Back to Journals » International Medical Case Reports Journal » Volume 14
Bardet–Biedl Syndrome in an Ethiopian
Received 1 January 2021
Accepted for publication 8 March 2021
Published 19 March 2021 Volume 2021:14 Pages 177—181
DOI https://doi.org/10.2147/IMCRJ.S299421
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Asamere Tsegaw,1 Tiliksew Teshome2
1Department of Ophthalmology, School of Medicine, University of Gondar, Gondar, Ethiopia; 2Department of Ophthalmology, Faculty of Medicine, Addis Ababa University, Addis Ababa, Ethiopia
Correspondence: Asamere Tsegaw
Department of Ophthalmology, University of Gondar, Gondar, Ethiopia
Tel +251 939912863
Email [email protected]
Abstract: Bardet–Biedl syndrome (BBS) is a rare familial and multi-system disorder with an autosomal recessive pattern of inheritance and wide range of clinical variability. Its main manifestations are progressive retinal dystrophy, renal dysfunction, post-axial polydactyly, central obesity, mental retardation, and hypogonadism. Renal failure is known to be the main cause of death in patients with BBS. Retinal dystrophy and other eye diseases seen in patients with BBS can cause severe visual impairment and blindness at an early age. After written consent was obtained from the patient, we report the clinical and laboratory data of the first case from Ethiopia of an 18-year-old boy with multi-system manifestations of the Bardet–Biedl Syndrome. We discuss the main clinical manifestations of the syndrome including its potentially blinding and fatal features. We emphasize the need for diagnosis of this syndrome at an early age as possible so that proper and multidisciplinary medical care can be given for such patients to prevent unnecessary morbidity and early mortality.
Keywords: Bardet–Biedl syndrome, retinal dystrophy, polydactyly, renal failure, Ethiopia
Introduction
The Bardet–Biedl syndrome (BBS) is a familial disorder with an autosomal recessive pattern of inheritance. It is a syndrome with multi-system features but characterized mainly by progressive retinal photoreceptor cells dystrophy, central obesity, postaxial polydactyly, mental retardation hypogonadism and renal dysfunction.1–3
BBS was first described in 1920 by Bardet4 and again in 1922 by Biedl.5 It was often confused with the Laurence-Moon syndrome which was first described in1866 by Laurence and Moon.6 Laurence and Moon described 4 cases of pigmentary retinal dystrophy, hypogonadism, obesity and spastic paraplegia. These syndromes are similar by being characterized by pigmentary retinopathy, mental retardation, obesity and hypogenitalism. But they differ in that BBS usually includes polydactyly and renal anomalies, whereas the Laurence-Moon syndrome is associated with spastic paraplegia.
Prevalence rates in North America and Europe range from 1:140,000 to 1:160,000 live births.7 An extensive search of literature revealed no published case report or large-scale case series study from Ethiopia or Africa. To our knowledge, this is the first case report of BBS from Ethiopia.
Case Report
Our patient was an 18 years old boy from Dire Dawa town in the eastern part of Ethiopia. He was seen at a private clinic in Addis Ababa, the capital, in September 2009 for poor vision. He was then referred to the Retina Clinic at Menelik II Hospital, Addis Ababa, for further evaluation. The chief complaint was progressive reduction of vision in both eyes which was noted since early childhood. His parents admitted that he had delayed developmental milestones like walking and speech when compared to his peers. Although he was currently attending regular school, he repeatedly failed to progress into higher grades and was just a grade 7 student at this age. History of consanguinity was not obtained.
Vital signs were within normal limits. His weight was 93kg with a body mass index (BMI) of 32kg/m2 which is in the obese category of WHO classification of BMI.8 Examination of the abdomen revealed central truncal obesity (Figure 1), but there was no organomegaly or other abnormalities. Examination of the extremities revealed postaxial extra-fingers or polydactyly (Figures 2 and 3) in all upper and lower extremities making the total number of his fingers twenty-four. His fingers, especially in the lower limbs were typically short, broad and stubby (Figure 2). Neurological examination did not reveal any abnormality.
 |
Figure 1 Truncal obesity in an 18-year-old Ethiopian patient with Bardet–Biedl syndrome presented to Menelik-II Hospital Ophthalmology Department, Addis Ababa, Ethiopia. |
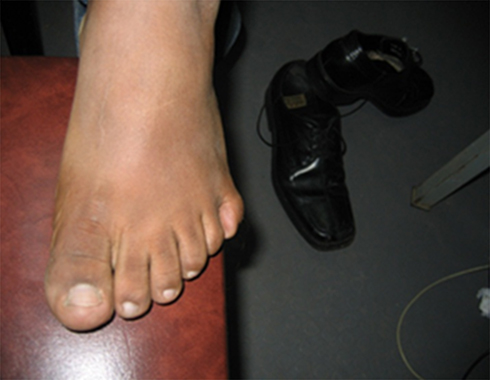 |
Figure 2 Sixth toe (polydactyly) in the left lower limb of an 18-year-old Ethiopian patient with Bardet–Biedl syndrome presented to Menelik-II Hospital Ophthalmology Department, Addis Ababa, Ethiopia. |
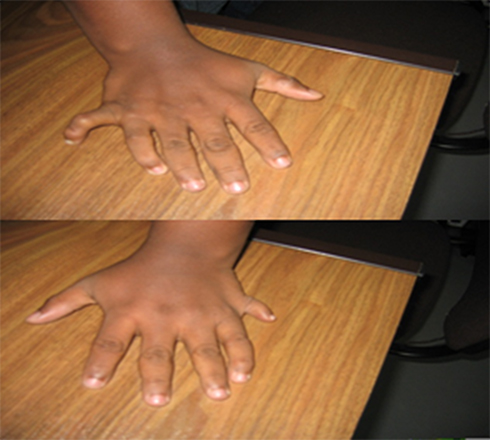 |
Figure 3 Six fingers (polydactyly) in the right and left upper limbs of an 18-year-old Ethiopian patient with Bardet–Biedl syndrome presented to Menelik-II Hospital Ophthalmology Department, Addis Ababa, Ethiopia. |
Ophthalmic examination showed that his Snellen visual acuity was 1/60 in both eyes. His visual acuity improved to 6/36 in both eyes after refractive correction with −5.0 diopter lens. He also had severe reduction of color vision which was detected on color vision test using Ishihara color plates. Funduscopic examination with the Volk double aspheric +90 diopter lens and fundus camera (Figures 4 and 5) showed that he had signs of bilateral retinal dystrophy whose features included pigmented macular scar, attenuated retinal arterioles, optic nerve head atrophy (“waxy disc pallor”) and patchy retinal pigment clumping. Electrophysiologic tests and genetic tests were not done because of lack of availability.
 |
Figure 4 Fundus picture of the right eye of an 18-year-old Ethiopian patient with Bardet–Biedl syndrome showing waxy optic disc pallor, myopicperi-papillary atrophy, retinal pigment clumping, and central macular scar. |
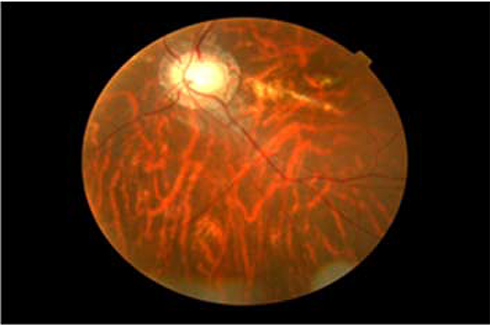 |
Figure 5 Fundus picture of the left eye of an 18-year-old Ethiopian patient with Bardet–Biedl syndrome showing waxy optic disc pallor, myopicperi-papillary atrophy, retinal pigment clumping, and central macular scar. |
Laboratory tests and other investigations done included complete blood cell count, Erythrocyte sedimentation rate (ESR), hemoglobin level, VDRL, Fasting blood sugar (FBS), chest x-ray and all were within normal limits. Renal function tests revealed abnormal renal function with elevated serum blood urea nitrogen (40mg/dl) and serum creatinine (3.0mg/dl) as well as proteinuria on urine dipstick analysis. Abdominal ultrasound examination was done for our patient to detect any structural abnormalities in the kidneys, but it revealed no such abnormalities. The patient was referred to Tikur Anbessa Hospital, Addis Ababa, for further evaluation of his renal status. After refractive correction of his myopia and proper documentation of his retinal and other ocular findings, appointment was given to him at the Retina Clinic of Menelik II Hospital for further follow-up of his eye problems.
Discussion
BBS is genetically heterogeneous and 26 different genes are identified to date whose mutation of each gene alone causes the syndrome.1,2,9,10, This great genetic heterogeneity is responsible for the observed variable expressivity or wide range of clinical variability.
Most symptoms and signs of the Bardet–Biedl syndrome (BBS) appear after several years of evolution and are not fully apparent during early childhood. Because of the slow development of the clinical features of BBS, the diagnosis remains difficult in young patients. The average age at diagnosis was 9 years in one large population-based survey done in the UK.3 This is late for such a debilitating condition and our patient was diagnosed at the age of 18 years, much later than the average age in the above report. Considering its adverse prognosis due to its life-threatening manifestations, a high index of suspicion is important for early diagnosis.
Central obesity is one of the main manifestations of BBS patients.1–3 Of the 109 patients in a series reported by Beales and Elcioglu, 72% of post pubertal subjects were overweight (BMI >25 kg/m2) of whom 16% had BMI>40 kg/m2 which is defined as morbidly obese according to the WHO classification of body mass index.72 Our patient had a BMI of 32kg/m2, within the obese category (BMI>30 kg/m2) of WHO classification.3,8,11
Renal functional impairment and structural abnormalities, such as renal parenchymal cysts or lobulations, dysplastic kidneys and unilateral agenesis were reported at a varying frequency in the literature.1–3,12,13 Renal functional impairment was recognized as a major feature of BBS phenotype recently. Beales and Elcioglu in their population-based survey used abdominal ultrasound and investigated 57 of the 109 BBS patients involved in their survey and detected one or more of those mentioned renal structural abnormalities in 26 (46%).3,12 They also found out that six (5%) had chronic renal failure at the time of the survey.3 On the other hand, O’Dea et al in Canada did a prospective cohort study on 38 confirmed BBS patients over a 6-year period and used both abdominal ultrasound and intravenous pyelogram (IVP) studies on 25 of 38 BBS patients with normal renal function and reported that 96% had some renal structural anomalies.11–13 Nine (25%) patients, initially with normal renal function, developed renal impairment over the course of the six-year follow-up. Eight patients died of renal failure over the course of the follow-up period.
Both authors concluded that renal failure is the main cause of death in BBS patients.3,13 Abdominal ultrasound examination done for our patient did not reveal any renal structural abnormality, but he had laboratory signs of renal functional impairment which clearly warranted further care and follow-up.
Retinal dystrophy is one of the main features of BBS.1–3 It is occasionally seen in children in the first decade of life, and electroretinogram (ERG) examination can be used to detect it in suspected children with no apparent signs of retinal dystrophy. Signs of retinal dystrophy are seen in all adult patients with BBS.1,3,7 Some of the commonest fundus features seen in such patients include a macular scar with or without epiretinal membrane, optic disc atrophy (“waxy” disc pallor), perivascular retinal pigmentary changes in “bonny spicule” pattern or pigment clumping and generalized retinal arterial narrowing.1–3,7 A part from the retinal dystrophy, refractive errors particularly myopia, divergent strabismus and color vision impairment are other ocular features of the syndrome.1–3,7 Our patient had all the mentioned ocular features of BBS except strabismus.
Patients with BBS usually have fully formed bony extra-fingers (polydactyly) situated on the lateral border of the hand or foot. It is also one of the major features of BBS, and 70% of the patients can have one or more extra-fingers at birth.1–3 Our patient had extra-fingers in all of his upper and lower extremities. Hypogonadism, characterized by mal-descended testes, small penis often buried in adipose tissues, is a universal finding in males and is a major feature of BBS.2 Irregular menstrual cycles may be seen in females.3,9,10
Mental retardation is one of the main features of the syndrome.3,9,10 But it is a more disputed feature of BBS as signs of mental retardation are not easily seen. Developmental delay, late in reaching milestones, more specifically delay in walking and speech development and learning difficulties are the other main features manifested in up to half of BBS patients.3,11
Clinical features of BBS are divided into primary and secondary features based on the frequency of their appearance in patients.1–3 The six primary features which were found to appear more frequently are retinal dystrophy, central obesity, polydactyly, mental retardation/learning difficulties, hypogonadism in males and renal dysfunction.1–3 There is also a long list of secondary features which include Oral/dental abnormalities such as hypodontia, microdontia, Neurologic abnormalities such as epilepsy, ataxia, speech abnormalities, Olfactory dysfunctions such as anosmia, hyposmia, Cardiovascular and thoraco-abdominal abnormalities such as congenital heart diseases, Hirschsprung disease, inflammatory bowel disease, Celiac disease, Endocrine and metabolic abnormalities such as type-II diabetes, hypothyroidism, polycystic ovary syndrome.1–3,14 To diagnose BBS we need to have at least four of the primary features or three primary features plus two of the secondary features.1–3,15 Our patient had five primary features which include retinal dystrophy, obesity, polydactyly, renal functional impairment and learning difficulties and additional secondary features such as myopia and color vision impairment which were more than enough to fulfill the diagnostic criteria.
After diagnosis, BBS patients need multidisciplinary medical care involving ophthalmologists, nephrologists, and endocrinologists, and base-line investigations which need to be done include blood sugar level, renal function tests, abdominal ultrasound, Intravenous pyelogram (IVP), urine analysis, refraction and treatment should be started accordingly. The condition should also be explained to parents or patients, and they should also be followed up regularly with frequency based on the severity of their problem.
Conclusion
The Bardet–Biedl Syndrome is an autosomal recessive disorder, and it is well known to cause severe visual disability due to retinal dystrophy as well as mortality mainly due to renal failure. Diagnosis of BBS at an early age may help with appropriate medical care to delay the occurrence of some life-threatening complications such as renal failure. This can help to improve patients’ quality of life and increase their lifespan. Ophthalmologists should be aware of this syndrome, its main ocular and systemic signs as well as the potential systemic complications so that they will be able to diagnose patients with BBS at an earlier age and give appropriate medical care in a multidisciplinary approach involving nephrologists.
Ethical Consideration
Ethical clearance was obtained from the Ethics Committee (Institutional Review Board) of University of Gondar to publish the case report and written consent was obtained from the patient to use his clinical data and pictures for the report. The patient understands that name and initials will not be published and due efforts will be made to conceal identity on the pictures.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Forsyth RL, Gunay-Aygun M. Bardet-Biedl syndrome overview. US National Library of Medicine; July 14, 2003 [updated July 23, 2020].
2. Green JS, Parfrey PS, Harnett JD, Farid NR. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989;321(15):1002–1009. doi:10.1056/NEJM198910123211503
3. Beales PL, Elcioglu N. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437–446.
4. Bardet G. Sur un syndrome d’obesitecongenitale avec polydactilie etretinitepigmentaire (contribution a l’etude des formescliniques de l’obesitehypophysaire) [On congental obesity syndrome with polydactyly and pigmentary retinitis (contributions to the study of clinical forms of pituitary obesity)]. Amedee Le Grand. 1920;470:407.
5. Biedl A. Ein Geschwister paarmit adiposo-genitaler Dystrophie [A pair of siblings with adipos-genital dystrophy]. Dtsch Med Woschenschr. 1922;48:1630.
6. Laurence JZ, Moon RC. Four cases of retinitis pigmentosa occurring in the same family, and accompanied by general imperfections of development. Ophthalmol Rev. 1866;2:3241.
7. Iannaccone A, De Propis G, Roncati S, Rispoli E, Del Porto G, Pannarale MR. The ocular phenotype of the Bardet-Biedl syndrome comparison to non-syndromic retinitis pigmentosa. Ophthalmic genetics. 1997;18(1):13-26.
8. NHLBI Obesity Education Initiative Expert Panel on the Identification, Evaluation, and Treatment of Obesity in Adults. Executive summary of the clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults. Arch Intern Med. 1998;158(17):1855–1867. doi:10.1001/archinte.158.17.1855
9. Ansley SJ, Badano JL, Blacque OE. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425(6958):628–633. doi:10.1038/nature02030
10. Blacque OE, Reardon MJ, Li C. Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev. 2004;18(13):1630–1642. doi:10.1101/gad.1194004
11. Katsanis N, Lupski JR, Beales PL. Exploring the molecular basis of Bardet-Biedl syndrome D. Hum Mol Genet. 2001;10(20):2293–2299. doi:10.1093/hmg/10.20.2293
12. Kwitek-Black AE, Carmi R, Geoffrey M. Linkage of Bardet-Biedl syndrome to chromosome 16q and evidence for non-allelic genetic heterogeneity. Nat Genet. 1993;5:392. doi:10.1038/ng1293-392
13. O’Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J. The importance of renal impairment in the natural history of Bardet-Biedl syndrome. Am J Kidney Dis. 1996;27:776–783. doi:10.1016/S0272-6386(96)90513-2
14. Marchese E, Ruoppolo M, Perna A, Capasso G, Zacchia M. Exploring key challenges of understanding the pathogenesis of kidney disease in Bardet–Biedl syndrome. Kidney Int Rep. 2020;5(9):1403–1415. doi:10.1016/j.ekir.2020.06.017
15. Kousi M, Söylemez O, Ozanturk A, et al. Evidence for secondary-variant genetic burden and non-random distribution across biological modules in a recessive ciliopathy. Nat Genet. 2020;52(11):1145–1150. doi:10.1038/s41588-020-0707-1
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.