
In patients presenting with a high viral load with known resistance to ganciclovir, guidelines recommend foscarnet as the first-line alternative treatment agent.21,22 Foscarnet is a pyrophosphate analog which acts as a noncompetitive inhibitor of many viral RNA and DNA polymerases. It is similar to (val)ganciclovir in that it is a virostatic agent. While foscarnet may be used in cases of ganciclovir resistance, its use can be limited by several factors. Foscarnet is only available intravenously and typically requires an inpatient admission for administration given both frequency of dosing as well as close monitoring required due to adverse effects. Treatment courses are often lengthy leading to increased costs for both the medication and hospital stay. Tolerability of foscarnet is limited by adverse effects including nephrotoxicity and electrolyte abnormalities. This nephrotoxicity can be graft ending in the setting of renal transplant.58
Cidofovir
Another alternative for treatment of ganciclovir resistant CMV is cidofovir, which works by inhibiting viral DNA synthesis. Historically, cidofovir has been considered as a potential treatment agent in patients with documented ganciclovir resistance. Cidofovir is available as an intravenous medication requiring inpatient hospital stay for administration. Its use is often limited by adverse effects, specifically renal toxicity and low barrier to genetic resistance. When utilized, it is recommended to administer probenecid pre- and post-infusion in an attempt to decrease the risk of nephrotoxicity. Resistance to cidofovir may occur via UL54 mutations. Cidofovir use has fallen out of favor as cidofovir has been shown to be ineffective for treatment of resistant or refractory CMV.59
Maribavir
Maribavir is an oral antiviral more recently approved for treatment of CMV infection or disease that is refractory to treatment with other antivirals (ganciclovir, valganciclovir, cidofovir or foscarnet).60 It exerts is action by competitively inhibiting the protein kinase activity of human CMV enzyme pUL97, resulting in inhibition of the phosphorylation of proteins. Approval of this agent has allowed for improved management of resistant and refractory CMV given it is an oral medication and has reduced associated toxicity. Due to its improved tolerability, current consensus guidelines recommend maribavir as a possible second line agent for treatment of CMV in patients intolerant to (val)ganciclovir.21,22 Specifically, maribavir may be an alternative or step down from foscarnet. When using maribavir, the manufacturer recommends completing a full 8-week course of therapy.
Use of maribavir is limited by a number of factors. High viral load is an obstacle to use. Maribavir is not recommended for use until CMV viral load is <50,000 IU/mL.21 Access to maribavir can be another rate limiting step as it can be costly and not always covered by prescription insurance. Takeda does offer a patient assistance program which may be utilized to help some patients obtain maribavir. Dysgeusia is a commonly reported adverse effect of maribavir, which in some patients can be dose limiting. Maribavir is metabolized hepatically via CYP3A4 and CYP1A2 and as such can interact with other medications metabolized via these pathways. Specifically, in transplant patients maribavir may increase tacrolimus area-under-the-curve and maximum concentrations by 1.5- and 1.4-fold, respectively.61 Tacrolimus doses should be adjusted accordingly when starting maribavir. Due to its mechanism of action, maribavir should not be used in conjunction with (val)ganciclovir as maribavir may antagonize and reduce the antiviral activity of ganciclovir.60 Finally, maribavir has a low genetic barrier to resistance that limits its use. Prolonged use of maribavir beyond the recommended 8 weeks is usually not possible and is not recommended.21 The most common maribavir resistance mutations are UL97 T409M and H411Y, as well as C480F in those previously exposed to ganciclovir.
Letermovir
In 2023, letermovir was FDA approved for prevention of CMV in high-risk renal transplant recipients. Letermovir has a novel mechanism of action as a terminase complex inhibitor. This new agent provides an alternative to ganciclovir for prevention of CMV following transplantation. Unlike the aforementioned agents, letermovir is well tolerated. Most importantly, it does not inhibit bone marrow like the other preventative agent, (val)ganciclovir. Consensus guidelines currently recommend letermovir as an alternative agent for CMV prophylaxis in those unable to tolerate valganciclovir due to lack of data in R+ patients and alternative organ types.21 However, studies have shown letermovir to be safe and effective for de novo CMV prophylaxis in other organ groups.62 Limitations to utilization of letermovir include access to medication and drug interactions. Letermovir can be costly and may require appeals to insurance to obtain coverage or utilization of Merck’s patient assistance program. Utilizing these approaches, studies show that 74% of patients can obtain letermovir for de novo prophylaxis.63 Letermovir is metabolized by CYP3A4 and can interact with other medications metabolized via this pathway, particularly tacrolimus. Tacrolimus levels should be adjusted when both starting and stopping letermovir. Letermovir is not approved for treatment, and resistance can develop quickly when used in the setting of active viral replication. Letermovir resistance develops via the UL56 codon 325 mutations. While it has been studied for use as secondary prophylaxis following treatment of CMV infection, use in this manner is not currently recommended.21
Predictive Potential of Viral Replication
Due to tolerability issues with currently available antivirals, and risk of resistance, mechanisms to limit exposure and relegate therapy to those who are at highest risk of progression to disease are needed. Given limitations associated with immune assays, and lack of clearly established absolute thresholds that are associated with negative outcomes,21,22 replication trends via frequent laboratory monitoring are currently the best available tool to achieve this. To utilize this tool, viral replication is necessary. While CMV disease has been associated with negative outcomes in the literature, the toxicity of viral replication itself is debated.64 The presence of virus in tissue and the intervascular space is inevitable It is the capacity of the host immune system to respond that is the key to the long-term ability to maintain virus in latency. It is here that predictive risk stratification and immune priming can occur. Therefore, replication avoidance is not the goal of CMV treatment and prophylaxis. Permissible replication allows stewardship of antiviral therapies and avoids resistance. Utilizing clues in replication trends can predict which require urgent treatment. These viral trends are predictable and therefore can be broadly applied.
Absolute Viral Load
There is no universal absolute viral load that necessitates treatment. This has been a point of much contention, particularly when utilizing the preemptive monitoring strategy for primary prophylaxis.65 Recently published consensus guidelines attempt to address this. (Table 2) However, thresholds are only suggested in one version of the guidelines, and the ranges suggested are wide. Additional risk stratification beyond serostatus and net immunosuppressive state is not specified.21 The absolute viral load is a single data point and cannot, in of itself, inform a clinician about the status of the host immune system and the ability to maintain in latency. That said, some absolute viral loads have been tied to clinical outcomes. In a landmark clinical trial evaluating the use of valganciclovir compared to IV ganciclovir a baseline viral load of <10,000 copies/mL was associated with an increased likelihood of viral clearance at day 21 and day 49 of therapy as compared to those with a viral load >10,000 copies/mL.66 These findings have been extrapolated to suggest 10,000 copies/mL as the viral load threshold for predicting symptomatic disease and is used at many centers as the threshold at which treatment is considered necessary.67 Furthermore, this threshold has been extrapolated utilizing the WHO international standard, and found to be similarly relevant (equivalent viral load of 8318 IU/mL).68 However, it is important to point out that the VICTOR study group did not find any correlation between absolute viral load and end organ disease. That said, there is literature which demonstrates viral load of >100,000 IU/mL to be tied to negative graft outcomes.69,70 When assessing absolute viral load, there are a number of different elements that must be considered. Absolute viral load in a high risk (D+/R-) recipient experiencing primary infection has different meaning than in those patients previously exposed (moderate risk, R+). Additionally, time from transplant is important as well, as the immunosuppressive burden influences the ability to maintain in latency. Replication at any quantitative value in a high risk (D+/R-) patient undergoing surveillance within the first post-transplant year is important. These patients are at high risk of progressive replication, with log rise as much as 1 log10 per week.71 Even in the presence of antiviral treatment many patients will continue to see increases for up to 3 weeks.72 Therefore, antiviral therapy should be provided at any quantitative value, even if the viral load is far below the threshold of 10,000 copies/mL, as these patients do not have preexisting immunity and will rapidly progress to symptomatic disease and the associated negative outcomes if treatment is not provided promptly. Conversely seropositive patients (R+) are known to reconstitute immunity at a median of 60 days post-transplant.73 Therefore, if an R+ patient develops quantifiable replication, a more conservative approach can be pursued as the host immune system may be able to control replication without treatment. By trending replication in this setting, avoidance of antiviral exposure and toxicity is possible. Furthermore, given the nature of a latent herpes viruses, replication can occur or be exacerbated in the setting of clinical stressors, irrespective of CMI. Indeed, the VICTOR study group determined that inflammation from any source was associated with increased and ongoing viral replication.68 Resolution of these clinical stressors will in turn resolve replication allowing return to latency.
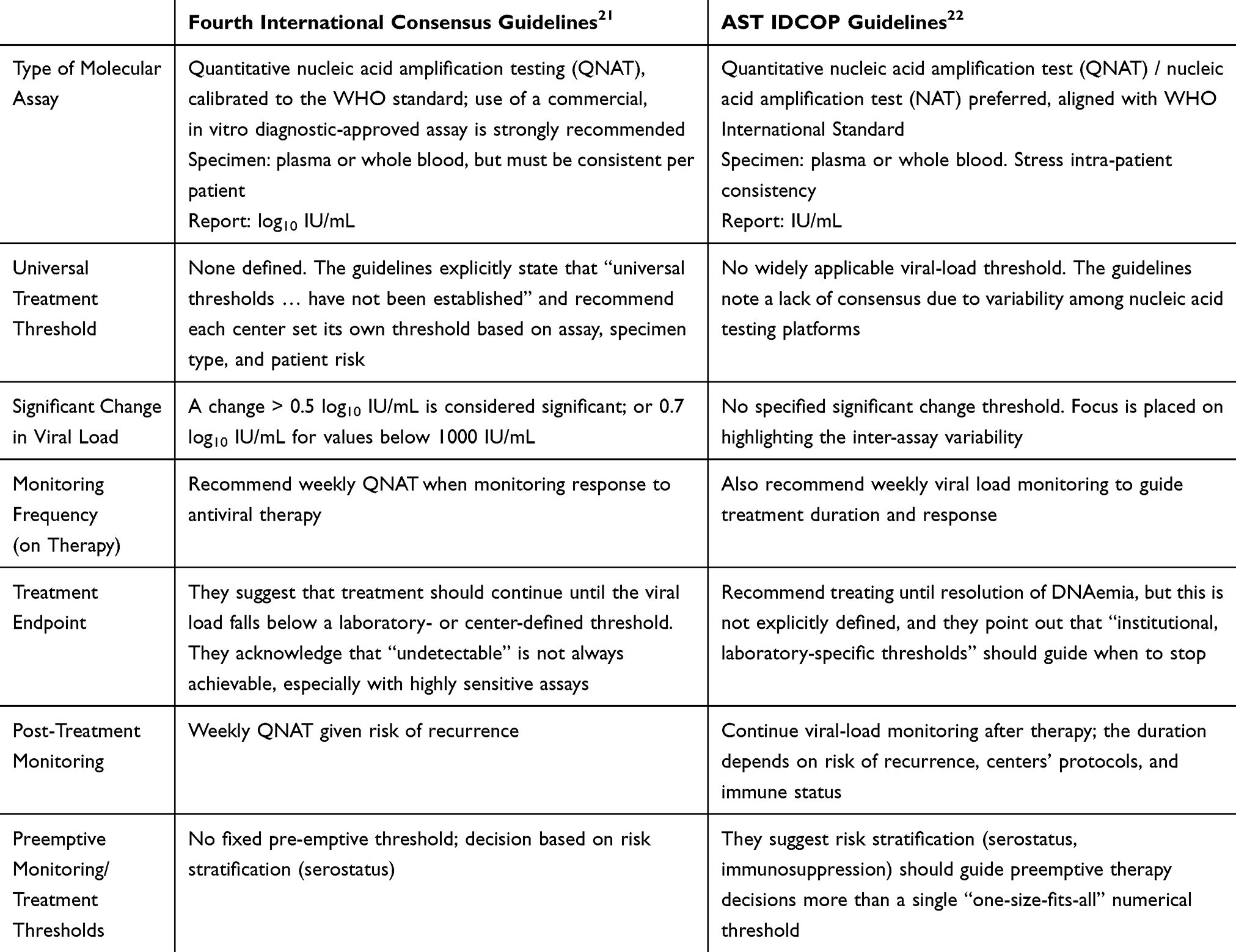 |
Table 2 Comparison Table: CMV Viral Load Threshold Guidance |
Replication Kinetics
Replication kinetics refers to the rate and trend of viral replication. Literature suggests that this trend is more clinically meaningful than any absolute viral load, which is subject to clinical overlap between asymptomatic and symptomatic disease based on patient-specific factors.30 Table 3 Replication kinetics are the window into the host immune response and are more reliable evaluation of CMV specific CMI than any available assay. Replication trends are measured in changes in log10 IU/mL of viral load. Given the wide range in absolute viral load using molecular diagnostics, when looking strictly at the numerical value there can be fluctuation that appears significant from point to point but is not clinically meaningful. Literature is available applying CMV replication kinetics in SOT recipients. Variation can be found in the currently available literature with regards to expected doubling times for CMV in this patient population, ranging from 1 to 4.3 days.74–76 Literature consistently reflects shorter doubling times in SOT recipients with high-risk (D+/R-) serostatuses.71,72 Shorter doubling time has also been shown to be an independent risk factor for CMV disease.77,78 Specifically, in lung transplant recipients, a 1 log10 IU/mL increase in viral load at any timepoint during the first 6 months post-transplant has 93% specificity for CMV pneumonitis. A change in viral load of >0.6 log10 IU/mL/week is considered progressive replication reflective of lack of CMV-CMI and often will necessitate treatment.30 Replication kinetics in SOT recipients are susceptible to changes related to iatrogenic immunosuppression; lymphocyte-depleting induction immunosuppression agent anti-thymocyte globulin has been shown to shorten doubling times, while maintenance immunosuppression agent everolimus has been shown to prolong doubling times.79
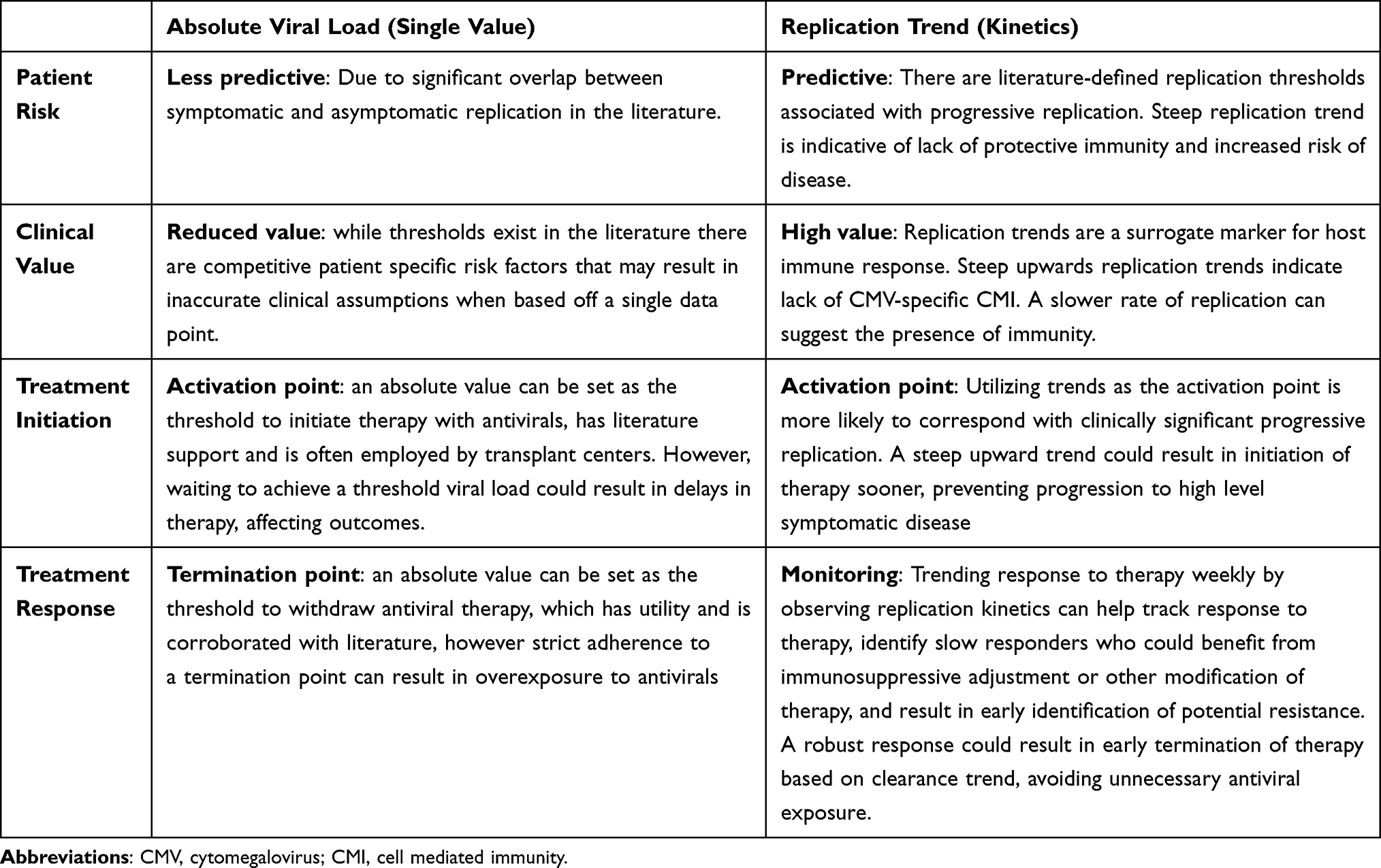 |
Table 3 Absolute Viral Load Vs Replication Trend |
Outside of the first replication event in a high-risk recipient mentioned above, low level replication can typically be observed and trended rather than immediately resorting to initiation of antiviral therapy. Similarly, trends can be observed to gauge response to therapy. Table 4 outlines our approach to utilizing trends clinically. When evaluating clearance kinetics literature suggests a typical favorable response as −0.6 log10 reduction per week.30 If weekly trends are <0.6 log10 IU/mL/week, this is below expected and suggests poor response to therapy and require investigation into adherence, immunosuppression and concomitant stress. If clearance kinetics are >0.6 log10 IU/mL/week, this suggests a robust response to therapy with likely presence of CMI and may allow for early termination of antivirals before clearance to negativity. When treating CMV end of therapy viral stagnation is typical. Withdrawal of treatment and observation of replication kinetics can be used to avoid ongoing exposure and resultant antiviral resistance. This is typically preferred over secondary prophylaxis given its lack of efficacy and ongoing antiviral exposure with associated risks.80 The replication trend after withdrawal can then be used to predict recurrence, which is also a common complication associated with treatment of CMV and leads to ongoing antiviral exposure. If replication recurs after antiviral withdrawal at a rate >0.6 log10 IU/mL/week, CMI is likely incompletely developed and return to therapy will be necessary. If recurrence with rapidly progressive viral replication occurs repeatedly, immunosuppressive adjustment is likely needed. Conversely, if replication recurs but change is <0.6 log10 IU/mL/week, then return to therapy and immunosuppressive adjustment is not necessary, and the patient can be observed off antivirals. These principles can also be applied to preemptive monitoring approach to prophylaxis. First replication in high-risk requires immediate treatment, as presumably CMV-specific CMI is not present. All others can be closely observed to avoid unnecessary antiviral exposure.
 |
Table 4 CMV Viral Replication Trend–Guided Clinical Response Algorithm |
Obstacles to Permissive Replication
There are several obstacles to this approach. Fear among the transplant team based on historical concerns that any replication is harmful to the patient and graft persist across centers. When utilizing this approach there needs to be free, open communication with the transplant team and the patient to provide reassurance and counseling. Furthermore, trending replication off antivirals requires close monitoring of labs. A method for consistent outreach and tabulating trends is necessary for successful outcomes. Laboratory errors and delays are common and limit the ability to trend replication and respond in timely manner, which has limited the applicability of this approach. These obstacles highlight the need for CMV antiviral stewardship (AVS) initiatives, to facilitate the utilization of viral replication trends therapeutically. CMV stewardship is an extension of antimicrobial stewardship (AMS) in the immunocompromised host.81 The major difference between AMS and CMV AVS is instead of stewarding antibiotics to prevent resistance and optimize outcomes, the CMV AVS stewards patients from transplant to development or reconstitution of CMV specific CMI.82 CMV antiviral stewardship is a multidisciplinary initiative comprised of transplant infectious infectious diseases physician, the transplant medical and surgical teams and transplant pharmacy and is uniquely poised to address the obstacles inherent to permissible replication and therapeutic use of replication trends, as it provides a dedicated service responsible for ensuring timely follow up, appropriate lab frequency and prompt delivery of treatment when treatment is indicated.83 The CMV AVS can serve as the liaison between the primary transplant provider and the transplant ID service, following predetermined protocols and evaluating appropriateness via targeted quality improvement.82 In this way, the obstacles inherent to permissible replication and utilizing replication trends therapeutically can be eliminated.
Conclusion and Future Perspectives
In conclusion, a designated viral load threshold above which to initiate treatment has not been definitively established. This highlights the importance of clinician expertise when interpreting the summative meaning of available data, most importantly viral load and trend(s) over time, but also taking into account other diagnostic testing such as cell-mediated immunity testing, clinical presentation, CMV serostatus, net state of immunosuppression and other relevant past medical history such as comorbid conditions and post-transplant course. Application of this strategy can avoid antiviral exposure, resistance and provides patient centric care. The CMV AVS is uniquely poised to take the role of evaluating replication trends along with unique patient-specific factors and apply these to appropriately utilize available therapeutics, ensuring success and improving outcomes. In the future, more sensitive and specific assays to measure CMV specific CMI could avoid lengthy treatment courses and prolonged monitoring periods. Better harmonization of molecular assays to avoid inter and intra-laboratory variability would streamline interpretation. Development of less toxic and more effective antiviral agents could also improve care. Antiviral agents that allow low-level replication but prevent progression to allow development or reconstitution of immunity in a protected state would also improve the current state. However, until these are available, the best option is to utilize the tools we do have. Replication trends and risk factor assessment allow the transplant clinician to predict outcomes and direct therapy more precisely than algorithms tied to an absolute viral load. Future research directions in this space should include prospective validation of kinetic thresholds as well as integration with immune biomarkers and standardization into CMV stewardship frameworks.
Acknowledgments
The authors would like to thank the Transplant Division for their ongoing support of CMV stewardship at UW Health.
Disclosure
MRJ has received research funding from and serves as a consultant for Merck & Co. The other authors report no conflicts of interest in this work.
References
1. Staras SA, Dollard SC, Radford KW, Flanders WD, Pass RF, Cannon MJ. Seroprevalence of cytomegalovirus infection in the United States, 1988-1994. Clin Infect Dis. 2006;43(9):1143–14. PMID: 17029132. doi:10.1086/508173
2. Fulkerson HL, Nogalski MT, Collins-McMillen D, Yurochko AD. Overview of human cytomegalovirus pathogenesis. Methods Mol Biol. 2021;2244:1–18. PMID: 33555579. doi:10.1007/978-1-0716-1111-1_1
3. Hidaka S, Tanabe K, Kobayashi S. Incidence of cytomegalovirus infection after kidney transplantation in the modern era of immunosuppression: the VINTAGE study. Ren Fail. 2025;47(1):2491658. PMID: 40260519; PMCID: PMC12016247. doi:10.1080/0886022X.2025.2491658
4. Bestard O, Kaminski H, Couzi L, Fernández-Ruiz M, Manuel O. Cytomegalovirus cell-mediated immunity: ready for routine use? Transpl Int. 2023;36:11963. PMID: 38020746; PMCID: PMC10661902. doi:10.3389/ti.2023.11963
5. Picarda G, Benedict CA. Cytomegalovirus: shape-shifting the immune system. J Immunol. 2018;200(12):3881–3889. PMID: 29866770; PMCID: PMC5992493. doi:10.4049/jimmunol.1800171
6. Verma S, Benedict CA. Sources and signals regulating type I interferon production: lessons learned from cytomegalovirus. J Interferon Cytokine Res. 2011;31(2):211–218. PMID: 21226618; PMCID: PMC3036178. doi:10.1089/jir.2010.0118
7. Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. 1989;320(26):1731–1735. PMID: 2543925. doi:10.1056/NEJM198906293202605
8. Schneider K, Loewendorf A, De Trez C, et al. Lymphotoxin-mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe. 2008;3(2):67–76. PMID: 18312841; PMCID: PMC2703178. doi:10.1016/j.chom.2007.12.008
9. Orange JS, Biron CA. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol. 1996;156(12):4746–4756. PMID: 8648121. doi:10.4049/jimmunol.156.12.4746
10. Farrell HE, Bruce K, Lawler C, Cardin RD, Davis-Poynter NJ, Stevenson PG. Type 1 interferons and NK cells limit murine cytomegalovirus escape from the lymph node subcapsular sinus. PLoS Pathog. 2016;12(12):e1006069. PMID: 27926941; PMCID: PMC5142805. doi:10.1371/journal.ppat.1006069
11. Podlech J, Ebert S, Becker M, Reddehase MJ, Stassen M, Lemmermann NA. Mast cells: innate attractors recruiting protective CD8 T cells to sites of cytomegalovirus infection. Med Microbiol Immunol. 2015;204(3):327–334. PMID: 25648117. doi:10.1007/s00430-015-0386-1
12. Stacey MA, Marsden M, Pham NTA, et al. Neutrophils recruited by IL-22 in peripheral tissues function as TRAIL-dependent antiviral effectors against MCMV. Cell Host Microbe. 2014;15(4):471–483. PMID: 24721575; PMCID: PMC3989063. doi:10.1016/j.chom.2014.03.003
13. Arens R, Wang P, Sidney J, et al. Cutting edge: murine cytomegalovirus induces a polyfunctional CD4 T cell response. J Immunol. 2008;180(10):6472–6476. PMID: 18453564; PMCID: PMC2587066. doi:10.4049/jimmunol.180.10.6472
14. Munks MW, Cho KS, Pinto AK, Sierro S, Klenerman P, Hill AB. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J Immunol. 2006;177(1):450–458. PMID: 16785542. doi:10.4049/jimmunol.177.1.450
15. Karrer U, Sierro S, Wagner M, et al. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J Immunol. 2003;170(4):2022–2029. PMID: 12574372. doi:10.4049/jimmunol.170.4.2022
16. Cicin-Sain L, Arens R. Exhaustion and inflation at antipodes of T cell responses to chronic virus infection. Trends Microbiol. 2018;26(6):498–509. PMID: 29249600. doi:10.1016/j.tim.2017.11.012
17. Quinn M, Turula H, Tandon M, Deslouches B, Moghbeli T, Snyder CM. Memory T cells specific for murine cytomegalovirus re-emerge after multiple challenges and recapitulate immunity in various adoptive transfer scenarios. J Immunol. 2015;194(4):1726–1736. PMID: 25595792; PMCID: PMC4684174. doi:10.4049/jimmunol.1402757
18. Sylwester AW, Mitchell BL, Edgar JB, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202(5):673–685. PMID: 16147978; PMCID: PMC2212883. doi:10.1084/jem.20050882
19. Verma S, Weiskopf D, Gupta A, et al. Cytomegalovirus-specific CD4 T cells are cytolytic and mediate vaccine protection. J Virol. 2015;90(2):650–658. Erratum in: J Virol. 2017 Aug 10;91(17):e00959-17. doi: 10.1128/JVI.00959-17. PMID: 26491148; PMCID: PMC4702662. doi:10.1128/JVI.02123-15
20. Dupont L, Reeves MB. Cytomegalovirus latency and reactivation: recent insights into an age old problem. Rev Med Virol. 2016;26(2):75–89. PMID: 26572645; PMCID: PMC5458136. doi:10.1002/rmv.1862
21. Transplantation Society International CMV Consensus Group; Kotton CN, Kumar D, Manuel O, et al. The fourth international consensus guidelines on the management of cytomegalovirus in solid organ transplantation. Transplantation. 2025;109(7):1066–1110. PMID: 40200403; PMCID: PMC12180710. doi:10.1097/TP.0000000000005374
22. Razonable RR, Humar A. Cytomegalovirus in solid organ transplant recipients-guidelines of the American society of transplantation infectious diseases community of practice. Clin Transplant. 2019;33(9):e13512. PMID: 30817026. doi:10.1111/ctr.13512
23. Lisnić B, Lisnić VJ, Jonjić S. NK cell interplay with cytomegaloviruses. Curr Opin Virol. 2015;15:9–18. PMID: 26208082. doi:10.1016/j.coviro.2015.07.001
24. Smith W, Tomasec P, Aicheler R, et al. Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell Host Microbe. 2013;13(3):324–335. PMID: 23498957; PMCID: PMC3601332. doi:10.1016/j.chom.2013.02.003
25. Lemmermann NA, Fink A, Podlech J, et al. Murine cytomegalovirus immune evasion proteins operative in the MHC class I pathway of antigen processing and presentation: state of knowledge, revisions, and questions. Med Microbiol Immunol. 2012;201(4):497–512. PMID: 22961127. doi:10.1007/s00430-012-0257-y
26. Poole E, Sinclair J. Sleepless latency of human cytomegalovirus. Med Microbiol Immunol. 2015;204(3):421–429. PMID: 25772624; PMCID: PMC4439429. doi:10.1007/s00430-015-0401-6
27. McSharry BP, Avdic S, Slobedman B. Human cytomegalovirus encoded homologs of cytokines, chemokines and their receptors: roles in immunomodulation. Viruses. 2012;4(11):2448–2470. PMID: 23202490; PMCID: PMC3509658. doi:10.3390/v4112448
28. Caliendo AM, St George K, Kao SY, et al. Comparison of quantita‐tive cytomegalovirus (CMV) PCR in plasma and CMV antigenemiaassay: clinical utility of the prototype AMPLICOR CMV MONITORtest in transplant recipients. J Clin Microbiol. 2000;38(6):
29. Pang XL, Chui L, Fenton J, LeBlanc B, Preiksaitis JK. Comparison ofLightCycler‐based PCR, COBAS amplicor CMV monitor, and pp65antigenemia assays for quantitative measurement of cytomegalo‐virus viral load in peripheral blood specimens from patients aftersolid organ transplantation. J Clin Microbiol. 2003;41(7):
30. Razonable RR, Hayden RT. Clinical utility of viral load in management of cytomegalovirus infection after solid organ transplanta‐tion. Clin Microbiol Rev. 2013;26(4):
31. Hirsch HH, Lautenschlager I, Pinsky BA, et al. An internationalmulticenter performance analysis of cytomegalovirus load tests. Clin Infect Dis. 2013;56(3):
32. Lisboa LF, Åsberg A, Kumar D, et al. The clinical utility of whole blood versus plasma cytomegalovirus viral load assays for moni‐toring therapeutic response. Transplantation. 2011;91(2):
33. Razonable RR, Brown RA, Wilson J, et al. The clinical use of various blood compartments for cytomegalovirus (CMV) DNA quantitation in transplant recipients with CMV disease. Transplantation. 2002;73(6):
34. Dioverti MV, Lahr BD, Germer JJ, Yao JD, Gartner ML, Razonable RR. Comparison of standardized cytomegalovirus (CMV) viral load thresholds in whole blood and plasma of solid organ and hematopoietic stem cell transplant recipients with CMV infection and disease. Open Forum Infect Dis. 2017;4(3):ofx143. doi:10.1093/ofid/ofx143
35. Fryer JF, Heath AB, Minor PD, et al. A collaborative study to establish the 1st WHO international standard for human cytomegalovirus for nucleic acid amplification technology. Biologicals. 2016;44(4):242–251. doi:10.1016/j.biologicals.2016.04.005
36. Pang XL, Fox JD, Fenton JM, et al. American society of transplantation infectious diseases community of practice. Interlaboratory comparison of cytomegalovirus viral load assays. Am J Transplant. 2009;9(2):258–268. doi:10.1111/j.1600-6143.2008.02513.x
37. Available from: https://www.qiagen.com/gb/products/diagnostics-and-clinical-research/transplant/quantiferon-transplant/quantiferon-cmv-assayits.
38. Available from: https://www.revvity.com/category/cytomegalovirusevvity.
39. Available from: https://www.eurofins-viracor.com/test-menu/30360-cmv-insight-t-cell-immunity.
40. Bestard O, Lucia M, Crespo E, et al. Pretransplant immediately early1-specific T cell responses provide protection for CMV infection after kidney transplantation. Am J Transplant. 2013;13(7):1793–1805. doi:10.1111/ajt.12256
41. Cantisan S, Lara R, Montejo M, et al. Pretransplant interferon-gamma secretion by CMV-specific CD8+ T cells informs the risk of CMV replication after transplantation. Am J Transplant. 2013;13(3):738–745. doi:10.1111/ajt.12049
42. Fernandez-Ruiz M, Gimenez E, Vinuesa V, et al. Regular monitoring of cytomegalovirus-specific cell-mediated immunity in intermediaterisk kidney transplant recipients: predictive value of the immediate post-transplant assessment. Clin Microbiol Infect. 2019;25(3):381.e1–381.e10. doi:10.1016/j.cmi.2018.05.010
43. TIMOVAL Study Group; Paez-Vega A, Gutierrez-Gutierrez B, Aguera ML, et al. Immunoguided discontinuation of prophylaxis for cytomegalovirus disease in kidney transplant recipients treated with antithymocyte globulin: a randomized clinical trial. Clin Infect Dis. 2022;74(5):757–765. doi:10.1093/cid/ciab574
44. Manuel O, Laager M, Hirzel C, et al. Swiss transplant cohort study (STCS). Immune monitoring-guided versus fixed duration of antiviral prophylaxis against cytomegalovirus in solid-organ transplant recipients: a multicenter, randomized clinical trial. Clin Infect Dis. 2024;78(2):312–323. doi:10.1093/cid/ciad575
45. Lisboa LF, Kumar D, Wilson LE, et al. Clinical utility of cytomegalovirus cell-mediated immunity in transplant recipients with cytomegalovirus viremia. Transplantation. 2012;93(2):195–200. doi:10.1097/TP.0b013e31823c1cd4
46. Reusing JO, Agena F, Kotton CN, et al. QuantiFERON-CMV as a predictor of CMV events during preemptive therapy in CMV-seropositive kidney transplant recipients. Transplantation. 2024;108(4):985–995. doi:10.1097/TP.0000000000004870
47. Manuel O, Husain S, Kumar D, et al. Assessment of cytomegalo‐virus specific cell‐mediated immunity for the prediction of cyto‐megalovirus disease in high‐risk solid‐organ transplant recipients:a multicenter cohort study. Clin Infect Dis. 2013;56(6):
48. Meesing A, Abraham R, Razonable RR. Clinical correlation of cytomegalovirus infection with CMV‐specific CD8+ T‐cell immunecompetence score and lymphocyte subsets in solid organ trans‐plant recipients. Transplantation. 2018;103(4):832–838.
49. Meesing A, Razonable RR. Absolute lymphocyte count thresh‐olds: a simple, readily available tool to predict the risk of cytomegalovirus infection after transplantation. Open Forum Infect Dis. 2018;5(10):ofy230. doi:10.1093/ofid/ofy230
50. Yoon M, Oh J, Chun KH, Lee CJ, Kang SM. Post-transplant absolute lymphocyte count predicts early cytomegalovirus infection after heart transplantation. Sci Rep. 2021;11(1):1426. PMID: 33446808; PMCID: PMC7809401. doi:10.1038/s41598-020-80790-4
51. El Helou G, Lahr B, Razonable R. Absolute lymphocyte count as marker of cytomegalovirus and allograft rejection: is there a “Safe Corridor” after kidney transplantation? Transpl Infect Dis. 2021;23(2):e13489. PMID: 33037728. doi:10.1111/tid.13489
52. Gardiner BJ, Nierenberg NE, Chow JK, Ruthazer R, Kent DM, Snydman DR. Absolute lymphocyte count: a predictor of recurrent cytomegalovirus disease in solid organ transplant recipients. Clin Infect Dis. 2018;67(9):1395–1402. PMID: 29635432; PMCID: PMC6927884. doi:10.1093/cid/ciy295
53. Chen SJ, Wang SC, Chen YC. Antiviral agents as therapeutic strategies against cytomegalovirus infections. Viruses. 2019;12(1):21. PMID: 31878068; PMCID: PMC7019738. doi:10.3390/v12010021
54. Danziger-Isakov L, Mark Baillie G. Hematologic complications of anti-CMV therapy in solid organ transplant recipients. Clin Transplant. 2009;23(3):295–304. PMID: 19191800. doi:10.1111/j.1399-0012.2008.00942.x
55. Matsumoto K, Shigemi A, Ikawa K, et al. Risk factors for ganciclovir-induced thrombocytopenia and leukopenia. Biol Pharm Bull. 2015;38(2):235–238. PMID: 25747982. doi:10.1248/bpb.b14-00588
56. Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev. 2010;23(4):689–712. doi:10.1128/CMR.00009-10
57. Emery VC, Griffiths PD. Prediction of cytomegalovirus load and resistance patterns after antiviral chemotherapy. Proc Natl Acad Sci. 2000;97(14):8039–8044. doi:10.1073/pnas.140123497
58. Rolling KE, Jorgenson MR, Descourouez JL, Mandelbrot DA, Redfield RR, Smith JA. Ganciclovir-resistant cytomegalovirus infection in abdominal solid organ transplant recipients: case series and review of the literature. Pharmacotherapy. 2017;37(10):1258–1271. PMID: 28699311. doi:10.1002/phar.1987
59. Chou S, Alain S, Cervera C, et al. Drug resistance assessed in a Phase 3 clinical trial of maribavir therapy for refractory or resistant cytomegalovirus infection in transplant recipients. J Infect Dis. 2024;229(2):413–421. PMID: 37506264; PMCID: PMC10873177. doi:10.1093/infdis/jiad293
60. Avery RK, Alain S, Alexander BD, et al. SOLSTICE trial investigators. maribavir for refractory cytomegalovirus infections with or without resistance post-transplant: results from a phase 3 randomized clinical trial. Clin Infect Dis. 2022;75(4):690–701. Erratum in: Clin Infect Dis. 2023 Feb 8;76(3):560. doi: 10.1093/cid/ciac970. PMID: 34864943; PMCID: PMC9464078. doi:10.1093/cid/ciab988
61. Pescovitz MD, Bloom R, Pirsch J, Johnson J, Gelone S, Villano SA. A randomized, double-blind, pharmacokinetic study of oral maribavir with tacrolimus in stable renal transplant recipients. Am J Transplant. 2009;9(10):2324–2330. PMID: 19663892. doi:10.1111/j.1600-6143.2009.02768.x
62. Kleiboeker HL, Descourouez JL, Saddler CM, Al-Adra D, Rice JP, Jorgenson MR. De novo letermovir for cytomegalovirus prophylaxis in high-risk liver transplant recipients. Clin Transplant. 2025;39(5):e70169. PMID: 40294127. doi:10.1111/ctr.70169
63. Descourouez JL, Kleiboeker H, Saddler CM, et al. The UW experience: feasibility of de novo letermovir for primary prophylaxis after abdominal solid organ transplant. Ann Pharmacother. 2025;59(8):730–735. PMID: 39815618. doi:10.1177/10600280241307383
64. Razonable RR, Humar A. Cytomegalovirus in solid organ transplantation. AJT. 2013;13(Supplement 4):93–106. doi:10.1111/ajt.12103
65. Eshraghi H, Hekmat R. Which CMV viral load threshold should be defined as CMV infection in kidney transplant patients? Transplant Proc. 2015;47(4):1136–1139. PMID: 26036538. doi:10.1016/j.transproceed.2014.11.066
66. VICTOR Study Group; Asberg A, Humar A, Rollag H, et al. Oral valganciclovir is noninferior to intravenous ganciclovir for the treatment of cytomegalovirus disease in solid organ transplant recipients. Am J Transplant. 2007;7(9):2106–2113. PMID: 17640310. doi:10.1111/j.1600-6143.2007.01910.x
67. Bennett D, Bergantini L, Ferrara P, et al. Cytomegalovirus infection is associated with development of chronic lung allograft dysfunction. Lung. 2022;200(4):513–522. PMID: 35794392; PMCID: PMC9360074. doi:10.1007/s00408-022-00551-0
68. Åsberg A, Humar A, Rollag H, et al. Lessons learned from a randomized study of oral valganciclovir versus parenteral ganciclovir treatment of cytomegalovirus disease in solid organ transplant recipients: the VICTOR trial. Clin Infect Dis. 2016;62(9):1154–1160. PMID: 26908810. doi:10.1093/cid/ciw084
69. Kleiboeker HL, Jorgenson MR, Leverson GE, et al. Risk factors for high level cytomegalovirus viremia in liver transplant recipients and associated outcomes. Transpl Infect Dis. 2022;24(4):e13898. PMID: 35780512. doi:10.1111/tid.13898
70. Jorgenson MR, Descourouez JL, Yang DY, et al. Impact and outcomes of primary cytomegalovirus disease in seronegative abdominal solid organ transplant recipients of cytomegalovirus unexposed donors (D-/R-). Transpl Infect Dis. 2021;23(3):e13564. PMID: 33449413. doi:10.1111/tid.13564
71. Atabani SF, Smith C, Atkinson C, et al. Cytomegalovirus replication kinetics in solid organ transplant recipients managed by preemptive therapy. Am J Transplant. 2012;12(9):2457–2464. PMID: 22594993; PMCID: PMC3510308. doi:10.1111/j.1600-6143.2012.04087.x
72. Singh N, Winston DJ, Razonable RR, et al. Unexpected cytomegalovirus (CMV) replication kinetics in CMV donor-seropositive, recipient-seronegative liver transplant recipients receiving preemptive antiviral therapy. J Infect Dis. 2022;225(3):436–442. Erratum in: J Infect Dis. 2023 Mar 1;227(5):737. doi: 10.1093/infdis/jiac125. PMID: 33755176; PMCID: PMC8807231. doi:10.1093/infdis/jiab132
73. Abate D, Saldan A, Fiscon M, et al. Evaluation of cytomegalovirus (CMV)-specific T cell immune reconstitution revealed that baseline antiviral immunity, prophylaxis, or preemptive therapy but not antithymocyte globulin treatment contribute to CMV-specific T cell reconstitution in kidney transplant recipients. J Infect Dis. 2010;202(4):585–594. PMID: 20594105. doi:10.1086/654931
74. Emery VC, Cope AV, Bowen EF, et al. The dynamics of human cytomegalovirus replication in vivo. J Exp Med. 1999;190(2):177–182. doi:10.1084/jem.190.2.177
75. Nebbia G, Mattes FM, Cholongitas E, et al. Exploring the bidirectional interactions between human cytomegalovirus and hepatitis C virus replication after liver transplantation. Liver Transpl. 2007;13(1):130–135. doi:10.1002/lt.21037
76. MATCH Programme Study Group; Lodding IP, Sengeløv H, da Cunha-Bang C, et al. Clinical application of variation in replication kinetics during episodes of post-transplant cytomegalovirus infections. EBioMedicine. 2015;2(7):699–705. PMID: 26288842; PMCID: PMC4534685. doi:10.1016/j.ebiom.2015.05.003
77. CMV Consensus Forum; Natori Y, Alghamdi A, Tazari M, et al. Use of viral load as a surrogate marker in clinical studies of cytomegalovirus in solid organ transplantation: a systematic review and meta-analysis. Clin Infect Dis. 2018;66(4):617–631. PMID: 29020339. doi:10.1093/cid/cix793
78. Emery VC, Sabin CA, Cope AV, Gor D, Hassan-Walker AF, Griffiths PD. Application of viral-load kinetics to identify patients who develop cytomegalovirus disease after transplantation. Lancet. 2000;355(9220):2032–2036. PMID: 10885354. doi:10.1016/S0140-6736(00)02350-3
79. Basso G, Felipe CR, Cristelli MP, et al. The effect of anti-thymocyte globulin and everolimus on the kinetics of cytomegalovirus viral load in seropositive kidney transplant recipients without prophylaxis. Transpl Infect Dis. 2018;20(4):e12919. PMID: 29797676. doi:10.1111/tid.12919
80. Gardiner BJ, Chow JK, Price LL, Nierenberg NE, Kent DM, Snydman DR. Role of secondary prophylaxis with valganciclovir in the prevention of recurrent cytomegalovirus disease in solid organ transplant recipients. Clin Infect Dis. 2017;65(12):2000–2007. PMID: 29020220; PMCID: PMC5850558. doi:10.1093/cid/cix696
81. So M, Hand J, Forrest G, et al. White paper on antimicrobial stewardship in solid organ transplant recipients. Am J Transplant. 2022;22(1):96–112. PMID: 34212491; PMCID: PMC9695237. doi:10.1111/ajt.16743
82. Jorgenson MR, Descourouez JL, Kleiboeker H, et al. Cytomegalovirus antiviral stewardship in solid organ transplant recipients: a new gold standard. Transpl Infect Dis. 2022;24(5):e13864. PMID: 35603982. doi:10.1111/tid.13864
83. Jorgenson MR, Descourouez JL, Schulz LT, et al. The development and implementation of stewardship initiatives to optimize the prevention and treatment of cytomegalovirus infection in solid-organ transplant recipients. Infect Control Hosp Epidemiol. 2020;41(9):1068–1074. PMID: 32456718. doi:10.1017/ice.2020.203
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Tacrolimus Trough Level Variation and Its Correlation to Clinical Outcomes and Consequences in Solid Organ Transplantation
Albilal S, Shawaqfeh MS, Albusaysi S, Fetyani L, Alnashmi F, Alshehri SD, Albekairy NA, Akhulaif A, Alzahrani L, Alwuhayde M, Obaidat AA, Al Bekairy AM
Transplant Research and Risk Management 2023, 15:1-11
Published Date: 8 August 2023