Back to Journals » Drug Design, Development and Therapy » Volume 12
Beraprost sodium preconditioning prevents inflammation, apoptosis, and autophagy during hepatic ischemia-reperfusion injury in mice via the P38 and JNK pathways
Authors Deng J, Feng J ![]() , Liu T, Lu X, Wang W, Liu N, Lv Y, Liu Q, Guo C
, Liu T, Lu X, Wang W, Liu N, Lv Y, Liu Q, Guo C ![]() , Zhou Y
, Zhou Y
Received 2 August 2018
Accepted for publication 17 October 2018
Published 29 November 2018 Volume 2018:12 Pages 4067—4082
DOI https://doi.org/10.2147/DDDT.S182292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Cristiana Tanase
Jingfan Deng,1 Jiao Feng,1 Tong Liu,1 Xiya Lu,1 Wenwen Wang,1 Ning Liu,2 Yang Lv,1 Qing Liu,1 Chuanyong Guo,1 Yingqun Zhou1
1Department of Gastroenterology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai 200072, People’s Republic of China; 2Department of Gastroenterology, Shanghai Tenth People’s Hospital, School of Clinical Medicine of Nanjing Medical University, Shanghai 200072, People’s Republic of China
Objective: The goal of this study was to determine the effects of beraprost sodium (BPS) preconditioning on hepatic ischemia-reperfusion (IR) injury and its underlying mechanisms of action.
Materials and methods: Mice were randomly divided into sham, IR, IR+BPS (50 µg/kg), and IR+BPS (100 µg/kg) groups. Saline or BPS was given to the mice by daily gavage for 1 week before the hepatic IR model was established. Liver tissues and orbital blood were collected at 2, 8, and 24 hours after reperfusion for the determination of liver enzymes, inflammatory mediators, apoptosis- and autophagy-related proteins, key proteins in P38 and c-Jun N-terminal kinase (JNK) cascades, and evaluation of liver histopathology.
Results: BPS preconditioning effectively reduced serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels, improved pathological damage, ameliorated production of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), and affected expressions of Bax, Bcl-2, Caspase-3, Caspase-8, and Caspase-9, microtubule-associated protein 1 light chain 3 (LC3), Beclin-1, and P62. The protective effects of BPS preconditioning were associated with reduced P38 and JNK phosphorylation.
Conclusion: BPS preconditioning ameliorated hepatic IR injury by suppressing inflammation, apoptosis, and autophagy, partially via inhibiting activation of the P38 and JNK cascades.
Keywords: liver injury, beraprost sodium, inflammation, apoptosis, autophagy, MAPK pathway
Introduction
Hepatic ischemia-reperfusion (IR) injury is an underlying complication implicated in cellular and tissue damage to the liver during clinical settings such as hepatic resection, liver transplantation, trauma, and hemorrhagic shock with subsequent resuscitation. It results in varying degrees of hepatic sequelae, liver failure, or even distant organ dysfunction with relatively high morbidity and mortality.1 Unfortunately, there is no effective strategy to clinically prevent hepatic IR injury. Thus, studies of hepatic IR injury in animal models can provide us with a deeper understanding of its pathogenesis and suggest possible preventive measures, including pharmacological preconditioning.2,3
Hepatic IR gives rise to inflammatory outbreak and oxidative-stress,4,5 which have been found to initiate a series of pleiotropic mitogen-activated protein kinase (MAPK) cascades.6 The P38 and c-Jun N-terminal kinase (JNK) cascades are most involved in hepatic IR injury.7–9 Activated P38 and JNK cascades are closely associated with aggravated inflammation and apoptotic or autophagic cell death during hepatic IR injury.10,11
The sterile inflammatory response induced by hepatic IR poses a great threat to the structure and function of the liver and distant organs.12 Several studies have reported that inhibiting P38 or JNK activation reduces these inflammatory cascades via inhibiting the expression of inflammatory mediators such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β).13,14
Apoptosis can be initiated through extrinsic or intrinsic pathways and is a prominent feature of the pathogenesis of hepatic IR injury.15 Inhibiting P38 or JNK phosphorylation has been investigated as a protective mechanism against apoptosis that could attenuate hepatic IR injury or other acute liver injuries.16–19 Autophagy is a multifunctional process that plays important roles in normal cellular homeostasis and human diseases, including hepatic IR injury.20 It was revealed that activated JNK induces Bcl-2 phosphorylation, which causes the dissociation of Beclin-1 from Bcl-2, leading to autophagic cell death.21 Thus, the inflammation, apoptosis, and autophagy that are involved in hepatic IR injury are at least partially mediated by P38 and JNK signaling.
Beraprost sodium (BPS) is an analog of prostacyclin that has a longer half-life, more stable chemical characteristics, and higher oral bioavailability.22 Apart from its arterial vasodilative and antiplatelet effects,23,24 BPS properties including anti-inflammatory and antioxidant activities have also been investigated.25–32 Blocking or inhibiting inflammatory cascades or oxidative stress has been demonstrated to protect against cell death in hepatic IR injury.33,34 Furthermore, BPS was also found to be protective against myocardial or cerebral IR injury.35,36 However, the effects of BPS have never been explored in animal models of hepatic IR injury. Therefore, the main goal of this study was to investigate the protective effects and potential mechanism of BPS for the prevention of hepatic IR injury. We hypothesized that the effects of BPS on hepatic IR injury would be achieved by inhibiting inflammation, apoptosis, and autophagy, partially via inhibiting P38 and JNK signaling.
Materials and methods
Reagents
BPS (purity: 98.9%) produced by Toray Industries Inc. (Tokyo, Japan) was stored at −20°C in the dark. It was diluted with sterile normal saline to different concentrations before use. The antibodies used in this study for Western blotting included anti-β-actin (60008-1-Ig), -Bcl-2 (26593-1-AP), -Bax (60267-1-Ig), -Caspase-3 (19677-1-AP), -Caspase-9 (66169-1-Ig), -Beclin-1 (11306-1-AP), -microtubule-associated protein 1 light chain 3 (LC3) (14600-1-AP), and -P62 (18420-1-AP) purchased from Proteintech (Rosemont, IL, USA); anti-cleaved Caspase-8 (8592), -IL-1β (12242), -TNF-α (11948), -JNK (9252), and -P38 (9212) purchased from Cell Signaling Technology (Danvers, MA, USA); anti-p-JNK (ARE6036; Antibody Revolution, Inc. San Diego, CA, USA); and anti-p-P38 (orb6578; Wuhan Goodbio Technology Co., Ltd., Wuhan, China). Except for the antibodies anti-Bcl-2 (orb10173; Wuhan Goodbio Technology Co., Ltd.) and -TNF-α (60291-1-Ig, Proteintech), the sources and identities of other antibodies including anti-Bax, -Beclin-1, -IL-1β, -p-JNK, and -p-P38 used for immunohistochemical staining were the same as those used for Western blotting. Apoptosis was evaluated by a TUNEL assay kit manufactured by Hoffman-La Roche Ltd. (Basel, Switzerland). Microplate test kits purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) were used for alanine aminotransferase (ALT) and aspartate aminotransferase (AST) detection. IL-1β and TNF-α levels were determined by ELISA kits purchased from Anogen-Yes Biotech Laboratories Ltd. (Ontario, Canada). Quantitative real-time (qRT) PCR kits purchased from TaKaRa Biotechnology Co., Ltd (Dalian, China) were applied to monitor target gene transcription.
Animals
Six-week-old male Balb/c mice weighing 20–22 g were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). Mice were kept in a clean environment under a 12/12 hour light or dark cycle and a temperature of 20°C–24°C. Mice were given access to food and water ad libitum. All experimental procedures involving mice were approved by the Animal Care and Use Committee of Shanghai Tongji University. Handling and care of mice conformed to the National Institute of Health Guidelines.
Experimental design
Pilot study
First, it was necessary to perform a pretest to investigate whether the selected dosages of BPS (50 and 100 μg/kg), which were based on other studies27,36 were safe and did not alter the normal function and structure of liver. Fifteen mice were randomly divided into the following three groups (n=5):
- Normal saline control (NC) group.
- Low dose BPS (50 μg/kg) group.
- High dose BPS (100 μg/kg) group.
The same volume of normal saline, low, or high dose BPS was given to mice by gavage daily for a week. Then the orbital blood was taken to determine ALT and AST levels, and the middle and left lateral liver lobes were removed for H&E staining.
Formal study
Seventy-two mice were randomized into the following four groups (n=18):
- Sham group: mice suffered a laparotomy, the first porta hepatis was dissociated without vascular clipping, and then the abdominal incision was sutured after normal saline preconditioning.
- IR group: hepatic IR models were established in mice after normal saline preconditioning.
- IR+BPS (50 μg/kg) group: mice were preconditioned with BPS (50 μg/kg) followed by establishing hepatic IR models.
- IR+BPS (100 μg/kg) group: mice were preconditioned with BPS (100 μg/kg) followed by establishing hepatic IR models.
The mice were given equal volumes of normal saline or BPS (50 or 100 μg/kg) by gavage daily for 7 days before hepatic IR models were established. Six mice were sacrificed to gather samples of the orbital blood and middle and left lateral liver lobes from each group at three predetermined time points: 2, 8, and 24 hours after reperfusion.
Mouse model of hepatic IR injury
A mouse model of 70% hepatic IR injury was established in this study. Mice were fasted for 16 hours before surgery, but had free access to water. The mice were intraperitoneally injected with 1.25% sodium pentobarbital (Nembutal; Sigma-Aldrich, St. Louis, MO, USA) at a dose of 40 mg/kg for anesthesia, and then placed on a sterile operating table with limbs fixed. After midline laparotomy, the liver lobes were turned over using a wet cotton swab, and the first porta hepatis, primarily composed of hepatic artery, portal vein, and common bile duct was blocked with a vascular clamp, which immediately caused hepatic segmental warm ischemia with the color of the hepatic lobes changing from reddish brown to light red. Next, abdominal viscera were restored to their natural anatomical position, the abdominal cavity was closed, and the incision was covered with a moist saline gauze. Finally, the mice were placed on an electric blanket to maintain body temperature until they revived. After 45 minutes of ischemia, the obstruction was relieved to restart reperfusion, and the abdominal incision was sutured.
Serum enzymes and inflammatory mediators
Serum samples were separated from blood by centrifuging at 4,600×g at 4°C for 10 minutes and preserved at −80°C after incubating the samples at 4°C for 5 hours. ALT and AST, two sensitive biochemical indexes of impaired liver function, were quantified through an automated chemical analyzer (AU1000; Olympus Corporation, Tokyo, Japan). Serums TNF-α and IL-1β were determined by ELISA kits.
Histopathological evaluation
Liver tissues collected from each mouse were fixed in 4% paraformaldehyde for at least 24 hours at room temperature. Small pieces of the tissues were embedded in paraffin after dehydration, clearing, and wax immersion. Then, serial 4-μm thick sections were cut and stained with H&E for assessment. The severity of liver damage at 8 hours post-reperfusion was blindly evaluated by Suzuki’s histological criteria.37
qRT-PCR
The total RNA from frozen liver tissues was extracted using TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) and was further isolated and purified. After determining the RNA concentration, samples were processed into cDNA using a reverse transcription kit. Gene expression at the mRNA level was detected by SYBR Green qRT-PCR through a 7900HT fast RT-PCR system (Applied Biosystems, Foster City, CA, USA). The primers used for qRT-PCR are shown in Table 1. The specificity of primers was verified by Sanger sequencing of the amplified PCR products. The relative mRNA expression levels were analyzed by the 2−ΔΔCt method and normalized by β-actin.38
 | Table 1 Oligonucleotide sequences of primers used for qRT-PCR |
Immunohistochemical staining
Paraffin sections from 8 hours after reperfusion were baked in an oven for 2 hours at 60°C, and then dewaxed and rehydrated. Antigen retrieval was performed by placing sections in citrate buffer, which was then heated to 95°C for 10 minutes; after cooling to room temperature, this process was repeated four times. Next, 3% hydrogen peroxide was added to the sections for 20 minutes to block endogenous peroxidase activity, and then 5% BSA was added to block nonspecific binding for 15 minutes (both at room temperature). Slices were then incubated overnight at 4°C with the following antibodies: anti-LC3 (1:100); anti-TNF-α, -IL-1β, -Bcl-2, -Bax, -p-JNK, and -p-P38 (all 1:200); and anti-Beclin-1 (1:1,000). The following day, secondary antibody was incubated with the slices for 1 hour at 37°C. A chromogenic substrate for peroxidase, 3,3′-diaminobenzidine, was added and oxidized by the peroxidase conjugated to the secondary antibody to produce observable brown color. Next, slices were counterstained with hematoxylin, dehydrated with a gradient alcohol series, cleared in xylene, sealed with neutral gum, and observed and imaged under a light microscope. Final evaluations were performed with Image-Pro Plus software 6.0 to calculate the integrated optical densities (IODs) of the positive staining area.
Western blotting
Fresh liver tissues were cut into small pieces and frozen immediately with liquid nitrogen and stored at -80°C. Liver tissues were ground into powder while in liquid nitrogen, and then were lysed in RIPA buffer containing protease inhibitors. Protein concentrations were measured using the bicinchoninic acid method. Then, protein samples were preserved in 5× loading buffer at -20°C before being separated by 10% or 12.5% SDS-PAGE and transferred onto polyvinylidene fluoride membranes. Membranes were blocked with 5% skimmed milk dissolved in PBS (5% BSA dissolved in PBS used for p-P38 and p-JNK) for at least 1 hour and subsequently incubated overnight at 4°C with the following primary antibodies: anti-TNF-α, -IL-1β, -P38, -JNK, -p-P38, and -p-JNK (all 1:500); anti-Caspase-3 and -cleaved Caspase-8 (both 1:800); and anti-β-actin, -Bcl-2, -Bax, -Beclin-1, -LC3, -p62 and -Caspase-9 (all 1:1,000). The next day, after washing thrice with PBS containing 0.1% Tween-20, the membranes were incubated with anti-rabbit or anti-mouse near-infrared fluorescent secondary antibodies. Finally, an Odyssey two-color infrared laser imaging system (LI-COR Biosciences, Lincoln, NE, USA) was used to detect excited fluorescent signal from the membrane.
TUNEL assay
After dewaxing with xylene and rehydration in a diminishing alcohol gradient, liver slices were processed with Proteinase K to increase cell and nuclear membrane permeability and to ensure full labeling with the TUNEL reaction mixture. An optical microscope and the Image-Pro Plus software 6.0 were used to observe and analyze TUNEL results.
Statistical analysis
All experiments were repeated three times. Experimental data were collected, processed, and presented as mean ± SD. One-way ANOVA using the Student-Newman-Keuls method was used to compare statistical differences among three or four groups using SPSS version 20.0 software (IBM, Armonk, NY, USA). A value of P<0.05 was regarded as statistically significant.
Results
BPS had no adverse effects on liver structure or function
No notable hepatocellular injury or structural tissue changes were observed among the three groups in the Pilot Study (Figure 1A). Additionally, there were no statistically significant differences in liver function, as determined by quantifying serum ALT and AST levels (Figure 1B). Therefore, toxic effects of BPS (50 or 100 μg/kg) on liver physiology and biochemistry were excluded.
 | Figure 1 BPS had no adverse effects on liver structure or function. |
BPS preconditioning ameliorated hepatic IR injury
Our overall evaluation of the effects of BPS on hepatic IR injury was determined by measuring serum ALT and AST levels and histological examinations at 2, 8, and 24 hours after reperfusion. Pathological changes of the liver were mainly manifested as varying degrees of sinusoidal congestion, hepatocyte degeneration or necrosis, inflammatory cell infiltration, and disturbances or destruction of the lobular architecture (Figure 2A). These changes were most serious in the IR group. The BPS pretreatment markedly improved liver pathology, and the effects increased with increasing BPS doses. Suzuki’s injury score for evaluating liver sections from 8 hours post-reperfusion also demonstrated this. Liver function changed accordingly; serum ALT and AST levels were significantly higher in the IR group than in the sham or BPS-preconditioned groups, and there was a greater reduction in the high-dose BPS group (Figure 2B). Thus, BPS preconditioning ameliorated the extent of functional and structural liver damage caused by hepatic IR in a dose-dependent manner.
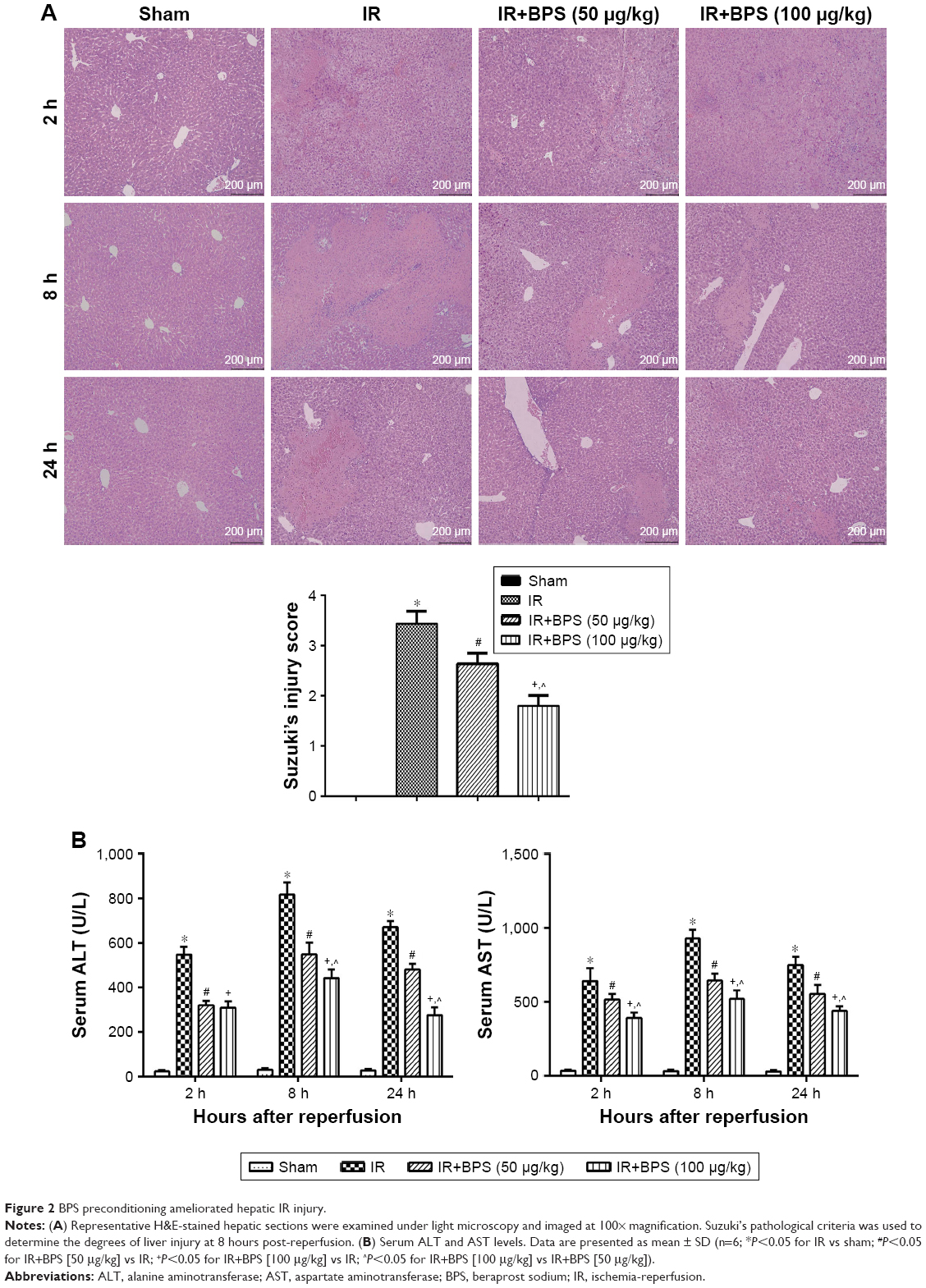 | Figure 2 BPS preconditioning ameliorated hepatic IR injury. |
BPS preconditioning inhibited inflammatory cascades involved in hepatic IR injury
Inflammation is a powerful detrimental factor that plays an important role in the damage caused by hepatic IR. TNF-α and IL-1β, two critical mediators of inflammation during hepatic IR injury, were detected by qRT-PCR, ELISA, immunohistochemical staining, and Western blotting. Hepatic IR significantly increased TNF-α and IL-1β expressions at mRNA levels compared to the sham group, and expressions of these cytokines peaked at 8 hours, and BPS preconditioning effectively reduced TNF-α and IL-1β expressions (Figure 3A). Similarly, TNF-α and IL-1β protein levels markedly increased in the IR group compared to the sham group, and BPS preconditioning markedly reduced TNF-α and IL-1β expressions as shown by ELISA (Figure 3B), immunohistochemical staining (Figure 3C), and Western blotting (Figure 3D); this effect was, especially, evident at the 100 μg/kg dose. Thus, BPS preconditioning inhibited inflammatory reactions by suppressing the production of inflammatory mediators such as TNF-α and IL-1β.
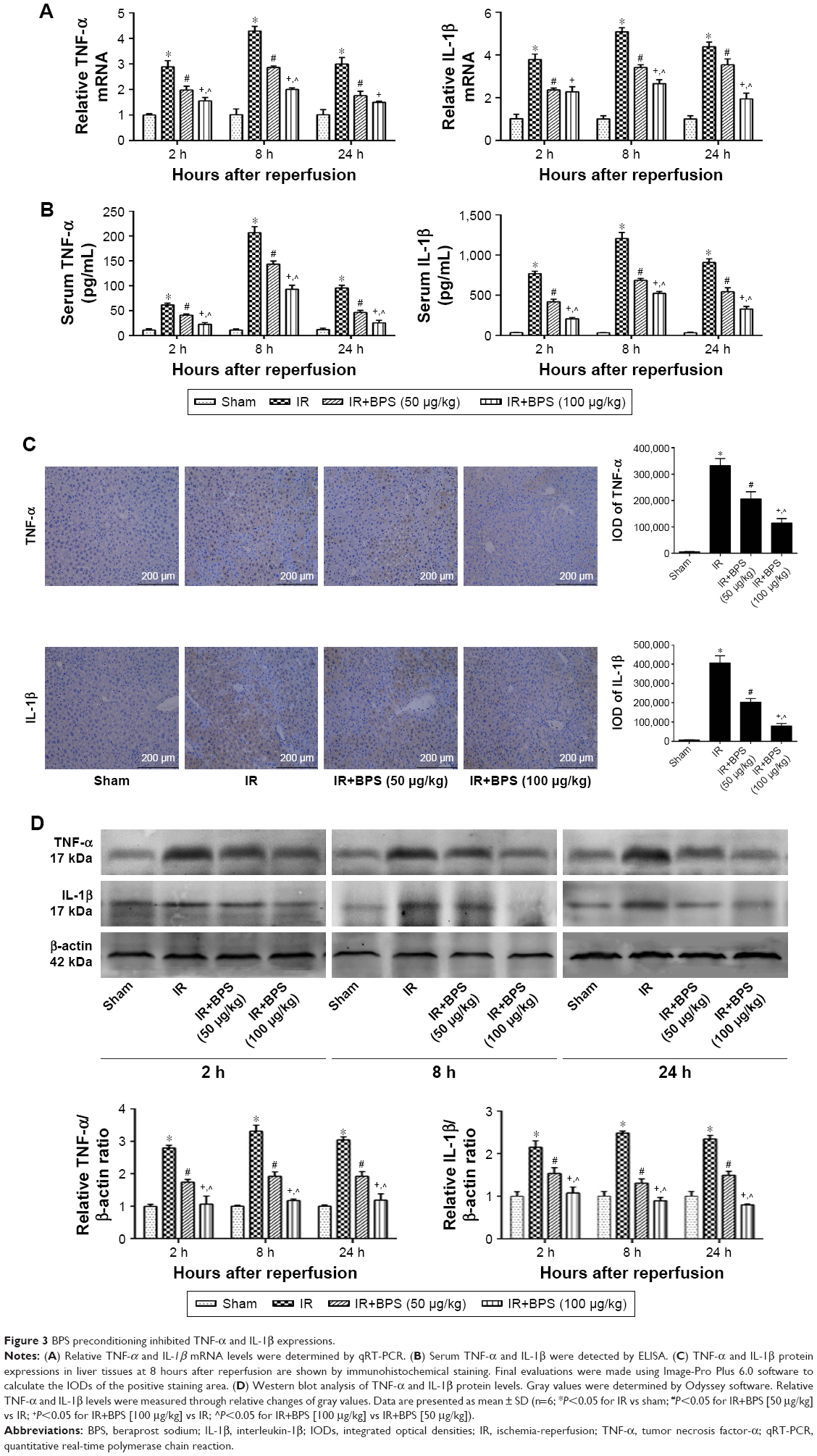 | Figure 3 BPS preconditioning inhibited TNF-α and IL-1β expressions. |
BPS preconditioning reduced both extrinsic and intrinsic apoptosis
Apoptosis is one of the consequences of liver injury caused by IR. Therefore, reduced apoptosis is presumably associated with better outcomes for hepatic IR injury. The TUNEL assay was used to analyze samples from 8 hours after reperfusion and revealed that only IR caused high levels of apoptosis; the most positive cells (dark brown nuclei) were found in the IR group. Apoptosis was significantly alleviated in the BPS-preconditioned groups, and the effect was dose-dependent; only a few apoptotic cells were observed in the sham group (Figure 4A). Analyses of qRT-PCR, immunohistochemical staining, and Western blot data from 2, 8, and 24 hours after reperfusion showed similar trends in apoptosis (Figure 4B–D). Hepatic IR caused increased Bax expression and a marked drop in Bcl-2 expression at the mRNA and protein levels; BPS preconditioning partially reversed these changes (Figure 4B–D). Caspase-3, Caspase-8, and Caspase-9 presented similar trends as Bax at the mRNA level (Figure 4B); similarly, cleaved Caspase-3, cleaved Caspase-8, and cleaved Caspase-9 levels also followed this trend (Figure 4D). Thus, BPS preconditioning attenuated apoptosis in a dose-dependent manner.
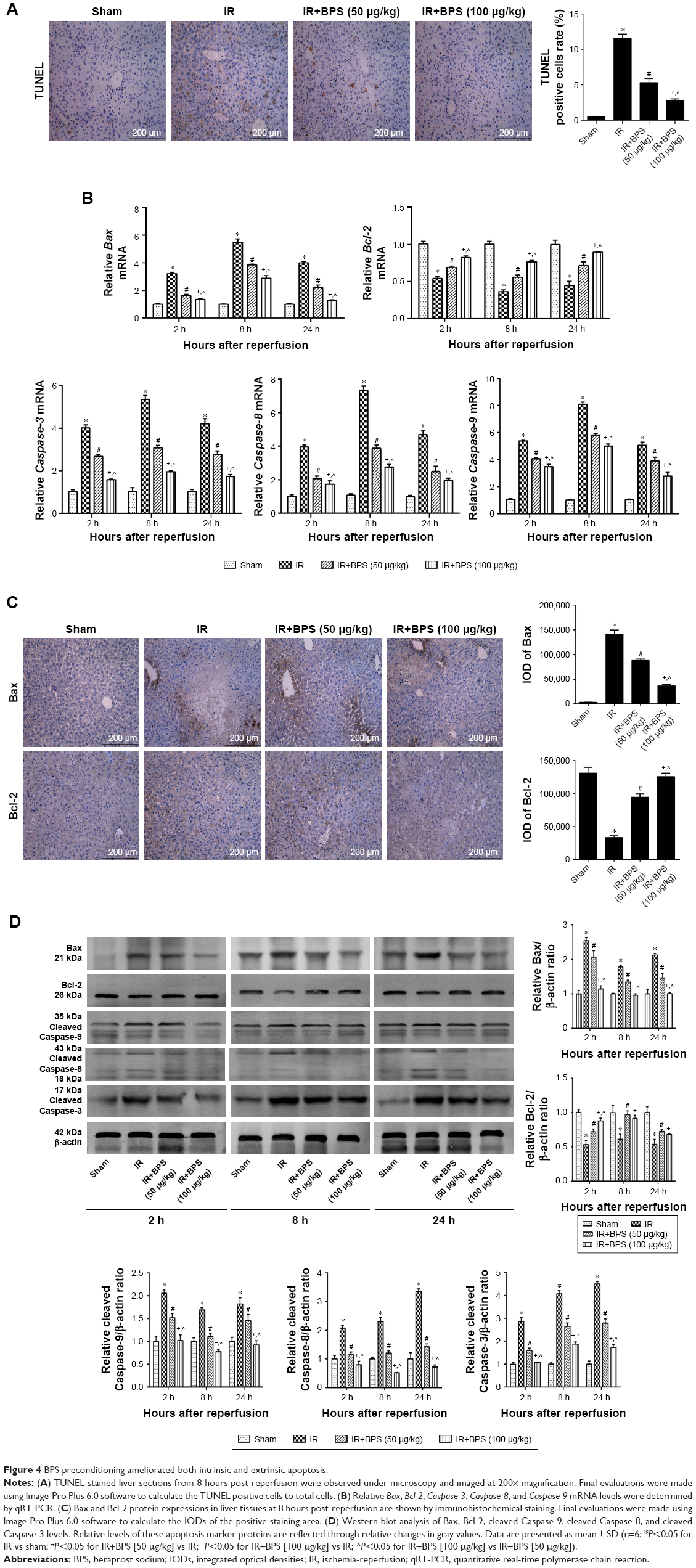 | Figure 4 BPS preconditioning ameliorated both intrinsic and extrinsic apoptosis. |
BPS preconditioning ameliorated hepatic IR-induced autophagy
Autophagy is another important process that contributes to hepatic IR injury. To explore the relationship between the protective effects of BPS pretreatment on hepatic IR injury and autophagy, autophagy-related proteins including LC3, Beclin-1, and P62 were detected. qRT-PCR showed that hepatic IR greatly activated LC3 and Beclin-1 transcription compared to the sham group (Figure 5A). Accordingly, Beclin-1 protein level was found to be increased by immunohistochemical staining (Figure 5B) and Western blotting (Figure 5C). The expression of LC3 II at protein level was increased as shown in Figure 5C. P62 mRNA and protein levels showed the opposite trend compared to Beclin-1 (Figure 5A and C). BPS preconditioning reduced LC3 II and Beclin-1 expressions and enhanced P62 expression, and the effect was markedly evident at the 100 μg/kg dose. Thus, BPS showed dose-dependent anti-autophagy activity in the hepatic IR model.
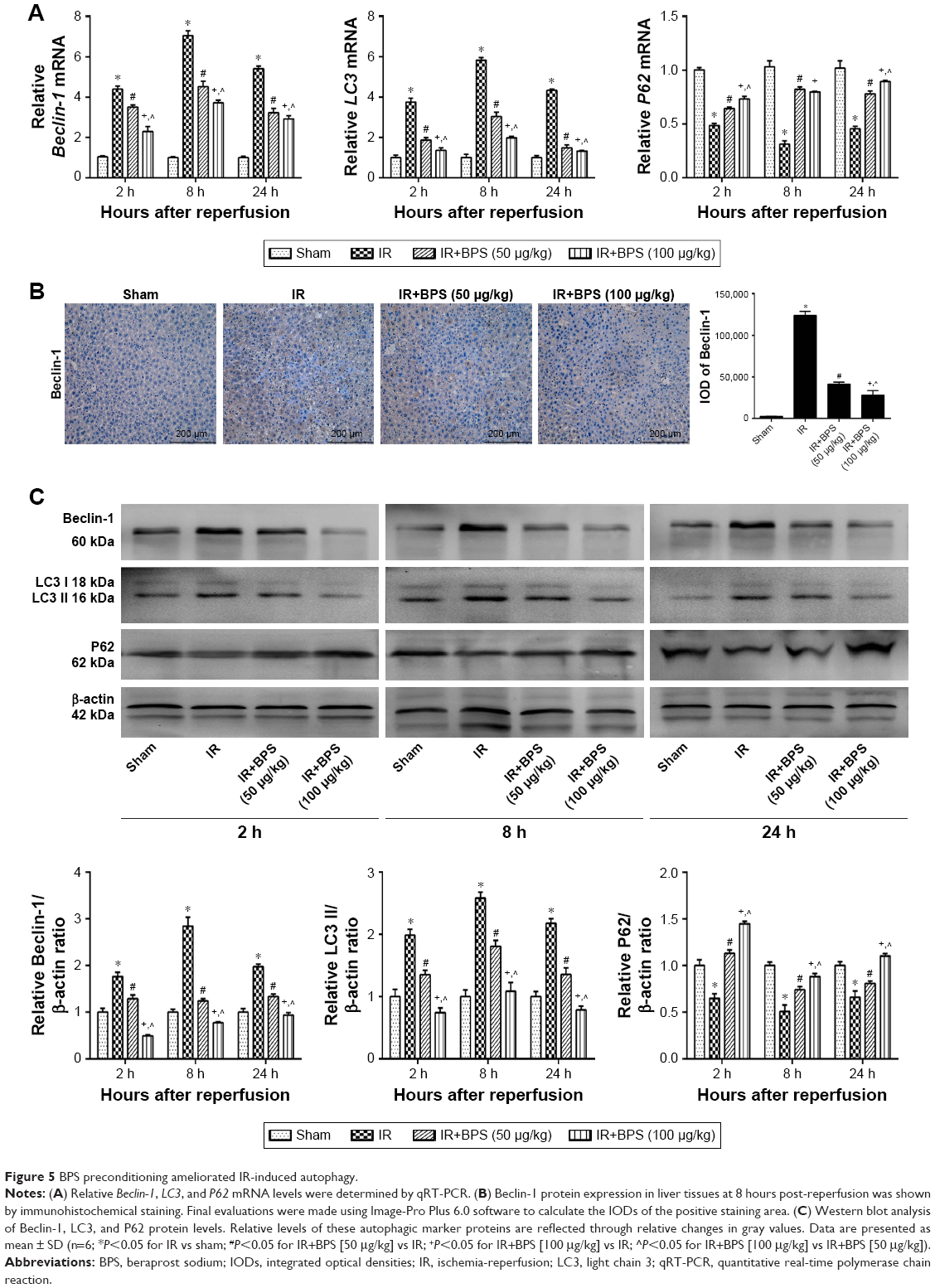 | Figure 5 BPS preconditioning ameliorated IR-induced autophagy. |
BPS suppressed the P38 and JNK cascades
From the above results, it was concluded that BPS preconditioning alleviated hepatic IR injury via inhibiting inflammation, apoptosis, and autophagy. However, it was uncertain which signaling pathway(s) was more affected by BPS. Through activating or inhibiting different signaling cascades, BPS could greatly inhibit inflammation, apoptosis, and autophagy. Activation of the P38 and JNK cascades is closely related to inflammation and cell death; therefore, we investigated whether the activity of BPS on hepatic IR injury was via inhibiting P38 and JNK activations. P38, JNK, and their phosphorylated versions, p-P38 and p-JNK, were evaluated at the protein levels. P38 and JNK expressions were not significantly different among the four groups; however, IR markedly activated P38 and JNK to p-P38 and p-JNK, respectively, compared to the sham group. BPS preconditioning inhibited P38 and JNK phosphorylation as shown by immunohistochemistry (Figure 6A) and Western blotting (Figure 6B). Thus, the protective effects of BPS against hepatic IR injury were at least partially mediated by inhibiting P38 and JNK phosphorylation.
 | Figure 6 BPS suppressed P38 and JNK phosphorylation. |
Discussion
Hepatic IR injury is a deleterious complication that greatly affects the postoperative survival and prognosis of patients who have undergone hepatic surgery, trauma, or shock. The precise cellular and molecular mechanisms underlying hepatic IR injury have not been clarified. Inflammation and oxidative stress are two powerful mechanisms that contribute to the progression of hepatic IR injury.4,5 In vitro studies have shown that BPS alleviated lipopolysaccharide-induced inflammation by suppressing TNF-α production in human monocytic cells by inhibiting P38, JNK, and ERK phosphorylation.25 In rat glomerular mesangial cells, BPS inhibited ROS production and increased antioxidant activity to probably protect against diabetic nephropathy.26 Three findings from in vivo studies have demonstrated that the liver is an important target of BPS intervention for acute or chronic progressive liver disease, and that the activity of BPS is associated with its anti-inflammatory or antioxidant properties.27–29 Therefore, BPS may be a promising drug against hepatic IR injury.
For this study, we established mouse models of hepatic IR injury to investigate whether BPS preconditioning protected against hepatic IR injury and their possible underlying mechanisms. Overwhelming sterile inflammation caused by hepatic IR imperils the viability of the liver and distant organs, which can severely threaten patient health.12 Inhibiting inflammation cascades could be a powerful tool to defend against hepatic IR injury. Kupffer cells ([KCs]; resident liver macrophages) are activated due to various stimuli including ROS during the early stages of reperfusion to initiate inflammatory cascades and subsequently release inflammatory mediators such as TNF-α and IL-1β, as well as produce ROS.4,39,40 TNF-α is a critical inflammatory mediator that plays key roles in further propagating KCs activation and the recruitment and activation of neutrophils, which consequently results in more intense inflammatory responses and the occurrence of other deleterious cellular events such as apoptosis.41 TNF-α and IL-1β also promote P38 and JNK phosphorylation,42 which further promotes the expression of inflammatory mediators such as TNF-α and IL-1β.43,44 Thus amplified inflammatory responses aggravate hepatic IR injury. Therefore, we explored whether BPS preconditioning could protect liver tissues by blocking inflammatory cascades. TNF-α and IL-1β are core inflammatory factors that were tracked in this study. Our study showed that BPS preconditioning significantly inhibited TNF-α and IL-1β expressions both at the mRNA and protein levels compared to the IR group at three different time points. In accordance with these results, liver pathology and function were both improved by BPS preconditioning, as shown by H&E staining, and serum ALT and AST levels. BPS preconditioning had protective effects against hepatic IR injury via suppressing expression of inflammatory mediators such as TNF-α and IL-1β; however, the underlying mechanisms remain unknown.
The MAPK family consists of three core kinases, which function sequentially in a cascade reaction that plays an important part in transmitting a wide array of extracellular signals intracellularly, thus regulating many cellular activities.45 Inhibition of phosphorylation and activation of P38 and JNK have been shown to be closely related to decreased inflammation in hepatic IR injury13,14 as well as inflammation caused by other stimuli.43,44 The anti-inflammatory effects of BPS have also been shown to be partially mediated by inhibiting P38 and JNK phosphorylation.25 Therefore, we explored P38 and JNK expressions as well as activation of their respective signaling cascades to explore potential mechanisms underlying the anti-inflammatory effects of BPS preconditioning on hepatic IR injury. Our results showed that BPS preconditioning significantly inhibited P38 and JNK phosphorylation. Furthermore, BPS preconditioning inhibited expression of the inflammatory cytokines TNF-α and IL-1β, as mentioned above. Therefore, we inferred that the anti-inflammatory effects of BPS that afforded protective effects against hepatic IR injury were at least partially mediated by suppressing P38 and JNK phosphorylation.
However, other mechanisms could be behind the protective effects of BPS preconditioning on hepatic IR injury. Apoptosis, which takes place through intrinsic or extrinsic pathways, contributes to hepatic IR injury. Bax is a pro-apoptotic protein that interacts with Bcl-2, a pro-survival protein, to determine cell survival or apoptosis fates through the intrinsic pathway.46 TNF-α is an important agonist that induces extrinsic apoptotic cell death of liver.47 P38 phosphorylation has been shown to cause the mitochondrial translocation of Bax.48,49 Studies have also found that activated JNK promotes Bcl-2 phosphorylation.50,51 The increased ratio of Bax/Bcl-2 contributes to mitochondrial outer membrane permeabilization, which releases cytochrome c and sequentially activates Caspase-9 and Caspase-3, thus causing apoptotic cell death through the intrinsic pathway.52,53 Additionally, hepatic IR induces inflammatory response, such as the release of TNF-α. Binding of TNF-α to corresponding death receptors causes allosteric activation of the receptor, followed by the formation of the death-inducing signaling complex, which results in the autocatalytic activation of Caspase-8, the subsequent activation of Caspase-3, and finally, apoptotic cell death through the extrinsic pathway.15 Furthermore, activated Caspase-8 can further intensify intrinsic apoptosis by cleaving Bid to tBid. tBid transiently interacts with and activates Bax, which then initiates intrinsic apoptosis.54 Therefore, in our study, BPS preconditioning was investigated for its ability to alleviate apoptosis as shown in TUNEL staining. Then, we further found that BPS preconditioning suppressed Bax expression and increased Bcl-2 expression, both at the mRNA and protein levels; Caspase-3, Caspase-8, and Caspase-9 showed similar changes as Bax. Taken together, we inferred that the inhibitory effects of BPS on intrinsic and extrinsic apoptosis were downregulating the Bax/Bcl-2 ratio and inhibiting TNF-α expression, both of which were at least partially mediated by inhibiting P38 and JNK phosphorylation.
The Bcl-2 family plays a key role in the balance between apoptosis and autophagy. Apoptosis and autophagy are linked through the Beclin-1/Bcl-2 complex. Bcl-2 is an anti-apoptotic protein with a BH3 binding groove that interacts with Beclin-1, a pro-autophagy protein with a BH3 domain, to inhibit autophagy.55 A subtle balance between free Beclin-1 and Beclin-1 complexed with Bcl-2 is critical for maintaining the basal level of autophagy that is required for normal cellular homeostasis;56 however, activated JNK induces Bcl-2 phosphorylation, which causes the dissociation of Beclin-1 from Bcl-2, leading to autophagic cell death.21 Thus, autophagy that is involved in hepatic IR injury is at least partially mediated by P38 and JNK signaling. Liu et al reported that beta-asarone attenuated IR-induced autophagy in the brain by upregulating Bcl-2 via inhibiting JNK phosphorylation,57 which leads to the dissociation of Bcl-2 from Beclin-1; free Beclin-1 subsequently induces autophagy. Beclin-1, LC3, and P62 are three autophagy markers. Our study showed that BPS preconditioning significantly alleviated autophagy, as indicated by the downregulation of Beclin-1 and LC3, and the upregulation of P62 at both the mRNA and protein levels. Therefore, the protective effects of BPS preconditioning against autophagy might be associated with a greater association between Bcl-2 and Beclin-1, which is achieved by inhibiting JNK phosphorylation to enhance Bcl-2 activity.
Based on our studies, we speculate that BPS preconditioning might be effective to alleviate severity of liver IR injury and improve prognosis for patients who underwent liver surgery. The protective mechanisms of BPS preconditioning on hepatic IR injury for humans might be similar to those observed from our studies on mice. The effects of BPS preconditioning against hepatic IR injury might be partially mediated through the inhibition of the P38 and JNK cascades, thus inhibiting inflammation and the release of inflammatory mediators, and alleviating apoptosis and autophagy as well, thereby reducing hepatocytes injury and liver enzymes release. Therefore, BPS preconditioning seems to be effective to alleviate hepatic IR injury and improve the survival and prognosis of patients. According to our studies, the possible human equivalent dose against liver IR injury could be roughly, preliminarily estimated based on body surface area. The equivalent dose is 4.05–8.1 μg/kg for humans who weigh 60 kg.58 Indeed, the dose conversion from animal to human is far from being so simple. Besides body surface area, there are many factors that need to be considered including weight, safety, pharmacokinetics, and physiological time and so on. Converting effective dose of BPS preconditioning used in mice against hepatic IR injury to pharmacologically active dose for humans needs further deep and extensive studies.
Hepatic IR injury has complicated and intersected mechanisms underlying its etiology that require further study. The protective effects of BPS on hepatic IR injury were found to be related to reduced inflammation, apoptosis, and autophagy via inhibiting the P38 and JNK cascades (Figure 7). Further efforts are required to more deeply explore the mechanisms involved in BPS preconditioning for hepatic IR injury.
 | Figure 7 Probable mechanisms of BPS preconditioning against hepatic IR injury. |
Conclusion
Hepatic IR induced inflammation, apoptosis, and autophagy, all of which contributed to liver injury progression and were partially ameliorated by BPS preconditioning. The activity of BPS in hepatic IR injury appeared to be inhibiting the P38 and JNK cascades.
Data availability
The data used to support the findings of this study are included within the article.
Acknowledgments
This study was supported by Shanghai Tenth Hospital’s improvement plan for National Natural Science Foundation of China (NO.SYGZRPY2017003). We thank Toray Industries Inc. (Tokyo, Japan) for kindly providing us with BPS.
Disclosure
The authors report no conflicts of interest in this work.
References
Nastos C, Kalimeris K, Papoutsidakis N, et al. Global consequences of liver ischemia/reperfusion injury. Oxid Med Cell Longev. 2014;2014(1):906965. | ||
Datta G, Fuller BJ, Davidson BR. Molecular mechanisms of liver ischemia reperfusion injury: insights from transgenic knockout models. World J Gastroenterol. 2013;19(11):1683–1698. | ||
Gracia-Sancho J, Casillas-Ramírez A, Peralta C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: a 2015 update. Clin Sci. 2015;129(4):345–362. | ||
Lentsch AB, Kato A, Yoshidome H, Mcmasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32(2):169–173. | ||
Elias-Miró M, Jiménez-Castro MB, Rodés J, Peralta C. Current knowledge on oxidative stress in hepatic ischemia/reperfusion. Free Radic Res. 2013;47(8):555–568. | ||
Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. | ||
Yoshinari D, Takeyoshi I, Kobayashi M, et al. Effects of a p38 mitogen-activated protein kinase inhibitor as an additive to university of wisconsin solution on reperfusion injury in liver transplantation. Transplantation. 2001;72(1):22–27. | ||
Uehara T, Bennett B, Sakata ST, et al. JNK mediates hepatic ischemia reperfusion injury. J Hepatol. 2005;42(6):850–859. | ||
King LA, Toledo AH, Rivera-Chavez FA, Toledo-Pereyra LH. Role of p38 and JNK in liver ischemia and reperfusion. J Hepatobiliary Pancreat Surg. 2009;16(6):763–770. | ||
Li J, Wang F, Xia Y, et al. Astaxanthin pretreatment attenuates hepatic ischemia reperfusion-induced apoptosis and autophagy via the ROS/MAPK pathway in mice. Mar Drugs. 2015;13(6):3368–3387. | ||
Xu S, Niu P, Chen K, et al. The liver protection of propylene glycol alginate sodium sulfate preconditioning against ischemia reperfusion injury: focusing MAPK pathway activity. Sci Rep. 2017;7(1):15175. | ||
van Golen RF, Reiniers MJ, Olthof PB, van Gulik TM, Heger M. Sterile inflammation in hepatic ischemia/reperfusion injury: present concepts and potential therapeutics. J Gastroenterol Hepatol. 2013;28(3):394–400. | ||
Kobayashi M, Takeyoshi I, Yoshinari D, Matsumoto K, Morishita Y. P38 mitogen-activated protein kinase inhibition attenuates ischemia-reperfusion injury of the rat liver. Surgery. 2002;131(3):344–349. | ||
Ocuin LM, Zeng S, Cavnar MJ, et al. Nilotinib protects the murine liver from ischemia/reperfusion injury. J Hepatol. 2012;57(4):766–773. | ||
Cao L, Quan XB, Zeng WJ, Yang XO, Wang MJ. Mechanism of hepatocyte apoptosis. J Cell Death. 2016;9:19. | ||
Li J, Xia Y, Liu T, et al. Protective effects of astaxanthin on ConA-induced autoimmune hepatitis by the JNK/p-JNK pathway-mediated inhibition of autophagy and apoptosis. PLoS One. 2015;10(3):e0120440. | ||
Liu T, Xia Y, Li J, et al. Shikonin attenuates concanavalin a-induced acute liver injury in mice via inhibition of the JNK Pathway. Mediators Inflamm. 2016;2016:2748367. | ||
Feng J, Zhang Q, Mo W, et al. Salidroside pretreatment attenuates apoptosis and autophagy during hepatic ischemia-reperfusion injury by inhibiting the mitogen-activated protein kinase pathway in mice. Drug Des Devel Ther. 2017;11:1989–2006. | ||
Wu L, Wang C, Li J, et al. Hepatoprotective effect of quercetin via TRAF6/JNK pathway in acute hepatitis. Biomed Pharmacother. 2017;96:1137–1146. | ||
Cursio R, Colosetti P, Gugenheim J. Autophagy and liver ischemia-reperfusion injury. Biomed Res Int. 2015;2015:417590. | ||
Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284(5):2719–2728. | ||
Saji T, Ozawa Y, Ishikita T, Matsuura H, Matsuo N. Short-term hemodynamic effect of a new oral PGI2 analogue, beraprost, in primary and secondary pulmonary hypertension. Am J Cardiol. 1996;78(2):244–247. | ||
Durongpisitkul K, Laoprasitiporn D, Layangool T, et al. Comparison of the acute pulmonary vasodilating effect of beraprost sodium and nitric oxide in congenital heart disease. Circ J. 2005;69(1):61–64. | ||
Okada Y, Marchevsky AM, Kass RM, Matloff JM, Jordan SC. A stable prostacyclin analog, beraprost sodium, attenuates platelet accumulation and preservation-reperfusion injury of isografts in a rat model of lung transplantation. Transplantation. 1998;66(9):1132–1136. | ||
Wang WL, Kuo CH, Chu YT, et al. Prostaglandin I(2) analogues suppress TNF-α expression in human monocytes via mitogen-activated protein kinase pathway. Inflamm Res. 2011;60(7):655–663. | ||
Zhang LY, Zou JJ, Liu ZM. Effects of beraprost sodium, a prostaglandin I(2) analog, on high glucose-induced proliferation and oxidative stress in a rat glomerular mesangial cell line. Pharmacology. 2011;87(5–6):350–358. | ||
Ohta S, Nakamuta M, Fukushima M, et al. Beraprost sodium, a prostacyclin (PGI) analogue, ameliorates concanavalin A-induced liver injury in mice. Liver Int. 2005;25(5):1061–1068. | ||
Misawa H, Ohashi W, Tomita K, Hattori K, Shimada Y, Hattori Y. Prostacyclin mimetics afford protection against lipopolysaccharide/d-galactosamine-induced acute liver injury in mice. Toxicol Appl Pharmacol. 2017;334:55–65. | ||
Kumei S, Yuhki KI, Kojima F, et al. Prostaglandin I2 suppresses the development of diet-induced nonalcoholic steatohepatitis in mice. Faseb J. 2018;32(5):2354–2365. | ||
Peng L, Li J, Xu Y, et al. The protective effect of beraprost sodium on diabetic nephropathy by inhibiting inflammation and p38 MAPK signaling pathway in high-fat diet/streptozotocin-induced diabetic rats. Int J Endocrinol. 2016;2016(4):1690474. | ||
Li J, Peng L, Du H, et al. The protective effect of beraprost sodium on diabetic cardiomyopathy through the inhibition of the p38 MAPK signaling pathway in high-fat-induced SD rats. Int J Endocrinol. 2014;2014(1):901437. | ||
Vicil S, Erdoğan S. Beraprost sodium, a prostacyclin (PGI) analogue, ameliorates lipopolysaccharide-induced cellular injury in lung alveolar epithelial cells. Turk J Med Sci. 2015;45(2):284–290. | ||
Chen K, Li JJ, Li SN, et al. 15-Deoxy-Δ12,14-prostaglandin J2 alleviates hepatic ischemia-reperfusion injury in mice via inducing antioxidant response and inhibiting apoptosis and autophagy. Acta Pharmacol Sin. 2017;38(5):672–687. | ||
Shen M, Lu J, Dai W, et al. Ethyl pyruvate ameliorates hepatic ischemia-reperfusion injury by inhibiting intrinsic pathway of apoptosis and autophagy. Mediators Inflamm. Epub 2013 Dec 25. | ||
Ueno Y, Miyauchi Y, Nishio S. Beraprost sodium protects occlusion/reperfusion injury in the dog by inhibition of neutrophil migration. Gen Pharmacol. 1994;25(3):427–432. | ||
Shakil H, Saleem S. Prostaglandin I2 IP receptor agonist, beraprost, prevents transient global cerebral ischemia induced hippocampal CA1 injury in aging mice. J Neurol Disord. 2014;2:1000174. | ||
Ge M, Yao W, Yuan D, et al. Brg1-mediated Nrf2/HO-1 pathway activation alleviates hepatic ischemia-reperfusion injury. Cell Death Dis. 2017;8(6):e2841. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. | ||
Wanner GA, Ertel W, Müller P, et al. Liver ischemia and reperfusion induces a systemic inflammatory response through Kupffer cell activation. Shock. 1996;5(1):34–40. | ||
Jaeschke H, Farhood A. Neutrophil and Kupffer cell-induced oxidant stress and ischemia-reperfusion injury in rat liver. Am J Physiol. 1991;260(3 Pt 1):G355–G362. | ||
Perry BC, Soltys D, Toledo AH, Toledo-Pereyra LH. Tumor necrosis factor-α in liver ischemia/reperfusion injury. J Invest Surg. 2011;24(4):178–188. | ||
Irwin MW, Woodgett JR, Penninger J, Parker T, Lu C, Liu P. TNF-α and IL-1β activate p38 mitogen-activated and C-jun N-terminal kinase signaling cascades in myocytes, and interacts with free radicals. J Card Fail. 1999;5(3):21. | ||
Nick JA, Young SK, Arndt PG, et al. Selective suppression of neutrophil accumulation in ongoing pulmonary inflammation by systemic inhibition of p38 mitogen-activated protein kinase. J Immunol. 2002;169(9):5260–5269. | ||
Morse D, Pischke SE, Zhou Z, et al. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J Biol Chem. 2003;278(39):36993–36998. | ||
Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta. 2011;1813(9):1619–1633. | ||
Fletcher JI, Meusburger S, Hawkins CJ, et al. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc Natl Acad Sci U S A. 2008;105(47):18081–18087. | ||
Yoon JH, Gores GJ. Death receptor-mediated apoptosis and the liver. J Hepatol. 2002;37(3):400–410. | ||
Ghatan S, Larner S, Kinoshita Y, et al. p38 MAP kinase mediates bax translocation in nitric oxide-induced apoptosis in neurons. J Cell Biol. 2000;150(2):335–348. | ||
Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281(30):21256–21265. | ||
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. | ||
Lee SH, Park SW, Pyo CW, Yoo NK, Kim J, Choi SY. Requirement of the JNK-associated Bcl-2 pathway for human lactoferrin-induced apoptosis in the Jurkat leukemia T cell line. Biochimie. 2009;91(1):102–108. | ||
Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18(4):157–164. | ||
Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11(9):621–632. | ||
Pei Y, Xing D, Gao X, Liu L, Chen T. Real-time monitoring full length bid interacting with Bax during TNF-alpha-induced apoptosis. Apoptosis. 2007;12(9):1681–1690. | ||
Levine B, Sinha SC, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–606. | ||
Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. | ||
Liu L, Fang YQ, Xue ZF, He YP, Fang RM, Li L. Beta-asarone attenuates ischemia-reperfusion-induced autophagy in rat brains via modulating JNK, p-JNK, Bcl-2 and Beclin 1. Eur J Pharmacol. 2012;680(1–3):34–40. | ||
Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7(2):27–31. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.