Back to Journals » Drug Design, Development and Therapy » Volume 20
Beyond Terminal Blockade: A Mechanism-Based Approach to Complement Inhibitor Selection in Paroxysmal Nocturnal Hemoglobinuria
Authors Chen C ![]() , Zhong J
, Zhong J ![]() , Xiong D
, Xiong D ![]()
Received 15 May 2026
Accepted for publication 18 June 2026
Published 25 June 2026 Volume 2026:20 624829
DOI https://doi.org/10.2147/DDDT.S624829
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Chang Chen, Jinman Zhong, Dan Xiong
Department of Hematology, The Eighth Affiliated Hospital, Southern Medical University (The First People’s Hospital of Shunde, Foshan), Foshan, Guangdong, 528300, People’s Republic of China
Correspondence: Jinman Zhong, Department of Hematology, The Eighth Affiliated Hospital, Southern Medical University (The First People’s Hospital of Shunde, Foshan), Foshan, Guangdong, 528300, People’s Republic of China, Email [email protected] Dan Xiong, Department of Hematology, The Eighth Affiliated Hospital, Southern Medical University (The First People’s Hospital of Shunde, Foshan), Foshan, Guangdong, 528300, People’s Republic of China, Email [email protected]
Abstract: Complement inhibitor selection in paroxysmal nocturnal hemoglobinuria (PNH) can no longer be reduced to a binary class-level decision. Terminal C5 inhibitors provide durable control of intravascular hemolysis (IVH) and the most mature evidence for thromboembolic risk reduction, supporting their continued primacy in patients with high thrombotic risk or established venous thromboembolism. Persistent anemia during C5 inhibition is mechanistically heterogeneous. Before it is ascribed to extravascular hemolysis (EVH) or bone marrow failure (BMF), the adequacy of terminal complement inhibition should be confirmed, as incomplete IVH suppression may contribute to residual hemolysis in some patients. Among patients with confirmed terminal suppression, persistent anemia is driven primarily by C3-mediated EVH in a subset of patients, whereas in others it arises from underlying BMF or a combination of both. Differentiating among these mechanisms is a prerequisite for escalation decisions rather than an optional refinement. The proximal complement inhibitors pegcetacoplan (C3), iptacopan (Factor B), and danicopan (Factor D) address EVH-driven anemia but have not been evaluated in trials powered for thrombosis prevention, creating an asymmetry in the evidence base that demands explicit clinical reasoning. This review proposes a phenotype-driven longitudinal management strategy stratifying treatment decisions by dominant disease mechanism, thrombotic risk, and practical treatment context. Diagnostic approaches to differentiating EVH‑dominant, BMF‑dominant, and overlap phenotypes in the relevant patient subsets, comparative evidence across inhibitor classes, and mechanism-based escalation strategies are addressed in sequence, alongside high-risk clinical scenarios and an evidence-gap analysis to guide future research.
Keywords: paroxysmal nocturnal hemoglobinuria, complement inhibitors, extravascular hemolysis, breakthrough hemolysis, phenotype-driven treatment
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired clonal disorder caused by a somatic mutation in PIGA, the gene encoding phosphatidylinositol glycan class A.1,2 Loss-of-function PIGA mutations impair glycosylphosphatidylinositol (GPI) anchor biosynthesis, producing partial or complete deficiency of GPI-anchored surface proteins on hematopoietic cells.2 The most clinically consequential deficit involves the complement regulators CD55 (decay-accelerating factor) and CD59 (membrane inhibitor of reactive lysis). Their absence renders PNH erythrocytes susceptible to spontaneous, low-level alternative pathway activation that can be substantially amplified during inflammation or infection, ultimately driving terminal complement activation and C5b-9 membrane attack complex (MAC) assembly on the erythrocyte surface.3,4
Unchecked MAC-mediated intravascular hemolysis (IVH) releases free hemoglobin into the circulation, where it scavenges nitric oxide and drives a cascade of vascular, renal, and inflammatory injury.5,6 Clinically, this manifests as fatigue, hemoglobinuria, smooth muscle dystonia, and elevated thrombotic risk, which represents the leading cause of mortality in hemolytic PNH. Pre-complement inhibition cohorts exhibited median survival of 15 to 20 years, with most deaths attributable to thromboembolism.3,7
The approval of eculizumab in 2007 established terminal C5 inhibition as the therapeutic cornerstone of PNH management.8 By blocking C5 cleavage, eculizumab prevents MAC formation, achieving durable IVH control and an approximately 80% relative reduction in thromboembolic events.9–11 Subsequent agents in this class have substantially reduced treatment burden without sacrificing efficacy: ravulizumab extends the intravenous dosing interval to eight weeks through enhanced neonatal Fc receptor recycling,12 while crovalimab enables low-volume subcutaneous self-injection every four weeks via a sequential antibody recycling mechanism.13,14 Both agents demonstrated non-inferiority to eculizumab for LDH normalization and transfusion avoidance in pivotal Phase III trials.15,16
Because C5 blockade cannot suppress upstream complement activation, PNH erythrocytes accumulate C3 opsonins despite controlled IVH, marking them for phagocytic clearance in the liver and spleen via extravascular hemolysis (EVH).17,18 The PEGASUS trial demonstrated this therapeutic ceiling, showing significant hemoglobin recovery when patients switched from eculizumab to the C3 inhibitor pegcetacoplan.19,20 The advent of proximal inhibitors targeting C3, Factor B, and Factor D expands the therapeutic armamentarium but introduces a clinical imperative: these agents can address EVH-driven anemia in patients with C3-mediated EVH as the dominant mechanism, and have no expected benefit in patients whose residual anemia reflects BMF rather than ongoing complement‑mediated destruction. Distinguishing EVH-dominant from BMF-dominant anemia in relevant patient subsets is therefore a prerequisite for any escalation decision.
This review proposes a phenotype-driven longitudinal management framework. By integrating mechanistic classification, thrombotic risk, and comparative evidence across inhibitor classes, we provide a structured clinical decision pathway for current practice and outline the evidence gaps that future research must resolve.
PNH Clinical Phenotypes and Treatment Goals
PNH as a Spectrum of Overlapping Phenotypes
PNH manifests as a spectrum defined by the relative contributions of complement-mediated hemolysis, thrombotic risk, and underlying BMF.21,22 Phenotypic heterogeneity dictates treatment priorities: uncontrolled IVH, elevated thrombotic risk, C3-mediated EVH during therapy, or insufficient erythropoiesis may predominate at different times in the same patient. Phenotype determination, rather than clone size alone, is the necessary starting point for clinical decision-making.18
IVH-Dominant PNH
The classical hemolytic phenotype arises from unchecked terminal complement activation and MAC-mediated intravascular erythrocyte lysis. Laboratory hallmarks include elevated LDH and hemoglobinuria. Clinical features reflect nitric oxide depletion and smooth-muscle dystonia, manifesting as fatigue, abdominal pain, dysphagia, and erectile dysfunction.3 The therapeutic objective is durable terminal complement suppression to normalize LDH, stabilize hemoglobin, and preserve organ function, particularly renal integrity.
Thrombotic-Risk Phenotype
Thromboembolism is the leading cause of mortality in hemolytic PNH. Risk is driven by granulocyte clone size (>50%), hemolytic activity (LDH >1.5x ULN), major PNH-related vascular symptoms, and coexisting prothrombotic factors such as infection or pregnancy.23–25 In patients with prior thrombosis or high-risk features, terminal C5 inhibition remains first-line to prevent recurrence, supported by an approximately 80% relative reduction in thromboembolic events compared with historical cohorts.10,23 Hemoglobin improvement is secondary to vascular protection in this population.
Mechanistic Classification of Persistent Anemia During C5 Inhibition
Persistent anemia during C5 inhibitor therapy requires mechanistic evaluation before any escalation decision is made.26 The first step is to confirm whether terminal complement suppression is genuinely adequate, as incomplete IVH control may itself account for residual hemolysis in a subset of patients.27,28 Pharmacokinetic suboptimality may result in transient loss of IVH control, most commonly associated with eculizumab end-of-interval trough vulnerability.28–30 A mechanistically distinct contributor involves surface-deposited C3b, which associates with Factor B to reconstitute alternative pathway convertase activity on the erythrocyte surface, competing with circulating C5 inhibitors for access to C5 and providing a pathway for residual MAC formation despite ongoing treatment.27,28 In patients where pharmacokinetic suboptimality is the suspected contributor, reassessment and optimization of the C5 inhibitor regimen should be the first step before class escalation is considered.30 In patients where persistent surface C3b accumulation is the suspected mechanism, the adequacy of terminal suppression may be more difficult to restore through regimen adjustment alone, and the threshold for considering proximal inhibition may be lower.30,31
Once adequate terminal blockade is established, persistent anemia should be further evaluated for its dominant mechanism. In patients with aplastic anemia-PNH overlap syndromes or long-standing marrow hypocellularity, residual anemia reflects primarily impaired erythropoiesis from underlying BMF, and complement inhibition of any class cannot be expected to improve hemoglobin,32 because the deficit arises from insufficient red-cell production rather than ongoing complement-mediated destruction.
Initiating a proximal inhibitor in a patient whose anemia is driven primarily by BMF will not improve hemoglobin and exposes the patient to breakthrough hemolysis risk during the transition.33 The diagnostic approach integrates reticulocyte count, indirect bilirubin, LDH on C5 inhibitor therapy, C3d erythrocyte opsonization, and bone marrow assessment.34,35 Table 1 details how these clinical and laboratory parameters support mechanistic classification of persistent anemia, distinguishing EVH-dominant, BMF-dominant, and overlap phenotypes from presentations in which incomplete terminal suppression warrants reassessment of the C5 inhibitor regimen rather than class escalation. A proportion of patients exhibit features of both mechanisms simultaneously. In this overlap population, partial rather than complete hemoglobin normalization should be anticipated even with effective proximal complement suppression, and bone marrow assessment is required before any proximal inhibitor is initiated.
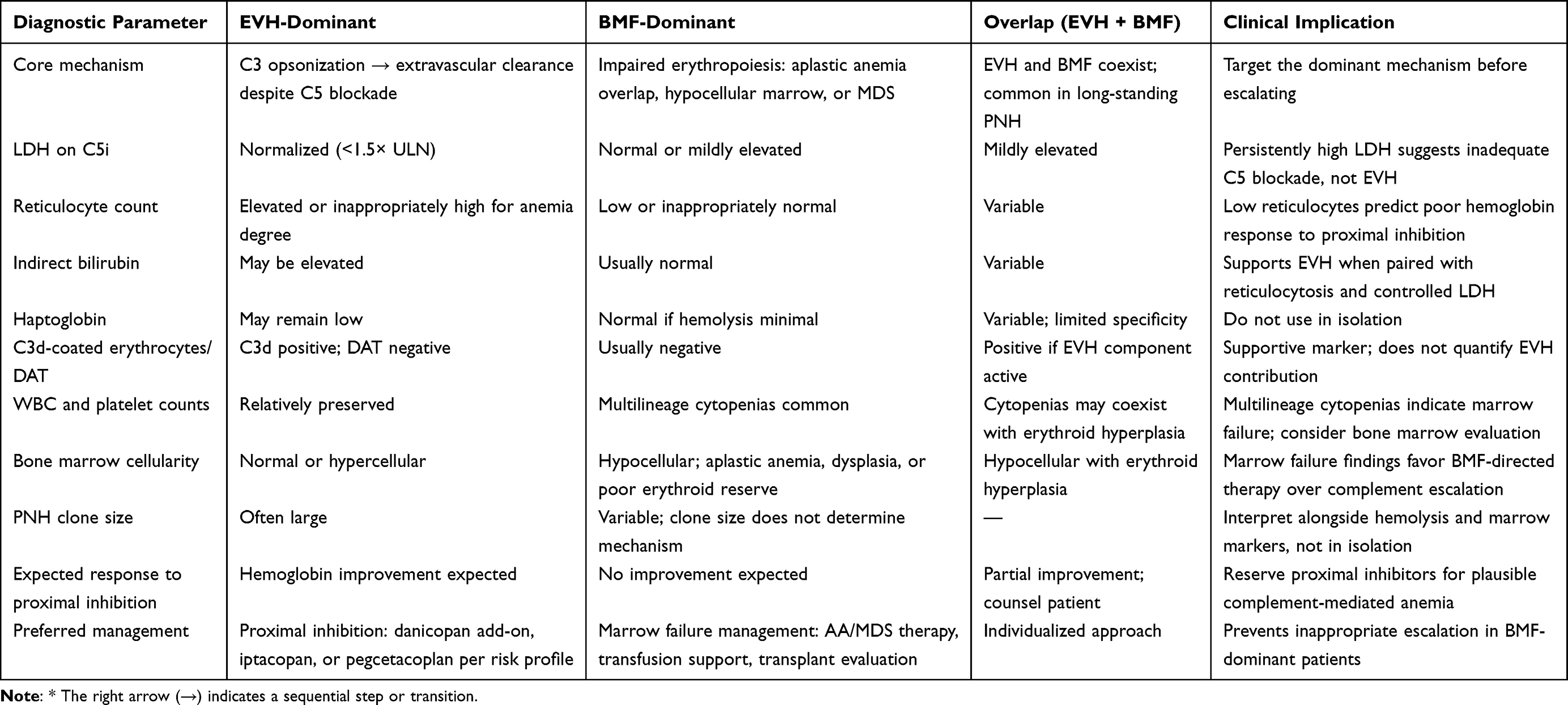 |
Table 1 Distinguishing EVH-Dominant from BMF-Dominant Anemia in PNH Patients on C5 Inhibitor Therapy |
BMF-Dominant and Subclinical PNH
Patients with small PNH clones or clinically predominant BMF (aplastic anemia or myelodysplastic syndrome) constitute a distinct group in which hematopoietic recovery is the primary therapeutic objective.36 Complement inhibitors may be adjunctive if concurrent hemolysis is clinically significant but cannot be expected to correct anemia arising from marrow insufficiency. In aplastic anemia–PNH overlap syndromes, many patients achieve clinically meaningful hematologic responses to immunosuppressive therapy (IST), and combining terminal C5 inhibition with IST is a rational approach that controls complement-mediated hemolysis while supporting marrow recovery.37
Therapeutic Goals Across Phenotypes
Effective PNH management requires attention to six clinical domains: control of IVH (LDH normalization, elimination of hemoglobinuria), hematologic improvement (hemoglobin stabilization, transfusion independence), thromboembolism prevention, organ protection (renal and pulmonary function), quality of life (reduction of treatment burden and fatigue), and long-term safety (infection vigilance, monitoring for clonal evolution, and management of therapeutic transitions). The relative priority of each domain shifts according to the patient’s dominant phenotype. In thrombotic-risk patients, vascular protection takes precedence. In EVH-confirmed inadequate responders, hematologic improvement through proximal inhibition becomes the principal goal. In BMF-dominant patients, marrow-directed therapy is required alongside or instead of complement inhibition.18 The extent to which each available agent can meet these phenotype-specific goals depends on its mechanism of action, the patient population in which it was studied, and its administration profile, considerations addressed in turn for each approved complement inhibitor class.
The Complement Inhibitor Armamentarium: Comparative Evidence
Complement inhibitor selection requires assessment of mechanism of action, trial evidence, route of administration, efficacy profile, breakthrough hemolysis (BTH) risk, and infection susceptibility. Approved agents fall into two mechanistic classes: terminal C5 inhibitors, which control IVH and carry the strongest evidence for thromboembolic risk reduction, and proximal inhibitors, which target upstream complement components to address EVH (Figure 1). These classes are not interchangeable, and the evidence supporting each derives from distinct patient populations with different anemia mechanisms.
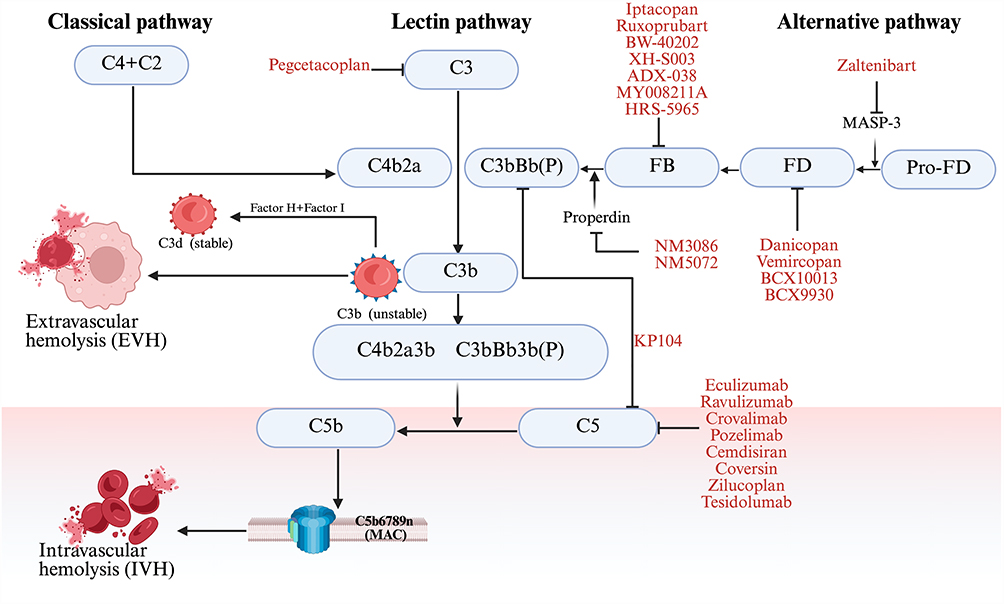 |
Figure 1 Mechanism of Complement Activation in PNH. Intravascular (IVH) and extravascular (EVH) hemolysis are depicted, as well as targets of current and new complement inhibitors (CI). On the opsonized red cell surface, unstable C3b is progressively processed to the stable C3d fragment under the regulatory control of complement Factors H (FH) and I (FI). C3d opsonization flags PNH erythrocytes for extravascular clearance (EVH), whereas C3b participates in C3/C5 convertase assembly, driving downstream membrane attack complex (MAC) formation and intravascular hemolysis (IVH). Abbreviations: MAC, membrane attack complex; MASP‑3, mannan‑binding lectin‑associated serine protease 3; CFB, complement component factor B; CFD, complement component factor D; C3, complement component 3; C5, complement component 5; FH, Factor H; FI, Factor I. |
Terminal C5 Inhibitors
Terminal C5 inhibitors block cleavage of C5 into its pro-inflammatory (C5a) and lytic (C5b) fragments, preventing MAC assembly and controlling IVH. This class carries the most extensive long-term efficacy and safety data in PNH, including the most mature evidence for thromboembolism prevention.
Eculizumab was the first approved agent and remains the clinical reference standard. In the TRIUMPH trial, it stabilized hemoglobin and reduced thromboembolic events by approximately 80% relative to historical cohorts.8 Nearly two decades of post-marketing experience have characterized its long-term safety profile and established its role in pregnancy and pediatric populations.
Ravulizumab was engineered with an extended half-life through enhanced neonatal Fc receptor recycling, extending the IV dosing interval from two to eight weeks.12 Pivotal Phase III trials, Study 301 in treatment-naive patients and Study 302 in patients switching from eculizumab, demonstrated non-inferiority for LDH normalization and transfusion avoidance, and six-year follow-up data confirm sustained efficacy.15,38,39 Ravulizumab is currently the most widely used first-line parenteral option.
Crovalimab employs sequential antibody recycling technology to enable low-volume subcutaneous self-injection every four weeks after an initial IV loading phase.13,14 The COMMODORE 2 trial in treatment-naive patients and COMMODORE 1 in patients switching from eculizumab both demonstrated non-inferiority on the primary endpoints of LDH control and transfusion avoidance.16,40
C5 inhibitors, which act downstream of C3 and therefore leave upstream opsonization unresolved, do not prevent EVH-driven residual anemia in patients who continue to accumulate C3 fragments on erythrocytes despite controlled IVH.17,41,42
Proximal Complement Inhibitors
Proximal inhibitors prevent C3 fragment deposition on PNH erythrocytes, addressing the EVH mechanism that C5 blockade leaves unresolved. Three agents are currently approved: pegcetacoplan (C3 inhibitor), iptacopan (Factor B inhibitor), and danicopan (Factor D inhibitor, approved exclusively as add-on therapy).41
Pegcetacoplan is a pegylated C3/C3b inhibitor (1080 mg administered subcutaneously twice weekly) that blocks all three complement activation pathways upstream of C5.43–45 Its pivotal evidence derives from the PEGASUS trial, which enrolled 80 patients with persistent anemia (hemoglobin below 10.5 g/dL) despite stable eculizumab therapy, an EVH-confirmed inadequate-responder population. At 16 weeks, pegcetacoplan produced a mean hemoglobin increase of +2.4 g/dL compared with a mean decline of −1.5 g/dL in patients continuing eculizumab (adjusted difference +3.84 g/dL, p<0.001), with 85% achieving transfusion independence versus 15% on eculizumab.19 In treatment-naive patients, the PRINCE trial, which compared pegcetacoplan against supportive care rather than an active C5 inhibitor, demonstrated hemoglobin improvement from 9.4 to 12.1 g/dL and a 91% transfusion-free rate versus 22% on supportive care.43 Pegcetacoplan carries the highest switch-associated BTH risk among approved proximal agents: severe hemolysis occurred in approximately 24% of switch patients during the PEGASUS transition period,46,47 requiring strict adherence to structured overlap protocols.
Iptacopan is an oral small-molecule inhibitor of complement Factor B, the catalytic subunit of the alternative pathway C3 convertase. In the APPLY-PNH trial, which enrolled patients with residual anemia (hemoglobin below 10.5 g/dL) despite C5 inhibitor therapy, 94% achieved a sustained hemoglobin increase of at least 2 g/dL without transfusion at week 24, and LDH normalized in 95%.48,49 In treatment-naive patients, the single-arm APPOINT-PNH trial reported that 92.2% met the primary endpoint at week 24, with a mean hemoglobin increase of 3.6 g/dL.49 The Phase IIIb APPULSE-PNH study addressed a distinct and previously understudied population: patients on stable anti-C5 therapy with a higher baseline hemoglobin (≥10 g/dL) who switched to iptacopan monotherapy. At week 24, 92.7% achieved hemoglobin of at least 12 g/dL, mean hemoglobin increased by +2.01 g/dL, no patient required transfusion, and no clinical BTH events were observed, meeting both noninferiority and superiority thresholds.50 Oral twice-daily dosing reduces administration burden substantially, but adherence is entirely patient-dependent and real-world adherence data under chronic disease conditions are not yet available.51
Danicopan is an oral Factor D inhibitor (150 mg three times daily) approved exclusively as add-on therapy to ravulizumab or eculizumab and is not effective as monotherapy.52,53 The ALPHA trial, which randomized EVH-confirmed patients on C5 inhibitor therapy to danicopan add-on versus placebo, demonstrated a mean hemoglobin increase of +2.94 g/dL at Week 12, with 60% versus 0% achieving a hemoglobin increase of at least 2 g/dL and 83% versus 38% achieving transfusion avoidance.52 Sustained efficacy was confirmed at 72 weeks, with a BTH rate of 6 events per 100 patient-years during long-term follow-up.54 Because the C5 inhibitor backbone is maintained throughout, danicopan carries the lowest switch-associated BTH risk of the available proximal strategies.41
All three approved proximal inhibitors share important limitations that must inform their use. No head-to-head randomized trial comparing any two proximal agents has been conducted, and cross-agent comparisons rely on indirect methods only. Follow-up does not exceed five years for any proximal agent, precluding meaningful comparison with the long-term safety record of C5 inhibitors on clonal evolution and thrombosis prevention. Real-world encapsulated bacterial infections have occurred in patients receiving proximal inhibitors despite completed vaccination, confirming that upstream complement inhibition does not eliminate infection risk.55–57
Evidence Summary
The comparative efficacy and safety data for all approved complement inhibitor strategies are summarized in Table 2. Trial populations, hemoglobin response, transfusion avoidance rates, BTH incidence, and key safety signals are presented alongside administration characteristics to support mechanism-guided and phenotype-driven agent selection.
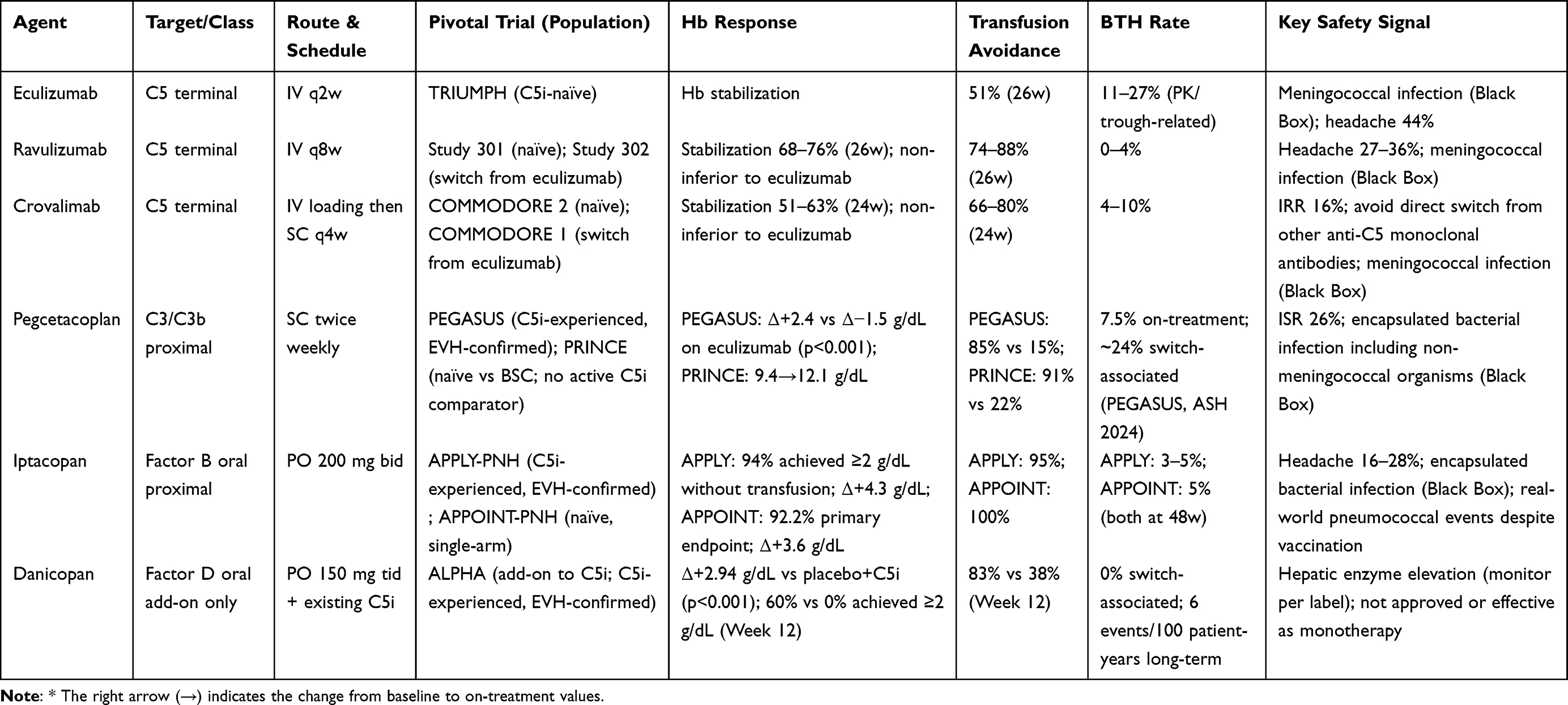 |
Table 2 Clinical Trial Evidence Summary for Approved Complement Inhibitor Strategies in PNH |
Complement Inhibitor Selection and Longitudinal Management
Complement inhibitor selection in PNH is not a class-level decision. The patient’s dominant disease mechanism, thrombotic risk profile, and individual clinical circumstances must be assessed in sequence.18,58 The seven steps of this decision process are illustrated in Figure 2 and addressed in the subsections below.
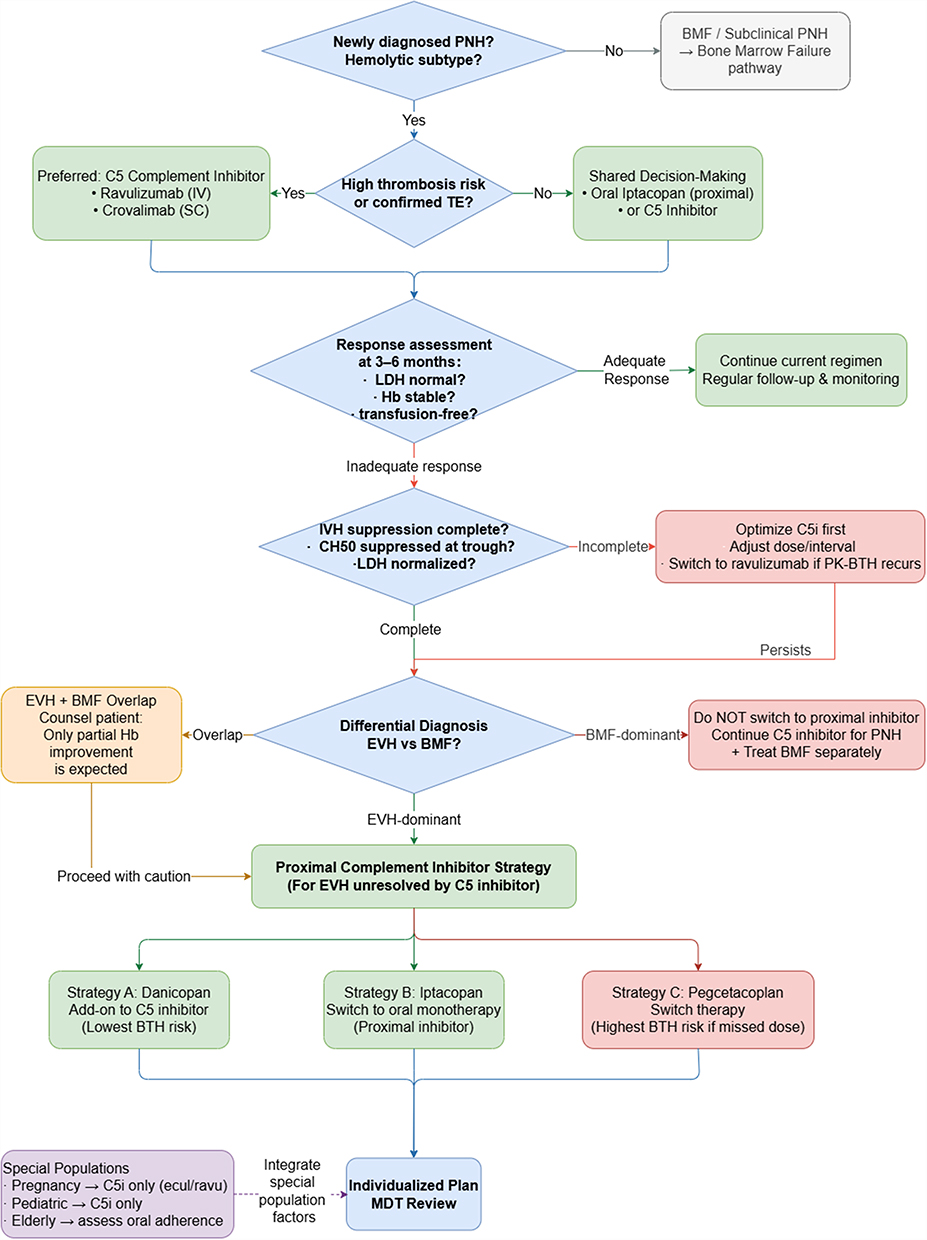 |
Figure 2 A stepwise clinical pathway for complement inhibitor selection and longitudinal management in paroxysmal nocturnal hemoglobinuria. Arrows (→) indicate sequential transitions between clinical decision nodes within the pathway. Abbreviations: BMF, bone marrow failure; C5i, C5 inhibitor; ecul, eculizumab; ravu, ravulizumab; IVH, intravascular hemolysis; EVH, extravascular hemolysis; PNH, paroxysmal nocturnal hemoglobinuria. |
Confirming the Indication for Complement Inhibition
The starting point is confirmation that the patient has hemolytic PNH for which complement inhibition is indicated. Clinical indicators warranting treatment include symptomatic hemolysis, significant or recurrent transfusion requirements, and end-organ complications such as renal insufficiency or thrombosis.59,60 Patients whose presentation is dominated by BMF with minimal hemolytic activity should be managed primarily through BMF-directed pathways. Complement inhibition may be appropriate if concurrent hemolysis is clinically significant, but it is not the primary intervention in this group.3 In patients with aplastic anemia–PNH overlap syndromes, combining IST with C5 inhibition is a rational therapeutic approach that can provide dual benefit by addressing both bone marrow insufficiency and active IVH, and treatment plans should be individualized according to the relative contributions of each mechanism.37
Thrombotic Risk Assessment
Thrombotic risk assessment is the primary determinant of first-line agent selection. Patients with a documented history of thromboembolism, large granulocyte clone size (>50%), elevated LDH (>1.5× ULN), or major PNH-related vascular symptoms should receive terminal C5 inhibition. This recommendation reflects the C5 inhibitor thrombosis prevention evidence, which demonstrates approximately 80% relative risk reduction across long-term cohort data, given that no currently approved proximal inhibitor has been evaluated in a trial powered for thrombosis endpoints.10,23 Notably, thrombotic events were systematically monitored and reported as major adverse cardiovascular events (MACE) in clinical trials of proximal complement inhibitors, and available safety data have not identified a notable signal of increased thrombotic risk, although these studies were not primarily powered to evaluate thromboembolic endpoints and follow-up remains limited. In high-thrombotic-risk patients, the absence of proximal inhibitor thrombosis outcome data is not a minor evidence gap;61 it is the central reason C5 inhibition remains the preferred strategy in this group.
First-Line Agent Selection in Treatment-Naive Patients
For treatment-naive patients with low-to-moderate thrombotic risk, both terminal C5 inhibitors and iptacopan are reasonable options, and the choice reflects shared discussion of route preference, anticipated EVH burden, and safety priorities. Ravulizumab achieves sustained terminal complement blockade through enhanced neonatal Fc receptor recycling, enabling eight-week IV dosing intervals.12 Crovalimab employs sequential antibody recycling technology, allowing subcutaneous self-injection every four weeks after an initial IV loading phase. Both agents have demonstrated non-inferiority to eculizumab for LDH normalization and transfusion avoidance.13,14,16,40 Iptacopan may be considered when the patient has a strong preference for oral therapy and thrombotic risk is assessed as low to moderate.62 However, neither APPOINT-PNH nor APPULSE-PNH included a concurrently randomized active C5 inhibitor control group: APPOINT-PNH was single-arm in complement inhibitor–naive patients, and APPULSE-PNH was single-arm in anti-C5-treated patients with baseline hemoglobin ≥10 g/dL,49 with efficacy assessed against each patient’s own anti-C5 baseline period. The absence of a randomized active comparator limits the strength of evidence for first-line iptacopan positioning and must be discussed explicitly during shared decision-making.61
Monitoring Treatment Response
Response assessment after starting complement inhibition should extend beyond hemoglobin. Durable IVH control is assessed through serial LDH measurements and resolution of hemoglobinuria. Reticulocyte count and indirect bilirubin provide insight into ongoing hemolysis, and C3d deposition on erythrocytes can confirm EVH in patients receiving C5 inhibitors.32,63 Adequate response is characterized by LDH normalization and acceptable hematologic stabilization; patients achieving this are continued on their current regimen. Those with persistent anemia despite controlled IVH require mechanistic classification before any escalation is initiated.31
Classifying Persistent Anemia on C5 Inhibitor Therapy
Evaluating persistent anemia during C5 inhibitor therapy requires first establishing the adequacy of terminal complement blockade through serial LDH monitoring and other markers of IVH. Once incomplete IVH suppression is ruled out (typically indicated by normalized or near-normal LDH levels), persistent anemia requires mechanistic classification before escalation is considered. Table 1 outlines the clinical and laboratory profiles that help differentiate EVH-dominant, BMF-dominant, and overlap phenotypes, linking these distinct features to their expected therapeutic responses and preferred management. Escalating to a proximal inhibitor based solely on low hemoglobin, without confirming the underlying mechanism, exposes BMF-dominant patients to transition-associated BTH without any prospect of hematologic benefit.18,47 If EVH is confirmed as the primary driver, proximal inhibition should be selected according to the strategy outlined below. If BMF is dominant, complement escalation should not be initiated and the anemia addressed through marrow-directed therapy, with C5 inhibition continued if hemolysis remains significant. In patients with mixed EVH and BMF features, proximal inhibition may produce a partial hemoglobin response, and this expectation should be communicated before treatment is changed.64
Escalation Strategies for Confirmed EVH
Three evidence-based strategies are available for patients with EVH-confirmed inadequate response to C5 inhibition, and the choice is governed by EVH severity, BTH risk tolerance, and patient preference regarding oral versus injectable administration.
Danicopan add-on (oral three times daily, added to existing ravulizumab or eculizumab) is preferred when maintaining the C5 inhibitor backbone is clinically desirable, particularly when thrombotic risk makes removing terminal complement protection unattractive.52 Because the C5 inhibitor is continued throughout, this strategy carries the lowest BTH risk of the three options.52,61
Iptacopan monotherapy switch (oral twice daily) is appropriate for patients with moderate-to-severe EVH who prefer oral monotherapy and in whom the C5 inhibitor backbone can be discontinued. Initiation must occur no later than one week after the last eculizumab dose, or no later than 7 days before the next scheduled ravulizumab dose.65 Given that iptacopan reaches steady-state concentrations in approximately 5 days with a half-life of approximately 25 hours, this transition window creates a pharmacological overlap period that ensures disease stability even in the absence of strict dosing overlap.66
Pegcetacoplan monotherapy switch (subcutaneous twice weekly) is an evidence-based option for patients with severe EVH but carries the highest transition-associated BTH risk: severe hemolysis occurred in approximately 24% of switch patients in the PEGASUS trial.67 Structured overlap protocols are mandatory, with eculizumab continued for at least four weeks after pegcetacoplan initiation.46
The comparative positioning of all approved agents, including transition-specific monitoring requirements, is summarized in Table 3, and the complete phenotype-driven clinical decision pathway is illustrated in Figure 2.
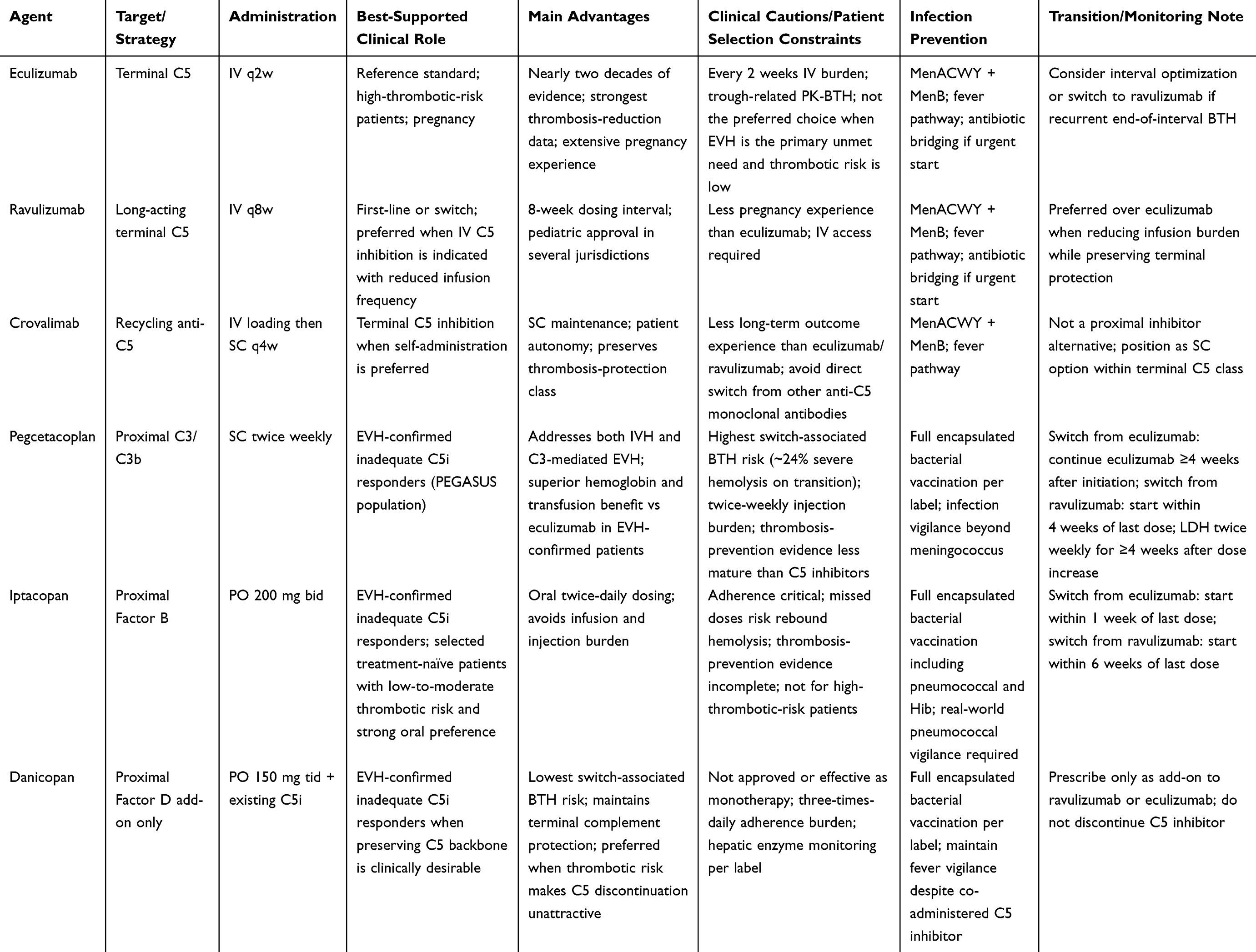 |
Table 3 Comparative Clinical Positioning of Approved Complement Inhibitors in PNH |
Management Adaptations for Special Populations
The phenotype-driven decision pathway described above requires modification in three clinical populations. In pregnancy, treatment selection is constrained by heightened thrombotic vulnerability and limited safety data for proximal inhibitors, making continuous terminal complement protection the preferred therapeutic principle.68–71 In pediatric PNH, mechanistic classification of persistent anemia assumes particular importance because bone marrow failure overlap is common and may dominate the hematologic picture.72,73 In elderly and frail patients, the feasibility of reliable oral adherence must be assessed before any strategy involving oral complement inhibitors is selected.58,74,75
Managing High-Risk Clinical Scenarios
Achieving stable hematologic control in PNH is necessary but not sufficient. A significant proportion of disease-related morbidity arises from acute hemolytic exacerbations, infectious complications, thromboembolic events, and the particular vulnerabilities of specific patient populations. Effective management requires anticipation of these high-risk scenarios, structured monitoring, and prompt intervention.32 The practical monitoring and transition checklist for all scenarios described in this section is provided in Table 4.
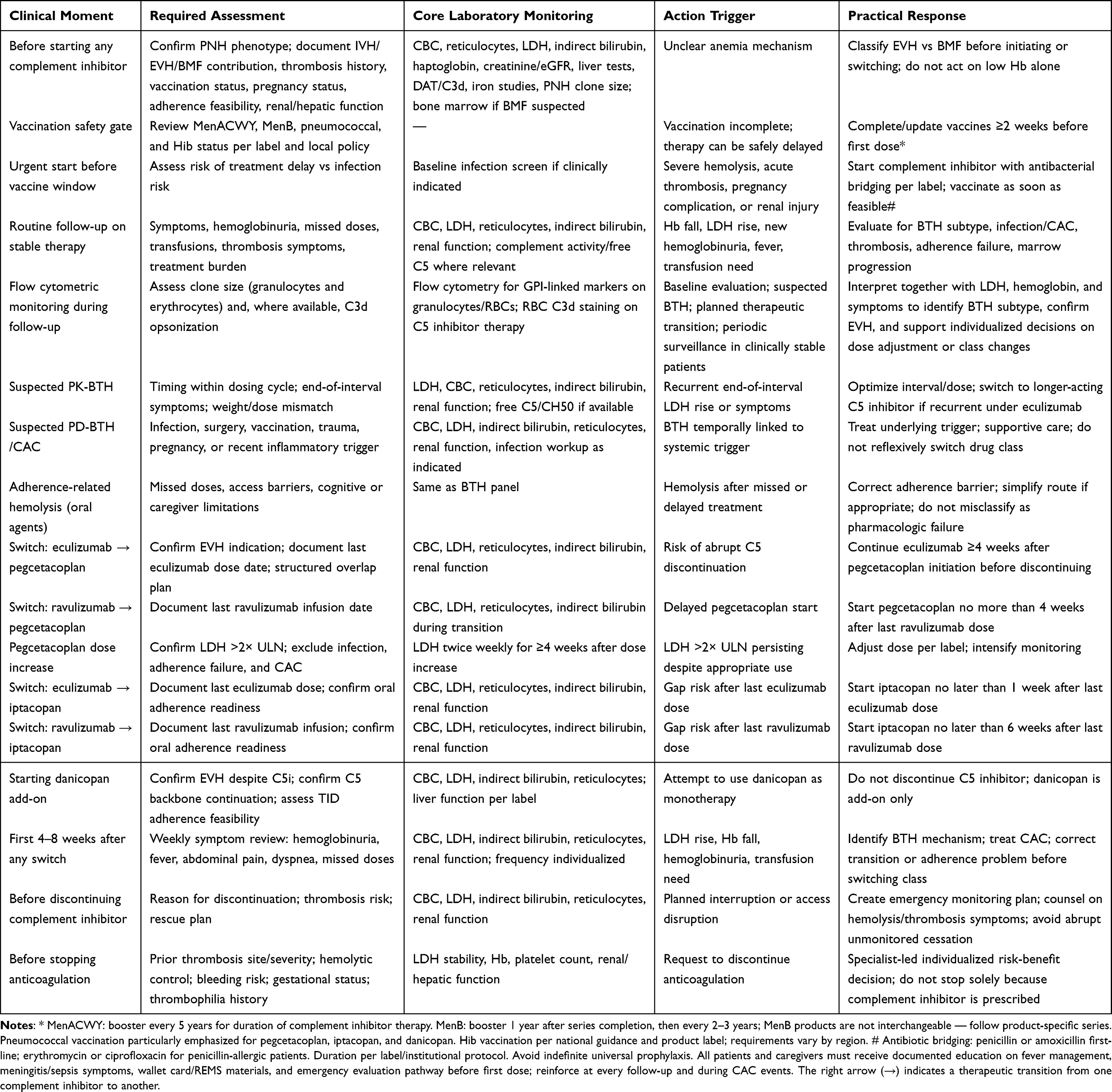 |
Table 4 Practical Monitoring and Transition Checklist for Complement Inhibitor Therapy in PNH |
Mechanistic Classification and Management of Breakthrough Hemolysis
BTH, defined as a hemolytic exacerbation occurring despite ongoing complement inhibitor therapy, should not be treated as a uniform event or interpreted as evidence of treatment failure without further evaluation. Effective management requires classifying BTH into one of three mechanistically distinct subtypes,62,67 because the appropriate response differs for each.
The vulnerability to, and clinical expression of, BTH differ across complement inhibitor strategies, reflecting both the level at which the cascade is blocked and the degree of erythrocyte-bound C3d opsonization. In patients receiving classical C5 inhibitor monotherapy, PNH erythrocytes typically accumulate substantial C3 fragments, most of which are ultimately converted to C3d, shielding them from MAC-mediated IVH but predisposing them to splenic clearance via EVH.76,77 When terminal complement blockade becomes incomplete because of pharmacokinetic trough levels or a severe pharmacodynamic trigger, these C3d-opsonized cells may experience abrupt increases in IVH markers and clinical symptoms. BTH episodes in this setting can therefore be clinically conspicuous, reflecting the large pool of sensitized erythrocytes that have accumulated under effective treatment.
By contrast, proximal complement inhibitor monotherapy blocks complement activation upstream of C3, markedly reducing or preventing C3 fragment deposition on PNH erythrocytes and yielding a predominantly C3d‑negative red cell population. In this setting, pharmacodynamic breakthrough that overwhelms proximal blockade is expected to manifest primarily as renewed IVH, because the terminal pathway is no longer shielded by a C5 inhibitor; such episodes can be severe,78 although available clinical data remain limited and are derived from selected trial populations. Combination regimens that add a proximal inhibitor to a C5 inhibitor backbone create a distinct mechanistic state. The addition of proximal blockade on top of C5 inhibition can substantially reduce erythrocyte C3d opsonization compared with C5 monotherapy, and emerging clinical and translational data suggest differential degrees of C3d reduction among proximal agents depending on their molecular target and dosing schedule,79–81 without permitting robust head‑to‑head comparisons at this time. Crucially, because terminal complement protection is maintained continuously by the C5 inhibitor backbone, add‑on strategies appear to minimize the risk of severe IVH‑type BTH and instead shift most residual events toward pharmacodynamic triggers that can often be managed without switching the therapeutic class.
Pharmacokinetic BTH occurs when drug exposure falls below the threshold required for sustained complement suppression, resulting in transient loss of IVH control. It is most commonly associated with eculizumab, whose fixed every-2-week dosing interval creates predictable trough-level vulnerability,30,82 and typically presents cyclically near the end of a dosing interval. Management consists of dose escalation or interval shortening rather than switching drug class; switching to ravulizumab is appropriate if end-of-interval BTH recurs under eculizumab.30
Pharmacodynamic BTH occurs when systemic inflammatory triggers (infection, surgery, trauma, or pregnancy) amplify complement activation beyond the suppressive capacity of the current regimen despite adequate drug levels. This is a class-wide vulnerability affecting both terminal and proximal inhibitors. The primary intervention is identification and treatment of the underlying trigger. The complement inhibitor should not be discontinued or switched reflexively during a complement-amplifying condition.30
Switch-associated BTH occurs during transition from C5 inhibition to proximal inhibitor monotherapy. Pegcetacoplan monotherapy transitions, which were associated with severe hemolysis in approximately 24% of switch patients in PEGASUS, carry the highest switch-associated BTH risk of currently available strategies.67 Structured overlap protocols are mandatory. When switching from eculizumab, pegcetacoplan must be initiated while eculizumab is continued, with eculizumab discontinued no earlier than four weeks after pegcetacoplan initiation.78 When switching from ravulizumab, pegcetacoplan must be started no more than four weeks after the last ravulizumab dose. For iptacopan, initiation must occur no later than one week after the last eculizumab dose or no later than six weeks after the last ravulizumab dose. Add-on strategies such as danicopan avoid switch-associated BTH entirely by maintaining continuous terminal complement protection throughout.46,47
Based on clinical experience and expert opinion, the size of the GPI-deficient red cell clone appears to influence vulnerability to BTH when complement blockade becomes suboptimal. Under sustained effective complement inhibition, large GPI-deficient clones can expand and exhibit prolonged survival. While this represents a successful therapeutic response, it also creates a sizeable reservoir of cells that are highly susceptible to hemolysis if protection is interrupted. When drug exposure falls or complement activation is markedly amplified, such patients may experience particularly abrupt and severe BTH, a phenomenon sometimes described as the “paradoxical consequence of effective PNH treatment”, and therefore warrant closer clinical vigilance.83
Flow cytometric monitoring of clone size and erythrocyte C3d opsonization provides the most direct means of tracking these risk factors longitudinally. Panels should be designed to evaluate both granulocyte and erythrocyte clone sizes using GPI-linked markers. In patients on C5 inhibitors, assessment of RBC C3d-positive cells may be considered to confirm active EVH and to support decisions about proximal inhibition. Flow cytometry is typically performed at baseline, during early treatment titration, and periodically during stable follow-up, with additional assessments in the setting of suspected BTH or major therapeutic transitions. The specific frequency and panel composition should be individualized, taking into account clinical status, prior results, and local resources, rather than following a rigid algorithm. Trends in clone size and C3d opsonization should be interpreted alongside LDH, hemoglobin, and symptoms to help distinguish BTH subtypes, confirm or refute EVH as a dominant mechanism, and inform tailored decisions on dose adjustment or class switches.84
Biomarker-guided assessment reduces the risk of reflexive switching. LDH and absolute reticulocyte count confirm hemolysis but do not establish its mechanism. To distinguish pharmacokinetic from pharmacodynamic BTH in patients on C5 inhibitors, free hemoglobin and terminal complement activity (CH50) should be measured.85,86 Pharmacokinetic BTH is characterized by a rise in free hemoglobin and recovery of CH50 activity at trough, coinciding with sub-therapeutic drug levels, whereas pharmacodynamic BTH typically shows elevated CRP and other inflammatory markers alongside hemolysis, with CH50 remaining suppressed despite adequate drug levels. In patients on proximal inhibitors, increased C3d-positive cells with declining haptoglobin despite stable LDH suggests that proximal blockade is being bypassed by alternative pathway amplification.32,87 The goal of this assessment is to implement targeted interventions rather than prematurely discontinuing a therapy that remains appropriate for the patient’s primary phenotype.
Infection Prevention
All complement inhibitors compromise innate immunity against encapsulated bacteria, and a structured infection prevention protocol is required regardless of which agent is used.56
Vaccination
Terminal C5 inhibitors impair serum bactericidal activity against Neisseria meningitidis by blocking MAC-mediated killing.88,89 Proximal inhibitors extend this vulnerability upstream, additionally impairing opsonization of Streptococcus pneumoniae, Haemophilus influenzae type b (Hib), and other encapsulated organisms. Before initiating any complement inhibitor, patients must complete vaccination against N. meningitidis (both MenACWY and MenB), S. pneumoniae, and Hib at least two weeks prior to the first dose,90–92 with boosters per current guidelines. MenACWY requires a booster every five years; MenB boosters should follow the product-specific schedule, as MenB vaccines are not interchangeable between products.93 Vaccination does not eliminate infection risk: real-world pneumococcal infections have occurred in patients receiving iptacopan despite completed vaccination,55 and a high index of suspicion for invasive bacterial disease must be maintained in all vaccinated patients on complement inhibition. Annual influenza vaccination is also recommended; where possible, vaccines should be administered within the first week after a complement inhibitor dose to reduce the risk of vaccination-associated complement amplification triggering BTH.57,94
Antibiotic Bridging in Urgent Initiation
When clinical urgency, including life-threatening hemolysis, acute thromboembolism, renal injury, or pregnancy complication, precludes the standard two-week pre-vaccination window, antibiotic bridging is required per prescribing label. Penicillin or amoxicillin is used as first-line prophylaxis; erythromycin or ciprofloxacin is reserved for penicillin-allergic patients.95 Bridging should be continued from treatment initiation until at least 15 days after completion of the second meningococcal vaccine dose.96 The decision to initiate bridging must be documented and communicated to all members of the treating team.
Ongoing Vigilance and Emergency Response
Vaccination and antibiotic bridging reduce but do not eliminate infection risk. All patients and caregivers must receive documented education on the symptoms of invasive bacterial infection before the first complement inhibitor dose,95 with emphasis on meningococcal disease, and this education should be reinforced at every follow-up visit. Every patient must have access to an emergency pathway that guarantees rapid evaluation and empirical parenteral antibiotic administration for febrile episodes regardless of vaccination status.97 In patients on proximal inhibitors, the emergency pathway must explicitly cover pneumococcal and Haemophilus influenzae type b infection presentations in addition to meningococcal disease. Patients should also be counseled that infection risk may be transiently elevated during therapeutic transitions, when complement protection is suboptimal.56
Thrombosis Management
Acute Thromboembolic Events
For acute thromboembolism in a patient not yet receiving complement inhibition, therapeutic anticoagulation and terminal C5 inhibitor therapy should be initiated concurrently. Complement inhibition should not be delayed pending anticoagulation stabilization: ongoing hemolytic drive and nitric oxide scavenging maintain a prothrombotic vascular state that anticoagulation alone does not resolve.7,25,98 For thromboembolic events occurring in patients already on complement inhibitor therapy, the workup should evaluate BTH, complement-amplifying conditions, and treatment non-adherence as contributing factors before attributing the event to treatment failure.
Duration of Anticoagulation
In patients with PNH-related thromboembolism, anticoagulation has historically been continued indefinitely given the perceived persistent recurrence risk.25 This approach is being reconsidered in patients who achieve complete, sustained IVH suppression on complement inhibitor therapy. Retrospective analyses suggest that anticoagulant discontinuation may be feasible without recurrence in highly selected patients with normalized LDH and radiologic resolution of the thrombotic event.23 This remains an area without prospective trial data,99 and the decision requires individualized specialist-led assessment weighing the reduction in prothrombotic drivers against the patient’s bleeding risk, comorbidities, and the site and severity of the event.100
Thrombosis Risk During Therapeutic Transitions
The thromboembolism prevention evidence in PNH is concentrated in the terminal C5 inhibitor class, which demonstrates approximately 80% relative risk reduction across long-term cohort data.10,23 Safety profiles from clinical trials of proximal inhibitors have not identified an increased risk of thrombotic events to date, but no currently approved proximal inhibitor has been evaluated in a trial powered to establish thromboembolism reduction as a primary endpoint,39,61 and addressing this gap remains a central research priority for the field. Until prospective thrombosis data for proximal agents mature, terminal C5 inhibitors remain the preferred strategy for patients with established thromboembolism, high-risk thrombotic features, or ongoing prothrombotic circumstances. When a monotherapy switch to a proximal inhibitor is planned, adherence to structured transition overlap protocols is required not only to prevent BTH but to minimize the interval of suboptimal complement protection that may contribute to thrombotic risk.94
Special Populations
Pregnancy and Family Planning
Pregnancy in PNH carries elevated risks of hemolytic exacerbation, venous thromboembolism, fetal loss, and maternal mortality,69,70,101,102 requiring co-management involving hematology, maternal-fetal medicine, and neonatology.103 Eculizumab has the most extensive published pregnancy safety evidence, with multiple case series reporting preserved hemolytic control and acceptable neonatal outcomes.70,104 Ravulizumab is being used with increasing frequency, supported by growing observational and registry data; however, its documented potential for transplacental transfer requires that neonatal teams be informed of maternal exposure and that neonates receive monitoring for complement activity after delivery.68,105 Crovalimab pregnancy data remain limited at this time. Proximal complement inhibitors lack adequate pregnancy safety and pharmacokinetic data. If pregnancy is confirmed while a patient is receiving a proximal complement inhibitor, urgent transition to a C5 inhibitor should be coordinated under expert guidance, with close hemolytic monitoring from the time of transition through delivery.103,106 For women of childbearing potential who are receiving proximal inhibitors, proactive contraceptive counseling and pre-conception transition planning to an established C5 inhibitor should therefore form part of routine follow-up.103,106 In exceptional circumstances where proximal inhibitor continuation during pregnancy cannot be avoided, management should be conducted under specialist-led supervision.103 Anticoagulation decisions in pregnant patients with PNH require individualized assessment of obstetric bleeding risk, gestational age, prior thrombotic history, and degree of hemolytic activity.107,108
Pediatric PNH
Pediatric PNH is uncommon and frequently arises in the context of aplastic anemia or hypoplastic marrow failure, requiring careful assessment of the relative contributions of hemolysis and impaired erythropoiesis. Bone marrow assessment and multilineage evaluation should therefore precede or accompany complement-directed treatment selection whenever the clinical picture suggests marrow failure overlap.72,73 Terminal C5 inhibition with weight-based dosing remains the standard of care.72 Ravulizumab has received regulatory approval for pediatric PNH in several jurisdictions, and its extended dosing interval reduces infusion frequency from every two weeks to every eight weeks, substantially reducing treatment burden for school-age children and adolescents.72,73 Proximal complement inhibitors should not be initiated in pediatric patients outside formal clinical trials, as age-specific pharmacokinetic, safety, and efficacy data are not yet available for any approved proximal agent.72 Age-appropriate vaccination against N. meningitidis (MenACWY and MenB), S. pneumoniae, and Hib is required before initiating complement inhibition,93,97 following pediatric immunization guidance. Long-term follow-up should include monitoring for clonal evolution, marrow progression, and transition planning to adult care.109
Elderly and Frail Patients
Elderly and frail patients require explicit pre-treatment assessment of oral complement inhibitor adherence feasibility.74 Cognitive impairment, polypharmacy, and social support limitations may compromise the consistent daily dosing required for iptacopan or the three-times-daily regimen of danicopan, and missed doses may precipitate hemolysis.75 If reliable adherence cannot be assured, parenteral C5 inhibitors with supervised administration may provide a more dependable therapeutic platform despite the associated treatment-access burden. Infection risk is amplified by immunosenescence and comorbid conditions, and caregiver education with documented action plans that account for potential cognitive or functional limitations should be incorporated into the treatment plan.56 Polypharmacy interactions require systematic review before initiating oral complement inhibitors, particularly because these agents are susceptible to clinically relevant CYP-mediated drug interactions. Anticoagulation decisions in this population require careful calibration of age-related hemorrhagic risk against PNH-related thrombotic burden.25 No age-specific dose adjustment has been formally established for any approved complement inhibitor, and the underrepresentation of elderly patients in clinical trials warrants heightened pharmacovigilance throughout treatment.58,59,61,62
Future Directions
The current evidence base supports individualized complement inhibitor selection but leaves several clinically significant questions unanswered. Future research must address these gaps while incorporating emerging therapeutic strategies that may further expand treatment options.
Evidence Gaps
Several clinically consequential gaps remain in the current evidence base, with two warranting particular discussion here given their direct bearing on treatment selection.
Of these, the absence of thrombosis outcome data for proximal complement inhibitors carries the most immediate clinical weight. Initial safety data from clinical trials of approved proximal complement inhibitors are encouraging and have not identified a clear signal of increased treatment‑emergent thrombotic events to date, but follow‑up remains relatively short. All three approved proximal agents were evaluated in trials powered for hemoglobin response; none was powered to establish thromboembolic event reduction as a primary endpoint.25,61 This limits their use in patients with high thrombotic risk and prevents direct comparison with the established C5 inhibitor thrombosis evidence base. Prospective trials with adjudicated thromboembolic endpoints and a minimum of three to five years of follow-up are required before this gap can be considered resolved.
Equally important, though less frequently acknowledged, is the lack of a prospectively validated biomarker panel for distinguishing C3‑mediated EVH‑driven anemia, BMF‑driven anemia, and residual IVH resulting from incomplete terminal complement inhibition. Current diagnostic practice relies on indirect surrogates, including reticulocyte count, indirect bilirubin, C3d opsonization, and LDH on C5 inhibitor therapy, none of which has been validated as a combined panel to differentiate these distinct phenotypes against hemoglobin response to proximal inhibition as the reference standard. Early machine‑learning models using clinical and sequencing variables have shown promising accuracy for predicting EVH in single‑center retrospective cohorts,33 but these findings remain preliminary and require external validation before they can inform routine clinical decision‑making. Without validated thresholds, the risk of misclassifying BMF-dominant patients as EVH candidates remains real, and prospective biomarker-response studies with pre-specified diagnostic thresholds are needed.
Beyond these primary concerns, other critical evidence gaps include head-to-head proximal inhibitor comparisons, long-term proximal inhibitor safety beyond five years, real-world oral adherence data, pharmacokinetic and pharmacodynamic personalization, pregnancy and pediatric proximal inhibitor safety, and health economic modeling under real-world access constraints.41
Emerging Strategies and Translational Challenges
The investigational pipeline may further refine complement inhibition, reduce treatment burden, or improve pathway selectivity, but future agents should be judged by clinically meaningful outcomes rather than mechanistic novelty alone. Selective alternative pathway regulators targeting MASP-3 (zaltenibart) or Factor Bb (ruxoprubart), bifunctional constructs combining C5 blockade with Factor H-mediated surface regulation (KP104), hepatocyte-targeted siRNA suppressing Factor B biosynthesis (BW-40202), and gene-based strategies enabling endogenous anti-C5 expression (HMI-104) each aim to address limitations of current therapy, including incomplete EVH control, transition-associated breakthrough hemolysis, frequent dosing, and long-term adherence burden.110–117 However, their eventual role will depend on whether they demonstrate durable hemolysis control, thromboembolic protection, infection safety, and practical feasibility under real-world conditions.
Experience with discontinued oral Factor D inhibitors illustrates the central translational challenge: mechanistic plausibility does not guarantee clinical success. Safety signals, insufficient efficacy, or limited durability can prevent promising approaches from reaching practice.118–120 Therefore, future therapeutic development should be assessed against the same standards that now guide approved inhibitor selection: phenotype-specific benefit, vascular protection, infection risk, transition safety, and long-term tolerability.
Access and Equity
Phenotype-driven complement inhibitor selection assumes access to clone quantification, marrow assessment, C3d testing, and multiple therapeutic options. In many settings, reimbursement restrictions, drug availability, and diagnostic infrastructure gaps limit implementation, meaning that real-world treatment often reflects access rather than optimal mechanistic fit.96 Biosimilar eculizumab may improve the affordability of terminal C5 inhibition, but its impact on global access and long-term comparability with originator therapy require continued post-market evaluation.121
Conclusion
Complement inhibitor selection in PNH must now be anchored in a phenotype-driven framework rather than binary class-level decisions. Terminal C5 inhibitors remain the imperative choice when thrombosis prevention is the dominant clinical priority, given the critical absence of comparative thromboembolic outcome data for proximal agents. Conversely, while proximal inhibitors offer profound hematologic improvement for confirmed EVH-driven anemia, distinguishing EVH from underlying BMF is a mandatory diagnostic prerequisite for escalation. Ultimately, optimizing this treatment algorithm requires future research to yield prospective thrombosis data for proximal inhibitors, validated biomarkers for mechanistic phenotyping, and long-term, real-world safety and adherence outcomes.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the following funding organizations: the Guangdong Basic and Applied Basic Research Foundation (Grant Nos. 2022A1515140022, 2024A1515140108), the Medical Scientific Research Foundation of Guangdong Province (Grant Nos. A2024577, A2025173), the Foshan Self-Funded Science and Technology Innovation Projects (Grant Nos. 2420001003676, 2520001002573), and the Scientific Research Start Plan of the Eighth Affiliated Hospital, Southern Medical University (Grant Nos. SRSP2023013, SRSP2023019, SRSP2024034, SRSP2024038).
Disclosure
The authors declare that they have no competing interests.
References
1. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood. 2014;124(18):2804–23. doi:10.1182/blood-2014-02-522128
2. Luzzatto L, Nakao S. Pathogenesis of paroxysmal nocturnal hemoglobinuria. Blood. 2025;145(26):3077–3088. doi:10.1182/blood.2024025975
3. Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2021;137(10):1304–1309. doi:10.1182/blood.2019003812
4. Panse JP, Höchsmann B, Schubert J. Paroxysmal nocturnal hemoglobinuria, pathophysiology, diagnostics, and treatment. Transfusion Med Hemother. 2024;51(5):310–320. doi:10.1159/000540474
5. Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293(13):1653–1662. doi:10.1001/jama.293.13.1653
6. Szlendak U, Budziszewska B, Spychalska J, Drozd-Sokołowska J, Patkowska E, Nowak J. Paroxysmal nocturnal hemoglobinuria: advances in the understanding of pathophysiology, diagnosis, and treatment. Pol Arch Intern Med. 2022;132(6). doi:10.20452/pamw.16271
7. Kokoris S, Polyviou A, Evangelidis P, et al. Thrombosis in paroxysmal nocturnal hemoglobinuria (PNH): from pathogenesis to treatment. Int J Mol Sci. 2024;25(22):12104. doi:10.3390/ijms252212104
8. Hillmen P, Young NS, Schubert J, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2006;355(12):1233–1243. doi:10.1056/NEJMoa061648
9. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol. 2007;25(11):1256–1264. doi:10.1038/nbt1344
10. Kelly RJ, Hill A, Arnold LM, et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011;117(25):6786–6792. doi:10.1182/blood-2011-02-333997
11. Plessier A, Esposito-Farèse M, Baiges A, et al. Paroxysmal nocturnal hemoglobinuria and vascular liver disease: eculizumab therapy decreases mortality and thrombotic complications. Am J Hematol. 2022;97(4):431–439. doi:10.1002/ajh.26474
12. Sheridan D, Yu Z, Zhang Y, et al. Design and preclinical characterization of ALXN1210: a novel anti-C5 antibody with extended duration of action. PLoS One. 2018;13(4):e195909. doi:10.1371/journal.pone.0195909
13. Fukuzawa T, Sampei Z, Haraya K, et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci Rep. 2017;7(1):1080. doi:10.1038/s41598-017-01087-7
14. Röth A, Nishimura J, Nagy Z, et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood. 2020;135(12):912–920. doi:10.1182/blood.2019003399
15. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood. 2019;133(6):540–549. doi:10.1182/blood-2018-09-876805
16. Röth A, He G, Tong H, et al. Phase 3 randomized COMMODORE 2 trial: crovalimab versus eculizumab in patients with paroxysmal nocturnal hemoglobinuria naive to complement inhibition. Am J Hematol. 2024;99(9):1768–1777. doi:10.1002/ajh.27412
17. Mastellos DC, Reis ES, Yancopoulou D, Risitano AM, Lambris JD. Expanding complement therapeutics for the treatment of paroxysmal nocturnal hemoglobinuria. Semin Hematol. 2018;55(3):167–175. doi:10.1053/j.seminhematol.2018.02.002
18. Bienz M, Patriquin CJ. The varieties of therapeutic experience: navigating treatment options for patients with PNH. Hematol Am Soc Hematol Educ Program. 2025;2025(1):154–163. doi:10.1182/hematology.2025000701
19. Hillmen P, Szer J, Weitz I, et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med. 2021;384(11):1028–1037. doi:10.1056/NEJMoa2029073
20. de Latour RP, Szer J, Weitz IC, et al. Pegcetacoplan versus eculizumab in patients with paroxysmal nocturnal haemoglobinuria (PEGASUS): 48-week follow-up of a randomised, open-label, phase 3, active-comparator, controlled trial. Lancet Haematol. 2022;9(9):e648–59. doi:10.1016/S2352-3026(22)00210-1
21. Pires da Silva BGP, Fonseca NP, Catto LFB, Pereira GC, Calado RT. The spectrum of paroxysmal nocturnal hemoglobinuria clinical presentation in a Brazilian single referral center. Ann Hematol. 2022;101(5):999–1007. doi:10.1007/s00277-022-04797-9
22. Babushok DV. When does a PNH clone have clinical significance? Hematol Am Soc Hematol Educ Program. 2021;2021(1):143–152. doi:10.1182/hematology.2021000245
23. Gurnari C, Awada H, Pagliuca S, et al. Paroxysmal nocturnal hemoglobinuria-related thrombosis in the era of novel therapies: a 2043-patient-year analysis. Blood. 2024;144(2):145–155. doi:10.1182/blood.2024023988
24. Dingli D, Maciejewski JP, Larratt L, et al. Relationship of paroxysmal nocturnal hemoglobinuria (PNH) granulocyte clone size to disease burden and risk of major vascular events in untreated patients: results from the International PNH Registry. Ann Hematol. 2023;102(7):1637–1644. doi:10.1007/s00277-023-05269-4
25. Ueda Y, Chou W, Goh Y, et al. Prevention and management of thromboembolism in patients with paroxysmal nocturnal hemoglobinuria in Asia: a narrative review. Int J Mol Sci. 2025;26(6):2504. doi:10.3390/ijms26062504
26. Shammo J, Gajra A, Patel Y, et al. Low rate of clinically evident extravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria treated with a complement C5 inhibitor: results from a large, multicenter, US real-world study. J Blood Med. 2022;13:425–437. doi:10.2147/JBM.S361863
27. Notaro R, Luzzatto L. Breakthrough hemolysis in PNH with proximal or terminal complement inhibition. New Engl J Med. 2022;387(2):160–166. doi:10.1056/NEJMra2201664
28. Harder MJ, Kuhn N, Schrezenmeier H, et al. Incomplete inhibition by eculizumab: mechanistic evidence for residual C5 activity during strong complement activation. Blood. 2017;129(8):970–980. doi:10.1182/blood-2016-08-732800
29. Nakayama H, Usuki K, Echizen H, Ogawa R, Orii T. Eculizumab dosing intervals longer than 17 days may be associated with greater risk of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Biol Pharm Bull. 2016;39(2):285–288. doi:10.1248/bpb.b15-00703
30. Brodsky RA, Peffault De Latour R, Rottinghaus ST, et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106(1):230–237. doi:10.3324/haematol.2019.236877
31. Fang B, Yu X, Zhou Y, et al. Case report: persistent anemia after eculizumab in paroxysmal nocturnal hemoglobinuria: non-dominantly active intravascular hemolysis. Hematology. 2026;31(1):2647316. doi:10.1080/16078454.2026.2647316
32. Tamdin T, Rodgers GM. Advances in complement inhibition therapies for paroxysmal nocturnal hemoglobinuria and autoimmune hemolytic disorders. J Blood Med. 2025;16:559–572. doi:10.2147/JBM.S543272
33. Duran MN, Tombul Z, Mete M, et al. Predicting extravascular hemolysis in paroxysmal nocturnal hemoglobinuria. Blood. 2024;144(Suppl 1):2695. doi:10.1182/blood-2024-204213
34. Debureaux PE, Kulasekararaj AG, Cacace F, et al. Categorizing hematological response to eculizumab in paroxysmal nocturnal hemoglobinuria: a multicenter real-life study. Bone Marrow Transpl. 2021;56(10):2600–2602. doi:10.1038/s41409-021-01372-0
35. Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. 2017;3(1):17028. doi:10.1038/nrdp.2017.28
36. Fattizzo B, Giannotta J, Zaninoni A, Kulasekararaj A, Cro L, Barcellini W. Small paroxysmal nocturnal hemoglobinuria clones in autoimmune hemolytic anemia: clinical implications and different cytokine patterns in positive and negative patients. Front Immunol. 2020;11:1006. doi:10.3389/fimmu.2020.01006
37. Hill A, de Latour RP, Kulasekararaj AG, et al. Concomitant immunosuppressive therapy and eculizumab use in patients with paroxysmal nocturnal hemoglobinuria: an international PNH registry analysis. Acta Haematol. 2023;146(1):1–13. doi:10.1159/000526979
38. Lee JW, Sicre De Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133(6):530–539. doi:10.1182/blood-2018-09-876136
39. Kulasekararaj A, Brodsky R, Schrezenmeier H, et al. Ravulizumab demonstrates long-term efficacy, safety and favorable patient survival in patients with paroxysmal nocturnal hemoglobinuria. Ann Hematol. 2025;104(1):81–94. doi:10.1007/s00277-025-06193-5
40. Scheinberg P, Clé DV, Kim JS, et al. Phase 3 randomized COMMODORE 1 trial: crovalimab versus eculizumab in complement inhibitor-experienced patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2024;99(9):1757–1767. doi:10.1002/ajh.27413
41. Hillmen P, Horneff R, Yeh M, Kolev M, Deschatelets P. Navigating the complement pathway to optimize PNH treatment with pegcetacoplan and other currently approved complement inhibitors. Int J Mol Sci. 2024;25(17):9477. doi:10.3390/ijms25179477
42. Risitano AM, Frieri C. Understanding pharmacological complement inhibition in paroxysmal nocturnal hemoglobinuria. Haematologica. 2024;109(3):704–708. doi:10.3324/haematol.2023.283805
43. Wong RSM, Navarro-Cabrera JR, Comia NS, et al. Pegcetacoplan controls hemolysis in complement inhibitor-naive patients with paroxysmal nocturnal hemoglobinuria. Blood Adv. 2023;7(11):2468–2478. doi:10.1182/bloodadvances.2022009129
44. Hoy SM. Pegcetacoplan: first Approval. Drugs. 2021;81(12):1423–1430. doi:10.1007/s40265-021-01560-8
45. Duval A, Roquigny J, Frémeaux-Bacchi V. Complement-targeting therapies in hemolytic diseases. Curr Opin Immunol. 2026;98:102686. doi:10.1016/j.coi.2025.102686
46. Peffault de Latour R, Griffin M, Kelly RJ, et al. Hemolysis events in the phase 3 PEGASUS study of pegcetacoplan in patients with paroxysmal nocturnal hemoglobinuria. Blood Adv. 2024;8(11):2718–2725. doi:10.1182/bloodadvances.2024012672
47. Fattizzo B, Pedone GL, Metafuni E, et al. Characterization of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria: an international multicenter experience. Am J Hematol. 2025;100(11):1963–1971. doi:10.1002/ajh.70032
48. Risitano AM, Kulasekararaj A, Roeth A, et al. Factor B inhibition with oral iptacopan monotherapy demonstrates sustained long-term efficacy and safety in anti-C5-treated patients (pts) with paroxysmal nocturnal hemoglobinuria (PNH) and persistent anemia: final 48-week results from the multicenter, phase III APPLY-PNH trial. Blood. 2023;142:571.
49. Peffault De Latour R, Röth A, Kulasekararaj AG, et al. Oral iptacopan monotherapy in paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2024;390(11):994–1008. doi:10.1056/NEJMoa2308695
50. Kulasekararaj A, Fontbrune FSD, Gaya A, et al. MDS-1141: APPULSE-PNH: oral iptacopan monotherapy demonstrates clinically meaningful hemoglobin (Hb) increases in patients with paroxysmal nocturnal hemoglobinuria (PNH) and Hb≥10 g/dL on anti-C5 therapy. Clin Lymphoma Myeloma Leukemia. 2025;25:S631–32. doi:10.1016/S2152-2650(25)02060-9
51. Holt M, Kelly RJ, Fermont JM, et al. Effectiveness of iptacopan versus C5 inhibitors in complement inhibitor-naive patients with paroxysmal nocturnal haemoglobinuria. Ejhaem. 2025;6(3):e270055. doi:10.1002/jha2.70055
52. Lee JW, Griffin M, Kim JS, et al. Addition of danicopan to ravulizumab or eculizumab in patients with paroxysmal nocturnal haemoglobinuria and clinically significant extravascular haemolysis (ALPHA): a double-blind, randomised, phase 3 trial. Lancet Haematol. 2023;10(12):e955–65. doi:10.1016/S2352-3026(23)00315-0
53. Kang C. Danicopan: first Approval. Drugs. 2024;84(5):613–618. doi:10.1007/s40265-024-02023-6
54. Kulasekararaj A, Griffin M, Piatek C, et al. Long-term efficacy and safety of danicopan as add-on therapy to ravulizumab or eculizumab in PNH with significant EVH. Blood. 2025;145(8):811–822. doi:10.1182/blood.2024026299
55. Zhong J, Chen C, Xu Y, He Y, Tan J, Xiong D. Viral infections and related fatal adverse events associated with complement inhibitors for PNH: a real-world pharmacovigilance analysis in FAERS. Front Pharmacol. 2025;16:1639685. doi:10.3389/fphar.2025.1639685
56. Subramanian AK, Java A, Gupta SK, Gupta S, Bomback AS. Strategies to mitigate infection risk in patients receiving complement inhibitor therapy. Clin Infect Dis. 2026. doi:10.1093/cid/ciag081
57. de Castro C, Cano Garcia V, Hillmen P. Low risk for meningococcal and other encapsulated bacteria infections with systemically administered pegcetacoplan in paroxysmal nocturnal hemoglobinuria and C3 glomerulopathies. Blood. 2025;146(Supplement 1):6719. doi:10.1182/blood-2025-6719
58. Huang W, Hou H, Chou W, et al. Consensus of the hematology society of Taiwan on the management of paroxysmal nocturnal hemoglobinuria (PNH). J Formos Med Assoc. 2025. doi:10.1016/j.jfma.2025.10.025
59. Kelly RJ, Holt M, Szer J. Pharmacological therapies in paroxysmal nocturnal haemoglobinuria: focus on complement inhibition. Drugs. 2025;85(11):1413–1428. doi:10.1007/s40265-025-02235-4
60. Tantravahi S, Latremouille-Viau D, Desai R, et al. MDS-658 real-world clinical outcomes among patients untreated with complement inhibitors (CIs) in paroxysmal nocturnal hemoglobinuria (PNH) – a retrospective claims database analysis in the United States. Clin Lymphoma Myeloma Leukemia. 2024;24:S401–02. doi:10.1016/S2152-2650(24)01373-9
61. Fahim SM, Makam AN, Suh K, et al. Iptacopan and danicopan for paroxysmal nocturnal hemoglobinuria. J Manag Care Spec Pharm. 2024;30(6):618–623. doi:10.18553/jmcp.2024.30.6.618
62. Versino F, Fattizzo B. Progress in the use of biological therapies to treat paroxysmal nocturnal hemoglobinuria: focus on patient profiling. Expert Opin Biol Ther. 2025;25(10):1071–1085. doi:10.1080/14712598.2025.2574983
63. Gavriilaki E, de Latour RP, Risitano AM. Advancing therapeutic complement inhibition in hematologic diseases: PNH and beyond. Blood. 2022;139(25):3571–3582. doi:10.1182/blood.2021012860
64. Latyshev V, Fidarova Z, Lukina E, Parovichnikova E. Direct switch from iptacopan to pegcetacoplan in a patient with paroxysmal nocturnal hemoglobinuria. Ejhaem. 2026;7(2):e70298. doi:10.1002/jha2.70298
65. Novartis Pharmaceuticals Corporation. FABHALTA (iptacopan) capsules: US prescribing information [Internet]. East Hanover (NJ): Novartis Pharmaceuticals Corporation; 2024. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/218276s001lbl.pdf.
66. Orland M, Gurnari C, Sanikommu S, Maciejewski J, Dingli D. Switching patients with PNH from pegcetacoplan to iptacopan: a case series. Hematology. 2026;31(1):2618403. doi:10.1080/16078454.2026.2618403
67. Fattizzo B, Versino F, Barcellini W. Breakthrough hemolysis in paroxysmal nocturnal hemoglobinuria throughout clinical trials: from definition to clinical practice. Blood. 2025;146(4):411–421. doi:10.1182/blood.2024027574
68. Patel BJ. Use of ravulizumab in a pregnant patient with PNH: case-based insights. Clin Ad Hematol Oncol. 2026;24(1 Suppl 2):1–12.
69. Manning JE, Ciantar E, Griffin M, Kelly RJ. Paroxysmal nocturnal haemoglobinuria in pregnancy-a systematic review with meta analysis. Ann Hematol. 2025;104(4):2517–2525. doi:10.1007/s00277-025-06353-7
70. Kelly RJ, Höchsmann B, Szer J, et al. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med. 2015;373(11):1032–1039. doi:10.1056/NEJMoa1502950
71. Füreder W, Granser S, Repa A, Farr A. Ravulizumab exposure in early pregnancy. Ann Hematol. 2025;104(11):6081–6084. doi:10.1007/s00277-025-06586-6
72. Yoo JJ, Chonat S. Evaluating ravulizumab for the treatment of children and adolescents with paroxysmal nocturnal hemoglobinuria. Expert Rev Hematol. 2022;15(5):385–392. doi:10.1080/17474086.2022.2073215
73. Chonat S, Kulagin A, Maschan A, et al. Pharmacokinetics, pharmacodynamics, efficacy, and safety of ravulizumab in pediatric paroxysmal nocturnal hemoglobinuria. Blood Adv. 2024;8(11):2813–2824. doi:10.1182/bloodadvances.2023012267
74. Adeyemi AH, Wiredu B, Okobi OE, Nebuwa P, Ezeani EI, Alozie AS. Challenges and strategies in medication management for patients with multiple comorbidities. Cureus. 2025;17(6):e85992. doi:10.7759/cureus.85992
75. Ngcobo NN. Silent dangers in elderly pharmacotherapy: the interplay of polypharmacy, multimorbidity, and drug interactions. J Eval Clin Pract. 2025;31(7):e70283. doi:10.1111/jep.70283
76. Risitano AM, Notaro R, Marando L, et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood. 2009;113(17):4094–4100. doi:10.1182/blood-2008-11-189944
77. Hill A, Rother RP, Arnold L, et al. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica. 2010;95(4):567–573. doi:10.3324/haematol.2009.007229
78. Griffin M, Kelly RJ, Panse J, et al. Management of acute breakthrough hemolysis with intensive pegcetacoplan dosing in patients with PNH. Blood Adv. 2024;8(7):1776–1786. doi:10.1182/bloodadvances.2023011691
79. Zhang L, Chen JY, Kerr C, Cobb BA, Maciejewski JP, Lin F. Reduced red blood cell surface level of Factor H as a mechanism underlying paroxysmal nocturnal hemoglobinuria. Leukemia. 2021;35(4):1176–1187. doi:10.1038/s41375-020-1008-5
80. Risitano AM, Kulasekararaj AG, Scheinberg P, et al. Oral iptacopan monotherapy in paroxysmal nocturnal haemoglobinuria: final 48-week results from the open-label, randomised, phase 3 APPLY-PNH trial in anti-C5-treated patients and the open-label, single-arm, phase 3 APPOINT-PNH trial in patients previously untreated with complement inhibitors. Lancet Haematol. 2025;12(6):e414–30. doi:10.1016/S2352-3026(25)00081-X
81. White TS, Arnall JR, Parish PC, Tolerico J, Tran T, Moore DC. Proximal complement inhibitors in paroxysmal nocturnal hemoglobinuria: an abundance of options in a rare disease. Expert Rev Hematol. 2025;18(1):5–09. doi:10.1080/17474086.2025.2449864
82. Risitano AM, Marotta S, Ricci P, et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front Immunol. 2019;10:1157. doi:10.3389/fimmu.2019.01157
83. Luzzatto L. Recent advances in the pathogenesis and treatment of paroxysmal nocturnal hemoglobinuria. F1000Res. 2016;5:209. doi:10.12688/f1000research.7288.1
84. Sutherland DR, Illingworth A, Marinov I, et al. ICCS/ESCCA consensus guidelines to detect GPI-deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 2 - reagent selection and assay optimization for high-sensitivity testing. Cytometry B Clin Cytom. 2018;94(1):23–48. doi:10.1002/cyto.b.21610
85. Cataland S, Ariceta G, Chen P, et al. Discordance between free C5 and CH50 complement assays in measuring complement C5 inhibition in patients with aHUS treated with ravulizumab. Blood. 2019;134(Supplement_1):1099. doi:10.1182/blood-2019-122421
86. Peffault de Latour R, Fremeaux-Bacchi V, Porcher R, et al. Assessing complement blockade in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Blood. 2015;125(5):775–783. doi:10.1182/blood-2014-03-560540
87. Lundberg P, de la Iglesia S, Kelly RJ, et al. Biomarker analyses in patients with paroxysmal nocturnal hemoglobinuria (PNH) treated with crovalimab and eculizumab: results from the Phase III randomized COMMODORE 2 Trial. Blood. 2023;142(Supplement 1):4088. doi:10.1182/blood-2023-178694
88. Zhang Z, Liu X, Zhang J, Zhang B. Real-world safety profile of eculizumab: an analysis of FDA adverse event reporting system and systematic review of case reports. Expert Opin Drug Saf. 2025;24(10):1157–1163. doi:10.1080/14740338.2024.2392885
89. Socié G, Caby-Tosi M, Marantz JL, et al. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haematol. 2019;185(2):297–310. doi:10.1111/bjh.15790
90. Bektas M, Copley-Merriman C, Khan S, Sarda SP, Shammo JM. Paroxysmal nocturnal hemoglobinuria: current treatments and unmet needs. J Manag Care Spec Pharm. 2020;26(12–b Suppl):S14–20. doi:10.18553/jmcp.2020.26.12-b.s14
91. Wang L, Liu Z, Yang C, Chen M, Han B. Eculizumab treatment for Chinese patients with hemolytic paroxysmal nocturnal hemoglobinuria (PNH): efficacy and safety - a single-center study. Hematology. 2025;30(1):2450575. doi:10.1080/16078454.2025.2450575
92. Syed YY. Iptacopan: first Approval. Drugs. 2024;84(5):599–606. doi:10.1007/s40265-024-02009-4
93. Centers For Disease Control And Prevention US. Meningococcal vaccine recommendations [Internet]. Atlanta (GA): CDC; 2026. Available from: https://www.cdc.gov/meningococcal/hcp/vaccine-recommendations/index.html.
94. Dingli D, De Castro Iii C, Koprivnikar J, et al. Expert consensus on the management of pharmacodynamic breakthrough-hemolysis in treated paroxysmal nocturnal hemoglobinuria. Hematology. 2024;29(1):2329030. doi:10.1080/16078454.2024.2329030
95. McNamara LA, Topaz N, Wang X, Hariri S, Fox L, MacNeil JR. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. Mmwr Morb Mortal Wkly Rep. 2017;66(27):734–737. doi:10.15585/mmwr.mm6627e1
96. Oliver M, Patriquin CJ. Paroxysmal nocturnal hemoglobinuria: current management, unmet needs, and recommendations. J Blood Med. 2023;14:613–628. doi:10.2147/JBM.S431493
97. Centers For Disease Control And Prevention US. Clinical guidance for managing meningococcal disease risk in patients receiving complement inhibitor therapy [Internet]. Atlanta (GA): CDC; 2026. Available from: https://www.cdc.gov/meningococcal/hcp/clinical-guidance/complement-inhibitor.html.
98. Bodó I, Amine I, Boban A, et al. Complement inhibition in paroxysmal nocturnal hemoglobinuria (PNH): a systematic review and expert opinion from central Europe on special patient populations. Adv Ther. 2023;40(6):2752–2772. doi:10.1007/s12325-023-02510-4
99. Emadi A, Brodsky RA. Successful discontinuation of anticoagulation following eculizumab administration in paroxysmal nocturnal hemoglobinuria. 2009:699–701.
100. Bienz M, Oliver M, Sperlich C, Patriquin CJ. Optimizing care in patients with paroxysmal nocturnal hemoglobinuria: managing suboptimal response and uncontrolled disease. J Blood Med. 2026;17(null):561117. doi:10.2147/JBM.S561117
101. Derzsy Z, Prohászka Z, Rigó Jr JJ, Füst G, Molvarec A. Activation of the complement system in normal pregnancy and preeclampsia. Mol Immunol. 2010;47(7–8):1500–1506. doi:10.1016/j.molimm.2010.01.021
102. de Guibert S, Peffault De Latour R, Varoqueaux N, et al. Paroxysmal nocturnal hemoglobinuria and pregnancy before the eculizumab era: the French experience. Haematologica. 2011;96(9):1276–1283. doi:10.3324/haematol.2010.037531
103. Chin-Yee B, Hedley BD, Su R, et al. Paroxysmal nocturnal hemoglobinuria in pregnancy treated with pegcetacoplan: case report and pharmacokinetic analysis. Ejhaem. 2025;6(6):e70179. doi:10.1002/jha2.70179
104. Zhang B, Chu R, Huang C, et al. Progress in the management of pregnancy with paroxysmal nocturnal hemoglobinuria: a review. J Womens Health. 2024;33(1):98–104. doi:10.1089/jwh.2023.0164
105. Jacob A, Mahmoud A, Eldweik L, Mubashir A. Transplacental transfer of ravulizumab in a pregnant woman with neuromyelitis optica: a case report. BMJ Neurol Open. 2025;7(2):e1286. doi:10.1136/bmjno-2025-001286
106. Du W, Mei L. A case report of pegcetacoplan use for a pregnant woman with paroxysmal nocturnal hemoglobinuria. Res Pract Thromb Haemost. 2024;8(4):102435. doi:10.1016/j.rpth.2024.102435
107. Morita Y, Nishimura J, Shimada T, et al. Successful anticoagulant therapy for two pregnant PNH patients, and prospects for the eculizumab era. Int J Hematol. 2013;97(4):491–497. doi:10.1007/s12185-013-1302-3
108. Kelly R, Arnold L, Richards S, et al. The management of pregnancy in paroxysmal nocturnal haemoglobinuria on long term eculizumab. Br J Haematol. 2010;149(3):446–450. doi:10.1111/j.1365-2141.2010.08099.x
109. Gurnari C, Pagliuca S, Maciejewski JP. Clonal evolution in aplastic anemia: failed tumor surveillance or maladaptive recovery? Leuk Lymphoma. 2023;64(8):1389–1399. doi:10.1080/10428194.2023.2215614
110. Karnabeda O, Moskalenko V, Lysak Z, et al. OMS906, a novel alternative pathway MASP-3 inhibitor, normalizes hemoglobin levels and increases clone size in treatment-naïve PNH patients. Blood. 2023;142(Supplement 1):573. doi:10.1182/blood-2023-177921
111. Griffin M, Kelly RJ, Gavillet M, et al. Monotherapy treatment with Zaltenibart (OMS906), an alternative pathway Masp-3 inhibitor, improved key hematologic parameters in patients with PNH with a suboptimal response to Ravulizumab: interim results from a Phase 2 proof-of-concept study. Blood. 2024;144(Supplement 1):4072. doi:10.1182/blood-2024-205866
112. ClinicalTrials.gov. Study of efficacy and safety of NM8074 in adult PNH patients who are naive to complement inhibitor therapy [Internet]. Bethesda (MD): National Library of Medicine (US); 2025. Available from: https://clinicaltrials.gov/study/NCT05646524.
113. Rare Disease Advisor. FDA awards orphan drug designation to NM5072 to treat anemia in PNH [Internet]. New York (NY): Rare Disease Advisor; 2024. Available from: https://www.rarediseaseadvisor.com/news/us-fda-awards-orphan-drug-designation-to-nm5072-to-treat-anemia-in-pnh/.
114. Zhang F, Zhang L, Yang C, et al. KP104, a bifunctional C5 antibody/factor H fusion protein, effectively controls both intravascular and extravascular hemolysis: interim results from a phase 2 study in complement inhibitor-naïve PNH patients. Blood. 2023;142(Supplement 1):572. doi:10.1182/blood-2023-186453
115. Center For Drug Evaluation NMPA. CTR20261241: a multicenter, randomized, open-label Phase II clinical trial to evaluate the efficacy and safety of multiple doses of BW-40202 in subjects with paroxysmal nocturnal hemoglobinuria. Beijing (China): CDE; 2026. Available from: https://www.chinadrugtrials.org.cn/clinicaltrials.searchlistdetail.dhtml.
116. Sharma Y, Scarpitti M, Rubin H, et al. Preclinical studies with HMI-104, an AAVHSC vectorized C5 monoclonal antibody, for the treatment of PNH. In: Molecular Therapy. CELL PRESS 50 HAMPSHIRE ST, FLOOR 5, CAMBRIDGE, MA 02139 USA; 2023:206.
117. Perry C, Von Buttlar X, Thota S. The advancing landscape of paroxysmal nocturnal hemoglobinuria treatment. Turk J Haematol. 2025;42(2):74–81. doi:10.4274/tjh.galenos.2025.2025.0054
118. Browett PJ, Kulasekararaj A, Notaro R, et al. Vemircopan (ALXN2050) monotherapy in paroxysmal nocturnal hemoglobinuria: interim data from a phase 2 open-label proof-of-concept study. Washington, DC: American Society of Hematology; 2022.
119. Kulasekararaj A, Malherbe JLR, McDonald A, et al. BCX9930, a potent, selective, oral factor D inhibitor, demonstrates proof-of-concept as monotherapy in patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2020;136(Supplement 1):14–15. doi:10.1182/blood-2020-138838
120. Kulasekararaj A, Füreder W, McDonald A, et al. P826: factor D inhibition with oral BCX9930 leads to sustained control of hemolysis and symptoms over 48 weeks in subjects with paroxysmal nocturnal hemoglobinuria inadequately controlled on C5 inhibitors. Hemasphere. 2022;6:720–721. doi:10.1097/01.HS9.0000846188.51290.75
121. Carrillo Infante C, Mujeebuddin A. Eculizumab and ravulizumab clinical trial and real-world pharmacovigilance of meningococcal infections across indications. PLoS One. 2025;20(9):e332073. doi:10.1371/journal.pone.0332073
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Advances in Complement Inhibition Therapies for Paroxysmal Nocturnal Hemoglobinuria and Autoimmune Hemolytic Disorders
Tamdin T, Rodgers GM
Journal of Blood Medicine 2025, 16:559-572
Published Date: 12 November 2025