Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Bacterial–viral load and the immune response in stable and exacerbated COPD: significance and therapeutic prospects
Authors D'Anna S, Balbi B, Cappello F ![]() , Carone M, Di Stefano A
, Carone M, Di Stefano A
Received 30 July 2015
Accepted for publication 13 October 2015
Published 1 March 2016 Volume 2016:11(1) Pages 445—453
DOI https://doi.org/10.2147/COPD.S93398
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Silvestro Ennio D’Anna,1 Bruno Balbi,2 Francesco Cappello,3,4 Mauro Carone,2 Antonino Di Stefano2
1Department of Rehabilitation, Cardiorespiratory Unit, Fondazione Istituto G. Giglio di Cefalù, 2Pneumology Unit and Laboratory of Cytoimmunopathology of Heart and Lung, Fondazione Salvatore Maugeri, IRCCS, Veruno (NO) and Cassano delle Murge (BA), 3Human Anatomy Section, Department of Experimental Biomedicine and Clinical Neuroscience, University of Palermo, Palermo, Italy; 4Euro-Mediterranean Institute of Science and Technology, Palermo, Italy
Abstract: Chronic obstructive pulmonary disease (COPD) is characterized by persistent airflow limitation and an abnormal inflammatory response of the lung. Bacteria and viruses are a major cause of COPD exacerbations and may contribute to COPD progression by perpetuating the inflammatory response in the airways. Bacterial variety diminishes with increasing COPD severity. Respiratory viruses can colonize the lower respiratory tract in stable COPD, altering the respiratory microbiome and facilitating secondary bacterial infections. In this review, we present the most updated information about the role of bacteria and viruses in stable and exacerbated COPD. In our opinion, to optimize therapeutic strategies, the dynamic events involving bacterial–viral infections and related immune response in COPD phenotypes need to be better clarified. Our paper would address these points that we consider of great importance for the clinical management of COPD.
Keywords: COPD phenotype, biomarkers, exacerbations, severity of COPD, microbiome
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by a persistent airflow limitation usually associated with an increased inflammatory response.1 Bronchial biopsies of stable mild COPD patients show increased levels of inflammatory cells compared with healthy nonsmokers; among these cells, CD8+ T-lymphocytes, neutrophils, and macrophages are prevalent.1,2 In severe COPD, while there is a decrease in the total number of inflammatory cells, a prevalence of inflammatory cells with phagocytic and proteolytic activity, such as neutrophils and macrophages, is reported.2 Acute exacerbations of COPD can cause a worsening of lung function that lasts many weeks, accelerating the rate of lung function decline.1 Viral and bacterial infections of the respiratory system are a major cause of COPD exacerbations. Microbiological examination of sputum from COPD patients hospitalized during an exacerbation shows that infections are mostly associated with bacterial pathogens of the sputum, followed by combined bacteria and viruses or sputum viruses.3 Another study,4 followed COPD patients for 1 year, observed a high rate of exacerbations in one subgroup – 37% of these were caused by bacteria, 10% by viruses, 12% by bacteria and viruses, while 14% were of undefined origin.4 Many investigators have observed a change of the inflammatory response in COPD patients during acute exacerbations, showing a general increase in the inflammatory state.2 These exacerbations are associated with an increase in neutrophils, T-lymphocytes, and, in some cases, eosinophils.4–6 Viruses and bacteria may cause acute exacerbations of COPD, or alternatively, they may amplify chronic inflammation in stable COPD.7
The aforementioned data, however, come from separate experimental sets using different methodological approaches, rendering difficult a direct comparison between bacterial–viral loads in the lungs of COPD patients and the related immune host responses. To date, few data are available on the dynamic mechanisms of bacteria and viruses colonizing the lungs, or the precise relationship between these events and disease progression during stable and exacerbated COPD. This review focuses on the present knowledge regarding the microbiome load (either bacteria or viruses) in stable and exacerbated COPD. We will not deal in depth here with the immune response related to bacterial–viral infections, since other reviews have already been published on this topic and also because there are few in vivo data available. We will focus instead on the dynamic events involved in COPD progression and touch on future prospects for therapeutic strategies.
Role of bacteria in COPD
Stable COPD patients
Pathogenic bacteria have been identified using culture techniques in 25%–50% of patients with stable COPD.8 A limitation of culture techniques, however, is the possibility of contamination from the upper airways. Moreover, more than 70% of bacterial species cannot be cultured by current techniques9,10 and many of the remaining species are very difficult to culture.11 The development of nonculture-based techniques, such as quantitative polymerase chain reaction (qPCR), has improved the capacity to detect bacteria, and has shown that the lungs are not sterile12,13 and that the mix of bacterial species composing the lung microbiome contributes to the disease state in chronic respiratory diseases.14
Lower airways in COPD patients are colonized by Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis and, in patients with more severe disease, by Pseudomonas aeruginosa (Figure 1).15–17 Some studies in stable-state COPD have observed a direct correlation between airway inflammation and the bacterial load, for example, before the advent of nonculture-based techniques, Hill et al17 performed sputum cultures in 160 stable COPD patients, 55 with normal blood α1-antitrypsin levels, 62 with severe α1-antitrypsin deficiency, and 43 with idiopathic bronchiectasis, and observed that the bacterial load is correlated to airway inflammation in patients with stable chronic bronchitis.17 Garcha et al18 studied sputum samples from 134 patients by qPCR and observed that in stable COPD, a higher bacterial load was correlated to more severe bronchial obstruction, higher dosage of inhaled corticosteroids, and a higher C-reactive protein (CRP) level. More recently, Barker et al19 studied sputum samples from 120 patients with stable COPD and paired sputum in 55 subjects with stable and exacerbated disease. Using qPCR, they observed that the majority of patients in a stable state (76%) had pathogenic bacteria in their airways. The detection of bacteria by means of nonculture-based techniques such as qPCR was also associated with increased sputum interleukin (IL)-1β, IL-10, and tumor necrosis factor α, and decreased Chemokine (C-C Motif) Ligand 13 (CCL13). In a stable state, the strongest relationship between bacterial load, inflammation, and symptoms was observed with H. influenzae, whether in codetection with other bacteria or as sole pathogen. Singh et al20 analyzed the sputum of 99 stable COPD patients by qPCR and observed a direct correlation between the load of H. influenzae, S. pneumoniae, and M. catarrhalis and airway inflammation and plasmatic fibrinogen. Erb-Downward et al21 analyzed bronchoalveolar lavage (BAL) fluid of three never smokers, seven healthy smokers, four COPD patients, and eight samples obtained from six patients undergoing lung transplantation for COPD. A significant bacterial load was found in all subjects without significant differences between groups. In some smokers with normal lung function, the authors found a lower diversity of lung microbiota and speculated that this relative reduction in the diversity could be persistent and could be either an “effect” of the lung inflammation or, in part, a “cause” of disease onset and progression.21
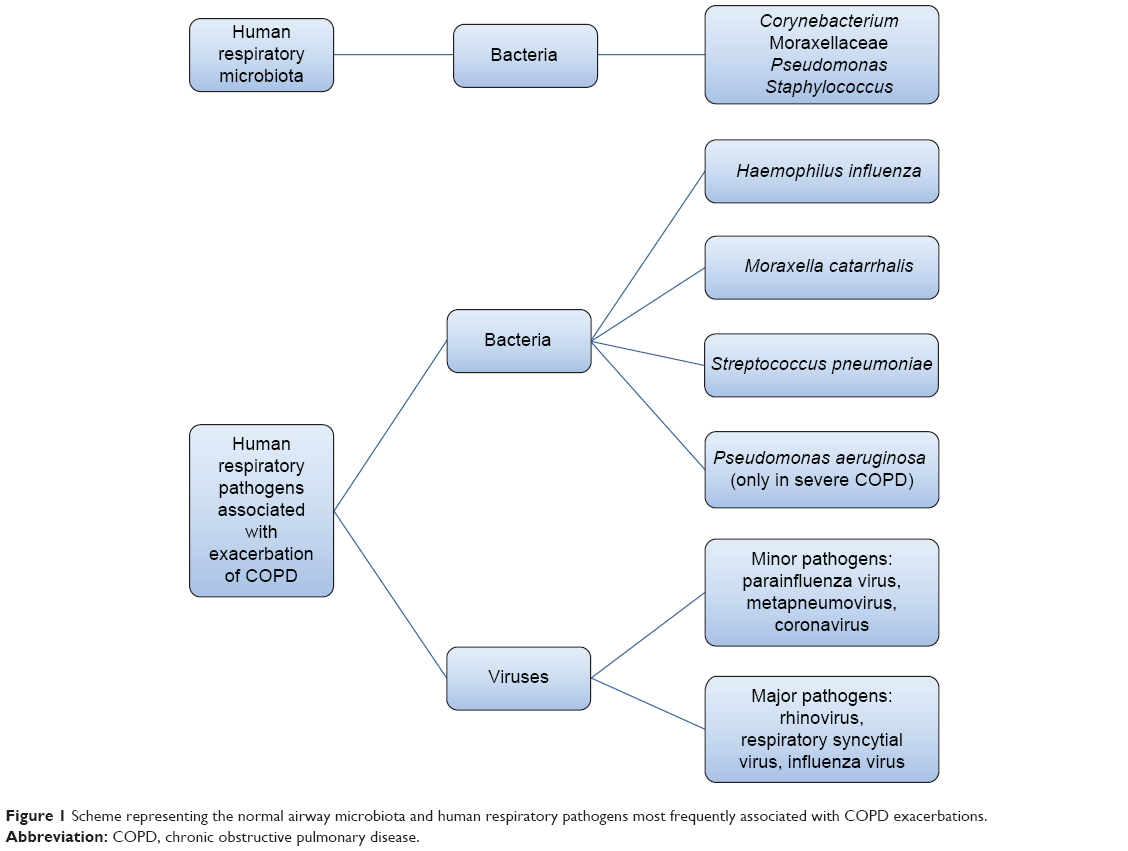 | Figure 1 Scheme representing the normal airway microbiota and human respiratory pathogens most frequently associated with COPD exacerbations. |
The molecular aspects of these dynamic modifications in the bacterial load and bacterial types in relation to the patient’s immune response need to be studied. It has been hypothesized that the appearance of new strains of foreign microbes initiates a cycle of infection, inflammatory response, and dysfunctional remodeling driving the progression of COPD.17,22
Sze et al23 analyzed samples from surgical lung resections and lung transplant donors of smokers (eight samples), nonsmokers (eight samples), COPD patients (eight samples), and cystic fibrosis patients (eight samples). They showed differences in the bacterial community of the COPD lung tissue compared with other groups. Actinobacteria were more abundant in the smoker group, while Lactobacillus were consistently present in the COPD Global Initiative for Chronic Obstructive Lung Disease (GOLD) 4 group. In the cystic fibrosis and COPD groups, the Burkholderia genus represented more than 5% of the bacterial community. The increased presence of Lactobacillus in the lung could be related to an inflammatory state associated to the formation of tertiary lymphoid follicles developing near the small airways.23 Lactobacillus could either be the target of the inflammation or, alternatively, it could act as an immune modulator and aid the inflammatory response. In their study, the authors showed that in the very severe COPD group, there was a shift in the relative abundance of a few bacterial populations without any one becoming dominant.
Relative abundance and an increasing “diversity” of the bacterial population in COPD was observed by Pragman et al24 who studied BAL samples from 22 patients with moderate-to-severe COPD and ten controls, and Wu et al25 who analyzed sputum of ten COPD and control subjects. Garcia-Nuñez et al26 studied with nonculture-based techniques the bronchial microbiome in the sputum of 17 stable COPD patients and observed that, in patients with more severe disease, the Proteobacteria phylum was overrepresented, together with a diminution of bacteria belonging to the Firmicutes phylum. The authors speculated that changes in the lung microbiome in more severe COPD patients could be due either to alterations of the airways typical of very severe diseased patients or to the repeated use of antibiotics. The alteration in microbiome composition observed may induce further lung inflammation contributing to the worsening of the disease.26 More recently, Sze et al27 examining 40 lung samples from five COPD (GOLD stage 4) subjects have confirmed the reduction in microbial diversity with a relative increase of Proteobacteria and Actinobacteria and a reduction of Firmicutes and Bacteroidetes phyla. The authors, moreover, reported a significant association between the alterations of the microbiome, the extent of emphysema, remodeling of bronchi and alveoli, and their infiltration by CD4+ T-cells. Sze et al hypothesize that the increased abundance of Proteobacteria and Actinobacteria in COPD (GOLD stage 4) airways could stimulate a more intense lung inflammation.27
These findings are in contrast to other studies.24,25 The discrepancy could be due to the small number of patients analyzed in these studies and differences in study design (Table 1). Furthermore, the study by Erb-Downward et al21 was based on samples obtained from BAL and bronchial brushes while in the study by Sze et al,23,27 the samples were obtained from lung tissue, rendering the findings less directly comparable. A limitation of the study by Sze et al23 is the absence of a moderate and severe COPD group (GOLD stage 2 and 3) that leaves open the question whether the emergence of Lactobacillus and Burkholderia in the lungs is gradually progressive or a sudden occurrence. Pragman et al24 studied BAL samples while Garcia-Nuñez et al26 analyzed sputum samples. Other facts influencing the findings are the heterogeneity of the COPD population: in some patients, there is a predominance of emphysema, while in others, chronic bronchitis is prevalent; moreover, the different drugs used to treat the disease in COPD patients could have altered the lung microbiome: some patients were treated with antibiotics and others with steroids or β2 agonists. A well-defined washout period from antibiotic and corticosteroid use is not reported in these studies. Some studies considered COPD patients in a stable state after 4 weeks had lapsed from the last exacerbation.19–30 One study indicated the antibiotics used during the exacerbation phase of the patients but no data are available about the dose of corticosteroid during that phase.30 One study reported the antibiotic dosage used.26 Since many of these studies use a quantitative approach quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) for microbiome evaluation, it is mandatory to define precisely the pharmacologic treatments and the washout duration for antibiotics and corticosteroids. We suggest that a 3-month washout period for antibiotic treatment, and at least 1-month washout for oral or inhaled corticosteroid use should be considered when evaluating COPD patients in stable conditions. Even with the limitations described and the need for studies with larger sample size and a well-defined pharmacological treatment regimen, it appears that the airways of COPD patients have a different microbiome and a larger number of bacteria compared with normal subjects.
 | Table 1 Breakdown of different bacterial microbiota studies in COPD |
There is rather concordance on the fact that with the progression of the disease, there is a reduction in the microbial diversity with an increase of some phyla (Proteobacteria and Actinobacteria) and a reduction of others that are normally present in COPD.
Exacerbated COPD patients
The role of bacterial pathogens during COPD exacerbations has recently been further investigated. Exacerbations of COPD are associated with changes in respiratory microbiota and airway inflammation.
In 2002, Sethi et al22 followed 81 COPD patients for 56 months, studying their sputum with culture-based techniques during stable disease and exacerbations, and observed that the isolation of new strains of pathogens was associated with an increased risk of exacerbations.22
The development of nonculture-based techniques for bacterial isolation has shed more light on the role of bacteria during an exacerbation process. Huang et al28 studied eight patients admitted to an intensive care unit for COPD exacerbation. Samples were obtained from endotracheal aspirates. Using nonculture-based techniques (qPCR), the authors observed a much greater diversity of bacteria than hitherto realized with culture techniques. Multiple oropharyngeal- and gut-associated bacterial species were isolated, suggesting a potential role of these strains in COPD exacerbations. Huang et al, hypothesized the presence of two bacterial microbiota related to exacerbated COPD, one characterized by a prevalence of Proteobacteria, the other by a prevalence of Firmicutes, which might be associated with more diversity.28 The patients in the group with prevalence of Firmicutes were those with a shorter intubation period before performing the endotracheal aspirate. Moreover, a decreased bacterial diversity was correlated with the length of intubation.28 It can be speculated that antibiotic treatment could have reduced the bacterial diversity. The limits of this study were the absence of control samples from non-intubated COPD and non-COPD patients and the lack of a longitudinal dimension. Bacteria in the oral cavity and gastrointestinal tract seed the airways in vulnerable patient populations.24 Millares et al29 analyzed paired sputum specimens from patients with COPD at baseline and during exacerbations, and found that exacerbations were associated with a selective increase in the relative abundance of bacteria typically associated with exacerbations (eg, Haemophilus, Pseudomonas, and Moraxella) despite inconsistent detection in culture. Huang et al30 observed the temporal changes in the airway microbiome before, at onset, and after an acute exacerbation. They studied bacterial infections in COPD sputum samples from 12 subjects enrolled in a longitudinal study: the microbiota members that resulted increased during exacerbations, were mainly of the Proteobacteria phylum. Treatment with antibiotics alone decreased the Proteobacteria, whereas treatment with corticosteroids alone enriched the Proteobacteria and other phyla. Neither of these two studies29,30 reported a decrease in bacterial community diversity, as would be expected after an acute infection or in more severe disease.
For a better understanding of the role of the microbiome in exacerbations of COPD, there is a need for longitudinal studies in exacerbated conditions after a sufficiently extended stabilization period, including appropriate washout from pharmacological treatments, as well as in nontreated controls. The parallel analysis of the immunological host response associated with these different clinical states in COPD patients also needs to be longitudinally conducted.
In the studies reported here, there is no grading of the severity of exacerbations. Indeed, some exacerbated COPD patients need only a change in medication at home, while others need to be admitted to the intensive care unit. Studies are required, in our opinion, to address this issue, that is, to classify the exacerbation process related to its severity.31 Also severity of bronchial obstruction during stable conditions could be considered in the grading scheme of exacerbations. In addition, more studies are needed to characterize the microbiome during COPD exacerbations. A list of the studies already carried out is reported in Table 1.
Role of viruses in COPD
Stable COPD patients
The use of PCR-based approaches to amplify virus-specific nucleic acid sequences has documented the invasion of respiratory viruses into the peripheral airways of COPD patients both during acute exacerbations and in stable conditions.32–36 Some authors suggest that, in stable COPD, virus colonization could play a role in maintaining the elevated inflammatory background.
Utokaparch et al35 with qPCR in frozen lung tissue from 20 GOLD stage 1 COPD patients in stable state and 20 healthy smoker controls, detected viral nucleic acid in 18/40 patients. The two most common viruses detected were influenza virus A and coronavirus 229E, each isolated in 10/40 samples.35 Influenza virus was more prevalent in COPD patients than in control subjects (40% vs 10%). COPD patients had a greater total viral load and a greater number of viruses per patient compared with healthy controls.37 Mallia et al37 also showed a significant inverse relationship between the forced expiratory volume in 1 second/forced vital capacity ratio and the number of macrophages and neutrophils in the small airways, and a direct relationship between inflammatory cells and total viral load. The authors postulated that viruses might act as a pathogen reservoir in COPD. COPD patients are considered more susceptible to virus infection and persistence of infection.
Again, the relationship between the degree of inflammation and viral load needs to be studied in patients under well standardized therapeutic conditions. Corticosteroid use could have favored the persistence of viruses in the airways, as observed by Utokaparch et al35 and Mallia et al.37 Yet only in the study by Mallia et al37 is antibiotics courses and exacerbations in the last year before the enrolment of patients documented. Specific information on antibiotics or corticosteroid use is infrequently available in other studies.35–37
Exacerbated COPD patients
Respiratory viral infections are traditionally those implicated in COPD exacerbations. Rhinovirus, coronavirus, influenza, respiratory syncytial viruses, parainfluenza, adenovirus, and metapneumovirus are the viruses that most often cause common cold in healthy individuals, and they play a role in acute exacerbations of COPD.35–38 These exacerbations can be more or less severe with more or less prolonged recovery times.
Numerous data from 1980 onward show an acute decline in forced expiratory volume in 1 second in COPD patients for up to 90 days after an acute infection of influenza virus.39
Recently, it was demonstrated that infection by respiratory viruses could influence the bacterial microbiome in patients with COPD, and vice versa. Molyneaux et al36 infected 14 COPD subjects and 17 healthy controls having normal lung function with rhinovirus and observed only in the sputum of COPD patients a rise in the bacterial load and a significant prevalence of H. influenzae with respect to the existing microbiota. The authors suggested that rhinovirus infection in COPD alters the respiratory microbiome and may induce secondary bacterial infections. There is a parallel increase in the inflammatory response after concomitant bacterial and viral infections in COPD. Wilkinson et al40 observed that COPD exacerbations due to a rhinovirus and H. influenzae coinfection are associated with an increased level of serum IL-6 compared with exacerbations where only one pathogen was isolated. Similar findings have been reported by Mallia et al:37 the authors observed that 60% of COPD patients experienced a secondary bacterial infection after being infected with rhinovirus.
These data have been confirmed by histologic analyses of infected lung samples from patients who died from 1918 type A and 2009 H1N1 influenza.41–43 In these patients, the predominant cause of death was a bacterial superinfection resulting in secondary bacterial pneumonia. In mice, the infection of influenza A virus increases the susceptibility to bacterial respiratory infections such as S. pneumoniae, Staphylococcus aureus, and H. influenzae.44,45 Influenza virus may cause this susceptibility to bacterial superinfection through various mechanisms. The damage of the epithelial cells and alterations of the epithelium by viral infection can facilitate the bacterial colonization of the airways.46 Another mode by which influenza virus can favor bacterial infection is by altering the function of the immune system, resulting in a failure of the control of bacterial replication (Figure 2).
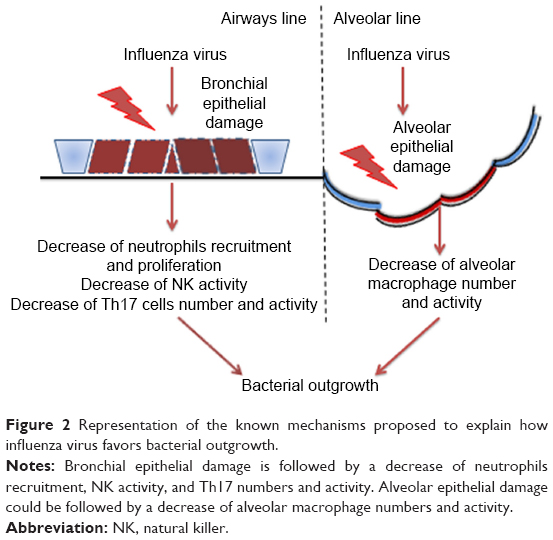 | Figure 2 Representation of the known mechanisms proposed to explain how influenza virus favors bacterial outgrowth. |
Influenza A virus causes apoptosis and alters the function of alveolar macrophages,47,48 suppressing their activation in response to secondary bacterial challenge and phagocytic capacity by increasing the expression of CD 200 receptor ligation antigen49 and reducing the expression in the alveolar macrophages of the scavenger macrophage receptor with collagenous structure (MARCO).48 The macrophages’ reduced production of cytokines and chemokines alters the response of other cells to pathogens, reducing for example the recruitment and activation of neutrophils. Neutrophil function is also altered by influenza A virus either through direct infection of the cells50 or through the induction of type I interferon secretion that leads to the inhibition of neutrophil infiltration and increased apoptosis.51 Natural killer cells are also affected by influenza virus: in an infected lung, they show a reduction of cytotoxic activity and the production of chemokines and cytokines52 such as the antibacterial effector cytokine tumor necrosis factor.53 There is evidence that the induction of a highly polarized type 1 T helper (Th1) immune response by respiratory viruses leads to a reduction of MARCO expression and impairs macrophages’ ability to ingest and kill bacteria.48 The fine balance of IL-10 production and Th17 response during a viral infection could explain the excessive susceptibility to the bacterial superinfection observed after influenza virus infection.54,55 van der Sluijs et al56 observed that blocking IL-10 production enhanced the clearance of a secondary infection of S. pneumoniae. Another effect due to increased type I interferon production following influenza virus infection is the suppression of the Th17 response during a secondary bacterial infection in mice.57
These data suggest that Th17 cells are important in organizing the immune response to bacteria such as S. pneumoniae and S. aureus. Viral inhibition of this pathway may form the basis of the increased secondary bacterial infections and the worst outcome in COPD patients.
Interestingly, Perera et al58 observed that higher serum CRP levels in COPD patients 14 days after the first exacerbation episode were correlated with a shorter time to the next exacerbation. Moreover, frequent exacerbators had higher serum levels of IL-6 and CRP in comparison to infrequent exacerbators. The authors suggested that these higher inflammatory indices in frequent exacerbators could be related to the faster forced expiratory volume in 1 second decline and increased mortality seen in this subgroup of patients.58 Nonrespiratory viruses such as human immunodeficiency virus can change the lung microbiome facilitating colonization by pathogenic gastrointestinal bacteria in infected patients.59,60
There are relatively few data available concerning viral infections in COPD patients caused by influenza virus A, coronavirus, Respiratory syncytial virus, and rhinovirus.35–37,39 Among nonrespiratory viruses involved in COPD exacerbations and lung microbiome alterations, data are limited only to human immunodeficiency virus infections.59,60 Moreover, these studies included only patients with mild COPD.35–37,39 As a consequence, further studies investigating the host immune response in relation to different stages and phenotypes of COPD patients are needed.
Specific data between viral and bacterial infections in stable and exacerbated COPD, in relation to the host immune response, need to be produced to gain a clearer picture on its interrelationship and related immune processes in COPD.
Conclusion
COPD is an illness characterized by an abnormal inflammatory response of the lung. The inflammatory cell infiltration in the airways of these patients varies according to the stage of the disease. Recent findings show that the immune response is impaired in the COPD lung.61
The microbiome in the airways of COPD patients is altered with increasing disease severity, and during the exacerbation process. Alterations of the normal bacterial populations in COPD patients with a reduction of bacterial variety are associated with an increased risk of exacerbation and severity, and bacteria and viruses play a pivotal causal role in COPD exacerbations and disease progression. Stable COPD patients can harbor respiratory viruses in their airways. Bacteria and viruses in the lungs could play a role in maintaining the inflammatory state in COPD patients during the stable phase. There is evidence that the bacterial and viral loads in the airways of stable COPD are directly correlated to the airways inflammation. Some studies evidence that some bacterial phyla such as Proteobacteria are more capable of stimulating inflammation in the airways of COPD patients.
To date, it is still not clear how some bacteria can influence the inflammatory response in the lungs and how lung virus infection/colonization can alter the local microbiome. In other words, the dynamic process involving bacterial and viral challenges that is able to change the clinical status of patients in relation to the host innate and adaptive immune response needs to be better explored.
The genetic changes probably present in COPD at different stages (severe vs mild) or having different phenotypes (frequent exacerbators vs nonfrequent) were outside the scope of the present review. Well-planned studies applying appropriate washout periods from therapeutic treatments of COPD patients in different clinical conditions are also needed for a correct quantitation of the microbiome in these different clinical states. Understanding the dynamics of the lung microbiome in different clinical conditions and the interactions with the related endogenous host immune response will improve our knowledge of the pathologic and molecular mechanisms underlying COPD, as a consequence enabling the development of new therapeutic strategies to limit the progression of the disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Global Initiative for Chronic Obstructive Lung Disease (GOLD): global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease. NHLBI/WHO workshop report. NIH Publication No 2701A. Last update 2015. Available from: http://www.goldcopd.com | ||
Di Stefano A, Caramori G, Ricciardolo FLM, Capelli A, Adcock IM, Donner CF. Cellular and molecular mechanisms in chronic obstructive pulmonary disease: an overview. Clin Exp Allergy. 2004;34:1156–1167. | ||
Papi A, Bellettato CM, Braccioni F, et al. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med. 2006;173:1114–1121. | ||
Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184:662–671. | ||
Bhowmik A, Seemugal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 2000;55:114–120. | ||
Saetta M, Di Stefano A, Maestrelli P, et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med. 1994;150:1646–1652. | ||
Brusselle G, Joos G, Bracke G. New insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378:1015–1026. | ||
Sethi S, Murphy T. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359:2355–2365. | ||
Suau A, Bonnet R, Sutren M, et al. Direct analysis of genes encoding 16s rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–4807. | ||
Hayashi H, Sakamoto M, Benno Y. Phylogenetic analysis of the human gut microbiota using 16s rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol Immunol. 2002;46:535–548. | ||
Sibley CD, Grinwis ME, Field TR, et al. Culture enriched molecular profiling of the cystic fibrosis airway microbiome. PLoS One. 2011;6:e22702. | ||
Charlson ES, Bittinger K, Haas AR, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. | ||
Cabrera-Rubio R, Garcia-Nunez M, Seto L, et al. Microbiome diversity in the bronchial tracts of patients with chronic obstructive pulmonary disease. J Clin Microbiol. 2012;50:3562–3568. | ||
Rosell A, Monsò E, Soler N, et al. Microbiologic determinants of exacerbations in chronic obstructive pulmonary disease. Arch Intern Med. 2005;165:891–897. | ||
Patel IS, Seemungal TA, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. Relationship between bacterial colonization and the frequency, character and severity of COPD exacerbations. Thorax. 2002;57:759–764. | ||
Miravitlles M, Espinosa C, Fernandez-Laso E, Martos JE, Maldonado JA, Gallego M. Relationship between bacterial flora in sputum and functional impairment in patients with acute exacerbations of COPD. Chest. 1999;116:40–46. | ||
Hill AT, Campbell EJ, Hill SL, Bayley DL, Stockley RA. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. J Clin Virol. 2000;109:188–195. | ||
Garcha DS, Thurston SJ, Patel ARC, et al. Changes in prevalence and load of airway bacteria using quantitative PCR in stable and exacerbated COPD. Thorax. 2012;67:1075–1080. | ||
Barker BL, Haldar K, Patel H, et al. Association between pathogens detected using quantitative polymerase chain reaction with airway inflammation in COPD at stable state and exacerbations. Chest. 2015;147:46–55. | ||
Singh R, Mackay AJ, Patel A, et al. Inflammatory thresholds and the species-specific effects of colonising bacteria in stable chronic obstructive pulmonary disease. Respir Res. 2014;15:114. | ||
Erb-Downward JR, Thompson DL, Han MK, et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One. 2011;6:e16384. | ||
Sethi S, Evans N, Grant BJ, Murphy TF. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347:465–471. | ||
Sze MA, Dimitriu PA, Hayashi S, et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:1073–1080. | ||
Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One. 2012;7:e47305. | ||
Wu D, Hou C, Li Y, et al. Analysis of the bacterial community in chronic obstructive pulmonary disease sputum samples by denaturing gradient gel electrophoresis and real-time PCR. BMC Pulm Med. 2014;14:179. | ||
Garcia-Nuñez M, Millares L, Pomares X, et al. Severity-related changes of bronchial microbiome in chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:4217–4223. | ||
Sze MA, Dimitriu PA, Suzuki M, et al. The host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192:438–445. | ||
Huang YJ, Kim E, Cox MJ, et al. A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS. 2010;14:9–59. | ||
Millares L, Ferrari R, Gallego M, et al. Bronchial microbiome of severe COPD patients colonised by Pseudomonas aeruginosa. Eur J Clin Microbiol Infect Dis. 2014;33:1101–1111. | ||
Huang YJ, Sethi S, Murphy T, Nariya S, Boushey HA, Lynch SV. Airway microbiome dynamics in exacerbations of chronic obstructive pulmonary disease. J Clin Microbiol. 2014;52:2813–2823. | ||
Mackay AJ, Donaldson GC, Patel AR, Singh R, Kowlessar B, Wedzicha JA. Detection and severity grading of COPD exacerbations using the exacerbations of chronic pulmonary disease tool (EXACT). Eur Respir J. 2014;43(3):735–744. | ||
Tan WC, Xiang X, Qiu D, Ng PT, Lam SF, Hegele RG. Epidemiology of respiratory viruses in patients hospitalized with near-fatal asthma, acute exacerbations of asthma, or chronic obstructive pulmonary disease. Am J Med. 2003;115:272–277. | ||
Wedzicha JA. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thor Soc. 2004;1:115–120. | ||
Singh M, Lee SH, Porter P, et al. Human rhinovirus 2A induces TH1 and TH2 immunity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2012;125:1369–1378. | ||
Utokaparch S, Sze MA, Gosselink JV, et al. Respiratory viral detection and small airway inflammation in lung tissue of patients with stable, mild COPD. COPD. 2012;2:197–203. | ||
Molyneaux P, Mallia P, Cox MJ, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188:1224–1231. | ||
Mallia P, Footitt J, Sotero R, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186:1117–1124. | ||
Greenberg SB, Allen M, Wilson J, Atmar RL. Respiratory viral infections in adults with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162:167–173. | ||
Smith CB, Kanner RE, Golden CA, Klauber MR, Renzetti AD. Effect of viral infections on pulmonary function in patients with chronic obstructive pulmonary diseases. J Infect Dis. 1980;141:271–280. | ||
Wilkinson TM, Hurst JR, Perera WR, Wilks M, Donaldson GC, Wedzicha JA. Effect of interactions between lower airway bacterial and rhinoviral infection in exacerbations of COPD. Chest. 2006;129:317–324. | ||
Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. | ||
Bautista E, Chotpitayasunondh T, Gao Z, et al. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Engl J Med. 2010;362:1708–1719. | ||
Centers for Disease Control and Prevention (CDC). Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A – United States, May–August 2009. MMWR Morb Mortal Wkly Rep. 2009;58:1071–1074. | ||
Ballinger MN, Standiford TJ. Postinfluenza bacterial pneumonia: host defenses gone away. J Interferon Cytokine Res. 2010;30:643–652. | ||
Jamieson AM, Yu S, Annicelli CH, Medzhitov R. Influenza virus-induced glucocorticoids compromise innate host defense against a secondary bacterial infection. Cell Host Microbe. 2010;7:103–114. | ||
Plotkowski MC, Puchelle E, Beck G, Jacquot J, Hannoun C. Adherence of type I Streptococcus pneumoniae to tracheal epithelium of mice infected with influenza A/PR8 virus. Am Rev Respir Dis. 1986;134:1040–1044. | ||
Didierlaurent A, Goulding J, Patel S, et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205:323–329. | ||
Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nature Med. 2008;14:558–564. | ||
Goulding J, Godlee A, Vekaria S, et al. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. J Infect Dis. 2011;204:1086–1094. | ||
Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, García-Sastre A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A. 2010;107:11531–11536. | ||
Shahangian A, Chow EK, Tian X, et al. Type I IFNs mediate development of post influenza bacterial pneumonia in mice. J Clin Invest. 2009; 119:1910–1920. | ||
Guo H, Kumar P, Moran TM, Garcia-Sastre A, Zhou Y, Malarkannan S. The functional impairment of natural killer cells during influenza virus infection. Immunol Cell Biol. 2009;87:579–589. | ||
Small CL, Shaler CR, McCormick S, et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J Immunol. 2010;184:2048–2056. | ||
Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. 2009;15:277–284. | ||
McKinstry KK, Strutt TM, Buck A, et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J Immunol. 2009;182:7353–7363. | ||
van der Sluijs KF, van Elden LJ, Nijhuis M, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. 2004;172:7603–7609. | ||
Kudva A, Scheller EV, Robinson KM, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. 2011;186:1666–1674. | ||
Perera WR, Hurst JR, Wilkinson TMA, et al. Inflammatory changes, recovery and recurrence at COPD exacerbation. Eur Respir J. 2007;29:527–534. | ||
Segal L, Rom W, Weiden M. Lung microbiome for clinicians. New discoveries about bugs in healthy and diseased lungs. Ann Am Thoracic Soc. 2014;11:108–116. | ||
Lozupone C, Cota-Gomez A, Palmer B, et al. Widespread colonization of the lung by Tropheryma whipplei in HIV infection. Am J Respir Crit Care Med. 2013;187:1110–1117. | ||
Di Stefano A, Caramori G, Barczyk A, et al. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax. 2014;69:516–524. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.