Back to Journals » Journal of Multidisciplinary Healthcare » Volume 10
Behçet’s syndrome: providing integrated care
Authors Esatoglu SN, Kutlubay Z ![]() , Ucar D, Hatemi I, Uygunoglu U
, Ucar D, Hatemi I, Uygunoglu U ![]() , Siva A
, Siva A ![]() , Hatemi G
, Hatemi G
Received 22 May 2017
Accepted for publication 29 June 2017
Published 14 August 2017 Volume 2017:10 Pages 309—319
DOI https://doi.org/10.2147/JMDH.S93681
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Sinem Nihal Esatoglu,1 Zekayi Kutlubay,2 Didar Ucar,3 Ibrahim Hatemi,4 Ugur Uygunoglu,5 Aksel Siva,5 Gulen Hatemi1
1Division of Rheumatology, Department of Internal Medicine, 2Department of Dermatology, 3Department of Ophthalmology, 4Division of Gastroenterology, Department of Internal Medicine 5Department of Neurology, Cerrahpasa Medical Faculty, Istanbul University, Istanbul, Turkey
Abstract: Behçet’s syndrome (BS) is a multisystem vasculitis that presents with a variety of mucocutaneous manifestations such as oral and genital ulcers, papulopustular lesions and erythema nodosum as well as ocular, vascular, gastrointestinal and nervous system involvement. Although it occurs worldwide, it is especially prevalent in the Far East and around the Mediterranean Sea. Male gender and younger age at disease onset are associated with a more severe disease course. The management of BS depends on the severity of symptoms. If untreated, morbidity and mortality are considerably high in patients with major organ involvement. Multidisciplinary patient care is essential for the management of BS, as it is for other multisystem diseases. Rheumatologists, dermatologists, ophthalmologists, neurologists, cardiovascular surgeons and gastroenterologists are members of the multidisciplinary team. In this study, we reviewed the epidemiology, etiology, diagnostic criteria sets, clinical findings and treatment of BS and highlighted the importance of the multidisciplinary team in the management of BS.
Keywords: Behçet disease, multidisciplinary care, diagnosis, treatment, vasculitis, epidemiology, management
Introduction
Hulusi Behçet, a Turkish dermatologist, first described two patients with a triple-symptom complex of relapsing oral and genital ulcerations and uveitis in 1937, and now this syndrome bears his name.1 Behçet’s syndrome (BS) is a complex multisystem vasculitis with an unknown etiology. Vasculitis can involve all sizes and types of vessels in BS, and several organ systems can be affected. Mucocutaneous lesions such as oral ulcers (OUs) and genital ulcers (GUs), erythema nodosum (EN) and papulopustular lesions (PPL) are the most common findings. Eyes, vascular, gastrointestinal (GI) and nervous system are the types of major organ involvement in BS.2 Among them, vascular and nervous system involvement are the most common causes of mortality in BS.3
Epidemiology
BS occurs worldwide. However, it is more prevalent among populations living along the Silk Road, an ancient network of trade routes that expands from the Mediterranean Sea to the Far East. Turkey has the highest prevalence of BS in the range of 20–421/100000 population.4 The surveys conducted in Turkey were quite similar in methodology, and all were cross-sectional and population based. The results of these surveys may be summarized as follows: 1) the highest prevalence was in the Asian part of Turkey, 2) pediatric cases were very rare, 3) the patients from urban areas seem to have a less severe disease, 4) the frequency of pathergy positivity differed across the regions, 5) there were less BS patients among the Armenians who live in Istanbul reflecting the genetic background and 6) HLA-B51 carriers were less common in these surveys than in hospital-based patients.4
Although it is less common in Europe and US, it is remarkable to point out that BS is much more common than the other vasculitides in France.5 An increase in prevalence over the decades observed in Japan, Germany and Italy may be explained by the increased disease recognition and migration from prevalent countries.6
Both genders are affected with an equal gender predilection; however, BS runs a more severe course among males.7 There is also a difference in disease expression between genders. A meta-analysis found that ocular involvement, folliculitis, PPL, skin pathergy test (SPT) positivity and vascular involvement were more common among males, whereas joint involvement, EN and GUs were more frequent among females. There was no gender difference for GI and neurologic involvement.8 Moreover, there is a considerable amount of observations regarding regional differences in clinical characteristics. GI involvement is reported as high as 30% among BS patients from Korea and Japan compared to 1–2% in those from the Mediterranean region.9 SPT positivity has been found to be prevalent in countries where BS is also prevalent.10 However, a systematic literature review found no evidence supporting ethnic variation in clinical expressions.11 Therefore, there is still a need for comparative studies using standardized criteria to clarify this issue.
BS usually occurs in the third decade of life and is rare in younger and older (>50) individuals. Younger age of onset is associated with severe disease, and course of BS becomes stable over time.12
Etiology
The cause of BS is still unknown; however, it is likely that both genetic and environmental factors play a role in the development of BS. Although BS is not a Mendelian disorder, familial aggregation supports a genetic background.13 HLA-B51 is the most established genetic risk factor for BS that was confirmed by the recent genome-wide association studies.14,15 A meta-analysis showed that HLA-B51 carriers had approximately six times increased risk of developing BS, regardless of ethnic origin.16 In another meta-analysis, assessing its role on disease expression, HLA-B51 positivity was more frequent among males and was somewhat correlated with GUs, ocular and skin lesions, whereas it had a protective role for GI involvement. Since the prevalence of vascular and neurologic involvement associated with increased mortality did not differ among HLA-B51-positive and -negative patients, the authors suggested that HLA-B51 did not have a prognostic role.17 Additionally, non-HLA associations such as ERAP1, IL23R and IL10 variations were also reported to be associated with BS susceptibility. A low frequency of BS among the Japanese who migrated to the US18 and among Turkish immigrants to Germany19 points out to the role of the environment. However, this finding has not been confirmed in a population-based study conducted in France.20 Other findings that support the role of environmental factors are a lower socioeconomic status in BS patients compared to patients with ankylosing spondylitis and inflammatory bowel diseases21 and a clear association of poor oral hygiene with OUs.22 Interestingly, recurrent aphthous stomatitis, which has a quite similar histopathology to BS ulcers, is more prevalent among high socioeconomic groups.23
Diagnostic/classification criteria
BS has no pathognomonic laboratory test for diagnosis. There are 17 sets of diagnostic criteria for BS.24 Among them, two were made by the collaboration of experts from several countries: The International Study Group (ISG) criteria for Behçet’s disease and The International Criteria for Behçet’s Disease (ICBD). ISG criteria for Behçet’s disease, developed in 1990, are the most widely used set (Table 1).25 It includes four clinical findings in addition to a positive SPT. Among the clinical findings such as oral, genital, skin and ocular lesions, recurrent OU is a mandatory item. Although the ISG criteria were published as diagnostic criteria, there was a later debate whether they should have been called classification criteria. This, however, was probably an unnecessary exercise since a diagnosis is nothing more or less than a classification in an individual patient.26 A new set of diagnostic criteria, ICBD, was proposed by an international team of Behçet’s experts in 200627 and then revised in 2010.28 In ICBD, vascular manifestations, that were used as a criterion in most of the previous sets of diagnostic criteria,29 and neurologic involvement were added to five items of ISG criteria (Table 1). Considering that OU may be absent in 1–10% of BS patients reported from 14 countries, especially at the beginning of the disease,30 OU was no longer a mandatory criterion. ICBD yielded not only higher sensitivity but also a decrease in specificity, leading to more misclassified patients. Since specificity is much more important than sensitivity in diagnosing rare diseases, the possibility of misdiagnosis of several patients with other diagnosis as BS is an important disadvantage for the ICBD criteria.31
 | Table 1 Diagnostic criteria sets for BS, ISG and ICBD Notes: aBS diagnosis is made in the presence of recurrent OUs and 2 additional criteria. OUs, GUs and EN may be reported by the patient or physician, whereas skin lesions other than EN must be observed by physician. bPoint score system >3 indicates BS diagnosis. Pathergy test is not involved in the primary scoring system. However, given its specificity for BS, when pathergy test is done, extra point may be assigned. Abbreviations: BS, Behçet’s syndrome; ISG, The International Study Group; ICBD, The International Criteria for Behçet’s Disease; OU, oral ulcer; GU, genital ulcer; EN, erythema nodosum; PPL, papulopustular lesions; CNS, central nervous system. |
Clinical findings
Mucocutaneous lesions
OUs
A comprehensive review on clinical characteristics of BS patients from 27 countries showed that 93–100% BS patients had OU.30 Differentiating OU of BS from those of recurrent aphthous stomatitis may be difficult especially when it occurs as the earliest manifestation. OUs are categorized according to their size and shape: major, minor and herpetiform ulcers (Figure 1A).32 Minor ulcers (<1 cm in diameter) are the most frequent type of OUs. Major ulcers (>1 cm in diameter) are seen less frequently but are more painful and deeper than minor ulcers. They also heal more slowly, often resulting in scarring. Herpetiform ulcers, the rarest type, manifest as numerous, shallow and small-pinpoint ulcers.33 All types of OUs can be present anywhere on the oral mucosa, and different types of OUs may also be seen at the same time.30 Unfortunately, most BS patients continue to have OUs during follow-up despite a decrease in frequency with aging.34 Moreover, frequent recurrence of OUs during early disease has been shown as a predictive factor for major organ involvement among males.35 They were also found to be the most common reason for work disability in BS in the UK.36 Hence, OUs remain to be an important clinical problem in BS.
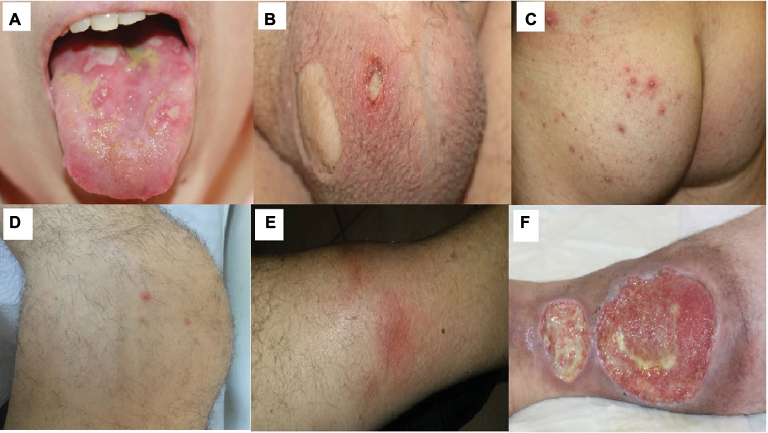 | Figure 1 Mucocutaneous lesions of Behçet’s syndrome. Notes: (A) OU; (B) GU with a scar; (C) PPL; (D) acne with arthritis; (E) EN and (F) leg ulcers. Abbreviations: OU, oral ulcer; GU, genital ulcer; PPL, papulopustular lesions; EN, erythema nodosum. |
GUs
GUs are the second most common clinical finding in BS.37 They occur mostly on the scrotum in men and vulva in women (Figure 1B). They usually begin as a papule that quickly ulcerates. They clinically resemble OUs, but healing is generally observed within 10–30 days, with scar formation in two-thirds of the patients.38 GU-like lesions, sometimes called extragenital ulcers, may be rarely observed in areas such as under the breasts, axilla and interdigital areas.39
Skin lesions
PPL and acneiform lesions are the most common skin lesions, occurring mostly on the trunk and the extremities (Figure 1C).40 They may not be differentiated from acne vulgaris especially among adolescents. Lesions located on the parts of the body other than the face were suggested to be more specific for BS.41 Moreover, PPL on legs were recommended to be a distinguishing feature for BS.42 There is controversy regarding the role of histopathological examination for differentiating PPL of BS from acne vulgaris.42 Hence, punch biopsy is not a part of clinical practice. In a controlled study, PPL were found to be more frequent in BS patients with arthritis (Figure 1D).43 Furthermore, coexistence of enthesopathy, arthritis and acne was reported as one of the disease clusters in BS.44
Nodular lesions are observed in 50% of BS patients, in the form of either EN or superficial thrombophlebitis (STM). EN is localized symmetrically on lower extremities, as well as thighs and sacral region (Figure 1E). They usually heal in 1–6 weeks, with post-lesional hyperpigmentation.45 Morphological and histopathological features of EN are similar in BS and other systemic conditions. However, neutrophilic vasculitis is more common in BS.46 STM usually presents with erythematous, tender, subcutaneous nodules arranged in a linear fashion. The vein can be palpated as a string-like hardening.
Sweet syndrome can be seen during the course of BS. It is characterized by fever, neutrophilia, and painful, erythematous, cutaneous lesions located on the face and extremities.
SPT
In patients with BS, a cutaneous inflammatory response may be induced by a sterile needle prick. We perform the SPT by applying three needle pricks to a hairless site on the flexor site of each forearm. After wiping the forearm with alcohol, 20 G needles are inserted in an oblique or perpendicular angle. Evaluation is done after 48 hours. The formation of a palpable erythematous papule or pustule of ~1–2 mm at the insertion point of at least one of the needles indicates a positive result.47 Men have a stronger pathergy reaction than women.48 However, its positivity was not shown to be associated with a more severe disease course.49 Although its sensitivity declines over time, it remains a specific diagnostic test for BS.50 There is only one randomized controlled study assessing the influence of immunosuppressive therapy on the pathergy reaction, and this study showed that etanercept did not suppress the pathergy reaction.51 Pathergy phenomenon may also develop in sites other than the skin, since any trauma or injury may cause an exaggerated inflammatory response in tissues in BS patients. Pathergy reaction triggered by vascular surgery, intraocular injections, dental procedures and venipuncture are well-known conditions.52
Ocular involvement
Ocular involvement is the most common major organ manifestation in BS. Around 50% of BS patients have ocular involvement; however, its frequency is up to 70% among young men whereas 30% among women and elderly BS patients.53 Males are more frequently affected and have an earlier disease onset and more severe disease.54 Intraocular inflammation may manifest as anterior, intermediate, posterior or panuveitis. Bilateral panuveitis with retinal vasculitis is the most common and severe form of ocular involvement. Ocular involvement usually occurs within 5 years after the disease onset.55 Mild ocular inflammation during early disease may be asymptomatic. Considering that ocular involvement can cause irreversible vision loss if left untreated, screening for ocular involvement at diagnosis is essential.53 Basic ophthalmologic examination include best corrected visual acuity, intraocular pressure measurement, slit-lamp examination to determine anterior chamber inflammation and dilated funduscopy of the retina and vascular structure. Ancillary techniques are ordered when we have any suspicion of vascular and macular involvement, which comprise fundus flourescein angiography and optic coherence tomography. It is important to differentiate BS uveitis from infectious and non-infectious uveitic conditions. First, ~20% of BS patients present with ocular involvement as an initial manifestation56 and systemic immunosuppressive treatment should not be delayed in such patients. Second, any patient with BS may also have infectious uveitis in which immunosuppressives are contraindicated.57 Ocular toxoplasmosis, a frequent cause of uveitis and a mimicker of ocular involvement of BS, is usually a unilateral disease with a characteristic active retinitis adjacent to an old scar. Fortunately, ocular inflammation due to BS has distinctive features. A masked study showed that up to 80% of Turkish uveitis specialists can differentiate BS and non-BS uveitis by evaluating ocular photographs.58
GI involvement
Gastrointestinal involvement of BS (GIBS) is one of the major organ involvements related with morbidity and mortality in BS. Its prevalence shows a distinct geographic variation. The frequency of GI involvement is higher in the Far East compared to Mediterranean countries.59 Men and women are equally affected. GIBS shares similar features with Crohn’s disease regarding bowel findings. The presence of other BS manifestations helps in differentiating these two diseases.60 Additionally, perforation and massive bleeding are more common in GIBS probably due to its vasculitic nature. The most common type of involvement is focal single ulceration in ileocecal area. Typical shape of ulcers in GIBS is round or oval (Figure 2). In histopathological examination, chronic active inflammation with or without vasculitis is present and granuloma is rarely observed. Mimicking conditions such as non-steroidal anti-inflammatory drug-induced enteropathy and tuberculosis must be ruled out before diagnosing GIBS.9
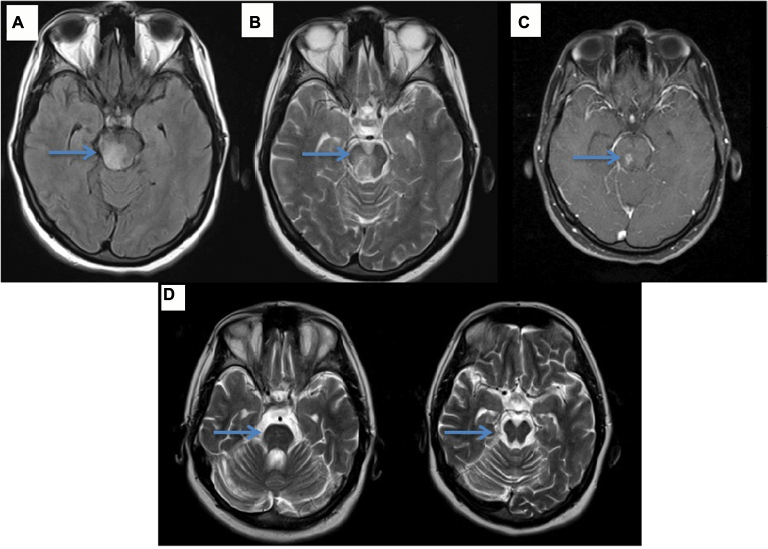 | Figure 2 (A, B) Axial FLAIR-T2 images reveal hyperintense lesion in the brainstem (arrow), (C) axial T1 gadolinium sequence shows gadolinium enhancement (arrow) and (D) prominent brainstem atrophy (arrow) on the follow-up MRI after 3 years. |
Musculoskeletal involvement
Arthritis and arthralgia are common manifestations among BS patients, with a prevalence of 40–70%.61 BS patients with joint involvement usually present with recurrent, inflammatory mono or oligoarthritis. Arthritis attacks tend to be self-limited, and erosive arthritis is rare.62 Although the larger joints such as knee, wrist, ankle and elbow are more frequently involved, hands may also be affected as seen in rheumatoid arthritis.63 PPL are the only extra-articular manifestation associated with arthritis attacks.64 There has been a debate regarding the association of BS with sacroiliitis. However, most of the controlled studies did not find a difference in the prevalence of sacroiliitis between BS patients and controls.65–68 Enthesopathy, another feature of seronegative spondyloarthropathies, is also common in a subgroup of BS patients with acne and arthritis.44 Fibromyalgia, a frequent musculoskeletal condition among rheumatologic disorders, is common in BS patients and does not seem to be associated with disease severity and manifestations.69
Nervous system involvement
Neuro-Behçet’s syndrome (NBS) occurs in ~5% of patients with BS; ~75–80% of NBS cases present with central nervous system involvement, which is called “parenchymal NBS” and affects the telencephalic–diencephalic junction, brainstem and spinal cord (Figure 3). These patients present with a subacute (or rarely acute) onset of severe headache, dysarthria, ataxia and hemiparesis. The second most common form of neurological involvement is cerebral venous sinus thrombosis (CVST), which is also called vascular NBS or extra-axial NBS. CVST occurs in up to 20% of patients with NBS in the adult BS population,70 whereas it is the most common form of neurological involvement seen in the pediatric NBS population.71 CVST will be detailed in the “Vascular involvement” section.
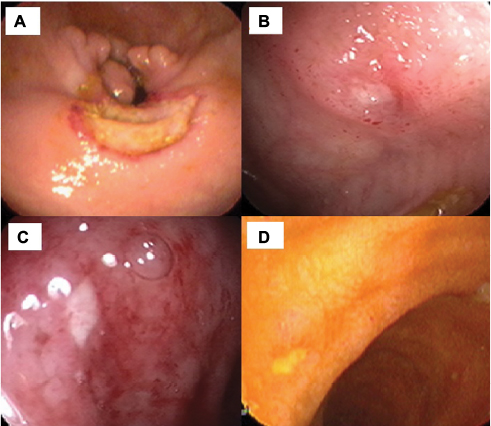 | Figure 3 Endoscopic appearance of round ulcers of GI involvement of BS. Notes: (A, B) Some of these ulcers can be deep with elevated edges and (C, D) some are shallow. Abbreviations: GI, gastrointestinal involvement; BS, Behçet’s syndrome. |
NBS is often included in the differential diagnosis of multiple sclerosis (MS) and in stroke in young adults, especially in the absence of its known systemic symptoms and signs. Optic neuritis, sensory symptoms and spinal cord involvement, which are common in MS, are rare in NBS. The magnetic resonance imaging (MRI) findings are distinct from those of NBS; more discrete, smaller brainstem lesions are seen in MS, as well as more periventricular and ovoid lesions in the hemispheres.72 Spinal cord involvement rarely extends more than a few vertebral segments in MS, compared with the more extensive lesions of NBS, similar to neuromyelitis optica spectrum disorder (NMOSD).73,74 However, the NMO-IgG Ab is negative in NBS myelitis. The CSF also reveals different patterns, with more prominent pleocytosis and a low rate of positivity for oligoclonal bands in parenchymal NBS as compared with MS.75
Vascular involvement
Vascular involvement has a more severe course especially among male patients with a younger age at disease onset. Venous vessels are affected more commonly than arterial vessels. The inflammatory process at arterial vessels usually leads to aneurysms due to acute and destructive vasculitis. Bleeding is a symptom of arterial aneurysms rather than venous thrombosis. On the other hand, extensive adherent thrombus formation is the typical finding of venous vessel involvement without an increase of thromboembolism. The pathogenesis of thrombosis in BS seems to be caused by inflammation of the vessel wall rather than a procoagulant state.76 Venous and pulmonary artery involvement usually occurs within 5 years of disease onset, and arterial involvement tends to develop later during the disease course. Ten percent of BS patients have their first vascular event before fulfilling ISG criteria.77 Elevated acute phase reactants and fever are common features of vascular involvement in BS.78
Venous thrombosis
Lower extremities are the most common localization of deep vein thrombosis (DVT). DVT on legs are usually the first vascular event of BS patients.77 DVT due to BS has some characteristics that are useful to differentiate it from DVT due to non-BS causes. Male gender, younger age at disease onset and bilateral continuous involvement of deep proximal and superficial veins are suggestive of BS. Its course is also different with more relapses and collateral formation and less recanalization of thrombus.79
STM is also common among males. Its frequency may be higher than anticipated, since differentiating it from EN may be quite impossible without performing Doppler ultrasonography.76 Accurate diagnosis of STM is essential as DVT was found to be associated with STM.80
All parts of inferior vena cava (IVC), infrahepatic, hepatic and suprahepatic parts may be involved. Patients with IVC thrombosis present with abdominal pain and swelling of lower extremities. In physical examination, abdominal collateral veins become visible.81 Symptoms in superior vena cava thrombosis include dyspnea, face, neck and arm swelling during the acute–subacute phase. It has a benign course due to a low relapse rate. However, complications such as pleural effusion, chylothorax and sleep apnea disorder may be seen during the chronic phase.81
Although hypercoagulable conditions are the most common risk factors for Budd–Chiari syndrome (BCS) in the general population, BS is an important cause of BCS, especially in endemic areas for BS. It is essential to diagnose BS in a patient with BCS since mortality is extremely high if treated with only anticoagulation without immunosuppressive treatment. Interestingly, sometimes asymptomatic patients may be diagnosed incidentally and such patients seem to have a better prognosis.82
CVST has a clear association with the other types of DVT.77 CVST is the initial presenting lesion in 2.2% of BS patients.83 Typical symptoms are headache, diplopia and nausea, resulting from intracranial hypertension. Eye examination reveals bilateral papilledema. Magnetic resonance venography is the preferred imaging modality. Male gender, subacute course, younger age at disease onset, less neurological deficit and less venous infarction are main features of CVST due to BS.84
Pulmonary artery involvement
Pulmonary artery involvement is the most feared manifestation of BS. Even in the era of novel immunosuppressives, the mortality and morbidity are considerably high.85 As mentioned earlier, arterial involvement manifests as aneurysms or in situ thrombosis in BS.86 Isolated pulmonary artery thrombosis (PAT) and pulmonary artery aneurysms (PAA) with or without in situ thrombosis have similar clinical features and prognosis. Hemoptysis and dyspnea are the most common initial symptoms but former is less seen in PAT. Pulmonary parenchymal diseases such as nodules, consolidations and cavities are present in up to 90% of BS patients with pulmonary artery involvement.87 These findings were considered to be due to microscopic vascular disease.88 While PAT or PAA may respond well to immunosuppressive therapy, some patients may be complicated with recurrent hemoptysis during their follow-up due to bronchial artery enlargement.89
Peripheral arterial involvement
Peripheral artery involvement is much less common than pulmonary artery involvement. This may be explained by the similarities in structure between venous vessels and pulmonary arteries.81 Abdominal aorta and arteries of lower extremities are most often affected, whereas upper limb, carotid and cerebral arteries as well as visceral arteries may be involved.90 The initial symptoms vary according to the involved artery. Peripheral artery aneurysms on extremities may be easily detected as painful and pulsatile mass.91
Cardiac involvement
Cardiac involvement is rare and manifests as intracardiac thrombus (ICT), pericarditis, myocarditis, endocarditis, endomyocardial fibrosis, coronary arteritis and valvular disease.92
ICT is rare but has a strong association with vascular involvement. Fever, dyspnea, hemoptysis and chest pain are common initial symptoms. The typical location of ICT is the right ventricle or atrium.93,94 Coronary aneurysms, frequently co-occurring with coronary stenosis, may be isolated or multiple. The clinical manifestations range from asymptomatic to acute coronary syndrome. Sinus valsalva aneurysms and the aortic root involvement may be complicated with acute or chronic aortic insufficiency.95
Post-thrombotic syndrome and leg ulcers
Post-thrombotic syndrome describes a set of clinical symptoms and signs associated with DVT. One study found that BS patients had higher post-thrombotic syndrome (PTS) risk when compared to patients with DVT due to non-BS causes.79 However, this finding was not confirmed in a later study.96
Leg ulcers in BS develop around the ankle, the area between knee and ankle or on the foot (Figure 1F). The underlying condition may be necrotizing vasculitis or DVT. Necrotizing vasculitis initially manifests as multiple EN-like lesions, and then these lesions ulcerate with well-demarcated margins. The margin of ulcers due to post-thrombotic syndrome is more likely to be irregular. Recurrences and refractoriness to therapy are common in both types.97
The multidisciplinary clinic for BS
Multidisciplinary patient care is essential for the diagnosis and management of multisystem diseases. It provides several benefits to the clinicians and the patients. Synergistic decisions for differential diagnosis and treatment can be developed, resulting from teamwork. This approach enables a faster and more accurate diagnosis and well-integrated treatment strategies that target all organs/organ systems that are involved at once. A standardized approach designed by the collaboration of clinicians from different departments allows clinicians to conduct clinical trials more easily. The process including consultation, diagnosis and treatment moves more rapidly.
A multidisciplinary team of rheumatologists, dermatologists, ophthalmologists, neurologists, cardiovascular surgeons and gastroenterologists, often led by rheumatologists, take part in the management of patients with BS.
Screening
Patients may be referred to our Behçet’s clinic due to various symptoms. Suspected cases are screened according to a standardized approach. Patients with recurrent OUs are questioned for other skin lesions such as GUs, EN and PPL. The frequency and duration of the lesions are also recorded. A dermatologist examines each patient for the presence of genital scars. The pathergy test, performed by dermatologists, is an important part of the screening procedure and helps the diagnosis when found positive, although a negative result does not rule out BS. Patients are questioned and examined for arthritis and major organ involvement such as ocular, vascular, nervous system and GI involvement. Finally, the ophthalmologists examine for the presence of ocular involvement. Alternatively, patients with uveitis or other major organ involvement may be referred with the suspicion of BS and the presence of mucocutaneous and other lesions may reveal the diagnosis.
Diagnosis
The diagnosis of BS may be considered relatively easy in prevalent countries. Primary care physicians have a higher awareness of BS and patients with BS symptoms are referred to rheumatologists at an early stage. Some patients with skin lesions directly present to the dermatologists. On the other hand, a considerable portion of BS patients are admitted to other departments for other types of organ involvement and then they are referred to rheumatologists. This process may be the source of a diagnostic delay in non-prevalent countries. A family history may also be helpful in the diagnosis since a history of BS in first-and/or second-degree relatives was reported as 6% among BS patients.98 On the other hand, inflammatory markers such as erythrocyte sedimentation rate, C-reactive protein, leukocytes, and various cytokine levels are not helpful in the diagnosis of BS.99 Acute phase reactants are only helpful for indicating active disease in vascular involvement and sometimes GI involvement.78
In our clinic, difficult cases are discussed regarding their diagnosis and management in a regular multidisciplinary meeting every week. Patients who do not fulfill ISG criteria but have major organ involvement with characteristic features for BS are followed in the same manner as patients fulfilling ISG criteria.
Treatment
In BS, clinical manifestations and their severity vary widely among patients. Moreover, the severity of the symptoms may differ during the disease course for any one patient. Mucocutaneous and joint involvement are associated with an impaired quality of life without irreversible damage, while ocular, GI, neurologic and vascular involvement may be complicated with loss of organ functions and death. Thus, the treatment should be individualized for each patient. The main principles of treatment are to suppress inflammation quickly during acute attacks in order to prevent damage and to prevent relapses with immunosuppressives if needed.2
The treatment of mucocutaneous lesions depends on the severity of each lesion. For isolated OUs and GUs, topical measures are usually sufficient. Antiseptic mouth washes, sucralfate suspensions, topical lidocaine- and corticosteroid-containing gels or triamcinolone acetonide (5 mg/mL) injections are used to reduce pain and healing time of oral aphthae. For genital lesions, topical antiseptics, mid-potent corticosteroid ointments and wet dressings are useful. In severe disease systemic therapy such as oral corticosteroids, azathioprine, colchicine, thalidomide, azathioprine, interferon-alpha and anti-tumor necrosis factor alpha agents (anti-TNFs) can be needed.100
For joint involvement, colchicine is the first-line treatment choice to prevent arthritis attacks. Azathioprine, interferon-alpha and anti-TNFs are the treatment options in patients with recurrent arthritis. Non-steroidal anti-inflammatory drugs and intra-articular corticosteroid injections may be tried during the attacks.101
Patients with posterior uveitis with or without retinal vasculitis should be treated with immunosuppressive agents such as azathioprine, cyclosporine, interferon-alpha and anti-TNFs. Systemic and/or intravitreal corticosteroid should be a part of the therapy in severe acute attacks.101
Patients with vascular involvement should be treated with immunosuppressives since they have been shown to reduce relapse rate of venous thrombosis when compared to solo anti-coagulation therapy.102 Azathioprine and interferon-alpha are usually the initial treatment modalities used in patients with other types of venous thrombosis. Anti-TNFs may be an option for resistant cases. Although there is no evidence to show the efficacy of anti-coagulation in preventing relapses of thrombosis due to BS, they may be tried in recurrent cases with the hope of reducing PTS risk, once PAA are ruled out. An aggressive approach including cyclophosphamide along with high-dose corticosteroids is essential in patients with vascular involvement such as BCS, pulmonary artery involvement and peripheral arterial aneurysms that are associated with a high morbidity and mortality. Surgery may be required in addition to immunosuppressives in patients with peripheral arterial aneurysms.81
The conventional treatment modalities for the management of GIBS are glucocorticoids, azathioprine, salazopyrine and other 5-aminosalicylic acid derivatives, similar to inflammatory bowel diseases. Anti-TNFs and thalidomide may be beneficial in refractory cases.103
The treatment of acute NBS includes high-dose intravenous methylprednisolone pulses for 7–10 days followed by gradual oral tapering over 3–6 months, depending on the relapse severity. Immunosuppressive treatment including azathioprine and infliximab is essential to avoid further relapses.104
Follow-up
Patients are usually seen every 3–6 months depending on the severity of their diseases. The frequency of mucocutaneous lesions and any new manifestations are recorded in each visit. Eye examination is repeated in every visit in patients presenting with ocular involvement. We screen vascular involvement in patients with elevated acute phase reactants and/or fever even if they do not have typical symptoms.
There is no consensus on when to stop the treatment in BS patients in remission. Moreover, remission has not been well defined in BS since it is difficult to conclude whether the stable disease is due to the treatment itself or due to relapsing remitting nature of the disease. Additionally, it is difficult to define a standard protocol for all BS patients for stopping treatment since men and younger patients have a higher risk of relapse. The decision to stop treatment for patients with only mucocutaneous involvement is somewhat easier and can be made together with the patient depending on how much these lesions impair the patient’s life quality. In patients with major organ involvement, immunosuppressives are tapered after 2–5 years of remission depending on the patient’s age, gender, severity of involvement and the amount of damage.
Acknowledgements
The authors thank all members of the multidisciplinary Behçet’s syndrome team at Istanbul University, Cerrahpasa Medical School, Rheumatology - Hasan Yazici, Sebahattin Yurdakul, Vedat Hamuryudan, Izzet Fresko, Melike Melikoglu, Emire Seyahi, Serdar Ugurlu, and Yesim Ozguler; Dermatology Cem Mat; Ophthalmology: Yılmaz Ozyazgan; Gastroenterology: Aykut Ferhat Celik; Neurology: Sebahattin Saip and Melih Tutuncu; Cardiovascular Surgery: Hasan Tuzun; Radiology: Canan Akman, Civan Islak, and Naci Kocer.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
Gulen Hatemi has received research grants, consulting fees or speaking fees from Abbvie, BMS, Celgene, MSD Pharmaceuticals, Mustafa Nevzat, UCB Pharma and Pfizer. The other authors report no conflicts of interest in this work.
References
Feigenbaum A. Description of Behcet’s syndrome in the Hippocratic third book of endemic diseases. Br J Ophthalmol. 1956;40(6):355–357. | ||
Hatemi G, Yazici Y, Yazici H. Behcet’s syndrome. Rheum Dis Clin North Am. 2013;39(2):245–261. | ||
Saadoun D, Wechsler B, Desseaux K, et al. Mortality in Behcet’s disease. Arthritis Rheumatol. 2010;62(9):2806–2812. | ||
Yurdakul S, Yazici Y. Epidemiology of Behçet’s syndrome and regional differences in disease expression. In: Yazici Y, Yazici H, editors. Behçet’s Syndrome. New York, NY: Springer; 2010:36. | ||
Yazici H, Seyahi E, Yurdakul S. Behcet’s syndrome is not so rare: why do we need to know? Arthritis Rheumatol. 2008;58(12):3640–3643. | ||
Calamia KT, Wilson FC, Icen M, Crowson CS, Gabriel SE, Kremers HM. Epidemiology and clinical characteristics of Behcet’s disease in the US: a population-based study. Arthritis Rheumatol. 2009;61(5):600–604. | ||
Yurdakul S, Yazici H. Behcet’s syndrome. Best Pract Res Clin Rheumatol. 2008;22(5):793–809. | ||
Bonitsis NG, Luong Nguyen LB, LaValley MP, et al. Gender-specific differences in Adamantiades-Behcet’s disease manifestations: an analysis of the German registry and meta-analysis of data from the literature. Rheumatology (Oxford). 2015;54(1):121–133. | ||
Hatemi I, Esatoglu SN, Hatemi G, Erzin Y, Yazici H, Celik AF. Characteristics, treatment, and long-term outcome of gastrointestinal involvement in Behcet’s Syndrome: a strobe-compliant observational study from a dedicated multidisciplinary center. Medicine (Baltimore). 2016;95(16):e3348. | ||
Yazici H, Chamberlain MA, Tuzun Y, Yurdakul S, Muftuoglu A. A comparative study of the pathergy reaction among Turkish and British patients with Behcet’s disease. Ann Rheum Dis. 1984;43(1):74–75. | ||
Lewis KA, Graham EM, Stanford MR. Systematic review of ethnic variation in the phenotype of Behcet’s disease. Scand J Rheumatol. 2007;36(1):1–6. | ||
Yazici H, Tuzun Y, Pazarli H, et al. Influence of age of onset and patient’s sex on the prevalence and severity of manifestations of Behcet’s syndrome. Ann Rheum Dis. 1984;43(6):783–789. | ||
Gul A, Inanc M, Ocal L, Aral O, Konice M. Familial aggregation of Behcet’s disease in Turkey. Ann Rheum Dis. 2000;59(8):622–625. | ||
Remmers EF, Cosan F, Kirino Y, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet’s disease. Nat Genet. 2010;42(8):698–702. | ||
Mizuki N, Meguro A, Ota M, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behcet’s disease susceptibility loci. Nat Genet. 2010;42(8):703–706. | ||
de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behcet’s disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheumatol. 2009;61(10):1287–1296. | ||
Maldini C, Lavalley MP, Cheminant M, de Menthon M, Mahr A. Relationships of HLA-B51 or B5 genotype with Behcet’s disease clinical characteristics: systematic review and meta-analyses of observational studies. Rheumatology (Oxford). 2012;51(5):887–900. | ||
Hirohata T, Kuratsune M, Nomura A, Jimi S. Prevalence of Behcet’s syndrome in Hawaii. With particular reference to the comparison of the Japanese in Hawaii and Japan. Hawaii Med J. 1975;34(7):244–246. | ||
Zouboulis CC, Kotter I, Djawari D, et al. Epidemiological features of Adamantiades-Behcet’s disease in Germany and in Europe. Yonsei Med J. 1997;38(6):411–422. | ||
Mahr A, Belarbi L, Wechsler B, et al. Population-based prevalence study of Behcet’s disease: differences by ethnic origin and low variation by age at immigration. Arthritis Rheumatol. 2008;58(12):3951–3959. | ||
Seyahi E, Ugurlu S, Seyahi N, et al. A survey of socioeconomic status in Behçet’s Syndrome. Clin Exp Rheumatol. 2004;22(suppl 34):88. | ||
Mumcu G, Ergun T, Inanc N, et al. Oral health is impaired in Behcet’s disease and is associated with disease severity. Rheumatology (Oxford). 2004;43(8):1028–1033. | ||
Crivelli MR, Aguas S, Adler I, Quarracino C, Bazerque P. Influence of socioeconomic status on oral mucosa lesion prevalence in schoolchildren. Community Dent Oral Epidemiol. 1988;16(1):58–60. | ||
Davatchi F, Sadeghi Abdollahi B, Chams-Davatchi C, et al. The saga of diagnostic/classification criteria in Behcet’s disease. Int J Rheum Dis. 2015;18(6):594–605. | ||
XXXX. Criteria for diagnosis of Behcet’s disease. International Study Group for Behcet’s Disease. Lancet. 1990;335(8697):1078–1080. | ||
Yazici H. Diagnostic versus classification criteria – a continuum. Bull NYU Hosp Jt Dis. 2009;67(2):206–208. | ||
International Team for the Revision of the International Criteria for Behcet’s Disease. Revision of the international criteria for Behcet’s disease (ICBD). Clin Exp Rheumatol. 2006;24(suppl 42):14–15. | ||
International Team for the Revision of the International Criteria for Behcet’s Disease. The International Criteria for Behcet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–347. | ||
Davatchi F. Diagnosis/classification criteria for Behcet’s disease. Patholog Res Int. 2012;2012:607921. | ||
Davatchi F, Shahram F, Chams-Davatchi C, et al. Behcet’s disease: from east to west. Clin Rheumatol. 2010;29(8):823–833. | ||
Yazici H, Yazici Y. Criteria for Behcet’s disease with reflections on all disease criteria. J Autoimmun. 2014;48-49:104–107. | ||
Rogers RS 3rd. Recurrent aphthous stomatitis in the diagnosis of Behcet’s disease. Yonsei Med J. 1997;38(6):370–379. | ||
Rotondo C, Lopalco G, Iannone F, et al. Mucocutaneous involvement in Behcet’s disease: how systemic treatment has changed in the last decades and future perspectives. Mediators Inflamm. 2015;2015:451675. | ||
Alibaz-Oner F, Mumcu G, Kubilay Z, et al. Unmet need in Behcet’s disease: most patients in routine follow-up continue to have oral ulcers. Clin Rheumatol. 2014;33(12):1773–1776. | ||
Hamuryudan V, Hatemi G, Sut N, Ugurlu S, Yurdakul S, Yazici H. Frequent oral ulceration during early disease may predict a severe disease course in males with Behcet’s syndrome. Clin Exp Rheumatol. 2012;30(3 suppl 72):S32–S34. | ||
Mehta P, Ambrose N, Haskard DO. Work-related disability in Behcet’s syndrome: a British case series. Clin Exp Rheumatol. 2014;32(4 suppl 84):S173–S174. | ||
Gurler A, Boyvat A, Tursen U. Clinical manifestations of Behcet’s disease: an analysis of 2147 patients. Yonsei Med J. 1997;38(6):423–427. | ||
Mat MC, Goksugur N, Engin B, Yurdakul S, Yazici H. The frequency of scarring after genital ulcers in Behcet’s syndrome: a prospective study. Int J Dermatol. 2006;45(5):554–556. | ||
Saylan T, Mat C, Fresko I, Melikoglu M. Behcet’s disease in the Middle East. Clin Dermatol. 1999;17(2):209–223. discussion 105–206. | ||
Alpsoy E, Zouboulis CC, Ehrlich GE. Mucocutaneous lesions of Behcet’s disease. Yonsei Med J. 2007;48(4):573–585. | ||
Alpsoy E, Aktekin M, Er H, Durusoy C, Yilmaz E. A randomized, controlled and blinded study of papulopustular lesions in Turkish Behcet’s patients. Int J Dermatol. 1998;37(11):839–842. | ||
Kutlubay Z, Mat CM, Aydin O, et al. Histopathological and clinical evaluation of papulopustular lesions in Behcet’s disease. Clin Exp Rheumatol. 2015;33(6 suppl 94):S101–S106. | ||
Diri E, Mat C, Hamuryudan V, Yurdakul S, Hizli N, Yazici H. Papulopustular skin lesions are seen more frequently in patients with Behcet’s syndrome who have arthritis: a controlled and masked study. Ann Rheum Dis. 2001;60(11):1074–1076. | ||
Hatemi G, Fresko I, Tascilar K, Yazici H. Increased enthesopathy among Behcet’s syndrome patients with acne and arthritis: an ultrasonography study. Arthritis Rheumatol. 2008;58(5):1539–1545. | ||
Lee ES, Bang D, Lee S. Dermatologic manifestation of Behcet’s disease. Yonsei Med J. 1997;38(6):380–389. | ||
Demirkesen C, Tuzuner N, Mat C, et al. Clinicopathologic evaluation of nodular cutaneous lesions of Behcet syndrome. Am J Clin Pathol. 2001;116(3):341–346. | ||
Zouboulis CC. Epidemiology of Adamantiades-Behcet’s disease. Ann Med Interne (Paris). 1999;150(6):488–498. | ||
Yazici H, Tuzun Y, Tanman AB, et al. Male patients with Behcet’s syndrome have stronger pathergy reactions. Clin Exp Rheumatol. 1985;3(2):137–141. | ||
Krause I, Molad Y, Mitrani M, Weinberger A. Pathergy reaction in Behcet’s disease: lack of correlation with mucocutaneous manifestations and systemic disease expression. Clin Exp Rheumatol. 2000;18(1):71–74. | ||
Davatchi F, Chams-Davatchi C, Ghodsi Z, et al. Diagnostic value of pathergy test in Behcet’s disease according to the change of incidence over the time. Clin Rheumatol. 2011;30(9):1151–1155. | ||
Melikoglu M, Fresko I, Mat C, et al. Short-term trial of etanercept in Behcet’s disease: a double blind, placebo controlled study. J Rheumatol. 2005;32(1):98–105. | ||
Sequeira FF, Daryani D. The oral and skin pathergy test. Indian J Dermatol Venereol Leprol. 2011;77(4):526–530. | ||
Ozyazgan Y, Ucar D, Hatemi G, Yazici Y. Ocular Involvement of Behcet’s syndrome: a comprehensive review. Clin Rev Allergy Immunol. 2015;49(3):298–306. | ||
Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, Huseyin Altunbas H, Urgancioglu M. Uveitis in Behcet disease: an analysis of 880 patients. Am J Ophthalmol. 2004;138(3):373–380. | ||
Kural-Seyahi E, Fresko I, Seyahi N, et al. The long-term mortality and morbidity of Behcet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore). 2003;82(1):60–76. | ||
Tugal-Tutkun I. Behcet’s uveitis. Middle East Afr J Ophthalmol. 2009;16(4):219–224. | ||
Tugal-Tutkun I, Gupta V, Cunningham ET. Differential diagnosis of Behcet uveitis. Ocul Immunol Inflamm. 2013;21(5):337–350. | ||
Tugal-Tutkun I, Onal S, Ozyazgan Y, Soylu M, Akman M. Validity and agreement of uveitis experts in interpretation of ocular photographs for diagnosis of Behcet uveitis. Ocul Immunol Inflamm. 2014;22(6):461–468. | ||
Skef W, Hamilton MJ, Arayssi T. Gastrointestinal Behcet’s disease: a review. World J Gastroenterol. 2015;21(13):3801–3812. | ||
Hatemi I, Hatemi G, Celik AF, et al. Frequency of pathergy phenomenon and other features of Behcet’s syndrome among patients with inflammatory bowel disease. Clin Exp Rheumatol. 2008;26(4 suppl 50):S91–S95. | ||
Bicer A. Musculoskeletal findings in Behcet’s disease. Patholog Res Int. 2012;2012:653806. | ||
Yurdakul S, Yazici H, Tuzun Y, et al. The arthritis of Behcet’s disease: a prospective study. Ann Rheum Dis. 1983;42(5):505–515. | ||
Yurtkuran M, Yurtkuran M, Alp A, et al. Hand involvement in Behcet’s disease. Joint Bone Spine. 2006;73(6):679–683. | ||
Fatemi A, Shahram F, Akhlaghi M, Smiley A, Nadji A, Davatchi F. Prospective study of articular manifestations in Behcet’s disease: five-year report. Int J Rheum Dis. 2017;20(1):97–102. | ||
Chang HK, Lee DH, Jung SM, et al. The comparison between Behcet’s disease and spondyloarthritides: does Behcet’s disease belong to the spondyloarthropathy complex? J Korean Med Sci. 2002;17(4):524–529. | ||
Chamberlain MA, Robertson RJ. A controlled study of sacroiliitis in Behcet’s disease. Br J Rheumatol. 1993;32(8):693–698. | ||
Yazici H, Tuzlaci M, Yurdakul S. A controlled survey of sacroiliitis in Behcet’s disease. Ann Rheum Dis. 1981;40(6):558–559. | ||
Kotevoglu N, Tasbas I, Bekiroglu N. Computed tomography does not support sacroiliitis as a feature of behcet disease: a metaanalytic review. J Clin Rheumatol. 2004;10(1):42–45. | ||
Lee SS, Yoon HJ, Chang HK, Park KS. Fibromyalgia in Behcet’s disease is associated with anxiety and depression, and not with disease activity. Clin Exp Rheumatol. 2005;23(4 suppl 38):S15–S19. | ||
Siva A, Saip S. The spectrum of nervous system involvement in Behcet’s syndrome and its differential diagnosis. J Neurol. 2009;256(4):513–529. | ||
Uluduz D, Kurtuncu M, Yapici Z, et al. Clinical characteristics of pediatric-onset neuro-Behcet disease. Neurology. 2011;77(21):1900–1905. | ||
Kocer N, Islak C, Siva A, et al. CNS involvement in neuro-Behcet syndrome: an MR study. AJNR Am J Neuroradiol. 1999;20(6):1015–1024. | ||
Yesilot N, Mutlu M, Gungor O, Baykal B, Serdaroglu P, Akman-Demir G. Clinical characteristics and course of spinal cord involvement in Behcet’s disease. Eur J Neurol. 2007;14(7):729–737. | ||
Uygunoglu U, Pasha M, Saip S, Siva A. Recurrent longitudinal extensive transverse myelitis in a neuro-Behcet syndrome treated with infliximab. J Spinal Cord Med. 2015;38(1):111–114. | ||
Saruhan-Direskeneli G, Yentur SP, Mutlu M, et al. Intrathecal oligoclonal IgG bands are infrequently found in neuro-Behcet’s disease. Clin Exp Rheumatol. 2013;31(3 suppl 77):25–27. | ||
Seyahi E, Yurdakul S. Behcet’s syndrome and thrombosis. Mediterr J Hematol Infect Dis. 2011;3(1):e2011026. | ||
Tascilar K, Melikoglu M, Ugurlu S, Sut N, Caglar E, Yazici H. Vascular involvement in Behcet’s syndrome: a retrospective analysis of associations and the time course. Rheumatology (Oxford). 2014;53(11):2018–2022. | ||
Seyahi E, Karaaslan H, Ugurlu S, Yazici H. Fever in Behcet’s syndrome. Clin Exp Rheumatol. 2013;31(3 suppl 77):64–67. | ||
Seyahi E, Cakmak OS, Tutar B, et al. Clinical and ultrasonographic evaluation of lower-extremity vein thrombosis in behcet syndrome: an observational study. Medicine (Baltimore). 2015;94(44):e1899. | ||
Tunc R, Keyman E, Melikoglu M, Fresko I, Yazici H. Target organ associations in Turkish patients with Behcet’s disease: a cross sectional study by exploratory factor analysis. J Rheumatol. 2002;29(11):2393–2396. | ||
Seyahi E. Behcet’s disease: how to diagnose and treat vascular involvement. Best Pract Res Clin Rheumatol. 2016;30(2):279–295. | ||
Seyahi E, Caglar E, Ugurlu S, et al. An outcome survey of 43 patients with Budd-Chiari syndrome due to Behcet’s syndrome followed up at a single, dedicated center. Semin Arthritis Rheum. 2015;44(5):602–609. | ||
Aguiar de Sousa D, Mestre T, Ferro JM. Cerebral venous thrombosis in Behcet’s disease: a systematic review. J Neurol. 2011;258(5):719–727. | ||
Kurt E, Kocer N, Ozguler Y, et al. An outcome survey of 100 patients with cerebral venous sinus thrombosis due to Behcet’s syndrome followed up at a single, dedicated center [abstract]. Arthritis Rheumatol. 2016;68(suppl 10):3966–3967. | ||
Hamuryudan V, Er T, Seyahi E, et al. Pulmonary artery aneurysms in Behcet syndrome. Am J Med. 2004;117(11):867–870. | ||
Seyahi E, Yazici H. Behcet’s syndrome: pulmonary vascular disease. Curr Opin Rheumatol. 2015;27(1):18–23. | ||
Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behcet disease: a series of 47 patients. Medicine (Baltimore). 2012;91(1):35–48. | ||
Uzun O, Erkan L, Akpolat I, Findik S, Atici AG, Akpolat T. Pulmonary involvement in Behcet’s disease. Respiration. 2008;75(3):310–321. | ||
Esatoglu SN, Seyahi E, Ugurlu S, et al. Bronchial artery enlargement may be the cause of recurrent haemoptysis in Behcet’s syndrome patients with pulmonary artery involvement during follow-up. Clin Exp Rheumatol. 2016;34(6 suppl 102):92–96. | ||
Saadoun D, Asli B, Wechsler B, et al. Long-term outcome of arterial lesions in Behcet disease: a series of 101 patients. Medicine (Baltimore). 2012;91(1):18–24. | ||
Ceyran H, Akcali Y, Kahraman C. Surgical treatment of vasculo-Behcet’s disease. A review of patients with concomitant multiple aneurysms and venous lesions. Vasa. 2003;32(3):149–153. | ||
Marzban M, Mandegar MH, Karimi A, et al. Cardiac and great vessel involvement in “Behcet’s disease”. J Card Surg. 2008;23(6):765–768. | ||
Emmungil H, Yasar Bilge NS, Kucuksahin O, et al. A rare but serious manifestation of Behcet’s disease: intracardiac thrombus in 22 patients. Clin Exp Rheumatol. 2014;32(4 suppl 84):S87–S92. | ||
Zhu YL, Wu QJ, Guo LL, et al. The clinical characteristics and outcome of intracardiac thrombus and aortic valvular involvement in Behcet’s disease: an analysis of 20 cases. Clin Exp Rheumatol. 2012;30(3 suppl 72):S40–S45. | ||
Demirelli S, Degirmenci H, Inci S, Arisoy A. Cardiac manifestations in Behcet’s disease. Intractable Rare Dis Res. 2015;4(2):70–75. | ||
Alibaz-Oner F, Aldag B, Aldag M, et al. Post-thrombotic syndrome and venous disease-specific quality of life in patients with vascular Behcet’s disease. J Vasc Surg Venous Lymphat Disord. 2016;4(3):301–306. | ||
Jung JY, Kim DY, Bang D. Leg ulcers in Behcet’s disease. Br J Dermatol. 2008;158(1):178–179. | ||
Karaca M, Hatemi G, Sut N, Yazici H. The papulopustular lesion/arthritis cluster of Behcet’s syndrome also clusters in families. Rheumatology (Oxford). 2012;51(6):1053–1060. | ||
Mumcu G, Yazici Y, Chamberlain AM. Disease assessment in Behçet’s disease. In: Yazici Y, Yazici H, editors. Behçet’s Syndrome. New York, NY: Springer; 2010:308. | ||
Mat C, Yurdakul S, Sevim A, Ozyazgan Y, Tuzun Y. Behcet’s syndrome: facts and controversies. Clin Dermatol. 2013;31(4):352–361. | ||
Hatemi G, Silman A, Bang D, et al. EULAR recommendations for the management of Behcet disease. Ann Rheum Dis. 2008;67(12):1656–1662. | ||
Ozguler Y, Leccese P, Christensen R, et al. A systematic literature review on the treatment of major organ involvement of Behçet’s syndrome informing the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis. 2016;75(suppl 2):800. | ||
Hatemi I, Hatemi G, Pamuk ON, Erzin Y, Celik AF. TNF-alpha antagonists and thalidomide for the management of gastrointestinal Behcet’s syndrome refractory to the conventional treatment modalities: a case series and review of the literature. Clin Exp Rheumatol. 2015;33(6 suppl 94):S129–S137. | ||
Akman-Demir G, Saip S, Siva A. Behcet’s disease. Curr Treat Options Neurol. 2011;13(3):290–310. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.