Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Behçet’s Disease in Children: Diagnostic and Management Challenges
Authors Costagliola G, Cappelli S, Consolini R
Received 12 April 2020
Accepted for publication 21 May 2020
Published 11 June 2020 Volume 2020:16 Pages 495—507
DOI https://doi.org/10.2147/TCRM.S232660
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Giorgio Costagliola, Susanna Cappelli, Rita Consolini
Laboratory of Immunology, Department of Clinical and Experimental Medicine, Division of Pediatrics, University of Pisa, Pisa, Italy
Correspondence: Giorgio Costagliola Email [email protected]
Abstract: Behçet’s Disease (BD) is an inflammatory disease of unknown etiology with multisystemic involvement, being the main clinical manifestations represented by recurrent oral and genital ulcerations and uveitis. The disease has typically a chronic-relapsing course and may cause significant morbidity and mortality due to eye, vascular and neurological involvement. Although BD is more frequently diagnosed in adulthood, the disease onset can also be in pediatric age. Pediatric-onset BD is commonly featured by an incomplete clinical picture, and therefore the diagnosis represents a considerable clinical challenge for the physicians. The first classification criteria for pediatric BD, based on a scoring system, have been proposed few years ago. This work focuses on the main difficulties concerning both the diagnostic approach and the treatment of BD in pediatric age. The recommendation for the treatment of pediatric BD has been recently updated and allowed a considerable improvement of the therapeutic strategies. In particular, the use of anti-TNFα drugs as a second-line option for refractory BD, and as a first-line treatment in severe ocular and neurological involvement, has demonstrated to be effective in improving the outcome of BD patients. The knowledge about the molecular pathogenesis is progressively increasing, showing that BD shares common features with autoimmune and autoinflammatory disorders, and thus leading to the use of new biologic agents targeting the main mediators involved in the determination of BD. Anti-IL-17, anti-IL-23, anti-IL-1 and anti-IL-6 agents have shown promising results for the treatment of refractory BD in clinical trials and will represent an important alternative for the therapeutic approach to the disease.
Keywords: Behçet’s disease, aphtosis, differential diagnosis, autoinflammatory diseases, autoimmunity, biologic drugs
Introduction
Behçet’s Disease (BD) is an inflammatory disease characterized by multisystemic involvement and featured by a chronic, relapsing disease course; histologically it appears as a vasculitis affecting both small and large vessels, with major involvement of the veins.1,2 The most prominent clinical manifestations of BD are recurrent oral ulcerations (ROU), genital ulcerations (GU) and ocular inflammation, but also neurological, gastrointestinal and articular involvement are reported with considerable frequency.3
The epidemiological distribution of BD is peculiar: the disease has a high prevalence among countries identifying the old “Silk Road”, a wide area between the Mediterranean countries and the eastern Asia.4 The higher prevalence of BD is demonstrated in Turkey and Nothern Jordan,5–7 and is also elevated in Korea, Northern China, Iran and Israel,3,8 while in the rest of the Europe and in the United States it is remarkably lower.5,9 This distribution suggested the existence of a predisposing factor diffused among the above-mentioned geographic area.4 In particular, it has been remarked that 50–70% of the patients suffering from BD resulted positive for the major histocompatibility allele HLA B-51.1,10 Despite the association with HLA-B51 increases the risk of developing BD of about 6 folds compared to general population,11 it accounts only for a small part of the genetic susceptibility to BD. Therefore, the etiology of BD is unknown, and several genetic and environmental factors seem to be implicated.12,13 The disease is more frequently diagnosed in adulthood, with a diagnostic peak formulated between the third and the fourth decades of life; however, an onset of BD in pediatric age, before the age of 16, is reported in a percentage that varies from 4% to 26% of the patients.1,14-16 Given its rarity in pediatric age and the latency between the disease onset and the expression of the entire clinical picture, both diagnosis and treatment of BD in children and adolescents still represent a difficult clinical challenge.
Herein, we review the current knowledge about the etiology and pathogenesis of BD and we analyze the different phenotypic manifestations of the disease in children, in order to provide some key issues useful to improve the diagnostic process and the clinical management of pediatric patients affected by BD.
Etiology and Pathogenesis: From Old to New Aspects
The etiology and the pathogenic mechanisms underlying the development of BD have been extensively studied, not least to identify targeted therapies for the disease.
An association between the disease and several non-HLA loci, mainly codifying genes involved in the immune response and in the production, function and signaling of cytokines, was identified. In particular, genome-wide association studies demonstrated the correlation between variants in the IL-10, IL-23R–IL-12RB2, ERAP1 and STAT genes and the development of BD.17–20 In pediatric BD the rate of familial aggregation is higher than in adults.15,21 This suggests that in pediatric age the individual genetic background might play a more pronounced role in the pathogenesis of BD, compared to adult patients, and can partly explain the difference in the clinical phenotype of adult and pediatric BD patients.
The role of environmental triggers for BD has also been analyzed: correlation between the development of BD and the immune response to Streptococcus sanguinis,12,22,23 and differences in the composition of the oral and gut microbiome24,25 have been highlighted.
The pathogenic mechanism of BD is complex and not completely elucidated; it involves multiple molecular pathways, showing common features with both autoimmune and autoinflammatory disorders.2,12 In fact, its association with both HLA-B51 and infectious agents, such as the above mentioned as Streptococcus sanguinis, suggested that BD could be the result of an uncontrolled activation of the immune system driven by an exogenous trigger, with a mechanism of molecular mimicry.26 On the other hand, the clinical phenotype, featured by relapsing episodes of inflammation, the high levels of pro-inflammatory cytokines and the absence of identified pathogenic autoantibodies sustain the hypothesis of BD as an autoinflammatory disease.2 Moreover, recently it has been suggested that BD could represent a subtype of spondyloarthropathy, basing on the involvement of common molecular mechanisms between BD and this group of conditions. BD and spondyloarthropathies share multiple aspects: the association with class I MHC alleles and their interaction with endoplasmic reticulum aminopeptidase 1, the enhanced Th-17 response, and the barrier dysfunction in the involved tissues, finally determining an aberrant immune response.12,27,28 Apart from the considerations about its inclusion in a specific category of diseases, we believe it is important to underline that BD is featured by the impairment of both innate and adaptive immunity. The relative weight of the single molecular pathways may be different in the single tissues and systems involved in the disease, thus explaining the difficulties of the treatment based on the different efficacy of the biologic agents on the heterogeneous clinical picture of the disease.
Molecular Mechanisms
Innate Immunity
Innate immune cells and soluble mediators play a key role in the pathogenesis of BD by contributing, both directly and indirectly, to the development of organ damage, trough the activation of the adaptive response. Neutrophil cell population is found in the vessels of patients with BD, determining a condition of neutrophilic vasculitis;29 neutrophils are the main cells identified in the histologic examination of the sites involved in BD, including oral and genital mucosa and the eye.29 The hyperactivation of neutrophils has been demonstrated by different studies, and it is partly related with HLA-B51.30 Activated neutrophils are able to enhance chemotaxis and effector response, with production of reactive oxygen species, phagocytosis, production of neutrophil-extracellular traps, and secretion of cytokines able to induce a Th1-mediated immune response.31,32 Moreover, the reactive oxygen species produced by neutrophils contribute to the endothelial dysfunction and, trough modification of the fibrinogen structure, to the development of thrombosis.32,33 Even NK cells are directly involved in the pathogenesis of BD, trough a complex modulation of other cellular components of the innate and adaptive immunity. Alterations both in number and function of peripheral NK cells, resulting in a Th1 response, have been demonstrated.12,34 Recently, the role of γδ T-cells in BD has been investigated,2,3 evidencing an enhanced activation of this cellular population in patients with BD.3,35 The activation of γδ T cells is partly depending from the circulating levels of IL-1β, central in the inflammatory response, and IL-23;36 γδ cells in turn become able to induce a TH17 immune response.37 Therefore, γδ cells may represent one of the multiple links between the inflammation and the activation of the adaptive immunity, finally determining the effector mechanism responsible of the organ damage in BD.
Adaptive Immunity
The balance of T-cells is altered in BD, showing reduced levels of T regulatory cells (T regs) and enhanced production of TH17 and TH1 cells.3,38 Consequently, a high Th17/T reg ratio, which influences the immune balance,39 resulting in the potential development of autoimmunity, has been reported. This immunological pattern, characterized by the prevalence of Th17 immune response, has been observed both in patients with cutaneous involvement (folliculitis) and with uveitis.40 Even Th22 cells, a subtype of CD4 cells, are elevated in patients with BD. Th22 cells participate in the pathogenesis of BD trough the production of TNF-α and IL-22, this pattern being associated with several autoimmune disorders.2,41
The role of B cells and autoantibodies in the pathogenesis of BD is controversial. Several autoantibodies have been analyzed in BD, without evidence of a conclusive correlation with the development of the disease. Anti-endothelial cells antibodies have been demonstrated in 18–50% of the patients and represent the autoantibody pattern with the most defined association with BD.26 However, their specificity is low, as they can be detected in other vasculitides, and their pathogenic role is uncertain. A theory is that they could contribute to the development of BD activating endothelial cells, therefore increasing the production of cytokines, or initiating the inflammatory response with their cytotoxic activity.3,8 Other antibody specificities have been detected in patients with BD, including anticardiolipin, anti-retinal and anti-Saccharomyces cerevisiae antibodies, but their causative role has not been demonstrated.26
Cytokines
The levels of several pro-inflammatory cytokines, produced by cells of the innate immune system, have been demonstrated in patients with BD. IL-1β, IL-6 and TNF-α have an important role in the induction of the immune response in BD, and therefore represent potential therapeutic targets for the disease.2,42,43 IL-1 and IL-6, together with IL-21 and IL-23, participate in the activation of TH-17 T cells, while TNF-α, mainly derived from the monocytic lineage, is important in the induction of autoimmunity.2,39 High levels of TNF-α and IL-6 have been detected in the aqueous humor and in the vitreous fluid of patients with active uveitis, respectively, and their pathogenic role has been demonstrated in the development of neuro-Behcet.42,44
As a result of the above-mentioned alterations of the adaptive immune response, the levels of cytokines related to Th1 and Th17 activations are elevated. Serum levels of IL-17, produced by TH17 cells, IFN-γ, IL-2, IL-12 and IL-18, produced by Th1 cells, together with a reduction of IL-10, produced by T regs, have been demonstrated in patients suffering from BD.12,45-48 This cytokine scenario underlies the complex pathogenesis and guides the future therapeutic strategy of BD.
Clinical Manifestations
Mucocutaneous Lesions
As described in Table 1, ROU is present in almost all children with BD (92–100%), similarlyto adult BD patients.15,16,21,49-53 In most patients it represents the first manifestation (80–98%),15,49,51 occurring at a mean age of 8–9 years.49 ROU can precede other symptoms by years and this time frame in children is even longer than in adults. The lesions tend to be widespread and multiple, but they may also be single. Both minor and major ulcers can be observed. They involve lips, tongue, cheeks and palate and disappear without scar. The mean healing time is 10 days but major ulcers may persist for weeks.1,14 ROU is a nonspecific sign and differential diagnosis includes a wide range of conditions, as summarized in Table 2. Increased number of ulcers (more than six at the same time), concurrent variation in size from that of herpetiform to major ulcers, diffuse erythematous surrounds and involvement of soft palate and oropharynx have been suggested to differentiate BD from conventional RAS.54,55
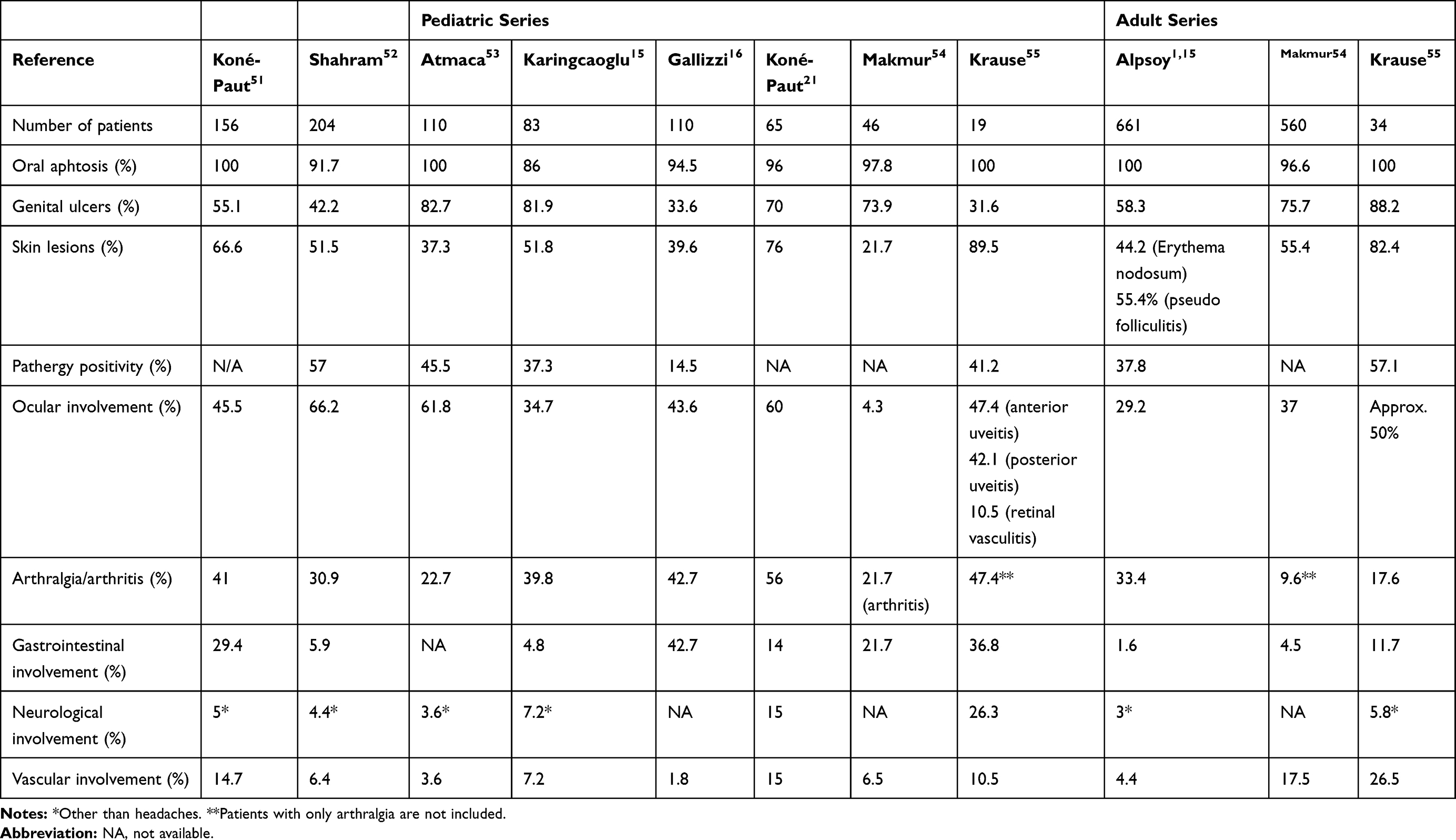 |
Table 1 Clinical Manifestations in Pediatric and Adult BD Cohorts |
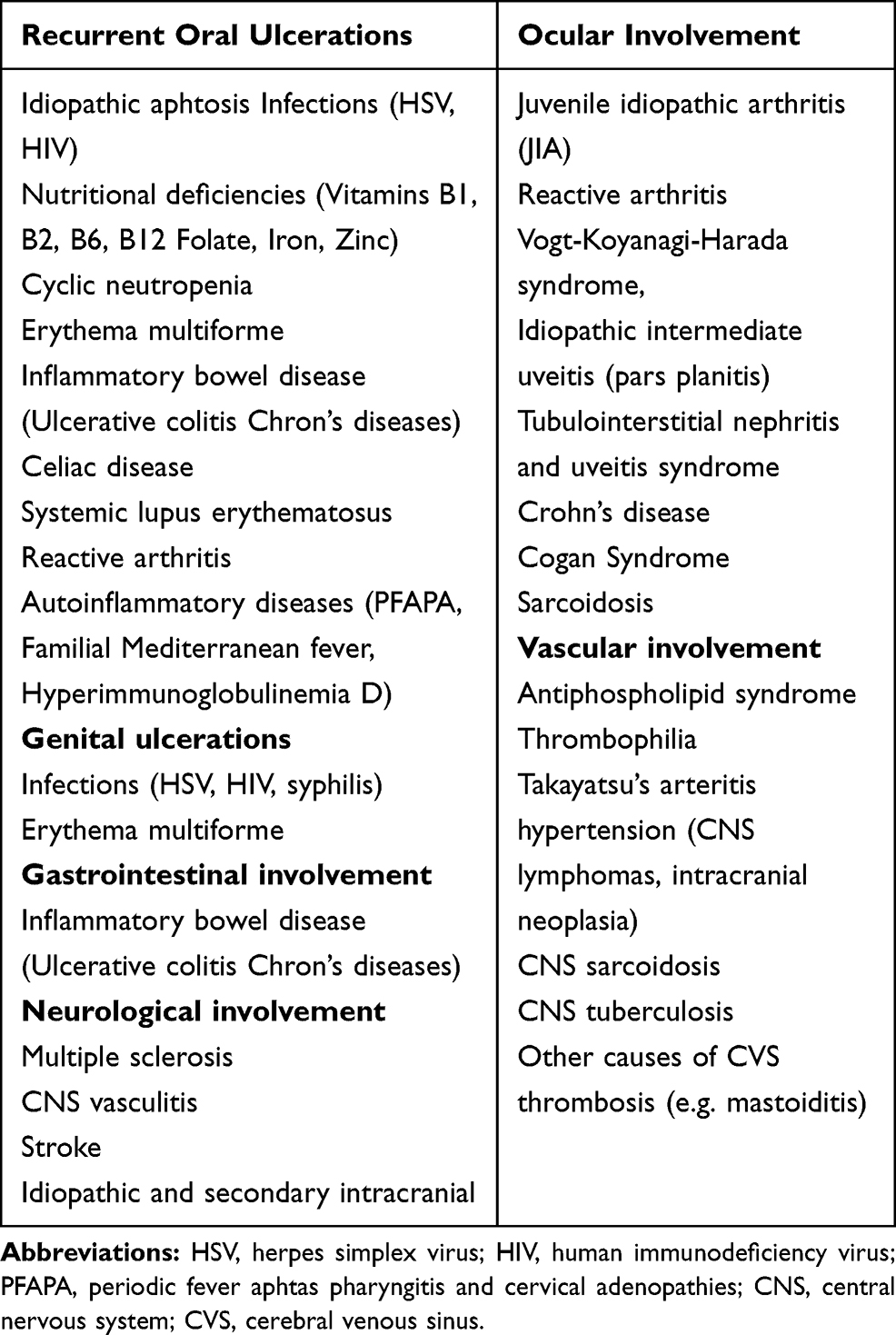 |
Table 2 Differential Diagnosis of Patients with BD According to Clinical Manifestations |
Although GU are reported to be less common than in adults (33–83% in children compared to 60–90% in adults),15,16,49-51,53 they are still the second-most common finding after ROU. In the pediatric populations, females have a higher rate of GU than males.52 GU are generally deeper and larger than oral ulcers, recur less frequently and can have a scarring tendency. They are typically located on the vulva and on the scrotum. Differential diagnosis includes HSV infection, erythema multiforme, fixed drug eruption, sexually transmitted diseases (syphilis and HIV infection).54 Skin aphthae over the perineal region have been described in 7% of children and should be differentiated from anal aphtosis that can be found in IBD.21
As in adult-onset disease, cutaneous lesions are very frequent in pediatric BD (40–90%). They appear later than OU, at a mean age of 10–13 years.16,21,49,50 Skin lesions may occur as erythema nodosum-like lesions, papulopustular lesions, folliculitis, superficial thrombophlebitis and cutaneous vasculitic lesions. The acne-like lesions are not only limited to the typical areas of acne (face and upper part of the trunk) but can also be found on lower limbs. Necrotic folliculitis and acne-like lesions are more common in males while erythema nodosum in females.49 The positivity of the pathergy reaction varies according to geographic distribution, being reported in 20–60% of the pediatric patients suffering from BD.15,16,21,49-51,53 However, it has been removed from the pediatric BD classification criteria51 and is no longer mandatory to define BD.
Musculoskeletal Involvement
The reported prevalence of joint involvement in pediatric population is slightly lower than in adults (30–40% vs 45–50%).15,16,21,49-51,53,56 Arthritis is typically recurrent, nonerosive and does not cause any deformity; knee and ankle are the most commonly involved joints. In the Pediatric Behçet’s disease (PEDBD) study, the rate of the axial involvement rate is reported to be 16.67% and the peripheral arthritis 47.44%.49 Enthesopathy may be observed while sacroiliac involvement is rare.
Eye Involvement
Around 30–70% of children suffer from uveitis.15,16,21,49-53 Atmaca et al found a similar rate of eye involvement in children and adults,51 while Koné-Paut et al reported that eye involvement in children is less frequent than in adults, but associated with worse prognosis.21 Uveitis is nearly two folds more frequent in males presenting with severe course.49,57,58 Eye involvement may lead to severe vision loss in 25% of cases.59 In most cases, ocular involvement occurs 2–4 years after disease onset,51 but in 10–20% of patients it represents the initial manifestation.49 The inflammation is typically nongranulomatous, accompanied by necrotizing obstructive vasculitis and affects anterior or posterior segment or both.60 The ocular disease may start unilaterally but bilateral posterior uveitis with retinal vasculitis is the most typical feature of ocular-BD. Involvement of the posterior segment is the most serious ocular manifestation and patients usually have a painless decrease in visual acuity.14,61 The most frequent posterior-segment complication is the macular edema which can either resolve with appropriate treatment or result in chronic structural changes such as scarring, atrophy, or formation of macular holes. Repeated episodes of posterior-segment flare-ups can result in end-stage ocular BD characterized by blindness with a clinical picture of optic atrophy, vascular attenuation, and diffuse retinal atrophy.60 The main symptoms of anterior uveitis are blurred vision, redness, periorbital or global pain, photophobia, and tearing. Slit-lamp exam may disclose conjunctival injection and hypopyon. Thanks to new therapeutic strategies the prognosis of eye involvement has improved in recent years.61 The differential diagnosis of a patient with inflammatory eye disease includes several conditions, as idiopathic juvenile arthritis (AIG), reactive arthritis, Vogt -Koyanagi-Harada syndrome, idiopathic intermediate uveitis (pars planitis), tubulointerstitial nephritis and uveitis syndrome, and Cogan Syndrome.61
Neurological Involvement
Both in children and adults the reported prevalence of neurologic involvement varies greatly according to studies. Neurologic manifestations occur in 5.3% to 59%62,63 of adult cases and in 3.6–36% of pediatric patients.15,16,21 In children the mean age at presentation is 11–12 years, with a male gender prevalence of 2–3:1.62,63 Neuro-Behçet disease (NB) affects predominantly the central nervous system (CNS), whereas the peripheral nervous system is rarely involved. Two major forms can be distinguished: the parenchymal and the vascular form. Clinical findings of parenchymal NB include headache, hemiplegia, cranial nerve palsies, aseptic meningitis, meningoencephalitis and neuropsychiatric disturbances. Vascular involvement has a better prognosis than the parenchymal form and is more common in children. The main manifestations of the vascular form include cerebral venous thrombosis (CVS) and pseudotumor cerebri.1,14,15 The neurological features of BD are non-specific; when they represent the first manifestations of BD, the differential diagnosis could be extremely difficult. Differential diagnosis includes multiple sclerosis, other CNS vasculitis, stroke, idiopathic and secondary causes of intracranial hypertension (CNS lymphomas, intracranial neoplasia), neuro-sarcoidosis, CNS-tuberculosis, other causes of CVS (eg, mastoiditis).63–65
Vascular Involvement
Vascular manifestations are reported in 5–40% of adults66,67 and 2–20% of children.15,16,21,49,50,52 The mean age at their onset is 11 years and a male prevalence has been observed.49,51 BD may affect any type and size of vessel, but venous involvement is prominent. The inflammation, as previously discussed, is predominantly driven by neutrophils, and thrombosis is secondary to the inflammatory process. Superficial venous thrombosis (SVT) and deep vein thrombosis (DVT) of the lower limbs are the most frequent vascular manifestations of BD. However, DVT can occur in atypical sites such as inferior and superior vena cava, suprahepatic veins (causing Budd-Chiari syndrome), portal vein, cerebral sinuses and right ventricle.68 Although less common, arterial involvement is a typical feature of BD. Pulmonary artery aneurysm is an important cause of mortality and morbidity. Arterial aneurism is the most common arterial finding, but occlusion and stenosis of aorta, femoral and pulmonary vessels may also be observed.26
Gastrointestinal Involvement
Gastrointestinal (GI) involvement is reported to be more common in children than in adults (4–40% vs 2–12%).15,16,21,49-53 The mean age at onset of GI symptoms is 8.9 years, as reported by the PEDBD.49 The most common manifestations are abdominal pain, nausea, vomiting, dyspepsia, diarrhea and gastrointestinal bleeding. Mucosal inflammation and ulcers can occur throughout the GI tract, more frequently in the ileocecal region. Deep aphthous and necrotic ulcerations may lead to abscess and perforation requiring surgery.1,14 Differential diagnosis should include IBD, in particular Chron’s Disease.
Miscellaneous Manifestations
Nearly half of pediatric BD patients have recurrent fevers.49 As in adult patients, fever can be associated with vascular and neurological disease but in children it can also be observed in association with attacks of oral aphtosis. This clinical presentation resembles PFAPA syndrome,69 which occurs frequently in childhood and should be considered in the differential diagnosis.
Other manifestations that have been reported in BD are pulmonary parenchymal lesions (nodules and cavities), pleural effusions, pericarditis, myocarditis and glomerulonephritis.70–72
Disease Course and Prognosis
BD onset is usually insidious, but acute life-threatening manifestations may represent the first symptom. Disease course is typically recurrent and unpredictable. Unlike adults, in whom symptoms usually decrease after a mean follow up of ten years, in children the disease often remains active and new symptoms appear over time.1 Younger patients and men generally have a more severe disease, showing an increasing frequency both of mortality and morbidity related to eye, vascular and neurologic disease.73
Diagnostic Approach
The diagnosis of BD relies substantially on the clinical features, but no specific symptoms and signs are described. The spectrum of the differential diagnosis is extremely wide, including autoimmune and autoinflammatory diseases. In children the diagnosis is even more challenging due to the long time interval between disease onset and the development of a clinical picture compatible with the BD diagnostic criteria. Consequently, the time to diagnosis is generally longer in pediatric patients (between 2 and 5 years) than in adults.15,49,53 The majority of children observed at the presentation of the disease have few suggestive symptoms to satisfy any BD classification criteria and the diagnosis is based on the physician’s expertise. In these cases, a diagnosis of “partial” BD is usually performed. Therefore, a detailed history and a systemic examination are recommended in the evaluation of a child suffering from oral and/or genital ulcers (the most typical BD presentation in children) together with a long critical monitoring during follow-up (Table 2). Until a few years ago the most commonly used criteria were the 1990 International Classification Criteria for BD defined by the International Study Group (ISG).74 According to these criteria, the diagnosis could be established by the concurrent presence of two of the following findings in addition to ROU (mandatory criterion): GU, skin lesions, ocular involvement and pathergy test positivity. These criteria displayed a sensitivity of 85% and a specificity of 96%.75 The relatively low sensitivity is explained by the high significance attributed to ROU, which can be absent only in 5% of the patients. In the year 2014, the International Team for the Revision of the International Criteria for BD (ICBD) proposed new criteria based on a scoring system, in order to increase the sensitivity of the previous ones.76 The new classification considered ROU as not mandatory criterion for the diagnosis, included vascular and neurologic findings, whereas pathergy test positivity was defined as an optional criterion. The sensitivity of these criteria is 93.9% and the specificity as 92.1% in adult patients.75 Both these criteria have been defined for adult patients and are not validated for pediatric BD. In 2015 the PEDBD study aimed to establish the criteria for pediatric patients using the largest cohort to date (Table 3).49 In contrast to the ISG criteria, ROU are not mandatory and pathergy test is not included. A recent study reported that PEDBD criteria show a greater sensitivity (73,5% vs 52.9%) but a lower specificity (97,7% vs 100%) than ISG criteria. The better sensitivity of PEDBD is particularly important in the pediatric area since it allows an early diagnosis.77
 |
Table 3 Consensus Classification of Pediatric Behçet Disease |
Therapeutic Strategies: From Old to New Drugs
The clinical phenotype and the system involved by the disease strongly influence the therapeutic strategies in adult and pediatric BD.78 The progressive knowledge of the pathogenic mechanisms underlying BD resulted in a considerable improvement in the disease management, with the introduction of new biologic drugs and an optimization of the use of conventional immunosuppressive agents.78,79 Table 4 summarizes the current recommendations78 and promising therapeutic strategies for the treatment of BD focusing on the different approaches suggested for the single disease manifestations. In this regard, it is important to remark that the clinical trials on BD are mostly directed to adult patients. Therefore, the recommendations for the treatment of BD in pediatric age are often derived from the guidelines used in the adult population.
 |
Table 4 Recommended Therapies for the Major Clinical Manifestations of BD |
Conventional Immunosuppressive Agents
Corticosteroids represent a valid option during both the acute phase and disease relapses and are used to treat a wide range of BD manifestations. The administration of topical corticosteroids has reported to be effective in the treatment of mucocutaneous manifestations and of monoarthritis, through infiltrative arthrocentesis.80 Systemic corticosteroids, frequently in combination with other immunosuppressive agents, are recommended for patients presenting with posterior uveitis, acute DVT, cerebral venous thrombosis, arterial involvement and severe gastrointestinal involvement.26,78 The use of colchicine is recommended as a preventive therapy to limit the recurrence of mucocutaneous manifestations, and as a first-line treatment for patients with arthritis.78,81 This drug is effective in reducing the frequency of genital ulcer exacerbations, while the efficacy on oral ulcers is controversial, since studies have shown conflicting results.82 Among immunosuppressive agents, azathioprine (AZA) is the most widely used drug, with the aim of sparing the administration of corticosteroid therapy. AZA is effective in patients with severe mucocutaneous manifestations, arthritis, active uveitis, DVT, CNS and gastrointestinal involvement.1,63,78,83,84 Cyclosporine, an agent with demonstrated efficacy in patients with uveitis and DVT, should be avoided in case of both active and inactive CNS involvement,78 as its administration has been linked to an increased risk of developing manifestations of neuro- Behçet.26,85 Cyclophosphamide is a therapeutic option for patients with severe vascular involvement,1,86 and is recommended in case of extended DVT, involving large vessels, and arterial aneurysms, in combination with corticosteroids.78 Methotrexate can be used in patients with ocular and mucocutaneous involvement, or in a combination therapy for neuro-Behçet,26 while mycophenolate mofetil represents an alternative in the treatment of CNS involvement,87 but showed poor results on mucocutaneous manifestations.88 The use of thalidomide, despite its effectiveness in mucocutaneous and gastrointestinal involvement, is strongly limited by the low safety profile and the adverse effects.26
Biologic Drugs and New Perspectives
Since their introduction, biologic drugs (mainly IFN-α and anti-TNFα) have markedly improved the management of patients suffering from BD. The first biological agent introduced for BD was IFN-alpha, for its well-known immunomodulatory properties. This cytokine is effective in inducing remission in patients affected by BD, with the best efficacy in those with severe uveitis.89
Based on the pathogenesis and, particularly, on the cytokine profiles of patients with BD, new biologic agents have been proposed for the treatment of BD (Table 5). As for conventional treatments, most of the studies are performed on adult patients with BD, and subsequently the drugs are used in pediatric population. The experience with the single agents in pediatric age is limited, mostly deriving from isolated case reports and series.16,90-92 Anti-TNFα agents have been used in a wide range of severe manifestations of BD, including uveitis, NB, gastrointestinal involvement, arthritis vascular and mucocutaneous manifestations.78,93 In particular, infliximab and adalimumab have been shown to be effective in patients with uveitis and severe GI disease,83,94-96 while randomized studies demonstrated the efficacy of etanercept on mucocutaneous involvement.97
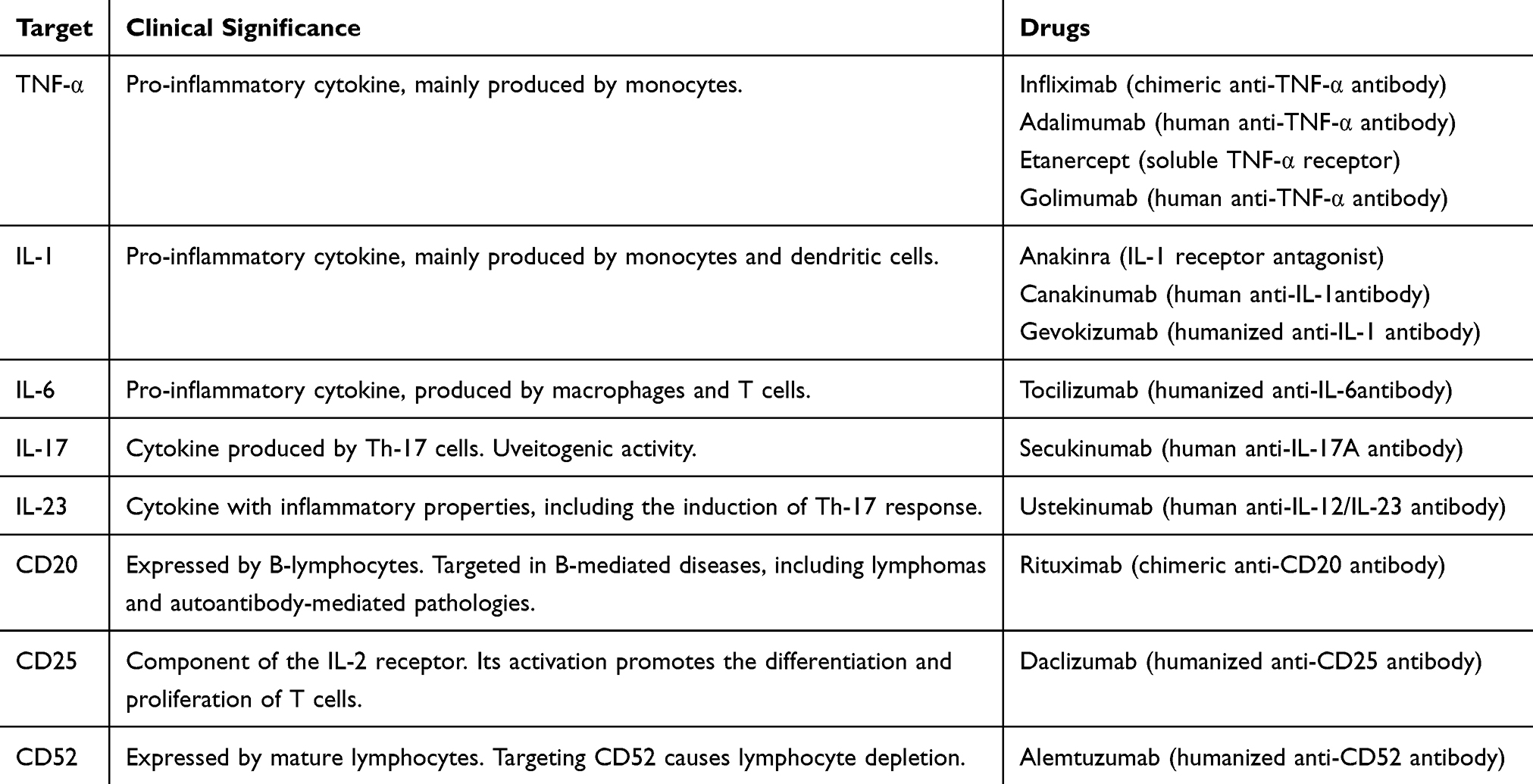 |
Table 5 Molecular Targets and Treatment Options |
The anti-IL-1 agents anakinra and canakinumab have been successfully used in adult and pediatric age for the treatment of refractory uveitis, retinal vasculitis and mucocutaneous manifestations, while gevokizumab showed conflicting results on ocular manifestations.16,92,98,99
The anti-IL-6 agent tocilizumab currently represents a valid therapeutic option for refractory BD, with a significant effect in patients with uveitis and promising effects on CNS involvement.93,100,101 However, it is known that this drug has scarce efficacy on mucocutaneous manifestations of BD,102 and that cases of drug-related cutaneous flares are reported.103,104 The recognized crucial role of the Th17-mediated immune response led to the introduction in the BD therapy of the anti-IL-17 drug secukinumab, whose efficacy has been demonstrated in preliminary studies for the treatment of mucocutaneous and articular manifestations, while its role in the management of uveitis is controversial.102,105 The anti-IL-12/IL-23 agent ustekinumab represents a promising therapeutic option, despite literature reports limited experience, mainly on patients with refractory oral ulcers.106,107 Beyond cytokine blockade, lymphocyte-directed treatments have been studies in BD. Alemtuzumab, an anti-CD52 agent, has been successfully used in patients with refractory BD, taking advantage of the lymphocyte depletion deriving from its administrations.108 However, the safety profile and the clinical applicability of this drug have yet to be defined.102 The ant-CD25 antibody daclizumab has been proposed for the treatment of refractory uveitis, for its effect of inhibition of the IL-2 signaling on T cells, but its use showed conflicting results.102,109 Despite the role of B-cells and autoantibodies has been shown not to be prominent in BD, the use of B-targeted therapies has been proposed, focusing on the vascular manifestations of the disease, as this condition may be associated with anti-endothelial autoantibodies. The anti-CD20 monoclonal antibody rituximab has proven to be effective in patients with retinal vasculitis, NB and uveitis, but the small number of patients evaluated is not sufficient to provide conclusions.102,110-112 Finally, apremilast, an inhibitor of phosphodiesterase 4, active on multiple mechanisms of the innate and adaptive immunity (including Th1 and Th17) represents a promising agent for the treatment of patients suffering from OU.113
Conclusion
BD is a heterogeneous disease, with multiform clinical presentation. In pediatric age the clinical picture may be frequently incomplete; therefore, differential diagnosis of BD is complex, and the latency between disease onset and the definitive diagnosis is common. The recent introduction of diagnostic criteria for BD in pediatric age will help to improve the diagnostic sensibility in this peculiar subpopulation of patients. The advances in the comprehension of the pathogenesis of BD allowed a significant improvement in the disease management, with the introduction of targeted therapies aiming to optimize the therapeutic approach of adult and pediatric patients suffering from BD.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yildiz M, Koker O. Pediatric Behcet’s disease - clinical aspects and current concepts. Eur J Rheumatol. 2019;5:1–10.
2. Tong B, Liu X, Xiao J, Su G. Immunopathogenesis of Behcet’s disease. Front Immunol. 2019;10:665. doi:10.3389/fimmu.2019.00665
3. Greco A, De Virgilio A, Ralli M, et al. Behcet’s disease: new insights into pathophysiology, clinical features and treatment options. Autoimmun Rev. 2018;17(6):567–575. doi:10.1016/j.autrev.2017.12.006
4. Verity DH, Marr JE, Ohno S, Wallace GR, Stanford MR. Behcet’s disease, the Silk Road and HLA-B51: historical and geographical perspectives. Tissue Antigens. 1999;54(3):213–220. doi:10.1034/j.1399-0039.1999.540301.x
5. Davatchi F, Chams-Davatchi C, Shams H, et al. Behcet’s disease: epidemiology, clinical manifestations, and diagnosis. Expert Rev Clin Immunol. 2017;13(1):57–65. doi:10.1080/1744666X.2016.1205486
6. Madanat WY, Alawneh KM, Smadi MM, et al. The prevalence of Behcet’s disease in the north of Jordan: a hospital-based epidemiological survey. Clin Exp Rheumatol. 2017;35 Suppl 108(6):51–54.
7. Bas Y, Seckin HY, Kalkan G, et al. Investigation of Behcet’s disease and recurrent aphthous stomatitis frequency: the highest prevalence in Turkey. Balkan Medl J. 2016;33(4):390–395. doi:10.5152/balkanmedj.2016.15101
8. Cho SB, Cho S, Bang D. New insights in the clinical understanding of Behcet’s disease. Yonsei Med J. 2012;53(1):35–42. doi:10.3349/ymj.2012.53.1.35
9. Akkoc N. Update on the epidemiology, risk factors and disease outcomes of Behcet’s disease. Best Pract Res Clin Rheumatol. 2018;32(2):261–270. doi:10.1016/j.berh.2018.08.010
10. Maldini C, Lavalley MP, Cheminant M, de Menthon M, Mahr A. Relationships of HLA-B51 or B5 genotype with Behcet’s disease clinical characteristics: systematic review and meta-analyses of observational studies. Rheumatology. 2012;51(5):887–900. doi:10.1093/rheumatology/ker428
11. de Menthon M, Lavalley MP, Maldini C, Guillevin L, Mahr A. HLA-B51/B5 and the risk of Behcet’s disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum. 2009;61(10):1287–1296. doi:10.1002/art.24642
12. Leccese P, Alpsoy E. Behcet’s disease: an overview of etiopathogenesis. Front Immunol. 2019;10:1067. doi:10.3389/fimmu.2019.01067
13. Pineton de Chambrun M, Wechsler B, Geri G, Cacoub P, Saadoun D. New insights into the pathogenesis of Behcet’s disease. Autoimmun Rev. 2012;11(10):687–698. doi:10.1016/j.autrev.2011.11.026
14. Kone-Paut I. Behcet’s disease in children, an overview. Pediatr Rheumatol Online J. 2016;14(1):10. doi:10.1186/s12969-016-0070-z
15. Karincaoglu Y, Borlu M, Toker SC, et al. Demographic and clinical properties of juvenile-onset Behcet’s disease: a controlled multicenter study. J Am Acad Dermatol. 2008;58(4):579–584. doi:10.1016/j.jaad.2007.10.452
16. Gallizzi R, Pidone C, Cantarini L, et al. A national cohort study on pediatric Behcet’s disease: cross-sectional data from an Italian registry. Pediatr Rheumatol. 2017;15(1):84. doi:10.1186/s12969-017-0213-x
17. Gul A. Genetics of Behcet’s disease: lessons learned from genomewide association studies. Curr Opin Rheumatol. 2014;26(1):56–63. doi:10.1097/BOR.0000000000000003
18. Mizuki N, Meguro A, Ota M, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behcet’s disease susceptibility loci. Nat Genet. 2010;42(8):703–706. doi:10.1038/ng.624
19. Hou S, Yang Z, Du L, et al. Identification of a susceptibility locus in STAT4 for Behcet’s disease in Han Chinese in a genome-wide association study. Arthritis Rheum. 2012;64(12):4104–4113. doi:10.1002/art.37708
20. Remmers EF, Cosan F, Kirino Y, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet’s disease. Nat Genet. 2010;42(8):698–702. doi:10.1038/ng.625
21. Kone-Paut I, Yurdakul S, Bahabri SA, et al. Clinical features of Behcet’s disease in children: an international collaborative study of 86 cases. J Pediatr. 1998;132(4):721–725. doi:10.1016/S0022-3476(98)70368-3
22. Kaneko F, Oyama N, Yanagihori H, Isogai E, Yokota K, Oguma K. The role of streptococcal hypersensitivity in the pathogenesis of Behcet’s Disease. Eur J Dermatol. 2008;18(5):489–498.
23. Yokota K, Hayashi S, Fujii N, et al. Antibody response to oral streptococci in Behcet’s disease. Microbiol Immunol. 1992;36(8):815–822. doi:10.1111/j.1348-0421.1992.tb02083.x
24. Consolandi C, Turroni S, Emmi G, et al. Behcet’s syndrome patients exhibit specific microbiome signature. Autoimmun Rev. 2015;14(4):269–276. doi:10.1016/j.autrev.2014.11.009
25. Coit P, Mumcu G, Ture-Ozdemir F, et al. Sequencing of 16S rRNA reveals a distinct salivary microbiome signature in Behcet’s disease. Clin Immunol. 2016;169:28–35. doi:10.1016/j.clim.2016.06.002
26. Bulur I, Onder M. Behcet disease: new aspects. Clin Dermatol. 2017;35(5):421–434. doi:10.1016/j.clindermatol.2017.06.004
27. McGonagle D, Aydin SZ, Gul A, Mahr A, Direskeneli H. ‘MHC-I-opathy’-unified concept for spondyloarthritis and Behcet disease. Nat Rev Rheumatol. 2015;11(12):731–740. doi:10.1038/nrrheum.2015.147
28. Giza M, Koftori D, Chen L, Bowness P. Is Behcet’s disease a ‘class 1-opathy’? The role of HLA-B*51 in the pathogenesis of Behcet’s disease. Clin Exp Immunol. 2018;191(1):11–18. doi:10.1111/cei.13049
29. Kobayashi M, Ito M, Nakagawa A, et al. Neutrophil and endothelial cell activation in the vasa vasorum in vasculo-Behcet disease. Histopathology. 2000;36(4):362–371. doi:10.1046/j.1365-2559.2000.00859.x
30. Eksioglu-Demiralp E, Direskeneli H, Kibaroglu A, Yavuz S, Ergun T, Akoglu T. Neutrophil activation in Behcet’s disease. Clin Exp Rheumatol. 2001;19(5 Suppl 24):S19–24.
31. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11(8):519–531. doi:10.1038/nri3024
32. Emmi G, Becatti M, Bettiol A, Hatemi G, Prisco D, Fiorillo C. Behcet’s syndrome as a model of thrombo-inflammation: the role of neutrophils. Front Immunol. 2019;10:1085. doi:10.3389/fimmu.2019.01085
33. Becatti M, Emmi G, Silvestri E, et al. Neutrophil activation promotes fibrinogen oxidation and thrombus formation in Behcet disease. Circulation. 2016;133(3):302–311. doi:10.1161/CIRCULATIONAHA.115.017738
34. Yamaguchi Y, Takahashi H, Satoh T, et al. Natural killer cells control a T-helper 1 response in patients with Behcet’s disease. Arthritis Res Ther. 2010;12(3):R80. doi:10.1186/ar3005
35. Freysdottir J, Hussain L, Farmer I, Lau SH, Fortune F. Diversity of gammadelta T cells in patients with Behcet’s disease is indicative of polyclonal activation. Oral Dis. 2006;12(3):271–277. doi:10.1111/j.1601-0825.2005.01185.x
36. Sutton EJ, Davidson JE, Bruce IN. The systemic lupus international collaborating clinics (SLICC) damage index: a systematic literature review. Semin Arthritis Rheum. 2013;43(3):352–361. doi:10.1016/j.semarthrit.2013.05.003
37. Nian H, Shao H, O’Brien RL, Born WK, Kaplan HJ, Sun D. Activated γδ T cells promote the activation of uveitogenic T cells and exacerbate EAU development. Invest Ophthalmol Vis Sci. 2011;52(8):5920–5927. doi:10.1167/iovs.10-6758
38. Deniz R, Tulunay-Virlan A, Ture Ozdemir F, et al. Th17- inducing conditions lead to in vitro activation of both Th17 and Th1 responses in Behcet’s disease. Immunol Invest. 2017;46(5):518–525. doi:10.1080/08820139.2017.1306865
39. Geri G, Terrier B, Rosenzwajg M, et al. Critical role of IL- 21 in modulating TH17 and regulatory T cells in Behcet disease. J Allergy Clin Immunol. 2011;128(3):655–664. doi:10.1016/j.jaci.2011.05.029
40. Kim J, Park JA, Lee EY, Lee YJ, Song YW, Lee EB. Imbalance of Th17 to Th1 cells in Behcet’s disease. Clin Exp Rheumatol. 2010;28(4 Suppl 60):S16–9.
41. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10(8):857–863. doi:10.1038/ni.1767
42. Akman-Demir G, Tuzun E, Icoz S, et al. Interleukin-6 in neuro-Behcet’s disease: association with disease subsets and long-term outcome. Cytokine. 2008;44(3):373–376. doi:10.1016/j.cyto.2008.10.007
43. Wakefield D, Lloyd A. The role of cytokines in the pathogenesis of inflammatory eye disease. Cytokine. 1992;4(1):1–5. doi:10.1016/1043-4666(92)90028-P
44. Santos Lacomba M, Marcos Martin C, Gallardo Galera JM, et al. Aqueous humor and serum tumor necrosis factor-α in clinical uveitis. Ophthalmic Res. 2001;33(5):251–255. doi:10.1159/000055677
45. Touzot M, Cacoub P, Bodaghi B, Soumelis V, Saadoun D. IFN-α induces IL-10 production and tilt the balance between Th1 and Th17 in Behçet disease. Autoimmun Rev. 2015;14(5):370–375. doi:10.1016/j.autrev.2014.12.009
46. Chi W, Zhu X, Yang P, et al. Upregulated IL-23 and IL-17 in Behcet patients with active uveitis. Invest Ophthalmol Vis Sci. 2008;49(7):3058–3064. doi:10.1167/iovs.07-1390
47. Oztas MO, Onder M, Gurer MA, Bukan N, Sancak B. Serum interleukin 18 and tumour necrosis factor-alpha levels are increased in Behcet’s disease. Clin Exp Dermatol. 2005;30(1):61–63. doi:10.1111/j.1365-2230.2004.01684.x
48. Aridogan BC, Yildirim M, Baysal V, Inaloz HS, Baz K, Kaya S. Serum Levels of IL-4, IL-10, IL-12, IL-13 and IFN-gamma in Behcet’s disease. J Dermatol. 2003;30(8):602–607. doi:10.1111/j.1346-8138.2003.tb00442.x
49. Kone-Paut I, Shahram F, Darce-Bello M, et al. Consensus classification criteria for paediatric Behcet’s disease from a prospective observational cohort: PEDBD. Ann Rheum Dis. 2016;75(6):958–964. doi:10.1136/annrheumdis-2015-208491
50. Shahram F, Nadji A, Akhlaghi M, et al. Paediatric Behcet’s disease in Iran: report of 204 cases. Clin Exp Rheumatol. 2018;36(6 Suppl 115):135–140.
51. Atmaca L, Boyvat A, Yalcindag FN, Atmaca-Sonmez P, Gurler A. Behcet disease in children. Ocul Immunol Inflamm. 2011;19(2):103–107. doi:10.3109/09273948.2011.555592
52. Makmur EL, Myers SH, Hanns L, Haskard DO, Brogan P, Ambrose N. Comparing the clinical profile of adults and children with Behcet’s syndrome in the UK. Clin Exp Rheumatol. 2019;37 Suppl 121(6):48–51.
53. Krause I, Uziel Y, Guedj D, et al. Childhood Behcet’s disease: clinical features and comparison with adult-onset disease. Rheumatology. 1999;38(5):457–462. doi:10.1093/rheumatology/38.5.457
54. Main DM, Chamberlain MA. Clinical differentiation of oral ulceration in Behcet’s disease. Br J Rheumatol. 1992;31(11):767–770. doi:10.1093/rheumatology/31.11.767
55. Alpsoy E, Zouboulis CC, Ehrlich GE. Mucocutaneous lesions of Behcet’s disease. Yonsei Med J. 2007;48(4):573–585. doi:10.3349/ymj.2007.48.4.573
56. Mendes D, Correia M, Barbedo M, et al. Behcet’s disease–a contemporary review. J Autoimmun. 2009;32(3–4):178–188. doi:10.1016/j.jaut.2009.02.011
57. Citirik M, Berker N, Songur MS, Soykan E, Zilelioglu O. Ocular findings in childhood-onset Behcet disease. J AAPOS. 2009;13(4):391–395. doi:10.1016/j.jaapos.2009.04.016
58. Tugal-Tutkun I, Urgancioglu M. Childhood-onset uveitis in Behcet disease:a descriptive study of 36 cases. Am J Ophthalmol. 2003;136(6):1114–1119. doi:10.1016/S0002-9394(03)00791-8
59. Hazleman BL. Rheumatic disorders of the eye and the various structures involved. Br J Rheumatol. 1996;35(3):258–268. doi:10.1093/rheumatology/35.3.258
60. Reiff A, Kadayifcilar S, Ozen S. Rheumatic inflammatory eye diseases of childhood. Rheum Dis Clin North Am. 2013;39(4):801–832. doi:10.1016/j.rdc.2013.05.005
61. Accorinti M, Pesci FR, Pirraglia MP, Abicca I, Pivetti-Pezzi P. Ocular Behcet’s disease: changing patterns over time, complications and long-term visual prognosis. Ocul Immunol Inflamm. 2017;25(1):29–36. doi:10.3109/09273948.2015.1094095
62. Metreau-Vastel J, Mikaeloff Y, Tardieu M, Kone-Paut I, Tran TA. Neurological involvement in paediatric Behcet’s disease. Neuropediatrics. 2010;41(5):228–234. doi:10.1055/s-0030-1269909
63. Mora P, Menozzi C, Orsoni JG, Rubino P, Ruffini L, Carta A. Neuro-Behcet’s disease in childhood: a focus on the neuro-ophthalmological features. Orphanet J Rare Dis. 2013;8(1):18. doi:10.1186/1750-1172-8-18
64. Noel N, Bernard R, Wechsler B, et al. Long-term outcome of neuro-Behcet’s disease. Arthritis Rheumatol. 2014;66(5):1306–1314. doi:10.1002/art.38351
65. Siva A, Saip S. The spectrum of nervous system involvement in Behcet’s syndrome and its differential diagnosis. J Neurol. 2009;256(4):513–529. doi:10.1007/s00415-009-0145-6
66. Seyahi E. Behcet’s disease: how to diagnose and treat vascular involvement. Best Pract Res Clin Rheumatol. 2016;30(2):279–295. doi:10.1016/j.berh.2016.08.002
67. Wu X, Li G, Huang X, et al. Behcet’s disease complicated with thrombosis: a report of 93 Chinese cases. Medicine. 2014;93(28):e263. doi:10.1097/MD.0000000000000263
68. Emmi G, Bettiol A, Silvestri E, et al. Vascular Behcet’s syndrome: an update. Intern Emerg Med. 2019;14(5):645–652. doi:10.1007/s11739-018-1991-y
69. Cantarini L, Vitale A, Bersani G, et al. PFAPA syndrome and Behcet’s disease: a comparison of two medical entities based on the clinical interviews performed by three different specialists. Clin Rheumatol. 2016;35(2):501–505. doi:10.1007/s10067-015-2890-5
70. Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behcet disease: a series of 47 patients. Medicine. 2012;91(1):35–48. doi:10.1097/MD.0b013e318242ff37
71. Geri G, Wechsler B, Thi Huong du L, et al. Spectrum of cardiac lesions in Behcet disease: a series of 52 patients and review of the literature. Medicine. 2012;91(1):25–34. doi:10.1097/MD.0b013e3182428f49
72. Ozen S. The “other” vasculitis syndromes and kidney involvement. Pediatr Nephrol. 2010;25(9):1633–1639. doi:10.1007/s00467-009-1327-2
73. Saadoun D, Wechsler B, Desseaux K, et al. Mortality in Behcet’s disease. Arthritis Rheum. 2010;62(9):2806–2812. doi:10.1002/art.27568
74. Disease ISGfBs. Criteria for diagnosis of Behcet’s disease. International Study Group for Behcet’s Disease. Lancet. 1990;335(8697):1078–1080.
75. Davatchi F, Assaad‐Khalil S, Calamia K.T,et al. The International Criteria for Behcet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–347. doi:10.1111/jdv.12107
76. Davatchi F. Diagnosis/classification criteria for Behcet’s disease. Patholog Res Int. 2012;2012:607921.
77. Batu ED, Sonmez HE, Sozeri B, Butbul Aviel Y, Bilginer Y, Ozen S. The performance of different classification criteria in paediatric Behcet’s disease. Clin Exp Rheumatol. 2017;35 Suppl 108(6):119–123.
78. Hatemi G, Christensen R, Bang D, et al. 2018 update of the EULAR recommendations for the management of Behcet’s syndrome. Ann Rheum Dis. 2018;77(6):808–818. doi:10.1136/annrheumdis-2018-213225
79. Caso F, Costa L, Rigante D. Biological treatments in Behcet’s disease: beyond anti-TNF therapy. Mediators Inflamm. 2014;2014:107421. doi:10.1155/2014/107421
80. Leccese P, Ozguler Y, Christensen R, et al. Management of skin, mucosa and joint involvement of Behcet’s syndrome: a systematic review for update of the EULAR recommendations for the management of Behcet’s syndrome. Semin Arthritis Rheum. 2019;48(4):752–762. doi:10.1016/j.semarthrit.2018.05.008
81. Yurdakul S, Mat C, Tuzun Y, et al. A double-blind trial of colchicine in Behcet’s syndrome. Arthritis Rheum. 2001;44(11):2686–2692. doi:10.1002/1529-0131(200111)44:11<2686::AID-ART448>3.0.CO;2-H
82. Scherrer MAR, Rocha VB, Garcia LC. Behcet’s disease: review with emphasis on dermatological aspects. An Bras Dermatol. 2017;92(4):452–464. doi:10.1590/abd1806-4841.20177359
83. Skef W, Hamilton MJ, Arayssi T. Gastrointestinal Behcet’s disease: a review. World J Gastroenterol. 2015;21(13):3801–3812. doi:10.3748/wjg.v21.i13.3801
84. Saadoun D, Wechsler B, Terrada C, et al. Azathioprine in severe uveitis of Behcet’s disease. Arthritis Care Res. 2010;62(12):1733–1738. doi:10.1002/acr.20308
85. Kotake S, Higashi K, Yoshikawa K, Sasamoto Y, Okamoto T, Matsuda H. Central nervous system symptoms in patients with Behcet disease receiving cyclosporine therapy. Ophthalmology. 1999;106(3):586–589. doi:10.1016/S0161-6420(99)90120-3
86. Ozguler Y, Leccese P, Christensen R, et al. Management of major organ involvement of Behcet’s syndrome: a systematic review for update of the EULAR recommendations. Rheumatology. 2018;57(12):2200–2212. doi:10.1093/rheumatology/key242
87. Shugaiv E, Tuzun E, Mutlu M, Kiyat-Atamer A, Kurtuncu M, Akman-Demir G. Mycophenolate mofetil as a novel immunosuppressant in the treatment of neuro-Behcet’s disease with parenchymal involvement: presentation of four cases. Clin Exp Rheumatol. 2011;29(4 Suppl 67):S64–7.
88. Adler YD, Mansmann U, Zouboulis CC. Mycophenolate mofetil is ineffective in the treatment of mucocutaneous adamantiades-Behcet’s disease. Dermatology. 2001;203(4):322–324. doi:10.1159/000051781
89. Kotter I, Gunaydin I, Zierhut M, Stubiger N. The use of interferon alpha in Behcet disease: review of the literature. Semin Arthritis Rheum. 2004;33(5):320–335. doi:10.1016/j.semarthrit.2003.09.010
90. Poddighe D, Mukusheva Z, Dauyey K, Assylbekova M. Adalimumab in the treatment of pediatric Behçet’s disease: case-based review. Rheumatol Int. 2019;39(6):1107–1112. doi:10.1007/s00296-019-04300-0
91. Nanthapisal S, Klein NJ, Ambrose N, Eleftheriou D, Brogan PA. Paediatric Behçet’s disease: a UK tertiary centre experience. Clin Rheumatol. 2016;35(10):2509–2516. doi:10.1007/s10067-016-3187-z
92. Pagnini I, Bondi T, Simonini G, Giani T, Marino A, Cimaz R. Successful treatment with canakinumab of a paediatric patient with resistant Behçet’s disease. Rheumatology. 2015;54(7):1327–1328. doi:10.1093/rheumatology/kev197
93. McNally TW, Damato EM, Murray PI, Denniston AK, Barry RJ. An update on the use of biologic therapies in the management of uveitis in Behcet’s disease: a comprehensive review. Orphanet J Rare Dis. 2017;12(1):130. doi:10.1186/s13023-017-0681-6
94. Tanida S, Inoue N, Kobayashi K, et al. Adalimumab for the treatment of Japanese patients with intestinal Behcet’s disease. Clin Gastroenterol Hepatol. 2015;13(5):940–8.e3. doi:10.1016/j.cgh.2014.08.042
95. Fabiani C, Vitale A, Emmi G, et al. Efficacy and safety of adalimumab in Behcet’s disease-related uveitis: a multicenter retrospective observational study. Clin Rheumatol. 2017;36(1):183–189. doi:10.1007/s10067-016-3480-x
96. Park J, Cheon JH. Anti-tumor necrosis factor therapy in intestinal Behcet’s disease. Gut Liver. 2018;12(6):623–632. doi:10.5009/gnl17462
97. Melikoglu M, Fresko I, Mat C, et al. Short-term trial of etanercept in Behcet’s disease: a double blind, placebo controlled study. J Rheumatol. 2005;32(1):98–105.
98. Bettiol A, Silvestri E, Di Scala G, et al. The right place of interleukin-1 inhibitors in the treatment of Behcet’s syndrome: a systematic review. Rheumatol Int. 2019;39(6):971–990. doi:10.1007/s00296-019-04259-y
99. Fabiani C, Vitale A, Emmi G, et al. Interleukin (IL)-1 inhibition with anakinra and canakinumab in Behcet’s disease-related uveitis: a multicenter retrospective observational study. Clin Rheumatol. 2017;36(1):191–197. doi:10.1007/s10067-016-3506-4
100. Essaadouni L, Ha-Ou-Nou FZ. Efficacy and safety of tocilizumab in neuro-Behcet’s disease: a case report. Revue Neurologique. 2017;173(3):171–172. doi:10.1016/j.neurol.2017.02.006
101. Kurtuncu M, Tuzun E, Akman-Demir G. Behcet’s disease and nervous system involvement. Curr Treat Options Neurol. 2016;18(5):19. doi:10.1007/s11940-016-0405-6
102. Sota J, Rigante D, Lopalco G, et al. Biological therapies for the treatment of Behcet’s disease-related uveitis beyond TNF-alpha blockade: a narrative review. Rheumatol Int. 2018;38(1):25–35. doi:10.1007/s00296-017-3775-5
103. Emmi G, Silvestri E, Squatrito D, Emmi L, Cantarini L, Prisco D. Tocilizumab-induced exacerbation of mucosal ulcers in a patient with multi-refractory Behcets disease. Semin Arthritis Rheum. 2016;46(1):e1–2. doi:10.1016/j.semarthrit.2016.03.006
104. Cantarini L, Lopalco G, Vitale A, et al. Paradoxical mucocutaneous flare in a case of Behcet’s disease treated with tocilizumab. Clin Rheumatol. 2015;34(6):1141–1143. doi:10.1007/s10067-014-2589-z
105. Di Scala G, Bettiol A, Cojan RD, Finocchi M, Silvestri E, Emmi G. Efficacy of the anti-IL 17 secukinumab in refractory Behcet’s syndrome: a preliminary study. J Autoimmun. 2019;97:108–113. doi:10.1016/j.jaut.2018.09.002
106. Mirouse A, Barete S, Desbois AC, et al. Long-term outcome of ustekinumab therapy for Behcet’s disease. Arthritis Rheumatol. 2019;71(10):1727–1732. doi:10.1002/art.40912
107. Lopalco G, Fabiani C, Venerito V, Lapadula G, Iannone F, Cantarini L. Ustekinumab efficacy and safety in mucocutaneous multi-refractory Behcet’s disease. Clin Exp Rheumatol. 2017;35 Suppl 108(6):130–131.
108. Mohammad AJ, Smith RM, Chow YW, Chaudhry AN, Jayne DR. Alemtuzumab as remission induction therapy in Behcet disease: a 20-year experience. J Rheumatol. 2015;42(10):1906–1913. doi:10.3899/jrheum.141344
109. Buggage RR, Levy-Clarke G, Sen HN, et al. A doublemasked, randomized study to investigate the safety and efficacy of daclizumab to treat the ocular complications related to Behcet’s disease. Ocul Immunol Inflamm. 2007;15(2):63–70. doi:10.1080/09273940701299370
110. Jade J, Chung K, Arendse M, Hussain Z, White D. Neuro-Behcet’s disease presenting with tumour-like lesions and responding to rituximab. J Clin Neurosci. 2016;32:139–141. doi:10.1016/j.jocn.2016.03.020
111. Kidd DP. Rituximab is effective in severe treatment-resistant neurological Behcet’s syndrome. J Neurol. 2015;262(12):2676–2677. doi:10.1007/s00415-015-7897-y
112. Davatchi F, Shams H, Rezaipoor M, et al. Rituximab in intractable ocular lesions of Behcet’s disease; randomized single-blind control study (pilot study). Int J Rheum Dis. 2010;13(3):246–252. doi:10.1111/j.1756-185X.2010.01546.x
113. Takeno M. Positioning of apremilast in treatment of Behcet’s disease. Modern Rheumatol. 2020;30(2):219–224. doi:10.1080/14397595.2019.1696504
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.